

Abstract
α-Melanocyte-stimulating hormone (α-MSH)-induced activation of the melanocortin-4 receptor in hypothalamic neurons increases energy expenditure and inhibits food intake. Active hypothalamic AMP-activated protein kinase (AMPK) has recently been reported to enhance food intake, and in vivo experiments suggested that intrahypothalamic injection of melanocortins decreased food intake due to the inhibition of AMPK activity. However, it is not clear whether α-MSH affects AMPK via direct intracellular signaling cascades or if the release of paracrine factors is involved. Here, we used a murine, hypothalamic cell line (GT1-7 cells) and monitored AMPK phosphorylation at Thr172, which has been suggested to increase AMPK activity. We found that α-MSH dephosphorylated AMPK at Thr172 and consequently decreased phosphorylation of the established AMPK substrate acetyl-coenzyme A-carboxylase at Ser79. Inhibitory effects of α-MSH on AMPK were blocked by specific inhibitors of protein kinase A (PKA) or ERK-1/2, pointing to an important role of both kinases in this process. Because α-MSH-induced activation of ERK-1/2 was blunted by PKA inhibitors, we propose that ERK-1/2 serves as a link between PKA and AMPK in GT1-7 cells. Furthermore, down-regulation of liver kinase B-1, but not inhibition of calcium-calmodulin-dependent kinase kinase-β or TGFβ-activated kinase-1 decreased basal phosphorylation of AMPK and its dephosphorylation induced by α-MSH. Thus, we propose that α-MSH inhibits AMPK activity via a linear pathway, including PKA, ERK-1/2, and liver kinase B-1 in GT1-7 cells. Given the importance of the melanocortin system in the formation of adipositas, detailed knowledge about this pathway might help to develop drugs targeting obesity.
The melanocortin system plays a pivotal role in controlling meal size and energy homeostasis. Adipose tissue-derived hormones, such as leptin, spark the activity of α-melanocyte-stimulating hormone (α-MSH)-releasing neurons, located in the arcuate nucleus of the hypothalamus. Secreted α-MSH activates melanocortin-4 receptor (MC4R)-expressing neurons of various hypothalamic nuclei, thus enhancing the release of anorexigenic stimuli (e.g. TRH and CRH) and inhibiting the liberation of orexigenic peptides (e.g. orexins and melanin-concentrating hormone) (1). Therefore, dysfunction of the melanocortin system inevitably leads to obese phenotypes in mammals. Accordingly, targeted disruption of the MC4R gene in mice causes an obesity/diabetes syndrome characterized by hyperphagia, hyperinsulinemia, and hyperglycemia (2). The importance of MC4R signaling for the regulation of human metabolism is highlighted by the finding that mutations in the MC4R gene are the most frequent monogenic cause of severe obesity (3–8).
AMP-activated protein kinase (AMPK) is a highly conserved serine/threonine kinase, consisting of a catalytic α-subunit and two regulatory β- and γ-subunits (9). AMPK is a key sensor and modulator of cellular energy levels. Activation of AMPK by increasing AMP/ATP ratios leads to inhibition of ATP-consuming, and activation of ATP-producing, processes (10, 11). In the hypothalamus, AMPK not only functions as a cellular energy sensor but also as a regulator of entire body energy balance by abating energy expenditure and enhancing food intake (12, 13).
Phosphorylation of threonine residue 172 (Thr172) in the activation loop of the α-subunit has been shown to play a major role in the regulation of AMPK activity (14–16). Calcium-calmodulin-dependent kinase kinase-β (CaMKKβ) (17–19), TGFβ-activated kinase-1 (TAK-1) (20, 21), and liver kinase B-1 (LKB-1) (22–25) are established AMPK kinases that have been shown to phosphorylate Thr172 and therefore to increase AMPK activity. cAMP-dependent protein kinase A (PKA) has been reported to increase phosphorylation of AMPK at Thr172 due to the activation of LKB-1 (26). On the other side, PKA has also been shown to decrease AMPK activity by increasing its phosphorylation at serine residue 173 or 485, indicating a dual role of PKA in the regulation of AMPK activity (27, 28). The molecular mechanism that either lead to PKA-mediated activation or inhibition of AMPK activity is not understood.
In vivo studies suggested that activation of PKA or inactivation of AMPK by melanocortins is involved in MC4R-mediated regulation of energy homeostasis via hypothalamic nuclei (13, 29). However, it is not clear whether melanocortin-induced signaling directly controls AMPK activity and, if so, whether melanocortin-induced signaling via PKA and AMPK converges into the same pathway.
Thus, we used hypothalamic GT1-7 cells to investigate α-MSH-induced functional interactions between PKA and AMPK on the cellular level. We observed that stimulation of GT1-7 cells with α-MSH resulted in dephosphorylation of AMPK at position Thr172 and, thus, most likely decreases AMPK activity in hypothalamic cells. In line with this observation, we found α-MSH-induced dephosphorylation of the established AMPK substrate acetyl-coenzyme A-carboxylase (ACC) at Ser79 in the same cells. Effects of α-MSH on the phosphorylation status of AMPK were blunted by two specific PKA inhibitors, suggesting that these two kinases are functionally linked. α-MSH-induced dephosphorylation of AMPK was also abrogated by inhibitors of MAPK kinases-1/2 (MEK-1/2), indicating that ERK-1/2 activity is required and that PKA is not sufficient for α-MSH-induced dephosphorylation of AMPK. α-MSH-induced phosphorylation of ERK-1/2 was also abolished by specific PKA inhibitors, suggesting that PKA acts upstream of ERK-1/2 and that ERK-1/2 are a functional link between PKA and AMPK. Finally, down-regulation of LKB-1 abolished α-MSH-induced dephosphorylation of AMPK, highlighting a role for LKB-1 in this process.
In summary, we show that α-MSH inhibits AMPK activity in GT1-7 cells and propose that PKA-mediated activation of ERK-1/2 and subsequent inhibition of constitutively active LKB-1 mediates the effects of melanocortins on AMPK activity.
Results
α-MSH-induced dephosphorylation of AMPK at Thr172 in GT1-7 cells
Murine, hypothalamic GT1-7 cells represent an established cell model to investigate signaling pathways related to melanocortins and energy homeostasis (30–34). Thus, we used GT1-7 cells and analyzed α-MSH-induced signaling in particular with regard to posttranslational modifications of AMPK that regulate its kinase activity. First, we examined α-MSH-induced Gs-mediated signaling, by measuring ligand-induced production of cAMP or activation of a cAMP-responsive element (CRE)-dependent reporter gene construct. As shown previously (30, 35), α-MSH increased both intracellular cAMP concentrations and CRE activity with comparable EC50 values of 4.3 ± 1.4 and 5.3 ± 1.2 nm, respectively (Fig. 1, B and D). Agouti-related protein (AGRP), however, an inverse agonist of MC4R (36), had opposite effects, indicating that endogenous MC4R modulate the cAMP/PKA pathway in GT1-7 cells (Fig. 1, A and C). A role of AMPK activity in melanocortin-induced effects on appetite control has been shown in vivo, but it is not clear whether α-MSH affects AMPK directly in MC4R expression neurons or whether the release of paracrine factors is involved. Thus, we took advantage of a phospho-specific antibody and monitored the phosphorylation status of AMPK at Thr172, which has been shown to correlate with AMPK activity (14–16). After serum starvation, α-MSH induced dephosphorylation of AMPK at Thr172 in a time-dependent (Fig. 2A) and concentration-dependent (Fig. 2B) manner, suggesting that α-MSH directly dephosphorylates AMPK in MC4R-expressing cells. To substantiate this finding, we established chemiluminescence-based whole-cell ELISA assays. In a first set of experiments, we stimulated GT1-7 cells with α-MSH for 10 min and monitored AMPK phosphorylation at Thr172 and total AMPK protein levels. Such a treatment of cells did no affect AMPK protein levels (Fig. 3B) but significantly decreased AMPK phosphorylation at Thr172 (Fig. 3, A and D), suggesting inhibitory effects of α-MSH on AMPK activity. Ser79 of ACC is an established substrate of AMPK (10, 37). To analyze whether or not α-MSH-induced dephosphorylation at Thr172 indeed reflects decreased AMPK activity, we also performed whole-cell ELISA assays with a phospho-specific antibody against Ser79 of ACC. As shown in Fig. 3, C and D, challenging GT1-7 cells with α-MSH significantly decreased ACC phosphorylation, indicating that α-MSH is a negative regulator of AMPK activity in a hypothalamic cell line.
Fig. 1.
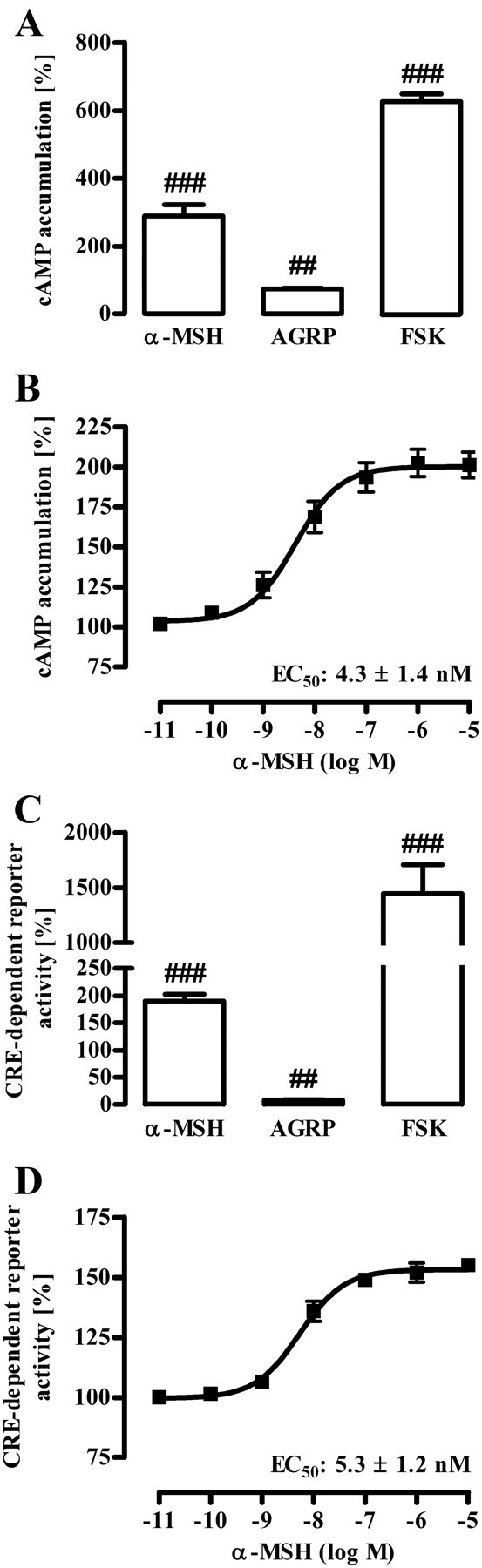
Gs-induced signaling by α-MSH in GT1-7 cells. A and B, cAMP accumulation was assessed after labeling of GT1-7 cells with [3H]adenine followed by the purification of [3H]cAMP and [3H]ATP by sequential chromatography. Cells were stimulated with 1 μm α-MSH, 100 nm AGRP, or 1 μm FSK (A) and with various concentrations of α-MSH for 60 min at 37 C (B). Five (A) and three (B) independent experiments were performed in triplicates, data compiled, normalized to basal (defined as 100%), and presented as the mean ± sem. Hashed signs indicate a significant (###, P < 0.001; ##, P < 0.01) difference to unstimulated cells. C and D, GT1-7 cells were transiently transfected with a reporter gene construct harboring the firefly luciferase gene under the control of a CRE-dependent promoter and as a control with a pcDNA3-YFP plasmid. Thirty-six hours after transfection (including 20 h of cell starvation), cells were stimulated with 1 μm α-MSH, 100 nm AGRP, or 1 μm FSK (C) and with various concentrations of α-MSH for 6 h (D). After lysis of cells, firefly luciferase activity and total YFP fluorescence were determined. Fluc/YFP ratios were calculated and then normalized by defining basal values as 100%. Results of five (C) and of three (D) independent experiments performed in triplicates are shown. Hashed signs indicate a significant (###, P < 0.001; ##, P < 0.01) difference to unstimulated cells.
Fig. 2.
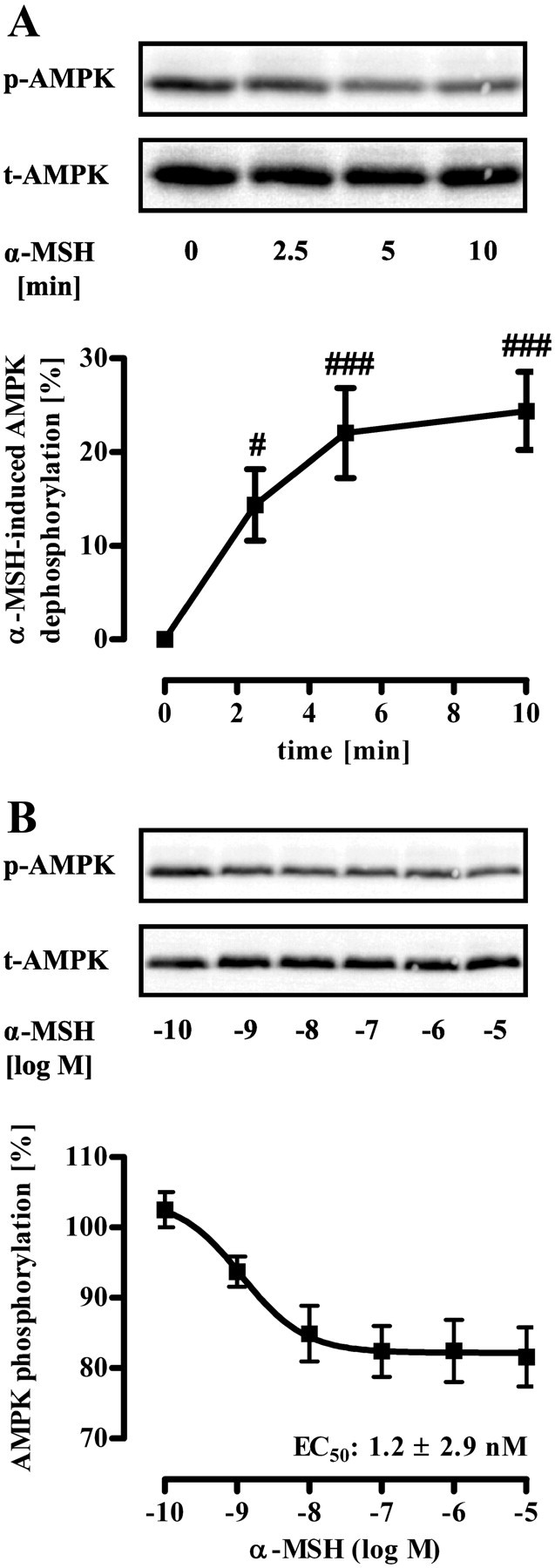
α-MSH-induced dephosphorylation of AMPK at Thr172 in GT1-7 cells monitored by Western blotting. Approximately 400,000 GT1-7 cells were grown on six-well plates, serum starved for 20 h, and stimulated with 1 μm α-MSH for various periods of time (A) or with various concentrations (B) for 10 min. Lysates were then analyzed by Western blotting using a phospho-specific antibody against Thr172 of AMPK. As a control of sample loading, the blot was stripped and reprobed with an antibody against t-AMPK-α. Representative blots are shown. Data of eight (A) and four (B) independent experiments were compiled and quantified. Data were normalized by setting values of unstimulated cells (basal) as 100%. A, α-MSH-induced dephosphorylation was calculated by subtracting values of stimulated cells from 100% and is shown as the mean ± sem. Hashed signs indicate a significant (#, P < 0.05; ###, P < 0.001) difference between treated and nontreated cells.
Fig. 3.

α-MSH-induced dephosphorylation of AMPK (Thr172) or of ACC (Ser79) in GT1-7 cells monitored by whole-cell ELISA. Approximately 25,000 GT1-7 cells were seeded into white 96-well dishes. After 20 h of serum starvation, cells were stimulated with 1 μm α-MSH for 10 min and phosphorylation of AMPK at Thr172 (A), total AMPK levels (B), or phosphorylation of ACC at Ser79 (C) monitored by specific antibodies in whole cells. Cell-bound, HRP-conjugated secondary antibody was detected by automatic injection of SuperSignal West Femto HRP substrate in a FLUOstar Omega plate reader at time point 1 sec (as indicated by the arrow) and total luminescence recorded for additional 8 sec. As a control, luminescence signals were measured in samples without any first antibody. A–C, Raw data (eight replicates) of one representative experiment are shown as the mean ± sem. D, Data of three independent experiments were compiled, control values subtracted and normalized by setting RLU values of unstimulated cells (basal) at time point 9 sec as 100%. α-MSH-induced dephosphorylation was than calculated by subtracting values of stimulated cells from 100%. Hashed signs (##, P < 0.01) indicate a significant difference to unstimulated cells (basal).
α-MSH-induced dephosphorylation of AMPK at Thr172 depends on LKB-1 activity
Dephosphorylation of AMPK requires either activation of a phosphatase or inhibition of a tonically active upstream kinase. So far, LKB-1, CaMKKβ, and TAK-1 have been described to phosphorylate AMPK at Thr172. Therefore, we used STO609 and oxozeaenol to block CaMKKβ and TAK-1 activity, respectively. Both inhibitors failed to affect AMPK phosphorylation, indicating that CaMKKβ and TAK-1 do not contribute to the regulation of AMPK activity in GT1-7 cells (Fig. 4A). As yet, direct chemical inhibitors of LKB-1 are not available. However, expression of the LKB-1 protein is selectively compromised by the LKB-1 destabilizer radicicol that binds to and inhibits heat-shock protein 90 (Hsp90), an endogenous essential chaperone of LKB-1 (38–40). As shown in Fig. 4B, treatment of GT1-7 cells with radicicol reduced both LKB-1 expression and phosphorylation of AMPK at Thr172, suggesting that LKB-1 is a constitutively active upstream kinase of AMPK in these cells. To lend further credence to this hypothesis, we took advantage of three distinct small interfering RNA (siRNA) designed to specifically inhibit LKB-1 expression. As shown in Fig. 4C, all specific, but not a randomly designed, siRNA inhibited LKB-1 expression and AMPK phosporylation at Thr172, indicating that AMPK is a substrate of LKB-1 in GT1-7 cells. Interestingly, radicicol or a specific siRNA against LKB-1, but not STO609 or oxozeaenol, also blunted α-MSH-induced dephosphorylation of AMPK in GT1-7 cells (Fig. 5, A–D), indicative for an involvement of LKB-1 in α-MSH-induced inhibition of AMPK.
Fig. 4.
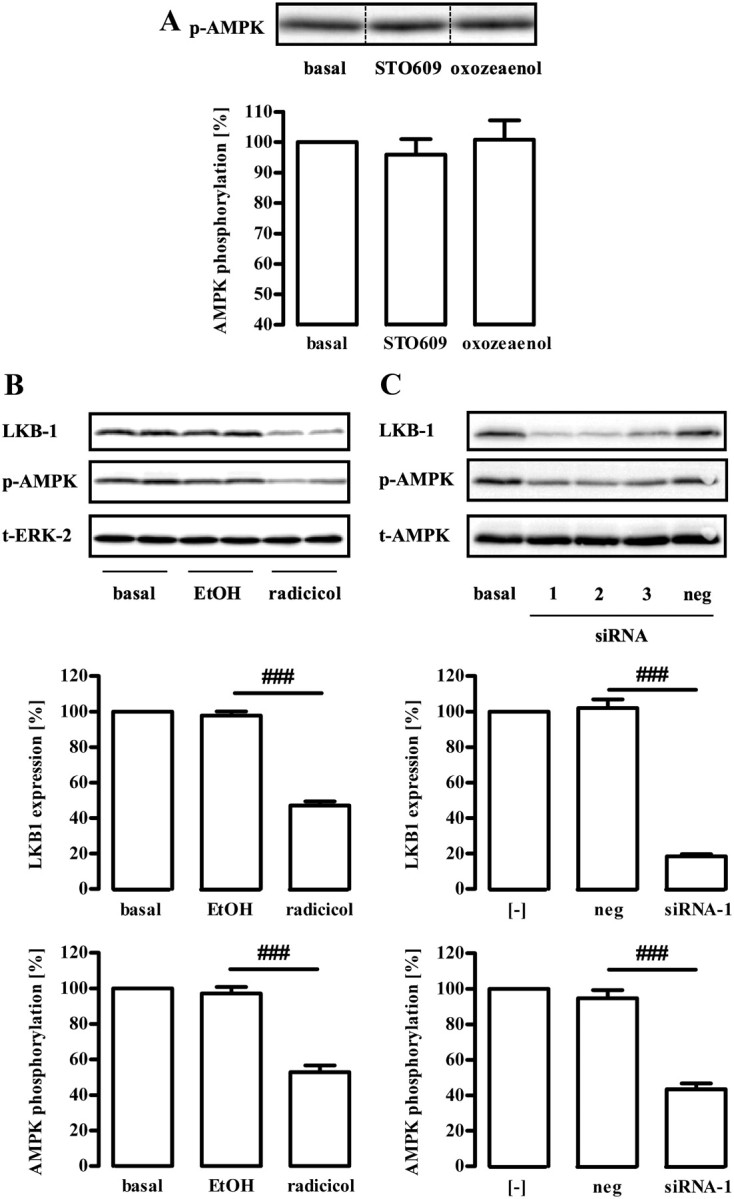
Role of LKB-1 for basal AMPK phosphorylation at Thr172. Approximately 400,000 GT1-7 cells were grown in six-well plates, serum starved for 20 h, and analyzed by Western blotting using a phospho-specific antibody against Thr172 of AMPK. A, Cells were treated for 30 min with the CaMKKβ inhibitor STO609 (1 μm) or the TAK-1 inhibitor oxozeaenol (500 nm), respectively. One representative blot is shown. The dotted line indicates grouping of different lanes from the same gel. B, Serum-starved cells were incubated with the Hsp90 inhibitor radicicol (10 μm) for 20 h or as a control with the carrier ethyl alcohol (EtOH) (1%). C, Cells were transfected with three distinct siRNA (1–3) designed to specifically down-regulate LKB-1 and one random siRNA (neg) by electroporation and serum starved for 20 h after 1 d of recovery. Samples were analyzed by Western blotting using an antibody either against AMPK-Thr172, LKB-1, ERK-2, or t-AMPK-α. One representative blot for each antibody is shown. Data of five (A) or four (B) independent experiments were compiled, normalized by setting basal values as 100%, and are shown in the bar graphs as the mean ± sem. A, Hashed signs indicate a significant (###, P < 0.001) difference between radicicol-treated and EtOH-treated cells or between siRNA-1 and the random siRNA in B.
Fig. 5.

Role of LKB-1 for α-MSH-induced dephosphorylation of AMPK at Thr172. Approximately 400,000 GT1-7 cells were grown in six-well plates, serum starved for 20 h, and analyzed by Western blotting using a phospho-specific antibody against Thr172 of AMPK. A, Cells were pretreated for 30 min with the CaMKKβ inhibitor STO609 (1 μm) or the TAK-1 inhibitor oxozeaenol (500 nm), respectively, and then stimulated or not with 1 μm α-MSH for 10 min. B, Serum-starved cells were incubated with the Hsp90 inhibitor radicicol (10 μm) for 20 h or as a control with the carrier ethyl alcohol (EtOH) (1%) and then stimulated or not with 1 μm α-MSH for 10 min. One representative blot is shown in each case. Data of five (A) or six (B) independent experiments were compiled, normalized by setting values of unstimulated cells (basal) or with inhibitor-treated cells as 100%. α-MSH-induced dephosphorylation was calculated by subtracting values of stimulated cells from 100% and is shown as the mean ± sem. Asterisks indicate a significant (***, P < 0.001; **, P < 0.01) difference to zero. Hashed signs indicate a significant (##, P < 0.01) difference between radicicol-treated and EtOH-treated cells. C, Cells were transfected with a siRNA designed to specifically down-regulate LKB-1 and a random siRNA (neg), serum starved for 20 h, and then stimulated or not with 1 μm α-MSH for 10 min. Samples were analyzed by Western blotting using an antibody either against AMPK-Thr172, LKB-1, p-ERK-1/2, ERK-2, or t-AMPK-α. Representative blots out of three independent experiments are shown. D, Data of three independent experiments were compiled, normalized by setting basal values of random siRNA (neg)-expressing cells or siRNA-1-expressing cells as 100%. α-MSH-induced dephosphorylation was calculated by subtracting values of stimulated cells of 100% and is shown as the mean ± sem. Asterisk indicates a significant (*, P < 0.05) difference to zero. Hashed sign indicates a significant (#, P < 0.05) difference between siRNA-1 and the control.
α-MSH-induced dephosphorylation of AMPK at Thr172 depends on ERK-1/2 activity
Next, we sought to identify a putative upstream kinase that mediates α-MSH-induced inhibition of AMPK via LKB-1. Recent studies suggested inverse correlations between decreased AMPK and increased ERK-1/2 activity (41). This is of particular interest, because recently, MC4R-mediated activation of ERK-1/2 in GT1-7 cells has been reported (42). To assess the role of ERK-1/2 in the regulation of AMPK activity, we analyzed effects of three different MEK inhibitors on phosphorylation of AMPK at Thr172. As shown in Fig. 6A, PD98059, U0126, and PD184352 reduced ERK-1/2 phosphorylation and inversely increased phosphorylation of AMPK, indicating that inhibition of ERK-1/2 leads to the phosphorylation of AMPK in GT1-7 cells. Having established that ERK-1/2 is an upstream regulator of AMPK, we wondered whether there is also a link between α-MSH-induced inhibition of AMPK activity on the one hand and increased ERK-1/2 activity on the other hand. Indeed, U0126 and PD184352 both prevented α-MSH-induced dephosphorylation of AMPK at Thr172 (Fig. 6B), indicating a functional link between α-MSH-induced dephosphorylation of AMPK and ERK-1/2 activity.
Fig. 6.
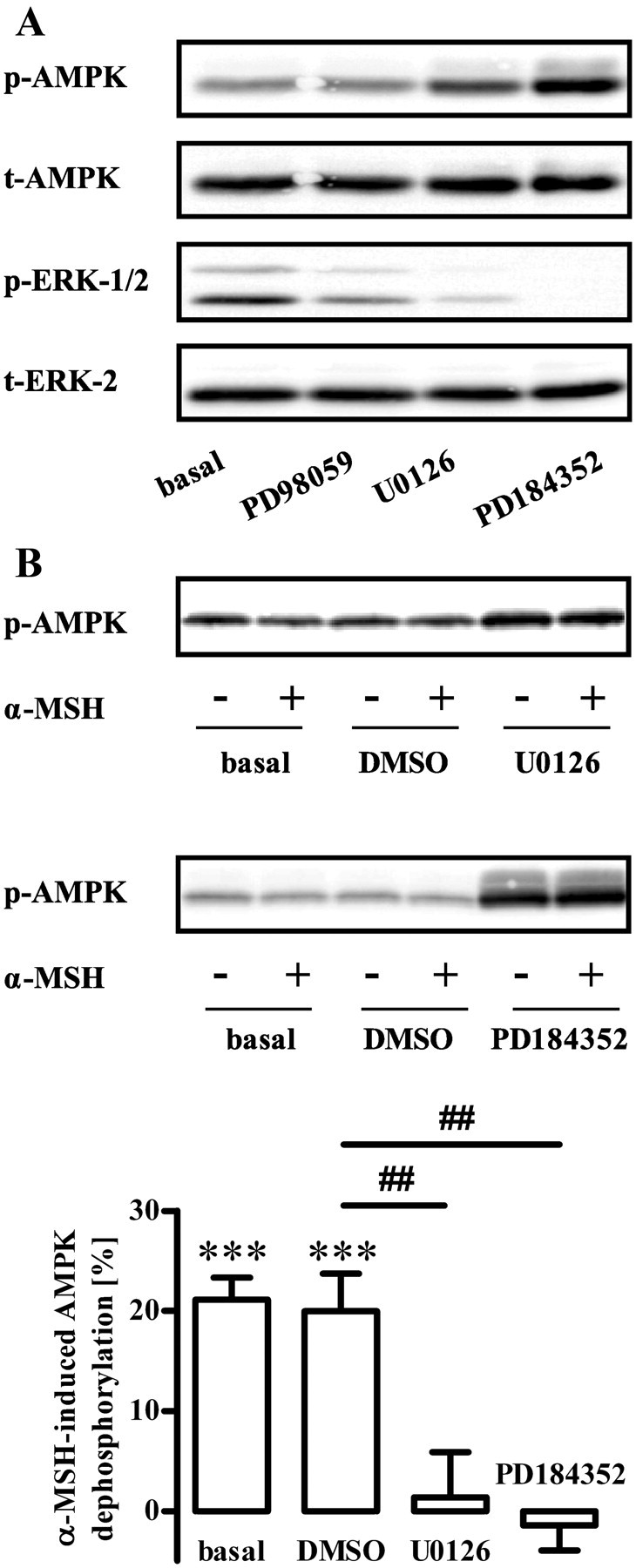
Role of ERK-1/2 for basal and α-MSH-induced dephosphorylation of AMPK at Thr172. A, Approximately 400,000 GT1-7 cells were grown in six-well plates, serum starved for 20 h, and stimulated with either PD98059 (50 μm), U0126 (10 μm), or PD184352 (10 μm) for 20 min. Lysates were then analyzed by Western blotting using either phospho-specific antibodies against Thr172 of AMPK and against phosphorylated ERK-1/2 or against unmodified proteins (t-ERK-2 or t-AMPK). One representative blot for each antibody is shown. B, Phosphorylation of AMPK was monitored as described in A. Additionally, cells were preincubated with 10 μm U0126 or PD184352 or carrier [0.5% dimethylsulfoxide (DMSO)] for 20 min and then stimulated or not with 1 μm α-MSH for 10 min. One representative blot is shown. Data of seven independent experiments were compiled, normalized by setting values of unstimulated cells (basal) or inhibitor-treated cells as 100%. α-MSH-induced dephosphorylation was calculated by subtracting values of stimulated cells from 100% and is shown as the mean ± sem. Asterisks indicate a significant (***, P < 0.001) difference to zero. Hashed signs indicate a significant (##, P < 0.01) difference between with U0126 or PD184352-treated and DMSO-treated cells.
α-MSH-induced phosphorylation of ERK-1/2 depends on PKA activity
So far, our results suggest a prominent role of ERK-1/2 and LKB-1 in α-MSH-induced inhibition of AMPK in GT1-7 cells. Next, we aimed at defining signaling components that functionally couple MC4R to ERK-1/2. In line with a previous study (42), we found transient phosphorylation of ERK-1/2 induced by α-MSH with a peak response at 3 min (Fig. 7A). Next, we analyzed the role of several potential upstream regulators of α-MSH-induced ERK-1/2 phosphorylation. We recently reported that MC4R are not exclusively coupled to Gs proteins but also interact with pertussis toxin (PTX)-sensitive G proteins in GT1-7 cells (35). Because PTX-sensitive G proteins have been reported to activate ERK-1/2 via the release of βγ-subunits, we tested effects of PTX and the specific βγ-blocker gallein on α-MSH-induced activation of ERK-1/2. Both inhibitors failed to block α-MSH-induced ERK-1/2 phosphorylation; thus, a role for PTX-sensitive G proteins in this process could be ruled out (Fig. 7, B and C). Similarly, specific inhibition of phosphatidylinositol 3 kinase (PI3K) (LY294002) or protein kinase C (PKC) bisindolylmaleimide-X [(BIM-X), Gö6983] had no effect on α-MSH-induced ERK-1/2 activation (Fig. 7, D and E). However, two structurally distinct inhibitors of PKA, Rp-cAMPs and KT5720, blocked about 70% of α-MSH-induced ERK-1/2 phosphorylation (Fig. 7, F–H), indicating that PKA is the major regulator of α-MSH-induced ERK-1/2 activation in GT1-7 cells.
Fig. 7.
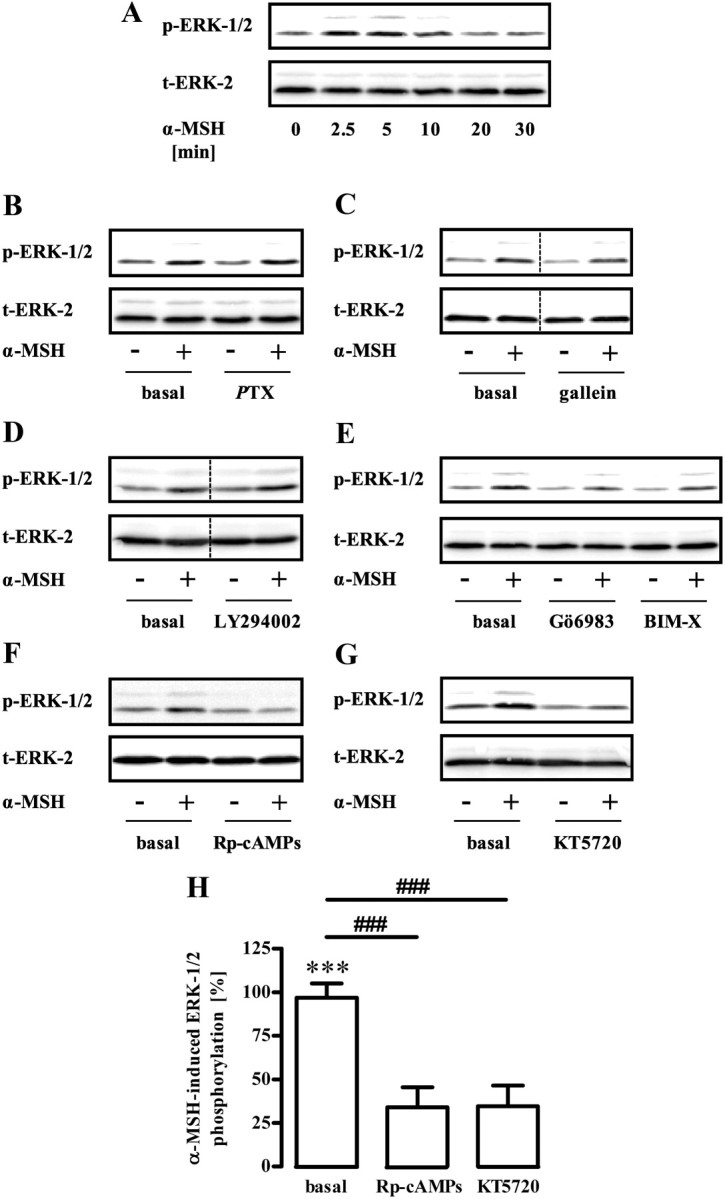
Role of PKA for α-MSH-induced phosphorylation of ERK-1/2. A, Approximately 400,000 GT1-7 cells were grown in six-well plates, serum starved for 20 h, and stimulated with 1 μm α-MSH for various periods of time. Lysates were then analyzed by Western blotting using a phospho-specific antibody against phosphorylated ERK-1/2. Afterwards, blots were stripped and reprobed with an antibody against ERK-2. One representative blot out of three experiments is shown. B–G, Phosphorylation of ERK-1/2 was monitored as described in A with the alteration that cells were stimulated with 1 μm α-MSH for 3 min and pretreated or not with (B) 100 ng/ml PTX over night, (C) with 10 μm gallein, (D) with 20 μm LY294002, (E) with 1 μm Gö6983 or BIM-X, (F) with 50 μm Rp-cAMPs, or (G) with 5 μm KT5720 for 30 min. One representative blot is shown for each condition. The dotted line indicates grouping of different lanes from the same gel. H, Data of 10 independent experiments with Rp-cAMPs or KT5720 were compiled, normalized by setting values of unstimulated cells (basal) or with inhibitor-treated cells as 100%. α-MSH-induced dephosphorylation was calculated by subtracting values of stimulated cells from 100% and are shown as the mean ± sem. Asterisks indicate a significant (***, P < 0.001) difference to zero. Hashed signs indicate a significant (###, P < 0.001) difference between Rp-cAMPs or KT5720-treated cells and basal.
α-MSH-induced dephosphorylation of AMPK at Thr172 depends on PKA activity
Up to this point, we accumulated data suggesting that α-MSH-induced dephosphorylation of AMPK occurs via PKA-mediated activation of ERK-1/2 and LKB-1. In such a scenario, inhibition of PKA activity should also abolish α-MSH-induced dephosphorylation of AMPK. In line with this prediction, selective inhibitors of PKA, but not of PI3K, PKC, or Gi/o signaling, blunted α-MSH-induced inhibition of AMPK (Fig. 8, A–G). These results strengthen our model, in which α-MSH-induced dephosphorylation of AMPK is mediated via PKA-promoted activation of ERK-1/2.
Fig. 8.

Role of PKA for α-MSH-induced dephosphorylation of AMPK at Thr172. Approximately 400,000 GT1-7 cells were grown on six-well plates, serum starved for 20 h, preincubated or not with (A) 100 ng/ml PTX over night, (B) with 10 μm gallein, (C) with 20 μm LY294002, (D) with 1 μm Gö6983 or BIM-X, (E) with 50 μm Rp-cAMPs, or (F) 5 μm KT5720 for 30 min, and stimulated with 1 μm α-MSH for 10 min. Lysates were then analyzed by Western blotting using a phospho-specific antibody against Thr172 of AMPK. In all cases, except from Rp-cAMPs, control cells were incubated with the carrier [0.1–0.5% dimethylsulfoxide (DMSO)]. One representative blot is shown. The dotted line indicates grouping of different lanes from the same gel. G, Data of seven independent experiments were compiled, normalized by setting values of unstimulated cells, DMSO, or inhibitor-treated cells as 100%. α-MSH-induced dephosphorylation was calculated by subtracting values of stimulated cells from 100% and is shown as the mean ± sem. Asterisks indicate a significant (***, P < 0.001) difference to zero. Hashed signs indicate a significant (##, P < 0.01) differences between Rp-cAMPs-treated and basal cells or between KT5720 and DMSO (0.5%)-treated cells.
Discussion
The goal of the present study was to show that melanocortins directly affect hypothalamic AMPK and to systematically dissect MC4R-induced signaling pathways leading to AMPK regulation in a hypothalamic cell line. We provide conclusive evidence that α-MSH induced dephosphorylation of AMPK in a PKA-, ERK-1/2-, and LKB-1-dependent manner (Fig. 9). The importance of the melanocortin system for maintaining energy homeostasis is beyond any doubt (2–8). Released in the hypothalamus by the peripheral satiety signal leptin, α-MSH forwards this peripheral stimulus to the central nervous system (43). Hypothalamic AMPK activity has recently been introduced as an important starvation signal whose activity is decreased in the hypothalamus after feeding and by anorexigenic stimuli, such as leptin, melanocortins, or glucose (13, 44). Although, it is not clear how altered AMPK activity in the hypothalamus modulates appetite, regulation of hypothalamic AMPK activity by melanocortins could be an important interface where appetite-controlling signals communicate and thus maintain a healthy energy homeostasis.
Fig. 9.

α-MSH-induced signaling that leads to the dephosphorylation of AMPK at Thr172 in GT1-7 cells. Signaling compounds required for α-MSH-induced dephosphorylation of AMPK at Thr172 in GT1-7 cells are depicted. Plus or minus signs next to the kinases indicate increased or decreased kinase activity, respectively.
Here, we show that in GT1-7 cells, α-MSH reduced phosphorylation at Thr172 in the activation loop of AMPK, which has been shown to correlate with decreased activity of this kinase (28). In line with this notion, we also demonstrate α-MSH-promoted dephosphorylation of the established AMPK substrate ACC at Ser79 (10, 37). Thus, in a hypothalamic cell line, similar functional interactions between AMPK and MC4R were observed, when compared with the aforementioned study using isolated hypothalamic neurons (13). In addition, MC4R-mediated AMPK dephosphorylation was reported for GT1-1 cells (45), which are closely related to GT1-7 cells used in our study. Therefore, negative regulation of AMPK activity by melanocortins appears to be a general property of hypothalamic cells, and in-depth knowledge of the signaling components linking both systems may help uncover key proteins that may serve as novel therapeutic targets.
PKA has also been shown to be important for MC4R-mediated regulation of energy homeostasis (29). However, downstream effectors of PKA in this process are not known, and a link between α-MSH-induced activation of PKA and inhibition of AMPK activity has not been described. Here, we show that two structurally distinct inhibitors of PKA significantly block α-MSH-induced dephosphorylation of AMPK, indicating that PKA- and AMPK-mediated effects induced by α-MSH converge in one signaling pathway in hypothalamic cells.
In other cell systems, opposing effects of PKA on AMPK phosphorylation have been demonstrated. In endothelial or liver cells, forskolin- or glucagon-induced PKA activation increases AMPK activity, in the case of glucagon due to LKB-1 activation (26, 46). In adipocytes, forskolin or catecholamines have been shown to activate AMPK via PKA (47, 48); however, increased AMP/ATP ratios by PKA-stimulated lipolysis were suggested as crucial underlying signaling events (49). In contrast, activation of PKA induced by forskolin or prostaglandin E2 has been reported to increase phosphorylation of AMPK at Ser485 and therefore to inhibit its kinase activity in Cos cells, mouse embryonic fibroblasts, or osteoblastic MG63 cells (28, 50). Thus, phosphorylation of AMPK at Ser485 initiated by PKA could also account for α-MSH effects on AMPK reported here. However, α-MSH-induced, PKA-dependent phosphorylation of AMPK at Ser485 was not detectable (data not shown), indicating that there is no direct interaction between PKA and AMPK in GT1-7 cells and that other signal components are involved.
Previous studies focusing on functional interactions between ERK-1/2 and AMPK have shown that ERK-1/2 is activated by AMPK and is therefore located downstream of the latter kinase (51). However, other researchers reported an inverse relationship of ERK-1/2 and AMPK, suggesting that ERK-1/2 may also act upstream of AMPK. Serum depletion of cardiac fibroblasts inhibited ERK-1/2 but increased AMPK phosphorylation at Thr172, and direct activation of AMPK by 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside decreased serum-induced ERK-1/2 activation (41), suggesting that ERK-1/2 and AMPK are regulated in a reciprocal fashion. Here, we report that three distinct MEK inhibitors decreased ERK-1/2 phosphorylation and simultaneously increased AMPK phosphorylation at Thr172 in the same population of GT1-7 cells, indicative for a functional link between ERK-1/2 and AMPK activity in a hypothalamic cell line. Further, α-MSH-induced and PKA-dependent dephosphorylation of AMPK requires ERK-1/2 activity. Thus, we suggest that PKA-mediated activation of ERK-1/2 underlies α-MSH-induced inhibition of AMPK.
Various signaling proteins have been reported to link MC4R and ERK-1/2 in different cell lines (52). After recombinant MC4R overexpression, α-MSH-induced activation of ERK-1/2 has been attributed to PTX-sensitive signaling pathways (42), to α-MSH-dependent cellular calcium transients (53), or to activation of PI3K (54). In GT1-1 cells that endogenously express MC4R, α-MSH-induced activation of ERK-1/2 was found to be resistant to PKA inhibitors and PTX but was blocked by specific PKC inhibitors (42). Thus, α-MSH-induced ERK-1/2 activation appears to strongly depend on the cellular context (52). As yet, α-MSH-triggered, PKA-mediated activation of ERK-1/2 in hypothalamic neurons has not been characterized at the cellular level. However, one study reported that after infusion into the hindbrain, melanocortins induced reduction in food intake of rats in an ERK-1/2 activity-dependent manner (55). Furthermore, in fresh hindbrain preparations, α-MSH-induced activation of ERK-1/2 was abrogated by a PKA inhibitor. These data indicate that PKA-dependent ERK-1/2 activation plays a major role in the regulation of food intake by melanocortins in hindbrain neurons (55). In line with these results, we found that two PKA inhibitors blocked α-MSH-induced activation of ERK-1/2. Thus, in analogy to the situation in hindbrain neurons, melanocortin-induced ERK-1/2 activation requires PKA activity in GT1-7 cells.
Although we cannot provide final proof, we suggest that ERK-1/2-mediated inhibition of constitutively active LKB-1 decreases the phosphorylation of AMPK at Thr172. This assumption is based on the observations that down-regulation of LKB-1 either by radicicol or specific siRNA decreased basal AMPK phosphorylation and blunted the inhibitory effects of α-MSH on AMPK. These data are in line with the observation that ERK-1/2-induced phosphorylation of LKB-1 at serine325 and 428 in melanoma cells hampered LKB-1-induced binding and activation of AMPK (56). Therefore, we propose a model in which α-MSH-induced activation of PKA leads to increased ERK-1/2 activity resulting in reduction of constitutive LKB-1-dependent phosphorylation of AMPK at Thr172 in GT1-7 cells (Fig. 9).
A recent study analyzed effects of the anorexigenic glucagon-like peptide-1 (GLP-1) on AMPK activity in hindbrain neurons (57). These authors put forward that coordinated PKA-mediated suppression of AMPK and activation of ERK-1/2 contributes to GLP-1-promoted reduction in food intake. Interestingly, GLP-1-promoted inhibition of AMPK and activation of ERK-1/2 was also detectable in GT1-7 cells, but no functional link between both kinases was reported. Another study has shown that the orexigenic adiponectin activated AMPK due to its inhibitory effects on ERK-1/2 activity in GT1-7 cells (58). Thus, it appears that activity of PKA, ERK-1/2, and AMPK is dynamically regulated by food intake-controlling hormones in hypothalamic GT1-7 cells or hindbrain neurons. Our data in concert with these previous results suggest that all three kinases along with LKB-1 merge into one major pathway of appetite control. Thus, the dissection of this signaling pathway not only sheds new light on the molecular events responsible for central melanocortin-induced effects on energy homeostasis but also opens up new avenues for pharmacological intervention on distinct levels.
Materials and Methods
Materials
DMEM, fetal bovine serum, and penicillin/streptomycin were purchased from Invitrogen (Darmstadt, Germany). The transfection reagent TurboFect was from Fermentas (St. Leon-Rot, Germany). Primary antibodies phospho (p)-ERK-1/2 or total (t)-ERK-2 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and t-AMPK-α, p-AMPK-Thr172, p-ACC-Ser79, or LKB-1, were from Cell Signaling (Danvers, MA). Antimouse or antirabbit horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Bio-Rad (München, Germany). All inhibitors were purchased from Enzo (Lörrach, Germany). PTX was obtained from Sigma-Aldrich (Deisenhofen, Germany). α-MSH was purchased from Biotrend (Destin, FL). The human AGRP fragment (86–132) was obtained from Peptides International (Louisville, KY). Luciferase assay substrate and buffer were from Promega (Madison, WI).
Cell culture and transfection
GT1-7 cells (kindly provided by Dr. Weiner, University of California, San Francisco, CA) were cultured in DMEM supplemented with fetal bovine serum (10%) and penicillin (100 U/ml) and streptomycin (100 μg/ml). For transfection of GT1-7 cells, 2 × 106 cells were seeded at a density of 70% in 10-cm dishes, cultured for 24 h, and then transfected with the appropriate amount of plasmid DNA using the TurboFect reagent from Fermentas as described by the manufacturer's protocol; 24 h after transfection, cells were detached and seeded in new dishes as required for the next experiment. For radicicol and PTX treatment, cells were serum starved for 20 h in the absence or presence of the inhibitor or the corresponding carrier control. To down-regulate the LKB-1 protein three specific siRNA (identification: s74497, s74498, and s74499) and one random siRNA construct (catalog no. 4390843) were purchased from Invitrogen. To obtain sufficient transfection efficacy, siRNA were introduced into GT1-7 cells by electroporation using the Neon transfection system also from Invitrogen according to the manufacturer's protocol. Briefly, for one pulse, 500,000 cells together with 50 nm of the corresponding siRNA were challenged with 1450 V for 30 msec and then seeded into six-well dishes.
cAMP accumulation
To determine agonist-promoted cAMP accumulation, approximately 200,000 GT1-7 cells were seeded in 12-well dishes coated with 0.1% poly-l-lysine 24 h before the experiment and labeled in serum-free DMEM containing 2 μCi/ml of [3H]adenine for 4 h. Cells were stimulated for 60 min at 37 C in DMEM containing 1 μm 3-isobutyl-1-methylxanthine along with various concentrations of ligand. The reaction was terminated by removing the medium and adding ice-cold 5% trichloroacetic acid to the cells. [3H]ATP and [3H]cAMP were then purified by sequential chromatography (dowex-resin/aluminium oxide columns), and the accumulation of [3H]cAMP was expressed as the ratio of [3H]cAMP/([3H]cAMP + [3H]ATP).
Firefly luciferase reporter gene assay
Four micrograms of pAD-CRE-Fluc plasmid containing the coding sequence of the firefly (Photinus pyralis) luciferase (Fluc) under the control of the CRE plasmid were transfected into approximately 2 × 106 GT1-7 cells on 10-cm dishes. As a control, 500 ng of a plasmid endcoding soluble yellow fluorescent protein (YFP) (pcDNA3-YFP) were cotransfected; 24 h after transfection, cells were seeded in 12-well dishes coated with 0.1% poly-l-lysine. After 12 h, cells were serum starved for 20 h and stimulated for 6 h in serum-free DMEM with the given ligands. Cells were lysed and Fluc activity determined using the luciferase substrate from Promega (Mannheim, Germany). Afterwards, total YFP emission (excitation, 480 ± 10 nm; emission, 515 ± 10 nm) was measured in the same lysates. Ratios of total YFP and Fluc emission were used to calculate concentration-dependent increases in reporter gene activity. Both recordings were performed in a FLUOstar Omega plate reader from BMG (Offenburg, Germany) using the luminescence or fluorescence mode, respectively.
Western blotting
Forty-eight hours before the experiment, approximately 400,000 GT1-7 cells were seeded in six-well plates. After 1 d, cells were serum starved for 20 h. Stimulation of the cells was carried out for the indicated time period at 37 C and stopped by cooling on ice. Cells were washed once with ice-cold PBS and lysed in Laemmli buffer. Lysate were homogenized by sonication, heated for 5 min at 95 C, and centrifuged (17,000 × g, 5 min). Proteins were resolved by SDS-PAGE and subsequent Western blotting using various antibodies: p-ERK-1/2 (1:2500; from Santa Cruz Biotechnology, Inc.) (E-4), t-ERK-2 antibody (1:10,000; from Santa Cruz Biotechnology, Inc.) (C-14), p-AMPK-Thr172 (1:10,000; from Cell Signaling) (40H9), LKB-1 (1:2000; from Cell Signaling) (D60C5), t-AMPK-α (1:10,000; from Cell Signaling) (F6), and the corresponding h HRP-coupled secondary antibody (1:10,000; from Bio-Rad), goat antimouse IgG (170–6516) and goat antirabbit IgG (170–6515). Immune reactivity was detected by monitoring the enhanced chemiluminescence-dependent light emission with a chemiluminescence detection system (Peqlab, Erlangen, Germany). Nitrocellulose membranes were stripped with 0.2 m glycine (pH 2) at 37 C for 48 h and then reprobed.
Chemiluminescence-based, whole-cell ELISA assays
Approximately 25,000 GT1-7 cells were seeded on white 96-well dishes, which were coated with 0.1% poly-l-lysine 1 h before the experiment. After 20 h of serum starvation, cells were stimulated with 1 μm α-MSH for 10 min, placed on ice, and washed with ice-cold PBS. After fixing the cells for 15 min with 4% PFA, they were permeabilized with ice-cold methanol/acteon (50:50) for 5 min. Next, cells were washed with PBS and then incubated with 0.5% of BSA in PBS for 15 min at 37 C to block unspecific binding sites. After an additional washing step, cells were incubated for 30 min at 37 C with the corresponding first antibody (p-AMPK-Thr172, 1:1000; t-AMPK-α, 1:1000; and p-ACC-Ser79, 1:500) in PBS/BSA. Primary antibodies were removed, cells washed and incubated with the corresponding HRP-conjugated secondary antibody (goat antimouse for t-AMPK-α and goat antirabbit for p-AMPK-Thr172 or p-ACC-Ser79 each 1:4000) in PBS/BSA for 30 min at 37 C. After several washing steps, 10 μl of pure PBS were added to the cells and the plate placed into a FLUOstar Omega plate reader from BMG; 50 μl of SuperSignal West Femto HRP substrate from Pierce (Rockford, IL) were automatically injected and total chemiluminescence immediately recorded.
Data analysis
Data obtained by Western blotting were analyzed using ImageJ. Statistical significance of differences was assessed by Student's t test (between two groups) or by one-way ANOVA and Tukey's honest significance post hoc test (between more than two groups).
Acknowledgments
This work was supported by the Friedrich-Baur-Stiftung Grant 11/10, Munich.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACC
- Acetyl-coenzyme A-carboxylase
- AGRP
- Agouti-related protein
- AMPK
- AMP-activated protein kinase
- BIM-X
- bisindolylmaleimide-X
- CaMKKβ
- calcium-calmodulin-dependent kinase kinase-β
- CRE
- cAMP-responsive element
- GLP-1
- glucagon-like peptide-1
- HRP
- horseradish peroxidase
- Hsp90
- heat-shock protein 90
- LKB-1
- liver kinase B-1
- MC4R
- melanocortin-4 receptor
- MEK-1/2
- MAPK kinases-1/2
- α-MSH
- α-melanocyte-stimulating hormone
- p
- phospho
- PI3K
- phosphatidylinositol 3 kinase
- PKA
- protein kinase A
- PKC
- protein kinase C
- PTX
- pertussis toxin
- siRNA
- small interfering RNA
- t
- total
- TAK-1
- TGFβ-activated kinase-1
- YFP
- yellow fluorescent protein.
References
- 1. MacNeil DJ , Howard AD , Guan X , Fong TM , Nargund RP , Bednarek MA , Goulet MT , Weinberg DH , Strack AM , Marsh DJ , Chen HY , Shen CP , Chen AS , Rosenblum CI , MacNeil T , Tota M , MacIntyre ED , Van der Ploeg LH. 2002. The role of melanocortins in body weight regulation: opportunities for the treatment of obesity. Eur J Pharmacol 450:93–109 [DOI] [PubMed] [Google Scholar]
- 2. Huszar D , Lynch CA , Fairchild-Huntress V , Dunmore JH , Fang Q , Berkemeier LR , Gu W , Kesterson RA , Boston BA , Cone RD , Smith FJ , Campfield LA , Burn P , Lee F. 1997. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141 [DOI] [PubMed] [Google Scholar]
- 3. Lee YS , Challis BG , Thompson DA , Yeo GS , Keogh JM , Madonna ME , Wraight V , Sims M , Vatin V , Meyre D , Shield J , Burren C , Ibrahim Z , Cheetham T , Swift P , Blackwood A , Hung CC , Wareham NJ , Froguel P , Millhauser GL , O'Rahilly S , Farooqi IS. 2006. A POMC variant implicates β-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab 3:135–140 [DOI] [PubMed] [Google Scholar]
- 4. Tao YX , Segaloff DL. 2003. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology 144:4544–4551 [DOI] [PubMed] [Google Scholar]
- 5. Hinney A , Bettecken T , Tarnow P , Brumm H , Reichwald K , Lichtner P , Scherag A , Nguyen TT , Schlumberger P , Rief W , Vollmert C , Illig T , Wichmann HE , Schäfer H , Platzer M , Biebermann H , Meitinger T , Hebebrand J. 2006. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adults from Germany. J Clin Endocrinol Metab 91:1761–1769 [DOI] [PubMed] [Google Scholar]
- 6. Hinney A , Hohmann S , Geller F , Vogel C , Hess C , Wermter AK , Brokamp B , Goldschmidt H , Siegfried W , Remschmidt H , Schäfer H , Gudermann T , Hebebrand J. 2003. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab 88:4258–4267 [DOI] [PubMed] [Google Scholar]
- 7. Govaerts C , Srinivasan S , Shapiro A , Zhang S , Picard F , Clement K , Lubrano-Berthelier C , Vaisse C. 2005. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides 26:1909–1919 [DOI] [PubMed] [Google Scholar]
- 8. Vaisse C , Clement K , Guy-Grand B , Froguel P. 1998. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 20:113–114 [DOI] [PubMed] [Google Scholar]
- 9. Woods A , Cheung PC , Smith FC , Davison MD , Scott J , Beri RK , Carling D. 1996. Characterization of AMP-activated protein kinase β and γ subunits. Assembly of the heterotrimeric complex in vitro. J Biol Chem 271:10282–10290 [DOI] [PubMed] [Google Scholar]
- 10. Hardie DG , Carling D. 1997. The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur J Biochem 246:259–273 [DOI] [PubMed] [Google Scholar]
- 11. Blanco Martínez de Morentin P , González CR , Saha AK , Martins L , Diéguez C , Vidal-Puig A , Tena-Sempere M , López M. 2011. Hypothalamic AMP-activated protein kinase as a mediator of whole body energy balance. Rev Endocr Metab Disord 12:127–140 [DOI] [PubMed] [Google Scholar]
- 12. Carling D. 2004. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci 29:18–24 [DOI] [PubMed] [Google Scholar]
- 13. Minokoshi Y , Alquier T , Furukawa N , Kim YB , Lee A , Xue B , Mu J , Foufelle F , Ferré P , Birnbaum MJ , Stuck BJ , Kahn BB. 2004. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569–574 [DOI] [PubMed] [Google Scholar]
- 14. Hawley SA , Davison M , Woods A , Davies SP , Beri RK , Carling D , Hardie DG. 1996. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271:27879–27887 [DOI] [PubMed] [Google Scholar]
- 15. Mitchelhill KI , Michell BJ , House CM , Stapleton D , Dyck J , Gamble J , Ullrich C , Witters LA , Kemp BE. 1997. Posttranslational modifications of the 5′-AMP-activated protein kinase β1 subunit. J Biol Chem 272:24475–24479 [DOI] [PubMed] [Google Scholar]
- 16. Gao G , Fernandez CS , Stapleton D , Auster AS , Widmer J , Dyck JR , Kemp BE , Witters LA. 1996. Non-catalytic β- and γ-subunit isoforms of the 5′-AMP-activated protein kinase. J Biol Chem 271:8675–8681 [DOI] [PubMed] [Google Scholar]
- 17. Woods A , Dickerson K , Heath R , Hong SP , Momcilovic M , Johnstone SR , Carlson M , Carling D. 2005. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2:21–33 [DOI] [PubMed] [Google Scholar]
- 18. Hawley SA , Pan DA , Mustard KJ , Ross L , Bain J , Edelman AM , Frenguelli BG , Hardie DG. 2005. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2:9–19 [DOI] [PubMed] [Google Scholar]
- 19. Anderson KA , Ribar TJ , Lin F , Noeldner PK , Green MF , Muehlbauer MJ , Witters LA , Kemp BE , Means AR. 2008. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab 7:377–388 [DOI] [PubMed] [Google Scholar]
- 20. Xie M , Zhang D , Dyck JR , Li Y , Zhang H , Morishima M , Mann DL , Taffet GE , Baldini A , Khoury DS , Schneider MD. 2006. A pivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci USA 103:17378–17383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Momcilovic M , Hong SP , Carlson M. 2006. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 281:25336–25343 [DOI] [PubMed] [Google Scholar]
- 22. Shaw RJ , Kosmatka M , Bardeesy N , Hurley RL , Witters LA , DePinho RA , Cantley LC. 2004. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101:3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lizcano JM , Göransson O , Toth R , Deak M , Morrice NA , Boudeau J , Hawley SA , Udd L , Mäkelä TP , Hardie DG , Alessi DR. 2004. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hawley SA , Boudeau J , Reid JL , Mustard KJ , Udd L , Mäkelä TP , Alessi DR , Hardie DG. 2003. Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hong SP , Leiper FC , Woods A , Carling D , Carlson M. 2003. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 100:8839–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kimball SR , Siegfried BA , Jefferson LS. 2004. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem 279:54103–54109 [DOI] [PubMed] [Google Scholar]
- 27. Djouder N , Tuerk RD , Suter M , Salvioni P , Thali RF , Scholz R , Vaahtomeri K , Auchli Y , Rechsteiner H , Brunisholz RA , Viollet B , Mäkelä TP , Wallimann T , Neumann D , Krek W. 2010. PKA phosphorylates and inactivates AMPKα to promote efficient lipolysis. EMBO J 29:469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hurley RL , Barré LK , Wood SD , Anderson KA , Kemp BE , Means AR , Witters LA. 2006. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem 281:36662–36672 [DOI] [PubMed] [Google Scholar]
- 29. Czyzyk TA , Sikorski MA , Yang L , McKnight GS. 2008. Disruption of the RIIβ subunit of PKA reverses the obesity syndrome of Agouti lethal yellow mice. Proc Natl Acad Sci USA 105:276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khong K , Kurtz SE , Sykes RL , Cone RD. 2001. Expression of functional melanocortin-4 receptor in the hypothalamic GT1-1 cell line. Neuroendocrinology 74:193–201 [DOI] [PubMed] [Google Scholar]
- 31. Shinyama H , Masuzaki H , Fang H , Flier JS. 2003. Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization. Endocrinology 144:1301–1314 [DOI] [PubMed] [Google Scholar]
- 32. Anderson RA , Zwain IH , Arroyo A , Mellon PL , Yen SS. 1999. The insulin-like growth factor system in the GT1-7 GnRH neuronal cell line. Neuroendocrinology 70:353–359 [DOI] [PubMed] [Google Scholar]
- 33. Sanz C , Vázquez P , Navas MA , Alvarez E , Blázquez E. 2008. Leptin but not neuropeptide Y up-regulated glucagon-like peptide 1 receptor expression in GT1-7 cells and rat hypothalamic slices. Metabolism 57:40–48 [DOI] [PubMed] [Google Scholar]
- 34. Mellon PL , Windle JJ , Goldsmith PC , Padula CA , Roberts JL , Weiner RI. 1990. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5:1–10 [DOI] [PubMed] [Google Scholar]
- 35. Büch TR , Heling D , Damm E , Gudermann T , Breit A. 2009. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J Biol Chem 284:26411–26420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ollmann MM , Wilson BD , Yang YK , Kerns JA , Chen Y , Gantz I , Barsh GS. 1997. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278:135–138 [DOI] [PubMed] [Google Scholar]
- 37. Davies SP , Carling D , Hardie DG. 1989. Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem 186:123–128 [DOI] [PubMed] [Google Scholar]
- 38. Hwang SL , Kim HN , Jung HH , Kim JE , Choi DK , Hur JM , Lee JY , Song H , Song KS , Huh TL. 2008. Beneficial effects of β-sitosterol on glucose and lipid metabolism in L6 myotube cells are mediated by AMP-activated protein kinase. Biochem Biophys Res Commun 377:1253–1258 [DOI] [PubMed] [Google Scholar]
- 39. Hwang SL , Yang BK , Lee JY , Kim JH , Kim BD , Kim BH , Suh KH , Kim DY , Kim DY , Kim MS , Song H , Park BS , Huh TL. 2008. Isodihydrocapsiate stimulates plasma glucose uptake by activation of AMP-activated protein kinase. Biochem Biophys Res Commun 371:289–293 [DOI] [PubMed] [Google Scholar]
- 40. Na LX , Zhang YL , Li Y , Liu LY , Li R , Kong T , Sun CH. 2011. Curcumin improves insulin resistance in skeletal muscle of rats. Nutr Metab Cardiovasc Dis 21:526–533 [DOI] [PubMed] [Google Scholar]
- 41. Du J , Guan T , Zhang H , Xia Y , Liu F , Zhang Y. 2008. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem Biophys Res Commun 368:402–407 [DOI] [PubMed] [Google Scholar]
- 42. Chai B , Li JY , Zhang W , Newman E , Ammori J , Mulholland MW. 2006. Melanocortin-4 receptor-mediated inhibition of apoptosis in immortalized hypothalamic neurons via mitogen-activated protein kinase. Peptides 27:2846–2857 [DOI] [PubMed] [Google Scholar]
- 43. Rahmouni K , Haynes WG. 2001. Leptin signaling pathways in the central nervous system: interactions between neuropeptide Y and melanocortins. Bioessays 23:1095–1099 [DOI] [PubMed] [Google Scholar]
- 44. Minokoshi Y , Shiuchi T , Lee S , Suzuki A , Okamoto S. 2008. Role of hypothalamic AMP-kinase in food intake regulation. Nutrition 24:786–790 [DOI] [PubMed] [Google Scholar]
- 45. Chai B , Li JY , Zhang W , Wu X , Zhang C , Mulholland MW. 2010. Melanocortin-4 receptor activation promotes insulin-stimulated mTOR signaling. Peptides 31:1888–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim JE , Song SE , Kim YW , Kim JY , Park SC , Park YK , Baek SH , Lee IK , Park SY. 2010. Adiponectin inhibits palmitate-induced apoptosis through suppression of reactive oxygen species in endothelial cells: involvement of cAMP/protein kinase A and AMP-activated protein kinase. J Endocrinol 207:35–44 [DOI] [PubMed] [Google Scholar]
- 47. Yin W , Mu J , Birnbaum MJ. 2003. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3-L1 adipocytes. J Biol Chem 278:43074–43080 [DOI] [PubMed] [Google Scholar]
- 48. Hutchinson DS , Chernogubova E , Dallner OS , Cannon B , Bengtsson T. 2005. β-Adrenoceptors, but not α-adrenoceptors, stimulate AMP-activated protein kinase in brown adipocytes independently of uncoupling protein-1. Diabetologia 48:2386–2395 [DOI] [PubMed] [Google Scholar]
- 49. Gauthier MS , Miyoshi H , Souza SC , Cacicedo JM , Saha AK , Greenberg AS , Ruderman NB. 2008. AMP-activated protein kinase is activated as a consequence of lipolysis in the adipocyte: potential mechanism and physiological relevance. J Biol Chem 283:16514–16524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Funahashi K , Cao X , Yamauchi M , Kozaki Y , Ishiguro N , Kambe F. 2009. Prostaglandin E2 negatively regulates AMP-activated protein kinase via protein kinase A signaling pathway. Prostag Oth Lipid Mediat 88:31–35 [DOI] [PubMed] [Google Scholar]
- 51. Chen HC , Bandyopadhyay G , Sajan MP , Kanoh Y , Standaert M , Farese RV , Farese RV. 2002. Activation of the ERK pathway and atypical protein kinase C isoforms in exercise- and aminoimidazole-4-carboxamide-1-β-D-riboside (AICAR)-stimulated glucose transport. J Biol Chem 277:23554–23562 [DOI] [PubMed] [Google Scholar]
- 52. Breit A , Büch TR , Boekhoff I , Solinski HJ , Damm E , Gudermann T. 2011. Alternative G protein coupling and biased agonism: new insights into melanocortin-4 receptor signalling. Mol Cell Endocrinol 331:232–240 [DOI] [PubMed] [Google Scholar]
- 53. Mountjoy KG , Kong PL , Taylor JA , Willard DH , Wilkison WO. 2001. Melanocortin receptor-mediated mobilization of intracellular free calcium in HEK293 cells. Physiol Genomics 5:11–19 [DOI] [PubMed] [Google Scholar]
- 54. Vongs A , Lynn NM , Rosenblum CI. 2004. Activation of MAP kinase by MC4-R through PI3 kinase. Regul Pept 120:113–118 [DOI] [PubMed] [Google Scholar]
- 55. Sutton GM , Duos B , Patterson LM , Berthoud HR. 2005. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology 146:3739–3747 [DOI] [PubMed] [Google Scholar]
- 56. Zheng B , Jeong JH , Asara JM , Yuan YY , Granter SR , Chin L , Cantley LC. 2009. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 33:237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hayes MR , Leichner TM , Zhao S , Lee GS , Chowansky A , Zimmer D , De Jonghe BC , Kanoski SE , Grill HJ , Bence KK. 2011. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab 13:320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cheng XB , Wen JP , Yang J , Yang Y , Ning G , Li XY. GnRH 2011 secretion is inhibited by adiponectin through activation of AMP-activated protein kinase and extracellular signal-regulated kinase. Endocrine 39:6–12 [DOI] [PubMed] [Google Scholar]