

Abstract
Fatty acids such as palmitic acid at high levels are known to induce endoplasmic reticulum (ER) stress and lipotoxicity in numerous cell types and thereby contribute to cellular dysfunctions in obesity. To understand the impact of high fatty acids on oocytes, ER stress and lipotoxicity were induced in mouse cumulus-oocyte complexes during in vitro maturation using the ER Ca2+ channel blocker thapsigargin or high physiological levels of palmitic acid; both of which significantly induced ER stress marker genes (Atf4, Atf6, Xbp1s, and Hspa5) and inositol-requiring protein-1α phosphorylation, demonstrating an ER stress response that was reversible with the ER stress inhibitor salubrinal. Assessment of pentraxin-3, an extracellular matrix protein essential for fertilization, by immunocytochemistry and Western blotting showed dramatically impaired secretion concurrent with ER stress. Mitochondrial activity in oocytes was assessed by 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide staining of inner mitochondrial membrane potential, and oocytes matured in thapsigargin or high-dose palmitic acid had significantly reduced mitochondrial activity, reduced in vitro fertilization rates, and were slower to develop to blastocysts. The deficiencies in protein secretion, mitochondrial activity, and oocyte developmental competence were each normalized by salubrinal, demonstrating that ER stress is a key mechanism mediating fatty acid-induced defects in oocyte developmental potential.
The lipotoxic effects of obesity are important contributing factors in diabetes, cardiovascular disease, and nonalcoholic liver disease (1–3). In obesity, lipid is deposited in adipose tissue and circulates at increased levels in bodily fluids but also accumulates in nonadipose tissues, such as skeletal muscle, liver, heart, and pancreas (4). This cellular uptake of circulating fatty acids, triglycerides, and cholesterol as well as de novo lipogenesis in response to elevated glucose leads to high intracellular levels of free fatty acids that trigger lipotoxicity, which is characterized by endoplasmic reticulum (ER) stress, linked to mitochondrial damage, and culminates in apoptosis.
Obese women often experience infertility due to anovulation (5–8) and decreased pregnancy rates (9–13) even after assisted reproduction (14, 15). We and others have reported that obesity affects ovulation and oocyte quality in association with lipotoxicity responses in cumulus-oocyte complexes (COC), namely, excess intracellular lipid accumulation and altered mitochondrial activity in oocytes (16, 17), as well as ER stress and increased apoptosis in the granulosa and cumulus cells surrounding oocytes (16, 18). Further, female mice with diet-induced obesity are less likely to ovulate, and when fertilized, oocytes exhibit impaired developmental competence such that they are slower to develop to the blastocyst stage and have altered ratios of inner cell mass and trophectoderm cells (19). Similar events appear to occur in women, namely, expression of ER stress marker Atf4 is increased in granulosa cells of obese women (16). Further, in obese women, the ovarian follicular fluid that surrounds the COC contains high levels of triglyceride and free fatty acid lipids (20), and treatment of mouse oocytes with this lipid-rich follicular fluid induces ER stress and blocks oocyte maturation (21). Thus, it is critical to understand the mechanisms by which lipotoxicity and ER stress impact oocyte quality and subsequent embryo development to understand how obesity causes reduced conception rates and infertility in women.
A key feature of lipotoxicity is the perturbation of ER homeostasis known as ER stress that triggers the unfolded protein response (UPR) and disrupts protein secretion pathways (22). The UPR functions through a three-branched pathway: protein kinase RNA-like ER kinase, inositol-requiring protein-1α (IRE1α), and transcription factor 6 (Atf6), to activate a series of compensatory reactions, including transient attenuation of protein synthesis, activation of ER-associated protein degradation (ERAD), and induction of chaperone proteins and folding catalysts (23, 24). ER stress can be detected by induction of the UPR pathway mediators. 1) Activation of the protein kinase RNA-like ER kinase pathway leads to phosphorylation of eukaryotic translation initiation factor 2α subunit (eIF2α) and enhanced mRNA expression of transcription factor Atf4, which regulates the promoters of several genes, such as the ER chaperone heat shock 70-kDa protein 5 (Hspa5) (also known as Grp78 or Bip) and others implicated in the UPR. 2) Phosphorylation of IRE1α enhances splicing form of X-box-binding protein-1 mRNA (Xbp1s), which also binds to gene promoters involved in the UPR and ERAD. 3) Up-regulation of Atf6 activates transcription of ER chaperones and ERAD genes. Extensive and prolonged exposure to high lipid eventually overwhelms these compensatory cascades, ultimately leading to apoptotic cell death (25). With the increasing recognition of ER stress pathways in cellular pathologies, ER stress becomes a valuable target for therapeutic intervention. Salubrinal, a selective eIF2α dephosphorylation inhibitor, has been found to protect cells from lipotoxicity induced ER stress (26–28). Salubrinal acts to maintain the increase in phospho-eIF2α and expression of Atf4, Hspa5, and CHOP, which promotes protein folding and the restoration of ER function and cellular homeostasis.
In the present study, we sought to directly determine whether high-lipid exposure causes ER stress in COC and whether ER stress is a key pathway mediating the effects of lipotoxicity responses on oocyte developmental competence outcomes. We investigated ER stress gene expression, cumulus cell protein secretion, oocyte mitochondrial activity, and developmental competence in mouse COC stimulated to mature in vitro and compared the effects of exposure to high levels of palmitic acid with those of a direct ER stress inducer thapsigargin. Most importantly, we determined whether the effects of lipotoxicity in COC are mediated by ER stress by testing whether the ER stress inhibitor salubrinal could reverse the cellular dysfunctions induced by palmitic acid exposure and improve oocyte developmental competence.
Results
Thapsigargin induces ER stress in mouse COC that is reversed with salubrinal
Expression of classic ER stress marker mRNA (Atf4, Atf6, Xbp1s, and Hspa5) was examined in COC exposed to the ER stress inducer thapsigargin, the ER stress inhibitor salubrinal, or both during maturation (Fig. 1). COC treated with thapsigargin expressed increased Atf4 (4.4-fold), Atf6 (5.3-fold), Xbp1s (6.0-fold), and Hspa5 (4.8-fold) compared with control COC matured in vitro. COC cultured in media containing both thapsigargin (100 nm) and the ER stress inhibitor salubrinal (100 nm) had normalized expression levels of each of the four ER stress markers similar to those of control COC matured in vitro. In vivo-matured COC, included as a control benchmark of normal gene expression associated with high oocyte competence, showed levels of each gene similar to in vitro controls or COC treated with salubrinal only. These results demonstrate that thapsigargin induces ER stress in COC and that this is reversible by cotreatment with the ER stress inhibitor salubrinal.
Fig. 1.

Thapsigargin induces ER stress in COC, which can be reversed by salubrinal. Mouse COC were matured in vitro for 16 h in the presence of 100 nm thapsigargin (Thap) or 100 nm thapsigargin plus 100 nm salubrinal (Thap+Sal), or 100 nm salubrinal (Sal) alone or in the absence of thapsigargin and salubrinal as control. Ovulated COC (in vivo matured) from eCG, hCG 16-h-treated mice were used for comparison with in vitro controls. Total RNA was extracted from COC, and expression of ER stress marker genes Atf4 (A), Atf6 (B), Xbp1s (C), and Hspa5 (D) was determined by RT-PCR. Values are mean ± sem expressed as fold change compared with calibrator sample; n = 3 pools of COC per treatment group. Different letters indicate significant differences by one-way ANOVA, Tukey post hoc test, A, P = 0.0016; B, P = 0.0022; C, P < 0.0001; D, P = 0.0184.
Thapsigargin impairs cumulus cell pentraxin-3 (PTX3) secretion, oocyte mitochondrial membrane potential (ΔΨm), and embryo development, defects that are reversed by salubrinal
COC morphology was evaluated after 16 h of maturation in control conditions compared with those treated with thapsigargin (100 nm), thapsigargin plus salubrinal (100 nm), and salubrinal alone. Poor expansion was evident in COC cultured in the presence of thapsigargin, with the outer layers of cumulus cells dissociating from the complex (Fig. 2A). These results quantified, with COC from the four treatment groups scored by independent blinded assessor in three independent experiments, showed significantly reduced expansion scores in thapsigargin-treated COC that were restored by salubrinal treatment (Fig. 2B). To determine whether the impaired COC expansion was associated with impaired protein production, the cumulus matrix protein PTX3 was measured. Control COC exhibited clear PTX3 protein localized extracellularly in the cumulus matrix. However, COC treated with thapsigargin displayed very little PTX3-positive staining in cumulus matrix, and the complexes were exceedingly fragile (Fig. 2C). PTX3 protein localization was further examined by Western blottings of cumulus extracellular matrix fractions vs. intracellular cumulus cell proteins. After 8 h of maturation, COC treated with thapsigargin or thapsigargin plus salubrinal had less PTX3 protein in the extracellular matrix fraction than control COC and those treated with salubrinal alone. Levels of PTX3 protein within cumulus cells was also reduced in COC treated with thapsigargin compared with the other groups. After 16 h of maturation, COC treated with thapsigargin had very low levels of PTX3 protein in the extracellular matrix extracts. However, cotreatment of thapsigargin plus salubrinal restored PTX3 protein secretion to similar levels as those in the controls (Fig. 2D). At this time point, PTX3 protein levels within cumulus cells were similar in all treatment groups, suggesting that thapsigargin may specifically interfere with PTX3 secretion.
Fig. 2.

Thapsigargin impairs cumulus expansion and protein secretion, which can be reversed by salubrinal. COC from eCG-treated mice were matured in vitro for 16 h in the presence of 100 nm thapsigargin (Thap), 100 nm thapsigargin plus 100 nm salubrinal (Thap+Sal), 100 nm salubrinal (Sal) alone, or in the absence of thapsigargin and salubrinal as control. A, The morphology of COC matured in vitro. B, COC expansion assessed according to a qualitative scoring system, 0 to +4, and presented as the mean ± sem, n = 3 experimental replicates with more than 25 COC per treatment group. Different letters indicate significant differences by one-way ANOVA, Tukey post hoc test; P < 0.0001. C, Matrix PTX3 (red fluorescence) was reduced in cumulus matrix of thapsigarin (Thap)-treated COC by immunohistochemistry. DAPI nuclear stain is shown as blue fluorescence. D, Western blot analysis of PTX3 from cumulus matrix extracts, and PTX3, phospho-IRE1α, and IRE1α proteins from cell pellets obtained from COC matured for 8 or 16 h.
The reduction in extracellular PTX3 protein was associated with the induction of ER stress in cumulus cells. At both 8 and 16 h of maturation, cumulus cells from thapsigargin and thapsigargin plus salubrinal COC had high levels of phospho-IRE1α compared with control COC or those treated with salubrinal alone. Total IRE1α protein, however, was similar in all treatment groups (Fig. 2D).
The ER physically interacts with mitochondria, and ER stress is linked to mitochondrial dysfunction (29, 30). To investigate whether thapsigargin can cause mitochondrial dysfunction in oocytes, ΔΨm was visualized by staining with the inner membrane potential dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1). Consistent with previous reports (16, 31), oocytes of ovulated COC (matured in vivo) exhibited red punctuate fluorescence localized to the pericortical region, indicating high ΔΨm, whereas green fluorescence indicating low ΔΨm localized to the deeper cytoplasm of oocytes (Fig. 3A). Oocytes from control COC matured in vitro had a similar pattern, although reduced in intensity compared with ovulated COC, similar to previous observations (32). In COC treated with thapsigargin, red fluorescence intensity and thus ΔΨm was reduced in the pericortical region (Fig. 3A). In COC treated with thapsigargin and salubrinal, red fluorescence intensity in the pericortical region of oocytes was similar to controls. The ratio of red/green fluorescence intensity provides an index of mitochondrial activity (33, 34) and when quantitatively analyzed further demonstrated significantly decreased mitochondrial activity in oocytes from COC matured in thapsigargin compared with controls (Fig. 3B). Importantly, the presence of salubrinal normalized mitochondrial activity to levels similar to untreated controls matured in vitro.
Fig. 3.

ER stress reduces oocyte ΔΨm, which is reversed by salubrinal. A, ΔΨm was assessed by JC-1 staining in oocytes matured in vitro for 16 h in the presence of 100 nm thapsigargin (Thap) or 100 nm thapsigargin plus 100 nm salubrinal (Thap+Sal), or 100 nm salubrinal (Sal) alone, or in the absence of thapsigargin and salubrinal as control. Ovulated COC (in vivo matured) from eCG, hCG 16-h-treated mice were used as a comparison for the in vitro controls. Red fluorescence indicates high ΔΨm, and green indicates low ΔΨm. B, The ratio of red to green fluorescence was quantified as an indicator of mitochondrial activity. Data are presented as mean ± sem, n = 25–35 oocytes from three independent experiments. Different letters indicate significant differences by one-way ANOVA, Tukey post hoc test; P < 0.05.
To determine the impact of ER stress on fertilization and oocyte developmental competence, COC matured in thapsigargin, or thapsigargin plus salubrinal, were fertilized in vitro and developmental endpoints compared with controls. Treatment of COC with thapsigargin (500 nm) resulted in COC that were morphologically healthy and identical to controls but that exhibited a significantly lower fertilization rate on d 2 (87.7% two-cell embryos) (Fig. 4). Cotreatment with salubrinal (200 nm) returned fertilization rates to normal (96.3%). The fertilized oocytes that had been exposed to thapsigargin also exhibited poorer embryo development rates with only 77.9% of two-cell embryos developing to four cells on d 3, which was a significant reduction compared with development rates of the controls. Cotreatment of COC with salubrinal during maturation normalized embryo development rates to 96.3% on d 2 and 98% on d 3.
Fig. 4.

COC matured in the presence of thapsigargin have significantly impaired embryo developmental competence after IVF, which is reversible with salubrinal. COC were matured in vitro for 16 h in the presence of 500 nm thapsigargin (Thap) or 500 nm thapsigargin plus 200 nm salubrinal (Thap+Sal), or 200 nm salubrinal (Sal) alone, or in the absence of thapsigargin and salubrinal as control. Ovulated COC (in vivo matured) from eCG, hCG 16-h-treated mice were used as a comparison with the in vitro controls. COC were then fertilized in vitro and the rate of embryo development assessed. Data presented as the mean % on time embryo development ± sem, n = 3 experimental replicates, representative of 90 oocytes per treatment. Day 3 and 5 development is from two-cell embryos on d 2. Different letters indicate significant differences by one-way ANOVA within each developmental stage, Tukey post hoc test; P < 0.05. IVF, in vitro fertilization.
Cumulatively, these results show that thapsigargin treatment of mouse COC causes reduced PTX3 protein production by cumulus cells, mitochondrial dysfunction in oocytes, and impaired future developmental potential. That a classical ER stress inhibitor salubrinal can normalize each of these processes demonstrates that ER stress is a key mechanism responsible for these defects.
Palmitic acid dose dependently induces ER stress in COC that is reversed by salubrinal
To determine the effects of a more physiological ER stressor on oocyte quality, COC were treated during their maturation with palmitic acid at 150, 275, 400, or 525 μm doses. These doses were chosen because they are within the range of palmitic acid concentrations found in human follicular fluid (Ref. 35 and our unpublished data).
All four ER stress marker genes (Atf4, Atf6, Xbp1s, and Hspa5) were dose dependently increased by palmitic acid. COC matured in 150 μm palmitic acid had similar mRNA expression levels to control COC (Fig. 5). However, Xbp1s mRNA was significantly increased at 275 μm, Atf4 mRNA was significantly increased at 400 μm, and Atf6 and Hspa5 mRNA were significantly increased at 525 μm palmitic acid. We next investigated the temporal pattern of palmitic acid-induced ER stress response in COC and whether salubrinal can reverse the ER stress induced at high doses. Mouse COC were matured in low (150 μm) or high (400 μm) concentrations of palmitic acid and ER stress marker genes were assessed at 8 and 16 h later. A dose of 400 μm palmitic acid, but not 150 μm, induced a clear response within 8 h with increased mRNA expression of each of the ER stress markers Atf4, Atf6, Xbp1s, and Hspa5 (Fig. 6). After 16 h, Atf4 and Xbp1s mRNA levels were still significantly higher in COC treated with high-dose palmitic acid compared with low dose or untreated controls. In COC treated with salubrinal for 8 h either in the presence or absence of palmitic acid, significantly elevated expression of Atf4 and Hspa5 was observed as expected based on the known action of salubrinal to increase eIF2α phosphorylation and induce Atf4 and Hspa5 (25, 36). Elevated expression of Xbp1s and Atf6 after 8 h of 400 μm palmitic acid treatment was reversed by 200 nm salubrinal. After 16 h of maturation, the induction of all four genes by palmitic acid was reduced to control levels by salubrinal cotreatment (Fig. 6). These data confirm that ER stress is induced by high concentration of palmitic acid in maturing mouse COC and that it can be reversed by the ER stress inhibitor salubrinal.
Fig. 5.

Palmitic acid dose dependently induces ER stress in COC. COC were matured in vitro for 16 h in 1% FCS as control or plus 150, 275, 400, or 525 μm palmitic acid. Total RNA was extracted from COC, and expression of ER stress marker genes Atf4 (A), Atf6 (B), Xbp1s (C), and Hspa5 (D) was analyzed by RT-PCR. Values are mean ± sem expressed as fold change compared with calibrator sample; n = 3 pools of COC per treatment group. Different letters indicate significant differences by one-way ANOVA, Tukey post hoc test; P < 0.05.
Fig. 6.

Induction of ER stress markers by high-dose (400 μm) palmitic acid is reversible by salubrinal. Immature COC from eCG-treated mice (0 h) COC were matured in vitro in 1% FCS as control or plus 150 or 400 μm palmitic acid or 400 μm palmitic acid and 200 nm salubrinal (Sal) for 8 or 16 h. Total RNA was extracted from COC and subjected to RT-PCR for analysis of ER stress marker genes mRNA expression. Atf4 (A), Atf6 (B), Xbp1s (C), and Hspa5 (D) mRNA expression levels were determined. Values are mean ± sem expressed as fold change compared with calibrator sample; n = 3 pools of cells per experiment. Different letters indicate significant differences by one-way ANOVA within each time course, Tukey post hoc test; P < 0.05.
High-dose palmitic acid impairs PTX3 secretion, oocyte ΔΨm, and embryo development, defects that are reversed by salubrinal
The previous experiments demonstrated that ER stress causes poor cumulus expansion and dramatically reduced PTX3 protein secretion to the cumulus matrix, because the ER stress inhibitor salubrinal was able to completely reverse these defects. We next investigated whether physiological high-dose palmitic acid causes similar functional defects in COC and whether this is reversible by salubrinal.
Immunocytochemistry showed abundant PTX3 in the matrix of in vivo and control in vitro-matured COC, whereas COC matured in high-dose palmitic acid had very little PTX3 protein in the cumulus matrix. Cotreatment of COC in high-dose palmitic acid plus salubrinal or treatment with salubrinal alone showed strong PTX3 staining in cumulus matrix, similar to the in vivo and in vitro controls (Fig. 7A). Western blotting of extracellular matrix also showed high PTX3 in control COC, which was reduced in COC treated with high-dose palmitic acid for 8 h or 16 h, whereas cotreatment with salubrinal increased PTX3 abundance in COC matrix after 16 h (Fig. 7B). In contrast, intracellular PTX3 protein was not affected by high-dose palmitic acid or salubrinal. The reduction in extracellular PTX3 protein was associated with increased levels of phosphorylated IRE1α, which was highest in COC matured in high-dose palmitic acid for 8 h (Fig. 7B). The reductions in PTX3 protein secretion were not reflective of changes in Ptx3 expression, because all treatment groups had Ptx3 mRNA levels that were identical to controls at 8 h of maturation and in fact higher than controls at 16 h of maturation (Fig. 7C). Together, these results indicate that protein secretion is impaired in COC in conjunction with ER stress induced by exposure to palmitic acid.
Fig. 7.
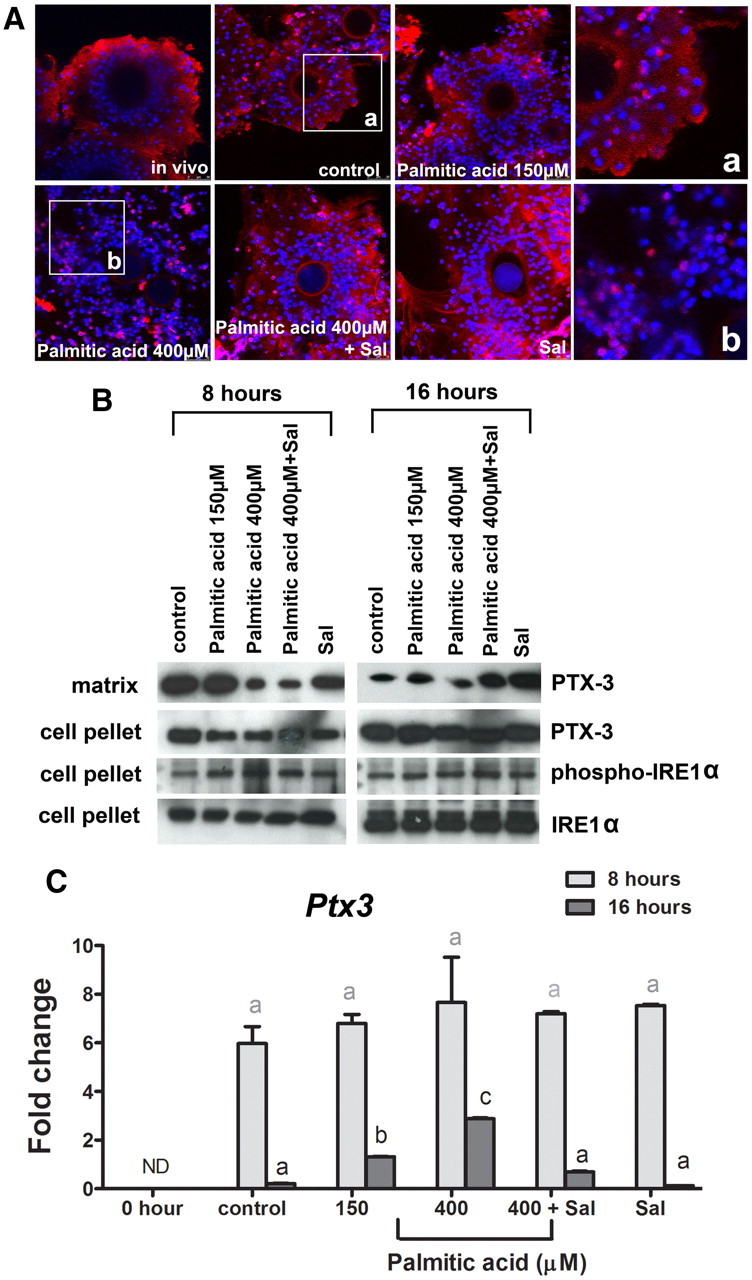
High-dose (400 μm) palmitic acid reduces PTX3 protein production in COC that is normalized with salubrinal. Immature COC from eCG-treated mice (0 h) COC were matured in vitro in 1% FCS as control or plus 150 or 400 μm palmitic acid or 400 μm palmitic acid and 200 nm salubrinal (Sal) for 8 or 16 h. A, Cumulus matrix PTX3 (red fluorescence) detected by immunocytochemistry was reduced in the cumulus matrix in 400 μm palmitic acid-treated COC. DAPI nuclear stain is shown as blue fluorescence. B, Western blot analysis of extracellular PTX3 from cumulus matrix extracts, and intracellular PTX3, phospho-IRE1α, and IRE1α proteins from cell pellets obtained from COC matured in vitro for 8 or 16 h. C, Ptx3 mRNA expression levels were determined by RT-PCR. Values are mean ± sem expressed as fold change compared with calibrator sample; n = 3 pools of cells per treatment group. Different letters indicate significant differences by one-way ANOVA within each time point, Bonferroni post hoc test; P < 0.05.
We next determined whether ΔΨm is reduced in oocytes matured in high physiological palmitic acid, similar to the effects seen in response to thapsigargen. Oocytes matured under control in vitro conditions and stained with JC-1 had less red fluorescence intensity, indicative of fewer high membrane potential mitochondria than in vivo-matured oocytes, as expected. However, in oocytes matured in high-dose (400 μm) palmitic acid but not low dose (150 μm) palmitic acid, red fluorescence intensity was further reduced in the pericortical region (Fig. 8A). In oocytes treated with high-dose palmitic acid plus salubrinal, red fluorescence intensity in the pericortical region was returned to levels similar to that of controls matured in vitro. Quantification of fluorescence intensity verified a significantly decreased mean ratio of red/green fluorescence intensity in oocytes matured in high-dose palmitic acid compared with controls or low-dose palmitic acid treatment (Fig. 8B). Cotreatment with salubrinal normalized mitochondrial activity in COC treated with high-dose palmitic acid, demonstrating that the mitochondrial dysfunction induced by high-dose palmitic acid is due to ER stress.
Fig. 8.

Oocyte ΔΨm is reduced in COC treated with high-dose palmitic acid and reversed by salubrinal but not by L-carnitine. ΔΨm was assessed by JC-1 staining in oocytes matured in vitro in the presence of 150 or 400 μm palmitic acid or without palmitic acid as control. High-dose (400 μm) palmitic acid-treated COC were also treated with either salubrinal (Sal; 200 nm) or L-carnitine (5 mm) as indicated. In vivo-matured oocytes from eCG, hCG 16-h-treated mice were used for comparison with in vitro controls (A and C). The ratio of red to green fluorescence was quantified as an indicator of mitochondrial activity. Data are presented as mean ± sem; n = 30–55 oocytes from three independent experiments. Different letters indicate significant differences by one-way ANOVA, Tukey post hoc test (B and D).
L-carnitine is a cellular metabolite that catalyzes fatty acid transportation from cytosol into mitochondria and increases mitochondrial activity (37). L-carnitine also reverses mitochondrial damage in multiple models of mitochondrial dysfunction, including nickel-treated neuroblastoma cells (38) and palmitoyl-coenzyme A-treated mitochondria from cardiocytes (39). We have reported that L-carnitine increases fatty acid metabolism in maturing COC and improves embryo development in vitro (40); and thus, we investigated whether the mitochondrial dysfunction induced by high-dose palmitic acid can be repaired by adding L-carnitine, similar to the effect of salubrinal. COC treated with L-carnitine (5 mm) alone had increased oocyte mitochondrial activity (0.33 ± 0.17) compared with oocytes from control COC (0.23 ± 0.18) but still less than in vivo-matured COC (0.44 ± 0.19) (Fig. 8, C and D). In contrast to the effects of salubrinal, L-carnitine treatment was not able to normalize oocyte mitochondrial activity in COC treated with high-dose palmitic acid. Thus, although L-carnitine can improve mitochondrial activity of oocytes matured in vitro, it cannot reverse the oocyte mitochondrial dysfunction seen in COC exposed to high-dose palmitic acid.
To determine the impact of palmitic acid induced ER stress on fertilization and embryo developmental competence, COC matured in high-dose (400 μm) palmitic acid, and high-dose palmitic acid plus salubrinal, were fertilized in vitro and compared with controls and those treated with low-dose palmitic acid (Fig. 9). On d 2, the fertilization rate of oocytes matured in high-dose palmitic acid was significantly lower (43.7%) than that of COC matured in low-dose palmitic acid or controls. The fertilized oocytes from COC treated with high-dose palmitic acid were also slower to develop to four cells on d 3 (87.9%) and to blastocysts on d 5 (50%), which was significantly lower than all other groups on d 3 and lower than the in vivo-matured group on d 5. Salubrinal treatment (200 nm) normalized the palmitic acid-induced decrease in fertilization rates and d 3 development rates to control levels.
Fig. 9.

COC treated with high-dose palmitic acid have significantly impaired embryo development that is reversed by salubrinal. COC from eCG-treated mice were matured in vitro for 16 h in the presence of 150 or 400 μm palmitic acid and/or 200 nm salubrinal (Sal) or without palmitic acid as control. In vivo-matured COC from eCG, hCG 16-h-treated mice were used for comparison with in vitro controls. COC were fertilized in vitro, and oocyte developmental competence was assessed by the rate of embryo development after in vitro fertilization. Data presented as the mean % on time embryo development ± sem, n = 3 independent experiments, representative of 90 oocytes per treatment. Day 3 and 5 development is from two-cell embryos on d 2. Different letters indicate significant differences by one-way ANOVA with Tukey post hoc test at each developmental stage.
Discussion
Obesity leads to anovulation and impaired oocyte developmental potential (17–19), which is associated with the induction of ER stress and lipotoxicity responses in ovarian cells (16). This study now elucidates cellular mechanisms by which lipotoxicity contributes to poor oocyte quality. Specifically, exposure of COC to the saturated fatty acid palmitic acid at concentrations mimicking the upper range of physiological levels induced ER stress; thereby reducing protein secretion, disrupting mitochondrial activity in oocytes, and impairing oocyte maturation and fertilization. Identical results were observed when COC were treated with a classical ER stress inducer thapsigargin. Most importantly, each of these defects was normalized with the ER stress inhibitor salubrinal, demonstrating that ER stress is a key mechanism responsible for these defects and that it can be reversed to restore oocyte developmental potential.
Palmitic acid treatment is a well-characterized standard model of lipotoxicity in other cell types, particularly β-cells and hepatocytes (3, 41–43). It is one of the most prevalent fatty acids in human ovarian follicular fluid and is at levels approximately half of those found in serum (35, 44). Circulating free fatty acid levels increase with body mass index (45), such that plasma total free fatty acids are approximately 350 μm with a moderate body mass index but increase to levels of 550-2000 μm with obesity (46–48), making it possible to estimate that palmitic acid concentrations in follicular fluid might range from 115 μm in moderate weight women up to 660 μm in obese women and would thus be similar to levels used in this study. Treatment of human granulosa cells with 300 μm palmitic acid induces apoptosis (49). Maturing bovine oocytes in the presence of palmitic acid reduces cumulus expansion increases cumulus cell apoptosis, impairs oocyte maturation and fertilization, and alters blastocyst development and metabolism (50–52). Similarly, brief exposure of mouse morula stage embryos to 250 μm palmitic acid resulted in altered metabolism and growth and lasting adverse effects postnatally (53). Our results now demonstrate that the detrimental effects of free fatty acids observed in these various models of physiological dyslipidemia may be mediated via alterations in cumulus matrix production and disruptions to oocyte mitochondrial activity arising from ER stress.
The ER has essential roles in multiple cellular processes, including the synthesis and folding of proteins that are then trafficked to the secretory pathway. COC expansion involves rapid production of large amounts of extracellular matrix proteins by cumulus cells and their exocytosis to form the viscous cumulus matrix (54), which is critical for oocyte maturation and ovulation (55). Specific matrix proteins, such as PTX3, are essential for the formation and organization of cumulus matrix and are required for female fertility, because PTX3−/− female mice develop normally but are infertile due to defects in ovulation and fertilization that are associated with poor cumulus matrix formation (56, 57). PTX3 is also expressed by human cumulus cells, and there is evidence suggesting that PTX3 levels are associated with improved outcomes after assisted reproduction (58, 59). Our study shows that perturbation of ER function by palmitic acid at concentrations similar to those reported in follicular fluid of infertile women (Ref. 35 and our unpublished data) results in reduced secretion of PTX3 (and probably other cumulus secreted proteins) and poor matrix formation. It is likely that in cumulus cells of obese women, lipotoxicity responses will be associated with similarly compromised cumulus matrix production that may be responsible for the anovulation and reduced pregnancy rates seen in these patients clinically. In support of this, elevated levels of free fatty acids in follicular fluid is associated with abnormal COC morphology, poor oocyte quality, and a trend to impaired in vitro fertilization outcome (35).
The functions of ER and mitochondria are tightly linked via modulations of intracellular Ca2+ signaling pathways (60, 61). The ER lumen stores a 1000-fold excess Ca2+ compared with cytosol and when stressed releases Ca2+ through inositol 1,4,5-trisphosphate receptor and the ryanodine receptor channels thereby provoking Ca2+ signaling, which critically affects mitochondrial function (62). ER Ca2+ release and subsequent Ca2+ uptake by mitochondria result in increasing reactive oxygen species production, uncoupling of oxidative phosphorylation, lowered ΔΨm, matrix swelling, and subsequent release of various apoptotic factors, including cytochrome C and effector caspases that lead to cellular apoptosis (25, 62). Consistent with our previous report that mice fed a high-fat diet for 4 wk exhibited both increased ER stress gene expression in COC and reduced ΔΨm in oocytes (16), we now show that high-dose palmitic acid exposure during oocyte in vitro maturation activates ER stress response pathways that are directly responsible for reduced mitochondrial activity, because this deleterious effect is reversed by salubrinal. However, the mitochondrial changes caused by high-dose palmitic acid were not normalized by stimulating mitochondrial activity with L-carnitine, indicating that the oocyte mitochondria are damaged rather than inactive and further establishing that mitochondrial dysfunction is a downstream response to ER stress. This is important, because mitochondria have numerous vital functions in oocytes (63) and has significant implications, because alterations in oocyte mitochondrial activity, due to in vitro manipulations (64) or in physiological mouse models of diet and metabolic disease (16, 17, 65–67), are linked to poor embryo developmental potential. Our results are now the first to show that mitochondrial dysfunction and poor developmental potential of oocytes maturing in a high-lipid environment can be improved by therapeutic targeting of ER stress.
Materials and Methods
Mice
Mice (CBA×C57Bl/6 F1) were maintained on a 12-h light, 12-h dark cycle with Standard Rat and Mouse Diet (Specialty Feeds, Glen Forrest, Australia) and water available ad libitum. All experiments were approved by the University of Adelaide Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Isolation of mouse COC
Immature unexpanded COC were isolated from 23-d-old prepubertal female mice by puncturing the antral follicles of ovaries collected 44 h after ip injection of 5 IU of equine chorionic gonadotropin (eCG) (Calbiochem, San Diego, CA). In vivo-matured expanded COC were obtained from oviducts by blunt dissection after 44-h eCG then and 16-h human chorionic gonadotropin (hCG) (5 IU of hCG; Calbiochem) administration ip. All COC were collected in HEPES-buffered α-MEM (Invitrogen, Carlsbad, CA) supplemented with 5% (vol/vol) fetal calf serum (FCS) (Invitrogen) or 3 mg/ml BSA (fatty acid free; Sigma, St. Louis, MO), as indicated.
In vitro maturation of COC
For in vitro maturation, immature COC (isolated from mice treated with eCG for 44 h) were cultured in groups of 30 in bicarbonate-buffered α-MEM supplemented with 5% (vol/vol) FCS, 50 mIU/ml recombinant human FSH (Sigma-Aldrich, St. Louis, MO), and 10 ng/ml epidermal growth factor (Sigma-Aldrich Pty. Ltd., Sydney, Australia) as control (Figs. 1–4), or with the addition of the indicated treatment, in drops of 100 μl overlaid with sterile mineral oil (Sigma-Aldrich) and incubated at 37 C in an atmosphere of 5% CO2 and 95% air for 16 h. Treatments consisted of control media supplemented with 100 nm/500 nm ER stress inducer thapsigargin (Merk, Whitehouse Station, NJ), or 100 nm/200 nm eIF-2α inhibitor salubrinal (Merck), or both (see Figs. 1–4).
In experiments assessing the effect of palmitic acid during in vitro maturation (see Figs. 5- 9), 1% (vol/vol) FCS was used in the culture media. Palmitic acid was solubilized and prepared by the method of Downs et al. (68) and added to culture media at 150, 275, 400, or 525 μm palmitic acid for dose-response experiments (see Fig. 5), or at 400 μm palmitic acid with or without 200 nm salubrinal (see Figs. 6–9).
Assessment of cumulus expansion
The degree of cumulus expansion of in vitro-matured COC was assessed after 16 h of culture by an independent assessor blinded to treatments, using the scale previously described (69). Briefly, a score of 0 indicates no expansion of cumulus cells, +1 the most outer layers of cumulus cells expanded, +2 expansion of the outer half of cumulus cells, +3 all layers expanded except the corona radiata, and +4 expansion of all layers of cumulus cells. For each treatment group, a mean cumulus expansion index (0.0–4.0) was calculated.
RNA isolation and real-time RT-PCR
Total RNA was isolated from COC using RNeasy Micro kit (QIAGEN, Valencia, CA) as per manufacturer's instructions. RNA concentration and purity were quantified using a Nanodrop ND-1000 Spectrophotometer (Biolab, Carmel, IN) before reverse transcribing 600 ng of RNA using random primers (Roche, Indianapolis, IN) and Superscript III Reverse Transcriptase (Invitrogen) according to the manufacturer's instructions. Ribosomal protein L19 was used as a validated internal control for every sample. Xbp1s primers were: Xbp1s reverse, 5′-AGG CTT GGT GTA TAC ATG G-3′ and Xbp1s forward, 5′-GGT CTG CTG AGT CCG CAG CAG G-3′ (70), and other primers were Quantitect Primer assays (QIAGEN). All Primers were shown to have comparable amplification efficiency against the internal control. Real-time PCR was performed in triplicate using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and a Rotor-Gene 6000 (Corbett, Valencia, CA) real-time rotary analyzer. Real-time RT-PCR data were analyzed using the 2−(ΔΔCT) method and expressed as the fold change relative to a calibrator sample, which was included in each run.
Immunocytochemistry
In each experiment, 10 COC per treatment group were fixed for 1 h in 4% paraformaldehyde (wt/vol) in PBS [80 mm Na2HPO4, 20 mm NaH2PO4, and 100 mm NaCl (pH 7.5)] with 1 mg/ml polyvinylpyrrolidone (Sigma) to prevent sticking, and then washed thoroughly in PBS and 1 mg/ml polyvinylpyrrolidone. The COC were incubated in blocking buffer containing 10% normal goat serum (Vector Laboratories, Burlingame, CA) in PBS for 1 h at room temperature. The COC were then incubated with rabbit polyclonal anti-PTX3 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), diluted 1:100 in blocking buffer, overnight at 4 C. After washing in PBS, COC were incubated with biotinylated goat-antirabbit (Millipore, Bedford, MA) IgG antibody, at 1:1000 in blocking buffer, for 1 h at room temperature. Finally, COC were washed in PBS and incubated with 1 ng/ml streptavidin-Alexa Flour 594 (Molecular Probes, Eugene, OR) and 1 mg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) in PBS for 1 h at room temperature. COC were visualized and images captured using a Leica (Heerbrugg, Switzerland) TCS SP5 spectral scanning confocal microscope system.
Protein isolation and Western blotting
To separate the extracellular matrix and cellular components of COC, 90 COC per treatment group were collected in 20 μl of culture media and incubated with 4 μl of 1000 IU/ml hyaluronidase (Invitrogen) and gentle rotation at room temperature for 1 min followed by centrifugation at 5000 × g for 1 min. The supernatant (extracellular matrix fraction) was collected and cell pellets resuspended in 24 μl of PBS. To extract the proteins, 6 μl of 5× reducing Laemmli buffer, 2% (wt/vol) sodium dodecyl sulfate, and 5% (vol/vol) β-mercaptoethanol were added to each fraction, and the samples were boiled at 100 C for 10 min.
Protein extracts (12 μl) from matrix fractions or cell pellet fractions were resolved by 10% SDS-PAGE gel and transferred to polyvinylidene fluoride membranes (Millipore). Membranes were blocked in Tris-buffered saline with Tween 20 (TBST) [10 mm Tris, 150 mm NaCl, and 0.05% Tween 20 (pH 7.5)] containing 3% (wt/vol) nonfat milk for 1 h at room temperature. Membranes were then incubated with primary antibodies for 2 h at room temperature in 3% milk/TBST. Primary antibodies used were rabbit monoclonal IRE1α antibody at 1:1000 (Cell Signaling, Beverly, MA), rabbit phosphor-IRE1α [Ser724] antibody at 1:1000 (Novus Biologicals, Littleton, CO) or rat monoclonal PTX3 antibody at 1:2000 (Enzo Life Sciences, New York, NY). Membranes were then washed in TBST and incubated with horseradish peroxidase-linked antirabbit IgG at 1:1000 (Millipore) or horseradish peroxidase-linked antirat IgG at 1:1000 (Millipore). Enhanced chemiluminescence detection (Amersham, Piscataway, NJ) was used as per manufacturer's instructions.
Analysis of inner ΔΨm
Oocytes were denuded by the addition of 10 μl of prewarmed 1000 IU/ml hyaluronidase (Invitrogen) to the culture drop for 1 min followed by gentle repeated pipetting with a fine glass pipette to remove remaining cumulus cells. Denuded oocytes were incubated with the inner mitochondrial membrane dye JC-1 (Invitrogen) at 1.5 μm for 15 min at 37 C in the dark (31). Oocytes were then imaged immediately in both green and red fluorescence channels using a Leica SP5 spectral scanning confocal microscope at identical magnification and gain settings throughout experiments. Using Analysis Pro software (Olympus, New York, NY), a square was placed to cover the oocyte image. Red or green fluorescence intensity was determined as the sum total of fluorescence in the boxed area.
In vitro fertilization and assessment of embryo development
COC matured in vitro and COC isolated from oviduct at 16 h after hCG (in vivo, ovulated) were used for in vitro fertilization. Sperm were collected from the caudal epididymis and vas deferens of 8-wk-old CBA×C57Bl/6 F1 male mice. Sperm were capacitated in bicarbonate-buffered α-MEM supplemented with 3 mg/ml of fatty acid-free BSA for 1 h at 37 C in an atmosphere of 5% CO2 and 95% air. After sperm capacitation, COC were washed twice, and COC and sperm (35,000 sperm/ml) were coincubated in 100 μl of bicarbonate-buffered α-MEM supplemented with 3 mg/ml of fatty acid-free BSA for 4 h at 37 C in an atmosphere of 5% CO2 and 95% air. All cumulus cell-free oocytes were then transferred to embryo development culture medium (Vitro Cleave; Cook Australia, Brisbane, Australia). Twenty-four hours after in vitro fertilization (d 2), the fertilization rate was assessed, and two-cell embryos were transferred to a fresh, 20-μl drop of culture medium. Embryo morphology was assessed on d 3 (44 h of embryo culture) and d 5 (the end of the culture period, 96–100 h after fertilization). Embryos were classified as appropriately developed (“on time”) using the following criteria: on d 2, embryos at the two-cell stage; on d 3, embryos at the four- to eight-cell stage; and on d 5, blastocysts or hatching blastocysts. The rate of development was assessed on d 2 as the percentage of embryos meeting the development criteria from the starting number of oocytes, whereas the rate of development was assessed on d 3 and 5 as the percentage of embryos meeting the development criteria from two-cell embryos on d 2.
Statistical analysis
All measures are reported as mean ± sem. Statistical significance was determined as indicated, by t test or one-way ANOVA with Tukey post hoc tests, as appropriate, using GraphPad Prism version 5.01 for Windows (GraphPad Software, Inc., San Diego, CA). A P value of less than 0.05 was considered statistically significant.
Acknowledgments
We thank the expert technical assistance of Kylie R. Dunning, Lisa K. Akison, and Laura N. Watson.
This work was supported by grants from the National Health and Medical Research Council of Australia and The Channel 7 Children's Research Foundation.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Atf6
- Transcription factor 6
- COC
- cumulus-oocyte complex
- DAPI
- 4′,6-diamidino-2-phenylindole
- eCG
- equine chorionic gonadotropin
- eIF2α
- eukaryotic translation initiation factor 2α subunit
- ER
- endoplasmic reticulum
- ERAD
- ER-associated protein degradation
- FCS
- fetal calf serum
- hCG
- human chorionic gonadotropin
- Hspa5
- heat shock 70-kDa protein 5
- IRE1α
- inositol-requiring protein-1α
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide
- ΔΨm
- mitochondrial membrane potential
- PTX3
- pentraxin-3
- TBST
- Tris-buffered saline with Tween 20
- UPR
- unfolded protein response
- Xbp1s
- X-box-binding protein-1 mRNA.
References
- 1. Cusi K. 2010. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr Diab Rep 10:306–315 [DOI] [PubMed] [Google Scholar]
- 2. Waterman CL , Kian-Kai C , Griffin JL. 2010. Metabolomic strategies to study lipotoxicity in cardiovascular disease. Biochim Biophys Acta 1801:230–234 [DOI] [PubMed] [Google Scholar]
- 3. Mei S , Ni HM , Manley S , Bockus A , Kassel KM , Luyendyk JP , Copple BL , Ding WX. 2011. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther 339:487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schaffer JE. 2003. Lipotoxicity: when tissues overeat. Curr Opin Lipidol 14:281–287 [DOI] [PubMed] [Google Scholar]
- 5. Rich-Edwards JW , Spiegelman D , Garland M , Hertzmark E , Hunter DJ , Colditz GA , Willett WC , Wand H , Manson JE. 2002. Physical activity, body mass index, and ovulatory disorder infertility. Epidemiology 13:184–190 [DOI] [PubMed] [Google Scholar]
- 6. Rich-Edwards JW , Goldman MB , Willett WC , Hunter DJ , Stampfer MJ , Colditz GA , Manson JE. 1994. Adolescent body mass index and infertility caused by ovulatory disorder. Am J Obstet Gynecol 171:171–177 [DOI] [PubMed] [Google Scholar]
- 7. Grodstein F , Goldman MB , Cramer DW. 1994. Body mass index and ovulatory infertility. Epidemiology 5:247–250 [DOI] [PubMed] [Google Scholar]
- 8. Wei S , Schmidt MD , Dwyer T , Norman RJ , Venn AJ. 2009. Obesity and menstrual irregularity: associations with SHBG, testosterone, and insulin. Obesity 17:1070–1076 [DOI] [PubMed] [Google Scholar]
- 9. van der Steeg JW , Steures P , Eijkemans MJ , Habbema JD , Hompes PG , Burggraaff JM , Oosterhuis GJ , Bossuyt PM , van der Veen F , Mol BW. 2008. Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod 23:324–328 [DOI] [PubMed] [Google Scholar]
- 10. Zaadstra BM , Seidell JC , Van Noord PA , te Velde ER , Habbema JD , Vrieswijk B , Karbaat J. 1993. Fat and female fecundity: prospective study of effect of body fat distribution on conception rates. BMJ 306:484–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jensen TK , Scheike T , Keiding N , Schaumburg I , Grandjean P. 1999. Fecundability in relation to body mass and menstrual cycle patterns. Epidemiology 10:422–428 [DOI] [PubMed] [Google Scholar]
- 12. Gesink Law DC , Maclehose RF , Longnecker MP. 2007. Obesity and time to pregnancy. Hum Reprod 22:414–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramlau-Hansen CH , Thulstrup AM , Nohr EA , Bonde JP , Sørensen TI , Olsen J. 2007. Subfecundity in overweight and obese couples. Hum Reprod 22:1634–1637 [DOI] [PubMed] [Google Scholar]
- 14. Wang JX , Davies M , Norman RJ. 2000. Body mass and probability of pregnancy during assisted reproduction treatment: retrospective study. BMJ 321:1320–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luke B , Brown MB , Stern JE , Missmer SA , Fujimoto VY , Leach R. 2011. Female obesity adversely affects assisted reproductive technology (ART) pregnancy and live birth rates. Hum Reprod 26:245–252 [DOI] [PubMed] [Google Scholar]
- 16. Wu LL , Dunning KR , Yang X , Russell DL , Lane M , Norman RJ , Robker RL. 2010. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology 151:5438–5445 [DOI] [PubMed] [Google Scholar]
- 17. Igosheva N , Abramov AY , Poston L , Eckert JJ , Fleming TP , Duchen MR , McConnell J. 2010. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One 5:e10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jungheim ES , Schoeller EL , Marquard KL , Louden ED , Schaffer JE , Moley KH. 2010. Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinology 151:4039–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minge CE , Bennett BD , Norman RJ , Robker RL. 2008. Peroxisome proliferator-activated receptor-γ agonist rosiglitazone reverses the adverse effects of diet-induced obesity on oocyte quality. Endocrinology 149:2646–2656 [DOI] [PubMed] [Google Scholar]
- 20. Robker RL , Akison LK , Bennett BD , Thrupp PN , Chura LR , Russell DL , Lane M , Norman RJ. 2009. Obese women exhibit differences in ovarian metabolites, hormones, and gene expression compared with moderate-weight women. J Clin Endocrinol Metab 94:1533–1540 [DOI] [PubMed] [Google Scholar]
- 21. Yang X , Wu LL , Chura LR , Liang X , Lane M , Norman RJ , Robker RL. 2012. Exposure to lipid-rich follicular fluid is associated with endoplasmic reticulum stress and impaired oocyte maturation in cumulus-oocyte complexes. Fertil Steril 10.1016/jfertnstert.2012.02.034 [DOI] [PubMed] [Google Scholar]
- 22. Shore GC , Papa FR , Oakes SA. 2011. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol 23:143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaufman RJ. 1999. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233 [DOI] [PubMed] [Google Scholar]
- 24. Rutkowski DT , Kaufman RJ. 2004. A trip to the ER: coping with stress. Trends Cell Biol 14:20–28 [DOI] [PubMed] [Google Scholar]
- 25. Kim I , Xu W , Reed JC. 2008. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7:1013–1030 [DOI] [PubMed] [Google Scholar]
- 26. Boyce M , Bryant KF , Jousse C , Long K , Harding HP , Scheuner D , Kaufman RJ , Ma D , Coen DM , Ron D , Yuan J. 2005. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307:935–939 [DOI] [PubMed] [Google Scholar]
- 27. Tian T , Zhao Y , Nakajima S , Huang T , Yao J , Paton AW , Paton JC , Kitamura M. 2011. Cytoprotective roles of ERK and Akt in endoplasmic reticulum stress triggered by subtilase cytotoxin. Biochem Biophys Res Commun 410:852–858 [DOI] [PubMed] [Google Scholar]
- 28. Kuo TF , Tatsukawa H , Matsuura T , Nagatsuma K , Hirose S , Kojima S. 2012. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J Cell Physiol 227–1130-1137 [DOI] [PubMed] [Google Scholar]
- 29. Pizzo P , Pozzan T. 2007. Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol 17:511–517 [DOI] [PubMed] [Google Scholar]
- 30. Giorgi C , De Stefani D , Bononi A , Rizzuto R , Pinton P. 2009. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol 41:1817–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Blerkom J , Davis P , Mathwig V , Alexander S. 2002. Domains of high-polarized and low-polarized mitochondria may occur in mouse and human oocytes and early embryos. Hum Reprod 17:393–406 [DOI] [PubMed] [Google Scholar]
- 32. Preis KA , Seidel GE , Gardner DK. 2007. Reduced oxygen concentration improves the developmental competence of mouse oocytes following in vitro maturation. Mol Reprod Dev 74:893–903 [DOI] [PubMed] [Google Scholar]
- 33. Vayssier-Taussat M , Kreps SE , Adrie C , Dall'Ava J , Christiani D , Polla BS. 2002. Mitochondrial membrane potential: a novel biomarker of oxidative environmental stress. Environ Health Perspect 110:301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilding M , Dale B , Marino M , di Matteo L , Alviggi C , Pisaturo ML , Lombardi L , De Placido G. 2001. Mitochondrial aggregation patterns and activity in human oocytes and preimplantation embryos. Hum Reprod 16:909–917 [DOI] [PubMed] [Google Scholar]
- 35. Jungheim ES , Macones GA , Odem RR , Patterson BW , Lanzendorf SE , Ratts VS , Moley KH. 2011. Associations between free fatty acids, cumulus oocyte complex morphology and ovarian function during in vitro fertilization. Fertil Steril 95:1970–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Diakogiannaki E , Welters HJ , Morgan NG. 2008. Differential regulation of the endoplasmic reticulum stress response in pancreatic β-cells exposed to long-chain saturated and monounsaturated fatty acids. J Endocrinol 197:553–563 [DOI] [PubMed] [Google Scholar]
- 37. Bieber LL. 1988. Carnitine. Annu Rev Biochem 57:261–283 [DOI] [PubMed] [Google Scholar]
- 38. He MD , Xu SC , Lu YH , Li L , Zhong M , Zhang YW , Wang Y , Li M , Yang J , Zhang GB , Yu ZP , Zhou Z. 2011. L-carnitine protects against nickel-induced neurotoxicity by maintaining mitochondrial function in Neuro-2a cells. Toxicol Appl Pharmacol 253:38–44 [DOI] [PubMed] [Google Scholar]
- 39. Oyanagi E , Yano H , Uchida M , Utsumi K , Sasaki J. 2011. Protective action of l-carnitine on cardiac mitochondrial function and structure against fatty acid stress. Biochem Biophys Res Commun 412:61–67 [DOI] [PubMed] [Google Scholar]
- 40. Dunning KR , Cashman K , Russell DL , Thompson JG , Norman RJ , Robker RL. 2010. β-Oxidation is essential for mouse oocyte developmental competence and early embryo development. Biol Reprod 83:909–918 [DOI] [PubMed] [Google Scholar]
- 41. Listenberger LL , Han X , Lewis SE , Cases S , Farese RV , Ory DS , Schaffer JE. 2003. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA 100:3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cnop M , Hannaert JC , Hoorens A , Eizirik DL , Pipeleers DG. 2001. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 50:1771–1777 [DOI] [PubMed] [Google Scholar]
- 43. Gentile CL , Frye MA , Pagliassotti MJ. 2011. Fatty acids and the endoplasmic reticulum in nonalcoholic fatty liver disease. Biofactors 37:8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pahuja SL , Kim AH , Lee G , Hochberg RB. 1995. Origin of estradiol fatty acid esters in human ovarian follicular fluid. Biol Reprod 52:625–630 [DOI] [PubMed] [Google Scholar]
- 45. Schiffelers SL , Saris WH , van Baak MA. 2001. The effect of an increased free fatty acid concentration on thermogenesis and substrate oxidation in obese and lean men. Int J Obes Relat Metab Disord 25:33–38 [DOI] [PubMed] [Google Scholar]
- 46. Iannello S , Bosco P , Camuto M , Cavaleri A , Milazzo P , Belfiore F. 2004. A mild form of Alstrom disease associated with metabolic syndrome and very high fasting serum free fatty acids: two cases diagnosed in adult age. Am J Med Sci 327:284–288 [DOI] [PubMed] [Google Scholar]
- 47. Iannello S , Camuto M , Cavaleri A , Milazzo P , Pisano MG , Bellomia D , Belfiore F. 2004. Effects of short-term metformin treatment on insulin sensitivity of blood glucose and free fatty acids. Diabetes Obes Metab 6:8–15 [DOI] [PubMed] [Google Scholar]
- 48. Belfiore F , Iannello S , Camuto M , Fagone S , Cavaleri A. 2001. Insulin sensitivity of blood glucose versus insulin sensitivity of blood free fatty acids in normal, obese, and obese-diabetic subjects. Metabolism 50:573–582 [DOI] [PubMed] [Google Scholar]
- 49. Mu YM , Yanase T , Nishi Y , Tanaka A , Saito M , Jin CH , Mukasa C , Okabe T , Nomura M , Goto K , Nawata H. 2001. Saturated FFAs, palmitic acid and stearic acid, induce apoptosis in human granulosa cells. Endocrinology 142:3590–3597 [DOI] [PubMed] [Google Scholar]
- 50. Leroy JL , Vanholder T , Mateusen B , Christophe A , Opsomer G , de Kruif A , Genicot G , Van Soom A. 2005. Non-esterified fatty acids in follicular fluid of dairy cows and their effect on developmental capacity of bovine oocytes in vitro. Reproduction 130:485–495 [DOI] [PubMed] [Google Scholar]
- 51. Aardema H , Vos PL , Lolicato F , Roelen BA , Knijn HM , Vaandrager AB , Helms JB , Gadella BM. 2011. Oleic acid prevents detrimental effects of saturated fatty acids on bovine oocyte developmental competence. Biol Reprod 85:62–69 [DOI] [PubMed] [Google Scholar]
- 52. Van Hoeck V , Sturmey RG , Bermejo-Alvarez P , Rizos D , Gutierrez-Adan A , Leese HJ , Bols PE , Leroy JL. 2011. Elevated non-esterified fatty acid concentrations during bovine oocyte maturation compromise early embryo physiology. PLoS One 6:e23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jungheim ES , Louden ED , Chi MM , Frolova AI , Riley JK , Moley KH. 2011. Preimplantation exposure of mouse embryos to palmitic acid results in fetal growth restriction followed by catch-up growth in the offspring. Biol Reprod 85:678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Russell DL , Salustri A. 2006. Extracellular matrix of the cumulus-oocyte complex. Semin Reprod Med 24:217–227 [DOI] [PubMed] [Google Scholar]
- 55. Russell DL , Robker RL. 2007. Molecular mechanisms of ovulation: co-ordination through the cumulus complex. Hum Reprod Update 13:289–312 [DOI] [PubMed] [Google Scholar]
- 56. Salustri A , Garlanda C , Hirsch E , De Acetis M , Maccagno A , Bottazzi B , Doni A , Bastone A , Mantovani G , Beck Peccoz P , Salvatori G , Mahoney DJ , Day AJ , Siracusa G , Romani L , Mantovani A. 2004. PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development 131:1577–1586 [DOI] [PubMed] [Google Scholar]
- 57. Varani S , Elvin JA , Yan C , DeMayo J , DeMayo FJ , Horton HF , Byrne MC , Matzuk MM. 2002. Knockout of pentraxin 3, a downstream target of growth differentiation factor-9, causes female subfertility. Mol Endocrinol 16:1154–1167 [DOI] [PubMed] [Google Scholar]
- 58. Zhang X , Jafari N , Barnes RB , Confino E , Milad M , Kazer RR. 2005. Studies of gene expression in human cumulus cells indicate pentraxin 3 as a possible marker for oocyte quality. Fertil Steril 83(Suppl 1):1169–1179 [DOI] [PubMed] [Google Scholar]
- 59. Gebhardt KM , Feil DK , Dunning KR , Lane M , Russell DL. 2011. Human cumulus cell gene expression as a biomarker of pregnancy outcome after single embryo transfer. Fertil Steril 96:47–52.e42 [DOI] [PubMed] [Google Scholar]
- 60. Berridge MJ. 2002. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32:235–249 [DOI] [PubMed] [Google Scholar]
- 61. Rizzuto R , Duchen MR , Pozzan T. 2004. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE 2004:re1. [DOI] [PubMed] [Google Scholar]
- 62. Decuypere JP , Monaco G , Bultynck G , Missiaen L , De Smedt H , Parys JB. 2011. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 1813:1003–1013 [DOI] [PubMed] [Google Scholar]
- 63. Dumollard R , Duchen M , Carroll J. 2007. The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol 77:21–49 [DOI] [PubMed] [Google Scholar]
- 64. Acton BM , Jurisicova A , Jurisica I , Casper RF. 2004. Alterations in mitochondrial membrane potential during preimplantation stages of mouse and human embryo development. Mol Hum Reprod 10:23–32 [DOI] [PubMed] [Google Scholar]
- 65. Wakefield SL , Lane M , Schulz SJ , Hebart ML , Thompson JG , Mitchell M. 2008. Maternal supply of omega-3 polyunsaturated fatty acids alter mechanisms involved in oocyte and early embryo development in the mouse. Am J Physiol Endocrinol Metab 294:E425–E434 [DOI] [PubMed] [Google Scholar]
- 66. Mitchell M , Schulz SL , Armstrong DT , Lane M. 2009. Metabolic and mitochondrial dysfunction in early mouse embryos following maternal dietary protein intervention. Biol Reprod 80:622–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Q , Ratchford AM , Chi MM , Schoeller E , Frolova A , Schedl T , Moley KH. 2009. Maternal diabetes causes mitochondrial dysfunction and meiotic defects in murine oocytes. Mol Endocrinol 23:1603–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Downs SM , Mosey JL , Klinger J. 2009. Fatty acid oxidation and meiotic resumption in mouse oocytes. Mol Reprod Dev 76:844–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vanderhyden BC , Caron PJ , Buccione R , Eppig JJ. 1990. Developmental pattern of the secretion of cumulus expansion-enabling factor by mouse oocytes and the role of oocytes in promoting granulosa cell differentiation. Dev Biol 140:307–317 [DOI] [PubMed] [Google Scholar]
- 70. Ozcan L , Ergin AS , Lu A , Chung J , Sarkar S , Nie D , Myers MG , Ozcan U. 2009. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9:35–51 [DOI] [PubMed] [Google Scholar]