

Abstract
ACTH is the most important stimulus of the adrenal cortex. The precise molecular mechanisms underlying the ACTH response are not yet clarified. The functional ACTH receptor includes melanocortin-2 receptor (MC2R) and MC2R accessory proteins (MRAP). In human embryonic kidney 293/Flp recombinase target cells expressing MC2R, MRAP1 isoforms, and MRAP2, we found that ACTH induced a concentration-dependent and arrestin-, clathrin-, and dynamin-dependent MC2R/MRAP1 internalization, followed by intracellular colocalization with Rab (Ras-like small guanosine triphosphate enzyme)4-, Rab5-, and Rab11-positive recycling endosomes. Preincubation of cells with monensin and brefeldin A revealed that 28% of the internalized receptors were recycled back to the plasma membrane and participated in total accumulation of cAMP. Moreover, certain intracellular Ser and Thr (S/T) residues of MC2R were found to play important roles not only in plasma membrane targeting and function but also in promoting receptor internalization. The S/T residues T131, S140, T204, and S280 were involved in MRAP1-independent cell-surface MC2R expression. Other mutants (S140A, S208A, and S202D) had lower cell-surface expressions in absence of MRAPβ. In addition, T143A and T147D drastically impaired cell-surface expression and function, whereas T131A, T131D, and S280D abrogated MC2R internalization. Thus, the modification of MC2R intracellular S/T residues may positively or negatively regulate its plasma membrane expression and the capacity of ACTH to induce cAMP accumulation. Mutations of T131, T143, T147, and S280 into either A or D had major repercussions on cell-surface expression, cAMP accumulation, and/or internalization parameters, pointing mostly to the second intracellular loop as being crucial for MC2R expression and functional regulation.
ACTH is the most important stimulus of the adrenal cortex by its capacity to stimulate steroidogenesis (1) but also in development through an important trophic action (for review, see Refs. 1–3). The effects of ACTH are mediated through its receptor called melanocortin-2 receptor (MC2R) (4), a G protein-coupled receptor (GPCR). MC2R knockout leads to neonatal lethality in three-quarters of the mutant mice with survivors exhibiting a marked atrophied zona fasciculata (5). In humans, mutations in MC2R or in MC2R accessory proteins (MRAP) are often the cause of familial glucocorticoid deficiency (6–9).
As in the case of all known melanocortin receptors (MCR), MC2R is coupled to guanine nucleotide-binding proteins, alpha subunit, thus stimulating adenylyl cyclase, resulting in cAMP production and protein kinase A (PKA) activation (10, 11). The regulation of MC2R expression and function is atypical in at least two areas. MC2R functional expression requires the coexpression of MRAP (9, 12; for review, see Refs. 13, 14), and MC2R is known for its high capacity for cAMP production, compared with other guanine nucleotide-binding protein, alpha subunit, coupled receptors (for review, see Ref. 15). Since the pioneering work of Lefkowitz et al. in 1970 (10), several studies have been conducted showing that several intracellular mediators may exert positive feedback loops to enhance ACTH-induced cAMP production (2, 3). Sustained activation of cAMP stimulation may also arise from absence of desensitization or internalization. However, we and others have shown that ACTH exposure induces MC2R desensitization and internalization (16–18) and that MC2R and arrestins colocalize during ACTH-induced internalization (18). Arrestins, namely arrestin2 (Arr2) and arrestin3 (Arr3) (also called β-Arr1 and β-Arr2, respectively), are ubiquitously expressed proteins that promote uncoupling of the GPCR from heterotrimeric G protein and promote endocytosis of arrestin-bound receptors internalization (19–21). After internalization, GPCR are known to traffic into early endosomes, late endosomes, and slow recycling perinuclear endosomes or can be targeted to lysosomes for degradation. Within the endosomes, the receptors may be dephosphorylated and recycled back to the cell-surface for additional rounds of signaling or sent to degradation (22). These endosomes are trafficked into cells with the help of Rab (Ras-like small guanosine triphosphate enzyme) GTPases (23, 24). Rab proteins are part of the subfamily of Ras-like small GTPases termed Rab GTPases, which are implicated in the regulation of intracellular trafficking. Over 60 members of the Rab GTPase family have been identified, and each is believed to be specifically associated with a particular organelle or pathway (25).
The time-lag regarding our knowledge on MC2R, the smallest human GPCR, as compared with other GPCR has basically been due to various difficulties linked to the specific functional expression of MC2R (26, 27). However, with the discovery of MRAP (9), much progress has now been achieved in this area (14, 28). In this context, our group has developed and characterized a cell line system that fully reproduces dose-response curves of ACTH-induced cAMP production similar to that described in primary cultures of human and rat adrenocortical cells (29, 30). The human embryonic kidney (HEK) 293/Flp recombinase target (FRT)/Myc-MC2R/MRAP-Flag cell lines, which endogenously express MRAP2, are characterized by the stable coexpression of Myc-tagged MC2R along with either of the Flag-tagged MRAP1 isoforms, MRAPα or MRAPβ, in an entirely human background (12, 31). Indeed, in humans, in comparison with rodents, two MRAP1 isoforms have been identified, namely MRAPα and MRAPβ, which can modulate the expression and functional properties of MC2R (12). MRAP1 promote intracellular MC2R targeting to the plasma membrane as well as high-affinity ACTH binding and signaling, whereas MRAP2 mostly serves in MC2R transport to the plasma membrane (31, 32).
In spite of much recent progress regarding MC2R and MRAP functionality (13, 14, 28), the precise molecular mechanisms explaining the sustained and high MC2R response to ACTH stimulation is not yet clarified. We hypothesized that after arrestin-dependent internalization, MC2R may be recycled back to the cell-surface for additional rounds of signaling and that the functional regulation of MC2R may involve its intracellular Ser and Thr (S/T) residues. The aims of the present study were thus first to characterize MC2R internalization and trafficking in cell lines expressing MC2R and MRAP1 isoforms and secondly to identify the important MC2R S/T residues involved in cell-surface expression and internalization of MC2R and in ACTH-induced cAMP accumulation.
Results
MC2R internalization is arrestin-, dynamin-, and clathrin dependent
To investigate whether MC2R interacts with arrestins during internalization in 293/FRT/Myc-MC2R/MRAPβ-Flag cells, cells were transiently transfected with enhanced green fluorescent protein (EGFP) (control) or with Arr2, Arr3, or their dominant negative forms Arr2 (319–418) (Arr2-DN) and Arr3 (201–409) (Arr3-DN) (33). Cells were then stimulated from 5 to 60 min in the presence of 100 nm ACTH, and cell-surface MC2R was measured by ELISA. Data fitted by an exponential function indicated that, after 60 min of incubation, ACTH induced a receptor loss of 26 ± 2% in control cells, 31 ± 2% in Arr2-overexpressing cells (P < 0.05, n = 3), and 36 ± 1% in Arr3-overexpressing cells (P < 0.001, n = 3). Moreover, this internalization was also significantly decreased in Arr2-DN-transfected cells (10 ± 2%; P < 0.01, n = 3) but not in Arr3-DN (22 ± 2%; P > 0.05) (Fig. 1A). This result indicates that endogenous Arr2 expressed in 293/FRT is involved in MC2R internalization and that MC2R internalization is affected by arrestin overexpression despite the high levels of endogenous arrestin in HEK293 cells (34).
Fig. 1.
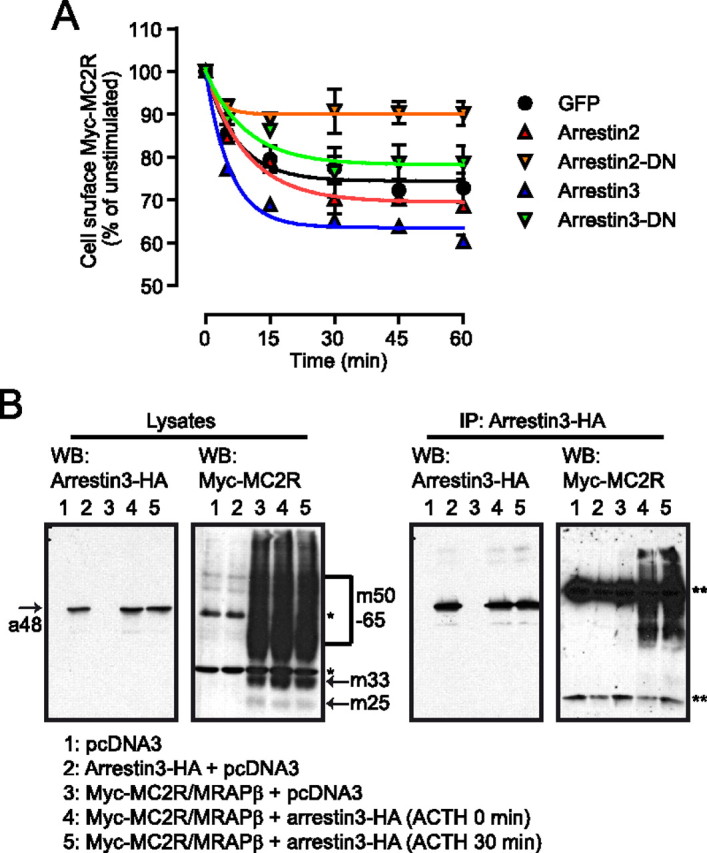
Interaction between arrestins and MC2R. A, Cells were transiently transfected with either control vector (GFP), Arr2, Arr2-DN, Arr3, or Arr3-DN and stimulated with ACTH 100 nm for the selected time periods after which cell-surface Myc-MC2R was measured by ELISA (n = 3). Data points were fitted with a decaying function. B, Native HEK293 cells were transiently cotransfected with pcDNA3, HA-Arr3, and Myc-MC2R/MRAPβ-Flag vectors as indicated. HA-Arr3 was immunoprecipitated and the resulting immunoprecipitates, and input lysates, were analyzed by reducing SDS-PAGE. Western blotting (WB) analyses against the HA and Myc tags are illustrated. a48 indicates Arr3 band, 48 kDa; m25, m33, and m50-65 designate the native, core-, and terminally glycosylated forms of MC2R, respectively; *, endogenous c-Myc (60 kDa); **, IgG light and heavy chains in the anti-Myc immunoblot after immunoprecipitation (IP).
To confirm the interaction between MC2R and Arr3, 293/FRT cells were cotransfected with Myc-MC2R/MRAPβ-Flag and -Arr3-hemagglutinin (HA) and subsequently stimulated or not with 100 nm ACTH for 30 min. In cell lysates, Myc-MC2R was present as multiple forms, either as native, core, or terminally glycosylated (m25, m33, and m50–65, respectively) (31), whereas in the arrestin-transfected cells, Arr3-HA appeared as a simple band of 48 kDa (Fig. 1B, left panel). MC2R was immunoprecipitated with Arr3 with and without ACTH stimulation but not in the absence of arrestin transfection (Fig. 1B, right panel). These data indicate that Arr3 and MC2R can be found in the same protein complex in both basal and ACTH-stimulated conditions.
ACTH-induced MC2R internalization was compared by confocal microscopy with isoproterenol-induced β2-adrenergic receptor (β2AR) and angiotensin II (Ang II)-induced Ang II type 1 receptor (AT1R) internalization in 293/FRT cells cotransfected with Arr3-EGFP. Cells were stimulated with 100 nm ACTH, 1 μm isoproterenol, or 1 μm Ang II, respectively, and cells were fixed at different time points. As shown in the merged images and in the fluorograms (Fig. 2, A–C), after 30 min of stimulation, MC2R and β2AR exhibited similar patterns of internalization characterized by a partial recruitment of Arr3 to the plasma membrane and into endosomes in the presence of their respective agonists (Fig. 2, A and B, arrows; entire time courses are presented in Supplemental Figs. 1 and 2, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org), whereas Ang II-stimulated AT1R accumulated and colocalized exactly with Arr3 into endosomes (Fig. 2C). These results are summarized in Fig. 2D.
Fig. 2.
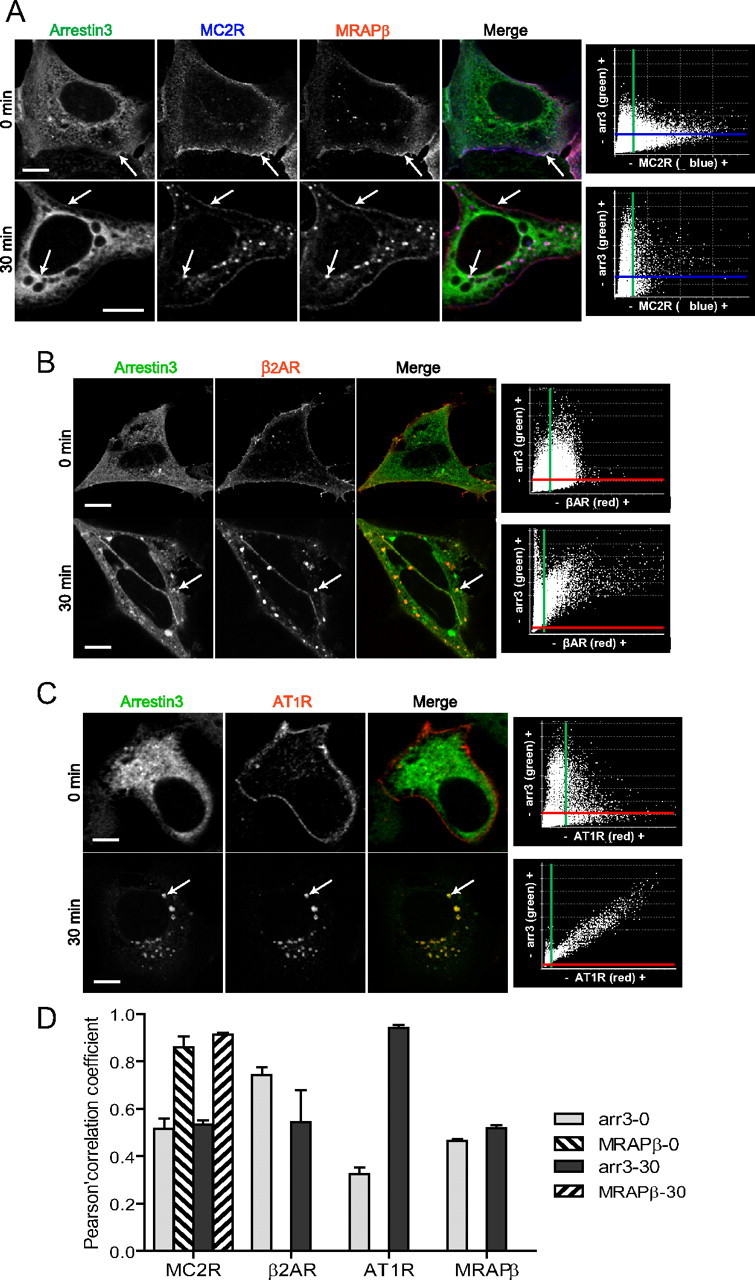
Interaction between arrestins and MC2R. Comparison with β2AR and AT1R. A–C, Confocal immunofluorescence microscopy images from 293/FRT/Myc-MC2R/MRAPβ-Flag cells transiently transfected with Arr3-EGFP and stimulated with ACTH 100 nm. Native 293/FRT cells were transiently cotransfected with Myc-β2AR (B) or AT1R-Flag (C) and Arr3-GFP and incubated for 30 min with or without 1 μm isoproterenol or 1 μm Ang II, respectively. Arr3 is labeled in green, MC2R in blue, and MRAP in red pseudocolors in merged images. β2AR and AT1R are labeled in red pseudocolor. In merged images, arrows point at triple colocalization (white color in the MC2R image sets and yellow color in the β2AR and AT1R image sets). Colocalization fluorograms based on three-dimensional reconstructions are shown in the right column. The x-axis of the fluorograms is representative of green labeling, and the y-axis is representative of red staining. Colocalization between green-labeling and red-labeling voxels is shown as being proportional to each other in fluorograms and corresponds to the yellow color in merged slices. Images are representative of three independent experiments where at least 50 cells were examined. Scale bars, 10 μm. D, Histogram representation of the mean ± sem of the Pearson's correlation coefficient of data from three to eight different cells; 0 indicates no significant correlation, and 1.0 indicates complete correlation.
Receptor internalization involves formation of vesicles (either clathrin-coated pits or caveolae), followed by their scission from the plasma membrane, an event mediated by the activity of dynamin (35). To verify whether MC2R internalization is dependent on dynamin activity, cells were transiently transfected with the dominant-negative dynamin-K44A mutant (GTPase inactive) and stimulated with vehicle, 0.1 nm, or 100 nm ACTH for 30 min. In contrast to control (GFP)-transfected cells, MC2R internalization was abrogated in dynamin-K44A-transfected cells (Fig. 3A). Similar data were also obtained in cells expressing MC2R and MRAPα (data not shown). Additionally, live-cell imaging was performed using the cell-impermeable HaloTag-ALEXAFluor488 ligand on 293/FRT cells stably expressing Halo-Myc-MC2R and MRAPβ (Fig. 3B). After 15 min of stimulation with 100 nm ACTH, Halo-Myc-MC2R internalized efficiently (Fig. 3C), whereas under similar conditions, there was no internalization in cells stimulated with 1 μm [Nle4, D-Phe7]α-melanocyte-stimulating hormone (NDP-MSH, a potent α-MSH analog) (Fig. 3C) nor in cells transfected with dynamin-K44A and then stimulated with 100 nm ACTH (Fig. 3C). Together, these results indicate that MC2R internalization is dynamin dependent.
Fig. 3.
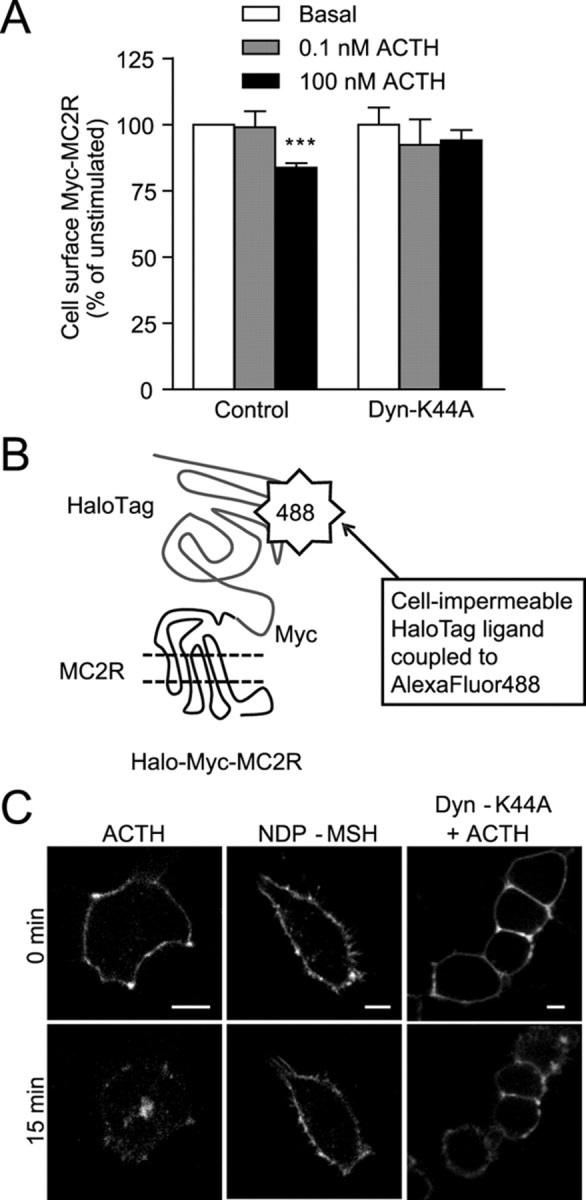
Dynamin-dependent MC2R internalization. A, Dynamin-K44A- or control (GFP)-transfected cells were stimulated with vehicle, 0.1 nm ACTH, or 100 nm ACTH for 30 min after which Myc-MC2R cell-surface expression in 293/FRT/Myc-MC2R/MRAPβ-Flag cells was measured by ELISA. Results are expressed as mean ± sem (n = 3). B, Schematic representation of the HaloTag system using the Halo-Myc-MC2R fusion protein. C, Live 293/FRT/Halo-Myc-MC2R/MRAPβ-Flag cells transfected or not with Dyn-K44A were incubated 15 min with the cell-impermeant HaloTag ligand, washed, and stimulated with ACTH 100 nm (left and right panels) or NDP-MSH 1 μm (center panels) and examined after 15 min with a spinning disk confocal microscope (representative images of three independent experiments). Scale bars, 10 μm. Statistical significance, compared with control: ***, P < 0.001.
MC2R internalization has previously been shown to be clathrin-, but not caveolin-, dependent in murine Y6 and M3 cells (16, 18). To verify whether MC2R internalization was also clathrin dependent in 293/FRT/Myc-MC2R/MRAPβ-Flag cells, cells were preincubated in hypertonic sucrose (450 mm) (known to disrupt clathrin adapters) and incubated with vehicle, 100 nm ACTH, or 100 nm ACTH + 1 mm 3-isobutyl-1-methylxanthine (IBMX) for 30 min. As shown in Fig. 4A, the presence of IBMX did not increase MC2R internalization compared with control conditions, whereas this internalization was abrogated in the presence of sucrose. In addition, increasing intracellular cAMP by preincubating the cells with forskolin (FSK), a direct activator of adenylyl cyclases, did not induce any internalization of MC2R. These results indicate that ACTH binding to MC2R is necessary to trigger MC2R internalization, that cAMP by itself is without effect, and that PKA activation alone is not sufficient to trigger receptor internalization. Similar data were also obtained in a cell line stably expressing MC2R and MRAPα (Supplemental Fig. 3). Immunofluorescence studies indicated that MC2R exhibited colocalization with clathrin triskelions after ACTH stimulation (Fig. 4B, arrow). In addition, results revealed that MC2R could colocalize with caveolin-1 in unstimulated, but not in stimulated, conditions (Fig. 4C).
Fig. 4.
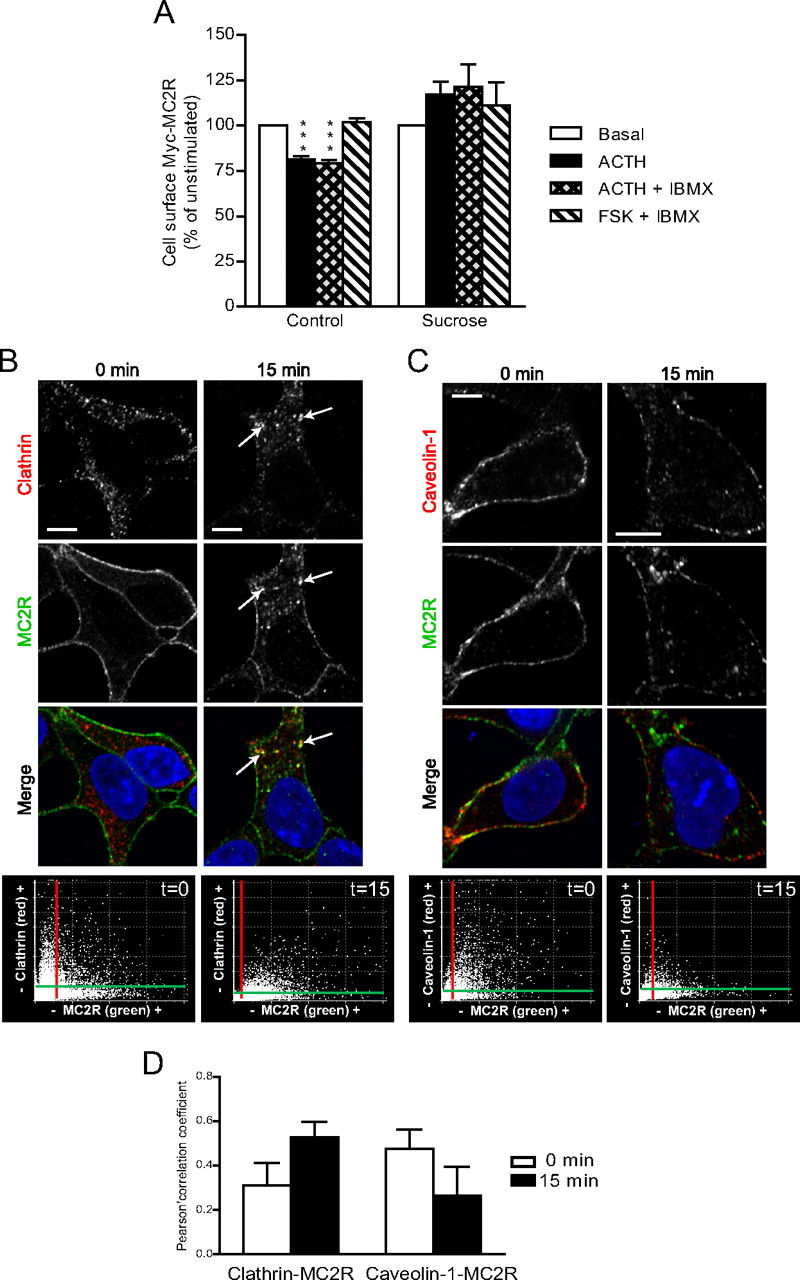
Clathrin-mediated MC2R internalization. A, 293/FRT/Myc-MC2R/MRAPβ-Flag cells were preincubated or not with 450 mm sucrose and stimulated with vehicle, 100 nm ACTH, 100 nm ACTH + 1 mm IBMX, or 10 μm FSK + 1 mm IBMX for 30 min followed by measurement of cell-surface MC2R by ELISA. Results are expressed as mean ± sem and are representative of three separate experiments. Statistical significance, compared with control: ***, P < 0.001. B and C, 293/FRT/Myc-MC2R/MRAPβ-Flag cells were stimulated with ACTH 100 nm for 15 min and processed for static immunofluorescence microscopy. Clathrin (B) or caveolin-1 (C) and Myc-MC2R were indirectly labeled with rabbit polyclonal antibodies against the native proteins or mouse monoclonal anti-Myc antibody and detected with appropriate Alexa Fluor-labeled secondary antibodies. Clathrin and Caveolin-1 are labeled in red and MC2R in green pseudocolors in merged images. Arrows point at colocalization (yellow color). Colocalization fluorograms based on three-dimensional reconstructions are shown in the right column, as described in Fig. 2. Images are representative of two independent experiments where at least 50 cells were examined. Scale bars, 10 μm. D, Histogram representation of the mean ± sem of the Pearson's correlation coefficient of data from three to eight different cells; 0 indicates no significant correlation, and 1.0 indicates complete correlation.
Internalized MC2R traffics into recycling endosomes
To determine the behavior of MC2R and MRAP after internalization, cells stably expressing MC2R and MRAPβ (or MRAPα in Supplemental data) were transiently transfected with Rab4-EGFP (recycling endosomes), Rab5-EGFP (early endosomes), Rab7-EGFP (late endosomes and lysosomes), or Rab11-EGFP (slow recycling perinuclear/Golgi endosomes) (23, 24, 36). Cells were incubated with vehicle or with 100 nm ACTH for different time periods (from 1 to 60 min), and behavior was followed by confocal microscopy. MRAPβ fluorescence colocalized with MC2R at the plasma membrane in control conditions and into endosomes upon ACTH stimulation, indicating a concomitant internalization of MC2R and MRAPβ. Internalized MC2R and MRAPβ colocalized with Rab4 (15 min; Fig. 5A), Rab5 (15 min; Fig. 5B), and Rab11 (30 min; Fig. 5D) but not with Rab7 (30 min; Fig. 5C) (see Supplemental Fig. 4 for correlation of intensity distribution as determined by fluorograms analyses and quantifications and Supplemental Figs. 5–8 for the entire 0- to 60-min time course). Results obtained using MRAPα, as an accessory protein for MC2R, were similar (Supplemental Figs. 5–8). These data indicate that, after ACTH-induced internalization, MC2R traffics in various types of endosomes, including Rab4, hence suggesting that MC2R and MRAP1 may be recycled back to the plasma membrane as a functional ACTH receptor complex.
Fig. 5.
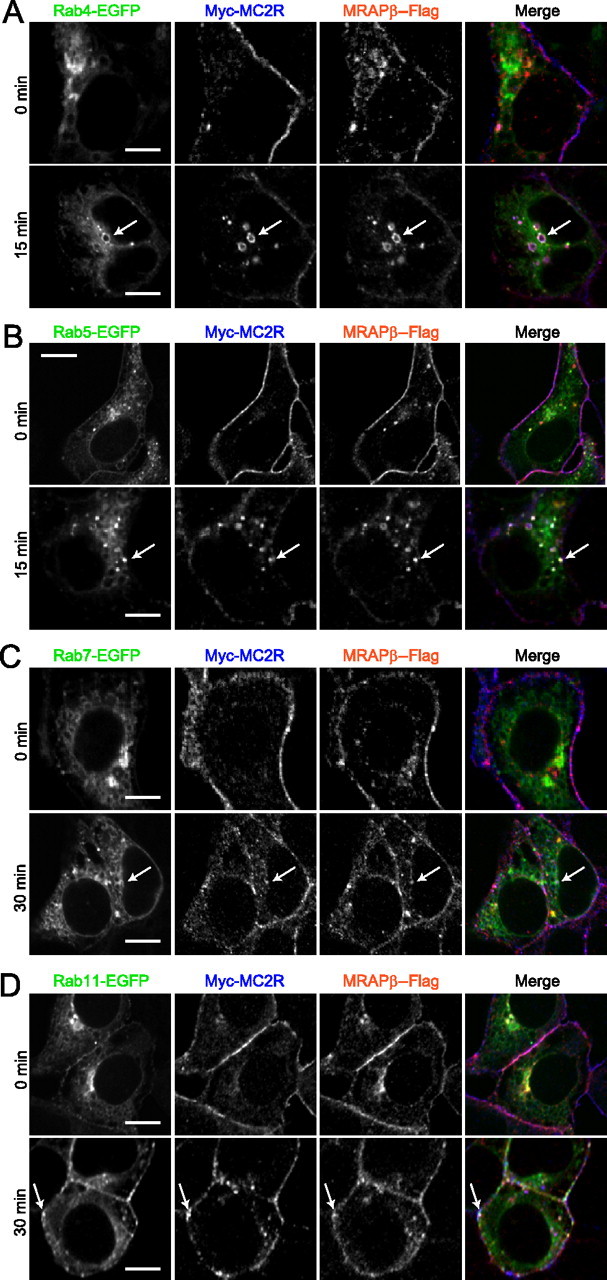
ACTH-stimulated endosomal trafficking of MC2R and MRAP. 293/FRT/Myc-MC2R/MRAPβ-Flag cells were transiently transfected with Rab4-EGFP (A), Rab5-EGFP (B), Rab7-EGFP (C), or Rab11-EGFP (D), stimulated with 100 nm ACTH for the indicated times, and fixed for immunofluorescence microscopy. Rab-EGFP proteins are labeled in green, MC2R in blue, and MRAP in red pseudocolors in merged images. Arrows point at triple colocalization (white color). Images are representative of two independent experiments where at least 50 cells were examined. Scale bars, 10 μm.
We next measured cell-surface expression levels in cells stimulated with increasing concentrations of ACTH (from 0.01 to 100 nm) and stimulated for 15, 30, 45, and 60 min. The maximal loss in cell-surface Myc-MC2R (MC2R internalization) after 60 min under 100 nm ACTH was evaluated at 28 ± 2% (t½, 100 nm = 6 min) (Fig. 6A). ACTH at concentrations lower than 0.3 nm did not induce any apparent internalization of MC2R. A characterization of MC2R internalization was also performed using MRAPα as an accessory protein (Supplemental Fig. 9). Furthermore, MC2R did not internalize in absence of MRAPα or MRAPβ (data not shown).
Fig. 6.

MC2R internalization and recycling. A, 293/FRT/Myc-MC2R/MRAPβ-Flag cells were washed and incubated with ACTH concentrations ranging from 0.01 to 100 nm, without IBMX, for the indicated times and fixed for detection of the extracellular Myc tag by cell-surface ELISA procedures (n = 3). B and C, 293/FRT/Myc-MC2R/MRAPβ-Flag cells were incubated with ACTH (0.1 or 100 nm) for 15, 30, 45, and 60 min in the presence or absence of 1 μg/ml BFA (B) or 25 μm monensin (C) and subsequently fixed for extracellular Myc tag detection by cell-surface ELISA procedures (n = 3). D, Basal cell-surface Myc-MC2R levels after a 90-min incubation with or without recycling inhibitors (n = 3). E, Effect of 1 μm FSK and recycling inhibitors on Myc-MC2R cell-surface expression (n = 2). F, Effect of 1 μg/ml BFA on 100 nm ACTH- and 1 μm FSK-induced cAMP accumulations in the presence of IBMX (n = 3). F, Effect of 100 nm ACTH in control cells (C) and in cells preincubated in 1 μg/ml BFA or 25 μm monensin without IBMX; cAMP accumulation was measured after a 60-min stimulation (n = 3). Results are expressed as mean ± sem. Statistical significance, compared with control: *, P < 0.05; ***, P < 0.001.
To determine whether MC2R was recycled back to the plasma membrane after ACTH stimulation, cells were preincubated in the absence or presence of the Golgi cisternae-disrupting agent brefeldin A (BFA) (1 μg/ml) (37, 38) or of the endosome recycling inhibitor monensin (25 μ[scap]m) (39, 40). Cells were then stimulated with low (0.1 nm) or high (100 nm) concentrations of ACTH for periods ranging from 15 to 60 min, after which MC2R internalizaion was measured by ELISA. As shown in Fig. 6, B and C, cells stimulated with 0.1 nm ACTH did not show any decrease in cell-surface receptor levels in the absence or presence of either BFA or monensin. In contrast, in the absence of BFA, 100 nm ACTH induced a time-dependent decrease in cell-surface expression of MC2R, which was amplified by coincubation with BFA. After a 60-min incubation, the loss of cell-surface MC2R increased from 29 ± 1 to 40 ± 1% in the presence of BFA (P < 0.001, n = 3). These differences indicated that 28 ± 2% of internalized receptors recycled back to the plasma membrane through a BFA-sensitive organelle (Fig. 6B). Similarly, in the absence of monensin, 33 ± 2% of initial cell-surface receptors were lost, and this amount increased to 51 ± 2% in the presence of monensin (P < 0.001), indicating that 35 ± 4% of internalized receptors were recycled via monensin-sensitive endosomes (Fig. 6C). As shown in Fig. 6, B and C, this recycling was not apparent at 5 min. Furthermore, after a 90-min incubation in BFA-containing medium, basal cell-surface levels of Myc-MC2R remained unchanged as compared with control cells (Fig. 6D), thus eliminating the possible contribution of newly synthesized receptors to the plasma membrane population analyzed. Moreover, as shown in Fig. 6E, FSK, alone or in combination with BFA or with monensin, did not affect and even slightly enhanced the expression of MC2R present at the cell-surface.
To investigate whether recycled receptors contributed to the sustained ACTH response, 100 nm ACTH- and 1 μm FSK-induced cAMP accumulation were measured in cells preincubated with or without BFA in the presence of IBMX. Throughout the 60-min stimulation with ACTH, cAMP accumulation was lower in BFA-treated cells compared with control cells (P < 0.05), although BFA had no effect on FSK-induced cAMP accumulation (Fig. 6F). In a complementary experiment, cells were preincubated with or without BFA or monensin in the absence of IBMX. After a 60-min stimulation with 100 nm ACTH, cAMP levels were lower in BFA- and monensin-treated cells compared with control cells (P < 0.001 each) (Fig. 6G). Together, these results indicate that MC2R undergoes recycling at the plasma membrane and that these recycled receptors are responsible for the accumulation of cAMP accumulation under ACTH stimulation.
Importance of MC2R S/T residues for MC2R cell-surface expression
To better understand the relationship between MC2R structure and MRAP interaction, the following experiments were designed to identify the intracellular S/T residues that may be important in regulating cell-surface expression and internalization of MC2R, as well as ACTH-mediated cAMP accumulation. To this end, all intracellular S/T residues of MC2R were mutated into a nonphosphorylatable residue (Ala) or a residue intended to mimic phosphorylation (Asp) (Fig. 7). Additional T143 mutants (T143S, T143G, and T143K) were generated and analyzed, because the phosphorylation of the homologous residue (T157) from the melanocortin-1 receptor (MC1R) has been shown to be critical for receptor export to the plasma membrane (41, 42). In the case of T143A/D/S/G/K, the lateral chain of A provides a hydrophobic, neutral, and apolar lateral chain; D is hydrophilic, negatively charged, and polar; S is hydrophilic, neutral, and polar; G has no lateral chain; and finally K is hydrophilic, positively charged, and polar. The position of these sites on the MC2R is illustrated in Fig. 7A and comparison with the others MCR is given in Fig. 7B.
Fig. 7.

Representation of the human MC2R indicating the S/T amino acid residues that have been mutated in this study A. Adapted from Ref. 63. B, Comparison of amino sequences alignment of the second intracellular of MCR 1 to 4. In yellow and blue, full or partial consensus sequences homology. Box 1 identified the T143 amino acid, box 2 amino acid frequently mutated in pathological situations, and box 3 identified the T147, which is unique to MC2R. Exo, Exoplasm; Cyto, cytoplasm.
All the above S/T mutant cDNA were stably transfected in 293/FRT cells via homologous recombination with the Flp-In system (12) and transfected with GFP (control), MRAPα (only for T143A/D/S/G/K), or MRAPβ. Using whole-cell-based ELISA, raw total expression and raw cell-surface expression (Supplemental Fig. 10, A–D) were measured (summarized in Supplemental Table 1). Because total expression levels varied for each mutant expressed in individual cell lines, cell-surface expression data were normalized to total expression levels (Fig. 8A and Supplemental Table 1). In the absence of MRAPβ, the T131A, T131D, S140D, T147A, T204A, and S280D mutants exhibited significantly higher normalized cell-surface expression than wild type (WT). Cotransfection of MC2R with MRAPβ decreased their cell-surface expression close to or slightly higher than normal (WT like) levels and increased the cell-surface expression of S202A above that of WT (Fig. 8, A and B, and Supplemental Table 1). In the absence of MRAP1, S140A, S202D, and S280A exhibited lower normalized cell-surface expression than WT (Fig. 8, A and B, and Supplemental Table 1). However, MRAPβ completely rescued the cell-surface expressions of S140A, S202D, and S208A mutants (Fig. 8A and Table 1). Although the basal level of cell-surface expression of all T143 mutants was very low, MRAPα or MRAPβ was able to partially rescue the cell-surface expressions of T143D, T143S, T143G, and T143K, except for T143A, whose expression remained very low (3% of WT) (Fig. 8B). In addition, the addition of MRAPα or MRAPβ failed to overcome the total absence of cell-surface expression of T147D.
Fig. 8.

Role of S/T residues in MC2R cell-surface expression. Each individual 293/FRT/Myc-mutant cell lines were transfected with control (GFP) or the indicated MRAP1 isoform and were processed through whole-cell ELISA procedures to determine normalized total and normalized cell-surface expression levels of MC2R mutants relatively to WT-MC2R. A and B, Cell-surface expression normalized over total expression of each Myc-tagged MC2R mutant, with or without MRAP1 isoform expression. Mutants with increased normalized cell-surface expressions without MRAP1 isoforms are indicated in blue, mutants with decreased normalized cell-surface expressions without MRAP1 isoforms are indicated in green, and mutants with low level of normalized cell-surface expressions with and without MRAP1 isoforms are indicated in red. Results are expressed as mean ± sem (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with WT-MC2R. C, Each Myc-tagged MC2R mutant was transiently transfected in stable 293/FRT/MRAPβ cells, and cell lysates were analyzed by SDS-PAGE in denaturing conditions. Western blotting (WB) was performed against the Myc tag using monoclonal antibodies; membranes were then stripped to assess the presence of total p44/p42mapk expression in cell lysates as loading controls (C). Native, core-, and terminally N-glycosylated forms of WT Myc-MC2R (25, 33, and 50–65 kDa) are indicated on the right. QQQQ is a mutant MC2R lacking all N-glycosylation sites, whereas untagged-MC2R does not contain the N-terminal Myc-epitope. *, Endogenous c-myc protein expressed in 293/FRT cells (60 kDa). This result was repeated twice with similar results.
Table 1.
Functional characteristics of Myc-tagged MC2R and mutants coexpressed with MRAPb
| Myc-MC2R | T1/2 cAMP (min) | Rmax cAMP (% of WT)& | R60, cAMP (% of initial rate) | Normalized surface expression with MRAPβ (% of WT) | R5, surface (% of initial rate) | RSR5 (% of initial) | RSR60 (% of WT)& |
|---|---|---|---|---|---|---|---|
| WT | 12.5 ± 1.4# | 100 | 3.19 ± 1.04# | 100 | 33.1 ± 5.4# | 85 ± 2# | 78 ± 1# |
| T131A | 13.2 (10.9, 16.9) | 91 ± 4 | 4.31 ± 0.04 | 127 ± 14a | <0.1c | 92 ± 4 | 95 ± 2c |
| T131D | 4.5 (3.2, 7.7)a | 22 ± 1c | <0.01c | 130 ± 14b | <0.1c | 97 ± 3c | ∼100c |
| S140A | 14.8 (13.2, 17) | 94 ± 3 | 6.05 ± 0.06 | 91 ± 8 | 25.5 ± 0.1 | 78 ± 1 | 72 ± 1c |
| S140D | 11.5 (9.5, 14.6) | 81 ± 3a | 2.71 ± 0.22a | 125 ± 18a | 43.2 ± 0.6b | 95 ± 2b | 92 ± 1c |
| T143A | ∼0.01 (wide) | 3 ± 1c | <0.01c | 1 ± 1c | 22.2 ± 1.6 | 68 ± 5 | 58 ± 5c |
| T143D | 8.61 (7.9, 9.4)a | 102 ± 2 | 0.80 ± 0.01 | 32 ± 4c | 16.3 ± 1.1a | 61 ± 4a | 54 ± 3c |
| T143S | 10.8 (7.8, 17.0) | 106 ± 6 | 2.09 ± 0.18 | 44 ± 5c | 27.7 ± 1.3 | 75 ± 4 | 67 ± 2c |
| T143G | N/D | N/D | N/D | 9 ± 3c | N/D | N/D | N/D |
| T143K | 5.14 (2.7, 45.3) (P = 0.07) | 35 ± 3c | 0.03 ± 0.01c | 8 ± 1c | N/D | N/D | N/D |
| T147A | 10.3 (7.8, 15.0) | 115 ± 5a | 1.79 ± 0.16 | 120 ± 12 | 23.6 ± 0.5 | 93 ± 2 | 91 ± 1c |
| T147D | N/A | <1c | N/A | <1c | <0.1a | N/A | N/A |
| S202A | 11.6 (9.3, 15.1) | 104 ± 4 | 2.74 ± 0.07 | 152 ± 44c | 40.6 ± 2.0 | 89 ± 3 | 83 ± 1a |
| S202D | 11.3 (7.6, 20.9) | 107 ± 8 | 2.48 ± 0.28 | 92 ± 8 | 60.7 ± 3.4 | 95 ± 2 | 86 ± 2b |
| T204A | 12.6 (9.4, 18.9) | 119 ± 7b | 3.67 ± 0.18 | 98 ± 11 | 18.8 ± 0.5 | 91 ± 3 | 89 ± 2b |
| T204D | 6.08 (4.1, 11.2)a | 101 ± 5 | 0.11 ± 0.02 | 69 ± 12 | 39.4 ± 0.2 | 87 ± 1 | 80 ± 2 |
| S208A | 12.7 (10.7, 15.6) | 105 ± 3 | 3.80 ± 0.15 | 93 ± 9 | 22.6 ± 1.5 | 82 ± 6 | 78 ± 2 |
| S208D | 11.3 (9.5, 13.9) | 95 ± 3 | 2.52 ± 0.06 | 96 ± 10 | 56.7 ± 0.4 | 95 ± 1 | 88 ± 2b |
| T209A | 12.1 (7.9, 24.8) | 109 ± 9 | 3.17 ± 0.05 | 101 ± 4 | 32.4 ± 1.5 | 86 ± 4 | 80 ± 2 |
| T209D | 11.9 (9.1, 17.0) | 89 ± 4a | 3.00 ± 0.23 | 87 ± 4 | 20.1 ± 0.4 | 88 ± 2 | 85 ± 1b |
| S280A | 13.3 (10.8, 17.4) | 96 ± 4 | 4.40 ± 0.07 | 70 ± 9a | 50.8 ± 1.0 | 90 ± 2 | 81 ± 2 |
| S280D | 10.4 (9.5, 11.4)a | 69 ± 1c | 1.83 ± 0.02b | 90 ± 10 | <0.1b | 97 ± 1b | ∼100c |
| S294A | 11.5 (8.1, 19.2) | 100 ± 6 | 2.67 ± 0.09 | 75 ± 9 | 36.4 ± 0.5 | 84 ± 2 | 76 ± 2 |
| S294D | 9.1 (5.3, 31.6) | 72 ± 7b | 1.04 ± 0.08a | 76 ± 13 | 49.0 ± 0.5 | 91 ± 1 | 81 ± 1 |
| QQQQ | N/D | 85 ± 3£ | N/D | 33 ± 2£ | 80.5 ± 2.9b | 74 ± 3 | 46 ± 10c |
For cAMP experiments, receptors were transiently transfected in 293/FRT/MRAPβ-cells. Cells were stimulated with 100 nm ACTH in the presence of IBMX for up to 60 min. For internalization experiments, receptors were stably expressed as recombinant 293/FRT/Myc-mutant cell lines and transiently transfected with MRAPβ before stimulation with 100 nm ACTH for up to 60 min. Results are shown as mean ± sem from three to five experiments. &, Determined by nonlinear regression. Data with £ were determined after 15 min of ACTH stimulation in a concentration-response experiment. QQQQ internalization parameters were measured in the same set of experiments as T143A, T143D, and T143S and in parallel with WT. sem assigned with # for WT were estimated based on the means from each experimental group, but statistical significances are based solely on the comparison with WT from a particular experimental group of mutants. The experimental groups are illustrated in Figs. 8 and 9. N/A, Not available; N/D, not determined; R60, cAMP, rate of cAMP accumulation at 60 min; R5, surface, rate of internalization at 5 min.
P < 0.05.
P < 0.01.
P < 0.001.
Because MC2R N-glycosylation is important for cell-surface expression of MC2R (31), proteins contained in cell extracts from 293/FRT/MRAPβ cells transiently transfected with the mutant MC2R were analyzed by reducing SDS-PAGE and Western blotting to verify if the mutants were N-glycosylated. As compared with WT and to the unglycosylated MC2R mutant (QQQQ) (31), all mutants were fully N-glycosylated and, hence, present in native (25 kDa), core-glycosylated (33 kDa), and terminally glycosylated (50–65 kDa) forms (Fig. 8C) (31). Thus, the absence or low level of cell-surface expression for T143/A/D/S/G/K, T147D, and S280A was not due to a lack of N-glycosylation.
Importance of S/T residues for MC2R cell-surface expression, internalization, and cAMP accumulation
Individual cell lines stably expressing MC2R mutants, including the QQQQ cell line (but not the T143G and T143K cell lines), were transfected with MRAPβ. The various cell lines were incubated with 100 nm ACTH for up to 60 min. Cell-surface Myc-MC2R was then measured as % of unstimulated cells (Fig. 9). Remaining cell-surface receptors after 5 min (RSR5) and RSR60, as well as the rate of internalization at 5 min (R5, surface) were calculated (Table 1). For cAMP, the percentages of 100 nm ACTH-induced cAMP accumulations over a 60-min period (Fig. 10) were transformed into time derivatives, whereas t½ (rate of cAMP accumulation at 60 min) and steady state of cAMP level [Rmax, cAMP (% of WT)] were used to characterize desensitization.
Fig. 9.

Role of intracellular S/T residues in MC2R cell-surface expression. A–F, Individual 293/FRT/Myc-mutant cell lines transiently transfected with MRAPβ were submitted to a time-course challenge with 100 nm ACTH, then processed for determination of cell-surface Myc-tagged receptors by ELISA. Studies were conducted in groups as shown in graphs and always in parallel with WT-MC2R. In C, T147D was not shown, because T147D was not expressed at the cell-surface. Results are expressed as mean ± sem (n = 3–5).
Fig. 10.

Importance of intracellular S/T residues in ACTH-induced cAMP accumulation. A–F, 293/FRT/MRAPβ cells were transfected separately with MC2R mutants and were submitted to a time-course challenge with 100 nm ACTH in the presence of 1 mm IBMX and were processed for cAMP measurements (n = 3). Studies were conducted as shown in graphs and always in parallel with WT MC2R. Results are expressed as mean ± sem.
To summarize the results illustrated in Figs. 9 and 10 and the properties of the various MC2R mutants (Table 1), three dimensional correlation analyses were performed between steady state of cAMP level (Rmax, cAMP), initial level of normalized cell-surface expression with MRAPβ (Table 1) and R5, surface. Clustering of the data was performed, and the depicted three-dimensional representations revealed four delimited clusters (Fig. 11A). Histogram representations of the three parameters are illustrated for each cluster in Fig. 11B. In the first cluster (Fig. 11, diamond), which included the WT-MC2R, both maximal cAMP accumulation after 60 min (Rmax, cAMP) and normalized initial surface expression were similar to WT (S140A, S202D, T204A, S208A, S208D, S209A, and T209D), except for S140D, T147A, and S202A (5, 9, and 11, in clusters; Fig. 11), which exhibited a higher initial cell-surface expression that the WT. The second cluster (Fig. 11, squares) included T143D, T143S, T204D, S280A, S294A, and S294D, for which the Rmax, cAMP was higher than expected from the initial cell-surface expression. This observation was particularly evident for T143D and T143S, which exhibited a low level of cell-surface expression. However, even if classified in this cluster, S294D had a profile somewhat similar to members of cluster 1. The third cluster (Fig. 11, stars) contained three members (T131A, T131D, and S280D) characterized by a Rmax, cAMP lower than expected from the initial cell-surface expression and by an absence of internalization (thus opposite to cluster 2). In this cluster, T131D had a low cAMP accumulation, however, in spite of a cell-surface expression higher than WT. Cluster 4 (Fig. 11, circles) contained two members, T143A and T147D, which were in fact nearly (T143A) or totally (T147D) nonfunctional, characterized by the near absence of cell-surface expression, with absence (T147D) or very low (T143A) cAMP generation upon ACTH stimulation. The R5, surface of QQQQ was much faster than WT, although it took at least 60 min to achieve its maximal internalization as opposed to roughly 30 min for WT (Table 1). In general, the faster the R5, surface, the slower the maximal internalization at 60 min.
Fig. 11.
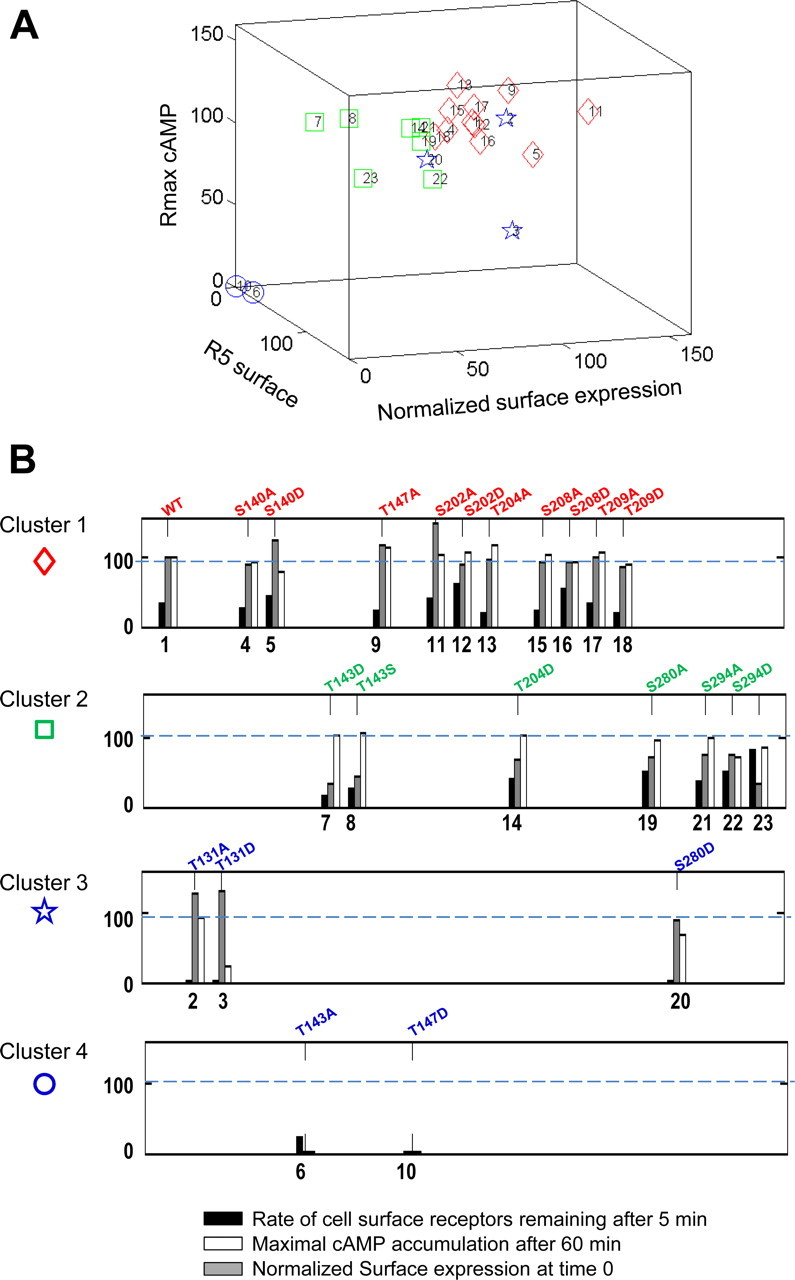
Clustering analysis of mutant MC2R functionality. Three parameters were considered: normalized cell-surface expression with MRAP1 at time 0 (gray column), rate of cell-surface receptors remaining after 5 min (R5, surface % of WT) (black column), and maximal cAMP accumulation after 60 min (Rmax cAMP, % of WT-MC2R) (white column). A, Three-dimensional representation of the four clusters, as identified from the k-means analysis, as detailed in Materials and Methods. B, Histogram representation of the four clusters. Comparative levels of MC2R cell-surface expression (gray), rate of cell-surface receptors remaining after 5 min (black), and maximal cAMP accumulation after 60 min (R60, cAMP) (white). Mutant MC2R, with corresponding numbers assigned on the graphs: WT-MC2R (1), T131A (2), T131D (3), S140A (4), S140D (5), T143A (6),T143D (7), T143S (8), T147A (9), T147D (10), S202A (11), S202D (12), T204A (13), T204D (14), S208A (15), S208D (16), T209A (17), T209D (18), S280A (19) S280D (20) S294A (21), S294D (22), and QQQQ mutant (23).
To further understand the behavior of the T143A/D/S mutants, ACTH responsiveness of these mutants was analyzed to specifically determine their ACTH potency (EC50). In stable MRAPα-expressing cells transiently transfected with the mutant receptors, EC50 values increased (thus ACTH potency decreased) from 60 (50, 72) pm for WT-MC2R to 150 (114, 199) pm for T143D (P < 0.001) and 96 (74, 123) pm for T143S (P < 0.01) (Fig. 12A). In MRAPβ-expressing cells, EC50 values increased from 315 (263, 377) pm for WT to 509 (409, 633) pm for T143D (P < 0.01) and 415 (333, 518) pm for T143S (P < 0.05) (Fig. 12B). As previously observed with QQQQ (31), even though T143D and T143S (cluster 2) displayed a reduced cell-surface expression (<50% of the WT), ACTH-induced cAMP accumulation remained virtually intact, indicating that a small fraction of cell-surface receptors is sufficient for achieving maximal efficacy, as previously observed in both rat and human cells (10, 12, 29, 30).
Fig. 12.
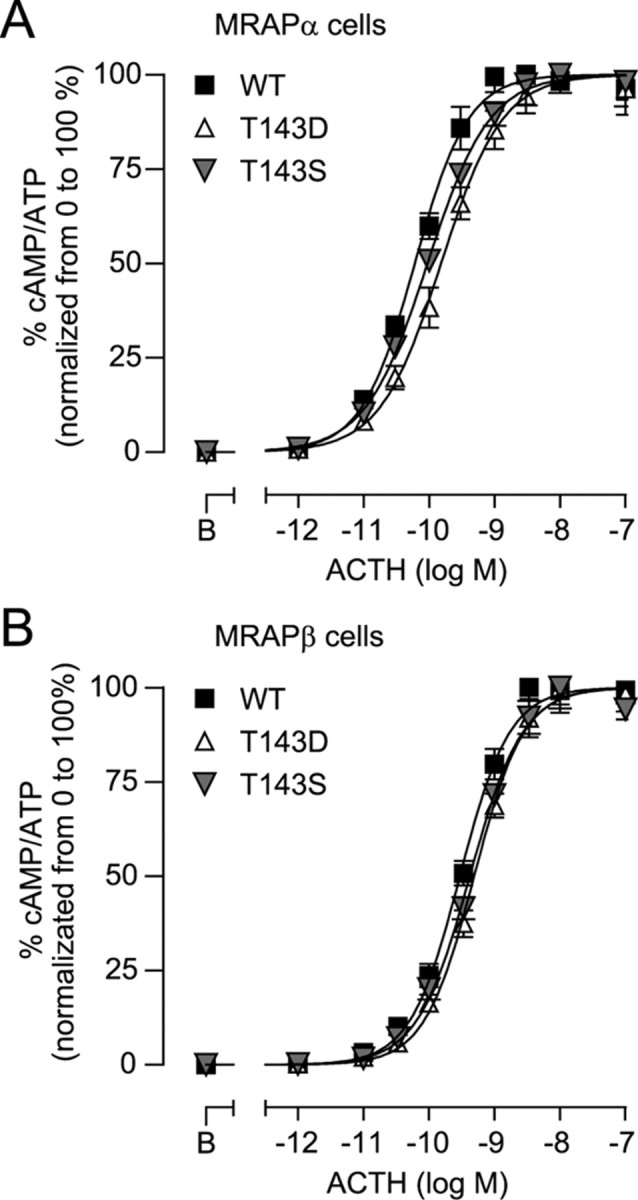
Functionality of T143 mutants. A and B, 293/FRT/MRAPα-Flag (A) or 293/FRT/MRAPβ-Flag (B) cells, transiently transfected with WT, T143D, or T143S, were washed and incubated with varying ACTH concentrations in the presence of IBMX for 15 min. The data obtained was normalized from 0 to 100% (basal to Rmax) for each transfection to visualize EC50 values. Curves were fitted with a log agonist vs. response nonlinear regression. Results are expressed as mean ± sem of three separate experiments, each performed in triplicate.
Discussion
Results here indicate that ACTH-induced MC2R internalization is concentration-dependent and accompanied by the concomitant trafficking of both MC2R and either MRAP1 isoform. Out of the population of internalized receptors, about one-third is recycled back to the plasma membrane to promote sustained cAMP signaling. Moreover, the substitution of five intracellular S/T residues of MC2R (T131, T143, T147, and S280) into either A or D had major repercussions on cell-surface expression, cAMP accumulation, and/or internalization parameters, pointing mostly to the second intracellular loop as being crucial for MC2R expression and functional regulation.
MC2R internalization and recycling in HEK293-transfected cells
The observed maximal extent of ACTH-induced MC2R internalization over a 60-min period, which varied from 20 to 30% depending on experiments and expression strategies, is consistent with a previous study performed in Y1 cells (16). This decrease in cell-surface expression of MC2R after ACTH exposure was found to be dependent on ACTH binding to MC2R, because increases in ACTH-independent cAMP levels with FSK or IBMX failed to alter MC2R internalization. Moreover, low ACTH concentrations did not induce MC2R internalization. Our results moreover indicate that internalization of MC2R is arrestin-dependent and suggest a greater affinity of MC2R for Arr3 than for Arr2. Furthermore, MC2R was immunoprecipitated with Arr3 both with and without ACTH stimulation. This is in keeping with the observations obtained by confocal microscopy, whereby both MC2R and Arr3 could be observed at the plasma membrane without any ACTH stimulation. This pattern of colocalization is similar to that observed for β2AR or for the chemoattractant receptor-homologous molecule expressed on Th2 cells (43). However, MC2R internalization was modest in comparison with β2AR expressed under the same conditions. ACTH-induced MC2R internalization was also dynamin-dependent, thus suggesting that endocytosis requires the scission of vesicles from the plasma membrane (44). Preincubation of cells in hypertonic sucrose (45) also confirmed that clathrin lattices are essential for MC2R internalization. Our results thus indicate that MC2R internalization in HEK293 cells is arrestin- and clathrin-dependent, as previously described in other MC-transfected cell types (16, 18, 46).
The observation by confocal microscopy that MC2R and MRAP isoforms cointernalized and trafficked conjointly in intracellular vesicles represents an important finding of this study. MRAP isoforms are essential for achieving MC2R functionality in most, if not all, cell types tested to date (6, 9, 12, 47). Thus, it could be hypothesized that if MRAP were not recycled with MC2R, one would not expect recycled MC2R to be ACTH responsive, and hence, such recycling would be irrelevant for ACTH signaling. Our results indicate that internalized MC2R traffics through Rab4-, Rab11-, and to a lesser extent Rab-recycling endosomes but much less markedly through Rab7-positive endosomes (late endosomes and lysosomes). The observation that BFA-mediated inhibition was 80% as efficient as the inhibition mediated by monensin suggests that a large proportion of internalized MC2R are likely recycled after trafficking into the Golgi apparatus. Moreover, at ACTH concentrations lower than 1 nm, MC2R did not internalize despite the presence of recycling inhibitors, indicating a concentration-dependent effect of ACTH on MC2R internalization. Importantly, only ACTH, but not FSK action, was sensitive to recycling inhibitors, clearly indicating that ACTH, but not cAMP itself, induced MC2R internalization. In addition, the observation that FSK, alone or in the presence of BFA or monensin, enhanced MC2R cell-surface expression is coherent with our previous results in primary cultures of adrenocortical cells (from both rat and human origin), in which PKA enhanced ACTH-induced cAMP production and accumulation (48, 49).
Importance of MC2R intracellular S/T residues
As recently reviewed by Dores (15) and by Fridmanis et al. (50), MC2R differs from the other MCR by its high level of expression and ligand binding properties. In spite of the significant progress achieved in the past 6 yr, there is still limited information as to the molecular mechanisms linking MC2R structure with its functional properties. As previously observed with QQQQ (31), it is interesting to observe that, even though T143D and T143S displayed a reduced cell-surface expression (<50% of the WT), ACTH-induced cAMP accumulation remained nearly intact, indicating that a small fraction of cell-surface receptors is sufficient for achieving maximal efficacy, as previously observed in both rat and human cells (10, 29–31).
T131A was found to be resistant to internalization and to desensitization. In addition, for T131D and S280D (cluster 3), there was a discrepancy between their lack of internalization (RSR60), in comparison with their reduced maximal ACTH-induced cAMP accumulations (R60, cAMP), which could not be attributed to lower cell-surface expression. These results may indicate that the mutation of the S/T into D has a major impact on MC2R structure, which may modify, for example, the affinity for ACTH. These mutations may also interfere with the nearby DRY motif (127-DRYIT-131) and create a major structural change in the receptor (51, 52). A S/T residue located in the carboxy terminus of the second intracellular loop is often conserved among GPCR (53), including all MCR. This conserved S/T residue may serve for G protein coupling or for proper folding of the receptor (53–55). For instance, similarly to MC2R-T143A, the MC1R-T157A mutant abolishes NDP-MSH binding and signaling to undetectable levels (41). Interestingly, the loss-of-function mutation MC2R-R146H located near MC2R-T143, identified in patients suffering from familial glucocorticoid deficiency (56), is linked to inefficient cell-surface expression and signaling of MC2R (6, 57). Similarly, MC1R-R160W is associated with MC1R loss-of-function, endoplasmic reticulum retention, and red hair in humans.
The T143A mutation, as well as T147D, provides evidence that a single amino acid can shut down receptor export and function almost completely, even if these receptors are well expressed intracellularly and N-glycosylated. According to the substitution analysis of T143 into A/D/S/G/K conducted here, an amino acid with a hydroxyl group or a negative charge at position 143 therefore appears essential for plasma membrane expression and normal function of MC2R in the presence of MRAPα or MRAPβ and likely more so than positively charged (K), lateral chain-deleted (G), or hydrophobic (A) substitutions. According to recent studies, MRAP2 may also take place at the cell-surface level, although it only has a very marginal role in cAMP production, even when overexpressed (31, 58, 59). Because 293/FRT cells endogenously express MRAP2 (31, 60), it is therefore plausible that, in basal conditions, up to 50% of WT-MC2R cell-surface receptors are under the control of MRAP2 (31, 32). In contrast to WT-MC2R, T143S and T143D (cluster 2) appeared to rely almost exclusively on MRAP1 expression for their export to the plasma membrane, increasing the ratio of functional (MRAP1-dependent) to nonfunctional (MRAP1-independent) receptors at the plasma membrane. The mutations of S140A, S202A, and S294A, as compared with their counterparts S140D, S202D, and S294D, also led to alterations in the rate of cell-surface receptors remaining after 5 min (R5, surface). The mutation of the remaining intracellular S/T residues (S202, T204, S208, and T209) (also in cluster 1) were considered of less importance for MC2R regulation, because the experimental data recorded with the A/D mutants were either less evident or without effect.
In a recent study, Fridmanis et al. (50) implemented the construction of a series of chimeric MC2R/MC4R to identify the regions of MC2R responsible for cell-surface expression and binding specificity. Results from the direct investigation of S/T residues in the present study are in agreement with their observations in that they found that the N terminus, as well as the third and fourth transmembrane domains, is involved in intracellular retention of MC2R. Indeed, MC2R N-glycosylation in its N terminus is important for its cell-surface expression (31) and internalization. Moreover, T143 and T147, located in the C-terminal segment of the second intracellular loop (in-between the third and fourth transmembrane domains), were largely deficient mutants due to intracellular retention.
In conclusion, this study provides evidence that after ACTH binding, MC2R is subjected to internalization, trafficking, and recycling through Rab4, Rab5, and Rab11 endosomes. In addition, analyses of MC2R cell-surface expression, cAMP production, and internalization performed with A/D-scanned intracellular S/T residues allowed to delineate key amino acids that are crucial determinants of MC2R cell-surface expression and function. Results here provide new and detailed insight into the mechanisms involved in relatively short-term regulation of MC2R activity and reveal some hallmarks of the human ACTH receptor.
Materials and Methods
Materials
The chemicals used in the present study were obtained from the following sources: fetal bovine serum, the Flp recombinase-mediated homologous recombination system (Flp-In), Lipofectamine, and PLUS Reagent were from Invitrogen Corp. (Burlington, Ontario, Canada); high glucose DMEM was from Wisent (Saint-Jean-Baptiste, Québec, Canada). PhosSTOP phosphatase inhibitor cocktail was from Roche (Laval, Québec, Canada). NuPAGE sample buffer was from Invitrogen Corp. (Carlsbad, CA) and reducing agent from Fermentas Canada (Burlington, Ontario, Canada). Phusion DNA polymerase and restriction enzymes were purchased from New England Biolabs (Ipswich, MA). The HaloTag technology was from Promega (San Luis Obispo, CA). Dynamin-K44A in pcDNA3 vector was a kind gift from Stephen Ferguson (Department of Physiology and Pharmacology, University of Western Ontario, London, Ontario, Canada). The mouse monoclonal anti-Myc-producing hybridoma clone 9E10 and the Myc-β2AR cDNA supplied in pcDNA3 were obtained from Michel Bouvier (Groupe de Recherche Universitaire sur le Médicament, Université de Montréal, Montréal, Québec, Canada). The pcDNA3/AT1R-Flag vector was from Richard Leduc (Département de Pharmacologie, Université de Sherbrooke). Rab4-, Rab5-, Rab7-, Rab11-, Arr2-, Arr3-GFP (in pEGFP vector), Arr2-HA, Arr2 (319-418) (Arr2-DN), Arr3-HA, and Arr3 (201-409) (Arr3-DN) were from J.-L. Parent (Service de Rhumatologie, Université de Sherbrooke). ACTH-(1-24) peptide (Cortrosyn) was purchased from Organon (Toronto, Canada). Ang II was from Bachem (Marina Delphen, CA). Alexa Fluor coupled secondary antibodies were from Invitrogen Corp. The enhanced chemiluminescence detection system was from GE Healthcare (Baie d'Urfe, Québec, Canada). FSK, ACTH-(1-24), IBMX, isoproterenol, monensin, and BFA were from Sigma-Aldrich (Oakville, Ontario, Canada). All other chemicals were of A-grade purity.
Expression vectors and site-directed mutagenesis
Myc-MC2R and MRAP-Flag both contained in the pcDNA5/FRT vector constructions that have been described previously (12). Myc-MC2R/MRAPα-Flag and Myc-MC2R/MRAPβ-Flag indicate that both MC2R and MRAP1 isoform are encoded in the same pcDNA5/FRT expression vector. To generate the pcDNA5/FRT/Halo-Myc-MC2R/MRAPβ-Flag expression vector, primers (forward, tcagtctcgagccaccatgggatccgaaatcggtacagc and reverse, gagagtactcgaggccggccagcccggggagccagc) were used to amplify the HaloTag cDNA by PCR from the pHT2 vector and to introduce XhoI restriction sites at the 5′ and 3′ ends of the PCR product using Phusion DNA polymerase. XhoI restriction was used to adjoin the HaloTag cDNA 5′ upstream and in frame with the Myc-MC2R cDNA in pcDNA3. Thereafter, the Halo-Myc-MC2R cDNA fusion was subcloned into pcDNA5/FRT, and the MRAPβ-Flag cistron was introduced using SphI to generate a bicistronic vector. The resulting expression vector was used to generate a stable cell line by Flp recombinase-mediated homologous recombination, as described previously (12). Halo-Myc-MC2R is a 606-amino acid fusion protein.
For MC2R site-directed mutagenesis, individual S/T → A/D mutants (T131A, T131D, S140A, S140D, T147A T147D, S202A, S202D, T204A, T204D, S208A, S208D, T209A, T209D, S280A, S280D, S294A, and S294D) and T143 → A/D/S/G/K mutants of MC2R were generated by PCR according to the Pfu turbo method (Stratagene, La Jolla, CA) using complementary primers barring the desired codons to replace the original S or T codons. To avoid any unwanted mutation in the template vector, mutagenesis was performed on the pcDNA3/Myc-MC2R template, after which the coding sequences were then subcloned in pcDNA5/FRT as described previously. Plasmids were sequenced at the Nanuq sequencing platform (Génome Québec, Université McGill, Montréal, Québec, Canada). All vector maps, cloning steps, restriction, and sequencing analyses were simulated and designed using Vector NTI Advance 10.3.0 (Invitrogen Corp.), for which appropriate files are available on request, as well as primers used.
Cell culture, transfection, and isogenic cell lines
The Flp recombinase-mediated homologous recombination system (Flp-In) was used to generate HEK 293/FRT (Flp Recombinase Target site) cell lines stably expressing both Myc-MC2R and MRAPα-Flag or MRAPβ-Flag as described previously. Unless stated otherwise, all experiments were performed with 293/FRT/Myc-MC2R/MRAPβ-Flag cells (se Results) or with 293/FRT/Myc-MC2R/MRAPα-Flag (Supplemental data). Cells were cultured in high-glucose DMEM with 7% fetal bovine serum, 2 mm GlutaMAX, and 100 μg/ml Zeocin. The establishment of other cell lines (i.e. stable MC2R mutant cell lines) was also performed with Flp-In recombination. When required, cells were transfected with 0.5 μg/35 mm dish or 0.125 μg/well (24-well plates) at 50% confluency using Lipofectamine and PLUS reagent. To maintain similar transcriptional and translational activity when required, transfection of pEGFP, or of an empty vector or of the untagged version of MC2R, was used as control when appropriate.
Time-course studies
Cells were washed once in Hanks' buffered saline (HBS) [130 mm NaCl, 3.5 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 2.5 mm NaHCO2, 5 mm HEPES (pH 7.4), containing 1 g/l dextrose] and subsequently stimulated with ACTH at concentrations ranging from 1 pm to 100 nm for 0, 5, 15, 30, 45, or 60 min, in the presence or absence of the phosphodiesterase inhibitor IBMX, at 37 C in HBS containing 0.1% BSA. At the end of the stimulations, cells were processed for cAMP measurements or whole-cell ELISA procedures. To investigate Myc-MC2R recycling, cells were preincubated for 15 min with or without the recycling inhibitor monensin (25 μm) (39, 40, 61) or the Golgi cisternae-disrupting agent BFA (1 μg/ml) (38) before ACTH stimulation. In experiments requiring transfection, such as Rab, arrestins, MC2R mutants, or MRAP, cells were transfected 24 h before the experiments.
cAMP measurements
Intracellular cAMP accumulation was performed in cells cultured in 35-mm dishes. Cells were loaded with tritiated adenine for 2 h, washed once in HBS, and subsequently stimulated with ACTH, in the presence or absence of the phosphodiesterase inhibitor IBMX, at 37 C in HBS containing 0.1% BSA. At the end of the stimulations, cells were lysed in 5% trichloroacetic acid, and intracellular cAMP accumulation was determined by measuring the conversion of [3H]-ATP into [3H]-cAMP eluted from Dowex and neutral alumina chromatography columns as described previously (12).
Measurements of cell-surface expression of MC2R by whole-cell ELISA procedures
The amount of cell-surface Myc-MC2R was measured in cells cultured in 24-well plates by detecting the extracellular Myc-tag fused to MC2R. Anti-Myc antibodies were applied after formaldehyde fixation (without cell membrane permeabilization). Alternatively, whole-cell Myc-tagged receptor expression was measured after MeOH fixation as described previously (31). Horseradish peroxidase-linked antibodies with 0.003% H2O2 and 1 μg/ml o-phenylenediamine were used to reveal receptor density as described previously (12). Nonspecific background levels were established with native 293/FRT cells (transiently transfected with MRAPβ). For internalization measurements, basal levels were considered as 100%. Loss in cell-surface receptor reflects receptor internalization (100% − receptor loss %). The maximal loss in cell-surface Myc-MC2R (MC2R internalization) was calculated as follows. Initial % of loss of cell-surface MC2R/loss of cell-surface in the presence of BFA or monensin × 100. Net internalization refers to MC2R internalization in the presence of monensin or BFA. Raw internalization refers to MC2R internalization in the absence of these reagents. The percentage of recycled receptors was calculated as follows: (net internalization − raw internalization)/net internalization. Because total expression levels varied for each mutant expressed in individual cell lines, cell-surface expression data were normalized to total expression levels (Fig. 7A and Supplemental Table 1). The advantage of using such an approach is that differences in the ability of each mutant to be expressed at the plasma membrane are more accurate than raw expression results, because total expression is taken into account (31). Raw total expression and raw cell-surface expression were measured (Supplemental Fig. 10, A–D, and summarized in Supplemental Table 1).
Immunofluorescence microscopy
Cells were seeded on poly-l-lysine glass coverslips placed in 35-mm dishes, transfected the next day with the indicated plasmids, and starved for 30 min with fresh DMEM 1 d later before the experiments. Thereafter, cells were stimulated with 100 nm ACTH for 0, 5, 15, 30, 45, or 60 min. After a single wash in cold HBS followed by fixation in MeOH, labeling for Myc-MC2R and MRAP-Flag was performed as described previously (12). Anti-Myc and anti-Flag primary antibodies were detected with goat antimouse or antirabbit secondary antibodies coupled to Alexa Fluor 647 and Alexa Fluor 568, respectively, whereas 4′,6-diamidino-2-phenylindole was used to stain the nuclei. In other instances, clathrin and caveolin-1 were detected with polyclonal antibodies against the endogenous proteins expressed in 293/FRT cells, followed by suitable secondary antibodies. Images were acquired on an Olympus Fluoview 1000 (FV1000) laser-scanning confocal microscope (Olympus, Tokyo, Japan) built around an IX81-ZDC inverted microscope fitted with a U Plan S-Apo 60× (1.35 NA) oil immersion objective (Olympus URFL-T, MAG Biosystems; Photometrics, Tucson, AZ). Emissions from each fluorophore were acquired sequentially to avoid fluorophore bleeding during acquisitions. All images were magnified two to three times with Photoshop CS3 (Adobe Systems, Inc., San Jose, CA). White color corresponds to color superposition of green, red, and blue colors, purple is the overlap between red and blue colors, and yellow is the overlap between red and green colors. The pixel fluorograms were obtained and analyzed according to the Pearson's correlation coefficient (62) by plotting pixel values of each component relative to the horizontal and vertical axes, respectively. Quadrant markers were placed forming background (lower left), red only (upper left), green only (lower right), and colocalizing (upper right) areas. The Pearson's correlation coefficient describes the correlation of the intensity distribution between channels, with a scale from 0 to 1, with 0 indicating no significant correlation, and 1.0 indicating complete correlation (62).
For live-cell imaging, stable 293/FRT/Halo-Myc-MC2R/MRAPβ-Flag cells were seeded in poly-l-lysine-coated MatTek 35-mm dishes containing a central glass plate 48 h before the experiments. Cells were washed with serum-free DMEM and incubated with HaloTag-Alexa Fluor 488 cell-impermeable ligand for 15 min at 37 C in a CO2 incubator. Subsequently, cells were washed thoroughly to label only cell-surface HaloTag enzyme fused at the extracellular N terminus of MC2R. Each dish was then mounted onto the confocal microscope, and cells were examined every 30 sec in the presence or absence of 100 nm ACTH or 1 μm NDP-MSH, added as 10× concentrates. In other experiments, cells were transiently transfected with dominant negative pcDNA3/Dynamin-K44A 24 h before the experiment. Confocal images were acquired with a U Plan S-Apo 60× oil objective using a CSU-XI confocal scanner unit (Yokogawa Electric, Sugar Land, TX), mounted onto an Olympus IX81-ZDC inverted microscope (Olympus) using a QuantEM:512SC EMCCD camera (Photometrics). Temperature was maintained at 37 C with a microincubator model TB-3 CCD (Warner Instruments, Hamden, CT) throughout the experiments. All acquisitions were controlled by MetaMorph 7.5 software (Molecular Devices, Downingtown, PA). Laser excitation was performed by a FRAP-3D laser launch from MAG Biosystems (Photometrics).
Immunoprecipitation and Western blotting
Immunoprecipitation was performed as described previously (61). Briefly, native 293/FRT cells transiently transfected with pcDNA5/FRT/Myc-MC2R/MRAPβ-Flag and Arr3-HA were stimulated or not with 100 nm ACTH for 30 min. Cell lysates were clarified and incubated with 2 μg of anti-HA monoclonal antibodies to immunoprecipitate arrestins, after which protein A-agarose was added, followed by an overnight incubation at 4 C. Initial lysates and immunoprecipitated proteins were resolved by 10% SDS-PAGE gels, and immunoblotting was performed using anti-HA or anti-Myc antibodies on nitrocellulose membranes (31).
Data analysis
Unless stated otherwise, results are presented as mean ± sem from at least three independent experiments, each conducted in duplicate or triplicate. GraphPad Prism 5.0 (GraphPad Software, San Diego, CA) was used for establishing all nonlinear regressions for the determination of EC50, halftimes (t½), maximal effects, sem, and 95% confidence intervals [given as mean (lower limit, higher limit)]. P values for nonlinear regression parameters (Rmax, t½, and EC50) were obtained with the extra sum-of-squares F test. Statistical analyses of other data presented in histograms were performed by ANOVA followed by Bonferoni's post hoc test. Time derivatives of cAMP accumulation curves were calculated using: rate (% cAMP/ATP × min−1) = k × (Rmax − Y0) × (exp(−kt)), with k and Rmax as variables. To eliminate Rmax as variables, these rates were also normalized to Rmax to establish the initial rates in each condition as 100%.
Clustering of the observations was performed with the k-means subroutine of the MATLAB software (The MathWorks, Inc., Natick, MA). Clustering algorithms are methods to divide a set of n observations in m groups so that members of the same group are more alike than members of different groups. The k-means function partitions data into k mutually exclusive clusters and returns the index of the cluster to which it has assigned each observation. Each cluster in the partition is defined by its number object and its centroid. The centroid for each cluster is the point to which the sum of distances from all objects in that cluster is minimized. The algorithm moves objects between clusters until the sum of distances of each object to its cluster centroid cannot be decreased further.
Acknowledgments
We thank Lucie Chouinard as well as the other members of the laboratory for their technical assistance in performing cAMP assays; Dr. Roger D. Cone (Vollum Institute, Oregon Health and Science University, Portland, OR) for providing the human MC2R cDNA; Dr. Michel Bouvier (Canada Research Chair in Signal Transduction and Molecular Pharmacology, Groupe de Recherche Universitaire sur le Médicament, Université de Montréal, Montréal, Québec, Canada) for providing the anti-Myc antibody; Dr. Stephen Ferguson (Department of Physiology and Pharmacology, The University of Western Ontario, London, Ontario, Canada) for the gift of dynamin-K44A; Dr. Robert Dumaine and Dr. Philippe Sarret for use of their confocal microscope and Jean Lainé for help with confocal studies; Dr. Marcel D. Payet for his kind assistance with data analyses as well as critical review of the manuscript; and to Pierre Pothier for critical reading of the manuscript.
This work was supported by Canadian Institutes of Health Research Grants MOP-82819 (to N.G.-P.) and MOP-69085 (to J.-L.P.) and by the Canada Research Chairs Program. N.G.-P. is a recipient of a Canada Research Chair in Endocrinology of the Adrenal Gland; J.-L.P. is a recipient of a Chercheur-boursier senior scholarship from the Fonds de la Recherche en Santé du Québec. N.G.-P. and J.-L.P. are members of the Fonds de la Recherche en Santé du Québec-funded Centre de Recherche Clinique Étienne-le Bel. S.R. is a recipient of a studentship from the Fonds de la Recherche en Santé du Québec.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ang II
- Angiotensin II
- β2AR
- β2-adrenergic receptor
- Arr2
- arrestin2
- AT1R
- Ang II type 1 receptor
- BFA
- brefeldin A
- EGFP
- enhanced green fluorescent protein
- FRT
- Flp recombinase target
- FSK
- forskolin
- GPCR
- G protein-coupled receptor
- HA
- hemagglutinin
- HBS
- Hanks' buffered saline
- HEK
- human embryonic kidney
- IBMX
- 3-isobutyl-1-methylxanthine
- MCR
- melanocortin receptor
- MC1R
- melanocortin-1 receptor
- MC2R
- melanocortin-2 receptor
- MRAP
- MC2R accessory protein
- NDP-MSH
- [Nle4, D-Phe7]α-melanocyte-stimulating hormone
- PKA
- protein kinase A
- Rab
- Ras-like small guanosine triphosphate enzyme
- Rmax, cAMP
- maximal steady state cAMP accumulation
- RSR5
- remaining cell-surface receptors after 5 min
- S/T
- Ser and Thr
- WT
- wild type.
References
- 1. Sewer MB , Waterman MR. 2003. ACTH modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc Res Tech 61:300–307 [DOI] [PubMed] [Google Scholar]
- 2. Gallo-Payet N , Payet MD. 2003. Mechanism of action of ACTH: beyond cAMP. Microsc Res Tech 61:275–287 [DOI] [PubMed] [Google Scholar]
- 3. Forti FL , Dias MH , Armelin HA. 2006. ACTH receptor: ectopic expression, activity and signaling. Mol Cell Biochem 293:147–160 [DOI] [PubMed] [Google Scholar]
- 4. Mountjoy KG , Robbins LS , Mortrud MT , Cone RD. 1992. The cloning of a family of genes that encode the melanocortin receptors. Science 257:1248–1251 [DOI] [PubMed] [Google Scholar]
- 5. Chida D , Nakagawa S , Nagai S , Sagara H , Katsumata H , Imaki T , Suzuki H , Mitani F , Ogishima T , Shimizu C , Kotaki H , Kakuta S , Sudo K , Koike T , Kubo M , Iwakura Y. 2007. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc Natl Acad Sci USA 104:18205–18210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung TT , Webb TR , Chan LF , Cooray SN , Metherell LA , King PJ , Chapple JP , Clark AJ. 2008. The majority of adrenocorticotropin receptor (melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab 93:4948–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clark AJ , Chan LF , Chung TT , Metherell LA. 2009. The genetics of familial glucocorticoid deficiency. Best Pract Res Clin Endocrinol Metab 23:159–165 [DOI] [PubMed] [Google Scholar]
- 8. Clark AJ , McLoughlin L , Grossman A. 1993. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet 341:461–462 [DOI] [PubMed] [Google Scholar]
- 9. Metherell LA , Chapple JP , Cooray S , David A , Becker C , Rüschendorf F , Naville D , Begeot M , Khoo B , Nürnberg P , Huebner A , Cheetham ME , Clark AJ. 2005. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet 37:166–170 [DOI] [PubMed] [Google Scholar]
- 10. Lefkowitz RJ , Roth J , Pricer W , Pastan I. 1970. ACTH receptors in the adrenal: specific binding of ACTH-125I and its relation to adenyl cyclase. Proc Natl Acad Sci USA 65:745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Penhoat A , Naville D , Begeot M. 2001. The adrenocorticotrophic hormone receptor. Current Opinion Endocrinol Diabetes 8:112–117 [Google Scholar]
- 12. Roy S , Rached M , Gallo-Payet N. 2007. Differential regulation of the human adrenocorticotropin receptor [melanocortin-2 receptor (MC2R)] by human MC2R accessory protein isoforms α and β in isogenic human embryonic kidney 293 cells. Mol Endocrinol 21:1656–1669 [DOI] [PubMed] [Google Scholar]
- 13. Cooray SN , Clark AJ. 2010. Melanocortin receptors and their accessory proteins. Mol Cell Endocrinol 331:215–221 [DOI] [PubMed] [Google Scholar]
- 14. Hinkle PM , Sebag JA. 2009. Structure and function of the melanocortin2 receptor accessory protein (MRAP). Mol Cell Endocrinol 300:25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dores RM. 2009. Adrenocorticotropic hormone, melanocyte-stimulating hormone, and the melanocortin receptors: revisiting the work of Robert Schwyzer: a thirty-year retrospective. Ann NY Acad Sci 1163:93–100 [DOI] [PubMed] [Google Scholar]
- 16. Baig AH , Swords FM , Szaszák M , King PJ , Hunyady L , Clark AJ. 2002. Agonist activated adrenocorticotropin receptor internalizes via a clathrin-mediated G protein receptor kinase dependent mechanism. Endocr Res 28:281–289 [DOI] [PubMed] [Google Scholar]
- 17. Clark AJ , Baig AH , Noon L , Swords FM , Hunyady L , King PJ. 2003. Expression, desensitization, and internalization of the ACTH receptor (MC2R). Ann NY Acad Sci 994:111–117 [DOI] [PubMed] [Google Scholar]
- 18. Kilianova Z , Basora N , Kilian P , Payet MD , Gallo-Payet N. 2006. Human MC2R expression and functionality. Effect of PKA and PKC on desensitization and internalization. Endocrinology 147:2325–2337 [DOI] [PubMed] [Google Scholar]
- 19. Laporte SA , Oakley RH , Holt JA , Barak LS , Caron MG. 2000. The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β 2-adrenergic receptor into clathrin-coated pits. J Biol Chem 275:23120–23126 [DOI] [PubMed] [Google Scholar]
- 20. Oakley RH , Laporte SA , Holt JA , Barak LS , Caron MG. 2001. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J Biol Chem 276:19452–19460 [DOI] [PubMed] [Google Scholar]
- 21. Lefkowitz RJ , Shenoy SK. 2005. Transduction of receptor signals by β-arrestins. Science 308:512–517 [DOI] [PubMed] [Google Scholar]
- 22. Marchese A , Paing MM , Temple BR , Trejo J. 2008. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol 48:601–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gáborik Z , Hunyady L. 2004. Intracellular trafficking of hormone receptors. Trends Endocrinol Metab 15:286–293 [DOI] [PubMed] [Google Scholar]
- 24. Seachrist JL , Ferguson SS. 2003. Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci 74:225–235 [DOI] [PubMed] [Google Scholar]
- 25. Schwartz SL , Cao C , Pylypenko O , Rak A , Wandinger-Ness A. 2007. Rab GTPases at a glance. J Cell Sci 120:3905–3910 [DOI] [PubMed] [Google Scholar]
- 26. Noon LA , Franklin JM , King PJ , Goulding NJ , Hunyady L , Clark AJ. 2002. Failed export of the adrenocorticotrophin receptor from the endoplasmic reticulum in non-adrenal cells: evidence in support of a requirement for a specific adrenal accessory factor. J Endocrinol 174:17–25 [DOI] [PubMed] [Google Scholar]
- 27. Rached M , El Mourabit H , Buronfosse A , Blondet A , Naville D , Begeot M , Penhoat A. 2005. Expression of the human melanocortin-2 receptor in different eukaryotic cells. Peptides 26:1842–1847 [DOI] [PubMed] [Google Scholar]
- 28. Webb TR , Clark AJ. 2010. Minireview: the melanocortin 2 receptor accessory proteins. Mol Endocrinol 24:475–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gallo-Payet N , Payet MD. 1989. Excitation-secretion coupling: involvement of potassium channels in ACTH-stimulated rat adrenocortical cells. J Endocrinol 120:409–421 [DOI] [PubMed] [Google Scholar]
- 30. Gallo-Payet N , Grazzini E , Côté M , Chouinard L , Chorvátová A , Bilodeau L , Payet MD , Guillon G. 1996. Role of calcium in the mechanism of action of ACTH in human adrenocortical cells. J Clin Invest 98:460–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roy S , Perron B , Gallo-Payet N. 2010. Role of asparagine-linked glycosylation in cell surface expression and function of the human adrenocorticotropin receptor (melanocortin 2 receptor) in 293/FRT cells. Endocrinology 151:660–670 [DOI] [PubMed] [Google Scholar]
- 32. Sebag JA , Hinkle PM. 2009. Regions of melanocortin 2 (MC2) receptor accessory protein necessary for dual topology and MC2 receptor trafficking and signaling. J Biol Chem 284:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Orsini MJ , Benovic JL. 1998. Characterization of dominant negative arrestins that inhibit β2-adrenergic receptor internalization by distinct mechanisms. J Biol Chem 273:34616–34622 [DOI] [PubMed] [Google Scholar]
- 34. Mundell SJ , Orsini MJ , Benovic JL. 2002. Characterization of arrestin expression and function. Methods Enzymol 343:600–611 [DOI] [PubMed] [Google Scholar]
- 35. Takei K , Slepnev VI , De Camilli P. 2001. Interactions of dynamin and amphiphysin with liposomes. Methods Enzymol 329:478–486 [DOI] [PubMed] [Google Scholar]
- 36. Jean-Alphonse F , Hanyaloglu AC. 2011. Regulation of GPCR signal networks via membrane trafficking. Mol Cell Endocrinol 331:205–214 [DOI] [PubMed] [Google Scholar]
- 37. Donaldson JG , Finazzi D , Klausner RD. 1992. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 360:350–352 [DOI] [PubMed] [Google Scholar]
- 38. Dinter A , Berger EG. 1998. Golgi-disturbing agents. Histochem Cell Biol 109:571–590 [DOI] [PubMed] [Google Scholar]
- 39. Lichtshtein D , Dunlop K , Kaback HR , Blume AJ. 1979. Mechanism of monensin-induced hyperpolarization of neuroblastoma-glioma hybrid NG108–15. Proc Natl Acad Sci USA 76:2580–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stein BS , Bensch KG , Sussman HH. 1984. Complete inhibition of transferrin recycling by monensin in K562 cells. J Biol Chem 259:14762–14772 [PubMed] [Google Scholar]
- 41. Sánchez-Laorden BL , Jiménez-Cervantes C , García-Borrón JC. 2007. Regulation of human melanocortin 1 receptor signaling and trafficking by Thr-308 and Ser-316 and its alteration in variant alleles associated with red hair and skin cancer. J Biol Chem 282:3241–3451 [DOI] [PubMed] [Google Scholar]
- 42. Sánchez-Laorden BL , Herraiz C , Valencia JC , Hearing VJ , Jiménez-Cervantes C , García-Borrón JC. 2009. Aberrant trafficking of human melanocortin 1 receptor variants associated with red hair and skin cancer: steady-state retention of mutant forms in the proximal golgi. J Cell Physiol 220:640–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roy SJ , Parent A , Gallant MA , de Brum-Fernandes AJ , Stanková J , Parent JL. 2010. Characterization of C-terminal tail determinants involved in CRTH2 receptor trafficking: identification of a recycling motif. Eur J Pharmacol 630:10–18 [DOI] [PubMed] [Google Scholar]
- 44. Sever S , Damke H , Schmid SL. 2000. Dynamin:GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J Cell Biol 150:1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hansen SH , Sandvig K , van Deurs B. 1993. Clathrin and HA2 adaptors: effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol 121:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cai M , Varga EV , Stankova M , Mayorov A , Perry JW , Yamamura HI , Trivedi D , Hruby VJ. 2006. Cell signaling and trafficking of human melanocortin receptors in real time using two-photon fluorescence and confocal laser microscopy: differentiation of agonists and antagonists. Chem Biol Drug Des 68:183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sebag JA , Hinkle PM. 2007. Melanocortin-2 receptor accessory protein MRAP forms antiparallel homodimers. Proc Natl Acad Sci USA 104:20244–20249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Côté M , Guillon G , Payet MD , Gallo-Payet N. 2001. Expression and regulation of adenylyl cyclase isoforms in the human adrenal gland. J Clin Endocrinol Metab 86:4495–4503 [DOI] [PubMed] [Google Scholar]
- 49. Tremblay E , Payet MD , Gallo-Payet N. 1991. Effects of ACTH and angiotensin II on cytosolic calcium in cultured adrenal glomerulosa cells. Role of cAMP production in the ACTH effect. Cell Calcium 12:655–673 [DOI] [PubMed] [Google Scholar]
- 50. Fridmanis D , Petrovska R , Kalnina I , Slaidina M , Peculis R , Schiöth HB , Klovins J. 2010. Identification of domains responsible for specific membrane transport and ligand specificity of the ACTH receptor (MC2R). Mol Cell Endocrinol 321:175–183 [DOI] [PubMed] [Google Scholar]
- 51. Moore SA , Patel AS , Huang N , Lavin BC , Grammatopoulos TN , Andres RD , Weyhenmeyer JA. 2002. Effects of mutations in the highly conserved DRY motif on binding affinity, expression, and G-protein recruitment of the human angiotensin II type-2 receptor. Brain Res Mol Brain Res 109:161–167 [DOI] [PubMed] [Google Scholar]
- 52. Yamano Y , Kamon R , Yoshimizu T , Toda Y , Oshida Y , Chaki S , Yoshioka M , Morishima I. 2004. The role of the DRY motif of human MC4R for receptor activation. Biosci Biotechnol Biochem 68:1369–1371 [DOI] [PubMed] [Google Scholar]
- 53. Lembo PM , Ghahremani MH , Morris SJ , Albert PR. 1997. A conserved threonine residue in the second intracellular loop of the 5-hydroxytryptamine 1A receptor directs signaling specificity. Mol Pharmacol 52:164–171 [DOI] [PubMed] [Google Scholar]
- 54. Prado GN , Mierke DF , Pellegrini M , Taylor L , Polgar P. 1998. Motif mutation of bradykinin B2 receptor second intracellular loop and proximal C terminus is critical for signal transduction, internalization, and resensitization. J Biol Chem 273:33548–33555 [DOI] [PubMed] [Google Scholar]
- 55. Kushwaha N , Harwood SC , Wilson AM , Berger M , Tecott LH , Roth BL , Albert PR. 2006. Molecular determinants in the second intracellular loop of the 5-hydroxytryptamine-1A receptor for G-protein coupling. Mol Pharmacol 69:1518–1526 [DOI] [PubMed] [Google Scholar]
- 56. Slavotinek AM , Hurst JA , Dunger D , Wilkie AO. 1998. ACTH receptor mutation in a girl with familial glucocorticoid deficiency. Clin Genet 53:57–62 [DOI] [PubMed] [Google Scholar]
- 57. Elias LL , Huebner A , Pullinger GD , Mirtella A , Clark AJ. 1999. Functional characterization of naturally occurring mutations of the human adrenocorticotropin receptor: poor correlation of phenotype and genotype. J Clin Endocrinol Metab 84:2766–2770 [DOI] [PubMed] [Google Scholar]
- 58. Chan LF , Webb TR , Chung TT , Meimaridou E , Cooray SN , Guasti L , Chapple JP , Egertová M , Elphick MR , Cheetham ME , Metherell LA , Clark AJ. 2009. MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. Proc Natl Acad Sci USA 106:6146–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sebag JA , Hinkle PM. 2009. Opposite effects of the melanocortin-2 (MC2) receptor accessory protein MRAP on MC2 and MC5 receptor dimerization and trafficking. J Biol Chem 284:22641–22648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hwang GW , Oh SE , Takahashi T , Lee JY , Naganuma A. 2010. siRNA-mediated knockdown of the melanocortin 2 receptor accessory protein 2 (MRAP2) gene confers resistance to methylmercury on HEK293 cells. J Toxicol Sci 35:947–950 [DOI] [PubMed] [Google Scholar]
- 61. Parent A , Hamelin E , Germain P , Parent JL. 2009. Rab11 regulates the recycling of the β2-adrenergic receptor through a direct interaction. Biochem J 418:163–172 [DOI] [PubMed] [Google Scholar]
- 62. Zinchuk V , Zinchuk O , Okada T. 2007. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem 40:101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Clark AJ , Cammas FM. 1996. The ACTH receptor. Baillieres Clin Endocrinol Metab 10:29–47 [DOI] [PubMed] [Google Scholar]