

Abstract
The C-terminal regions of glucagon-like peptide-1 (GLP-1) bind to the N terminus of the GLP-1 receptor (GLP-1R), facilitating interaction of the ligand N terminus with the receptor transmembrane domain. In contrast, the agonist exendin-4 relies less on the transmembrane domain, and truncated antagonist analogs (e.g. exendin 9–39) may interact solely with the receptor N terminus. Here we used mutagenesis to explore the role of residues highly conserved in the predicted transmembrane helices of mammalian GLP-1Rs and conserved in family B G protein coupled receptors in ligand binding and GLP-1R activation. By iteration using information from the mutagenesis, along with the available crystal structure of the receptor N terminus and a model of the active opsin transmembrane domain, we developed a structural receptor model with GLP-1 bound and used this to better understand consequences of mutations. Mutation at Y152 [transmembrane helix (TM) 1], R190 (TM2), Y235 (TM3), H363 (TM6), and E364 (TM6) produced similar reductions in affinity for GLP-1 and exendin 9–39. In contrast, other mutations either preferentially [K197 (TM2), Q234 (TM3), and W284 (extracellular loop 2)] or solely [D198 (TM2) and R310 (TM5)] reduced GLP-1 affinity. Reduced agonist affinity was always associated with reduced potency. However, reductions in potency exceeded reductions in agonist affinity for K197A, W284A, and R310A, while H363A was uncoupled from cAMP generation, highlighting critical roles of these residues in translating binding to activation. Data show important roles in ligand binding and receptor activation of conserved residues within the transmembrane domain of the GLP-1R. The receptor structural model provides insight into the roles of these residues.
Processing of proglucagon within L cells of the intestine results in the formation of a number of peptides including glucagon-like peptide-1 (GLP-1), which is secreted after nutrient ingestion as a consequence of both neuroendocrine activity and direct contact of luminal nutrients with L cells. Along with gastric inhibitory peptide released from intestinal K cells, GLP-1 is a major incretin hormone, substantially enhancing the postprandial insulin response through its ability to enhance glucose-dependent insulin release from pancreatic β-cells.
Intestinal GLP-1 exists as truncated versions of the full-length peptide with fasting plasma levels of GLP-1 7–36 amide and GLP-1 7–37 being approximately equivalent. However, in response to a meal, the GLP-1 response is predominantly a result of an increase in GLP-1 7–36 amide (1). The GLP-1 peptides mediate their biological effects via a single receptor type belonging to family (or class) B of the G protein-coupled receptor (GPCR) superfamily. Typical of receptors within family B, the GLP-1 receptor (GLP-1R) couples predominantly to Gαs, thereby mediating its cellular effects through the production of cAMP, although coupling to Gαi, Gαo, and Gαq/11 has been reported (2–4). Given the ability of GLP-1 to enhance glucose-dependent insulin release, the GLP-1R is an especially attractive target for the treatment of type 2 diabetes mellitus, particularly as the risk of drug-induced hypoglycemia is substantially less than with many current therapeutic approaches. Furthermore, GLP-1 exerts a range of additional pancreatic and extrapancreatic antidiabetogenic effects that have the potential to enhance its clinical efficacy. These include an ability to increase insulin biosynthesis and pancreatic β-cell mass, while suppressing glucagon secretion and appetite (5, 6).
GLP-1 is rapidly degraded in vivo by the serine protease dipeptidyl peptidase-IV (7, 8), resulting in a plasma half-life of only 1–2 min for the biologically active peptide, and this has driven the search for more stable analogs for therapeutic use. One such compound is exendin-4 (Fig. 1), a 39-amino acid peptide from the venom of the Gila monster Heloderma suspectum (9), which shares 53% sequence identity with GLP-1, is not a substrate for dipeptidyl peptidase-IV, and has proven efficacy in the regulation of blood glucose levels in diabetic patients (10). However, peptides provide far from ideal therapeutics, and this has focused the search for small molecule, orally active agonists of the GLP-1R. This, in part, has driven the need for greater understanding of the structure-function relationships between GLP-1 and the GLP-1R and also more generally for a better understanding of ligand-receptor interactions and activation mechanisms in family B GPCRs.
Fig. 1.

Amino acid sequences of ligands of the GLP-1R. The aligned amino acid sequences of the GLP-1R agonists GLP-1 7–36 amide, GLP-1 7–37, and exendin-4 are shown alongside that of the antagonist exendin 9–39. The residues highlighted in bold are conserved between GLP-1 and exendin.
Binding of peptide ligands to family B GPCRs is currently described by a two-domain model (11, 12) in which the C terminus of the peptide binds to the extracellular N-terminal domain of the receptor with high affinity. This acts as an affinity trap, promoting the interaction of the N terminus of the ligand with lower affinity sites within the transmembrane domain and/or extracellular loops (EC) of the receptor, which leads to receptor activation. Consistent with this, the N-terminal domain of the GLP-1R is critical in GLP-1 binding (13–15). However, binding to the isolated N-terminal domain of the receptor occurs with relatively low affinity, and full-length GLP-1R is required for high-affinity binding (16, 17). Thus, high-affinity binding of GLP-1 would seem to require interactions not only with the N terminus of the receptor but also with other sites including those with charged residues at the extracellular boundary of the second and fourth transmembrane helices and in EC1 (15, 18, 19). Here we have identified the contribution to ligand binding and receptor activation of a number of residues lying within the transmembrane domain of the GLP-1R as predicted by Swiss-Prot (http://www.uniprot.org/; entry P43220). These residues are conserved in mammalian GLP-1Rs, and the majority show strong conservation across family B GPCRs (see Table 1 and Fig. 2) suggesting important structural and/or functional roles. The consequences of these mutations have been used to inform the structural model of the receptor, which, in turn, has been used to understand how the mutagenesis affected ligand binding and receptor function.
Table 1.
Comparison of mutation sites in the GLP-1R with the equivalent sites in family B GPCRs with known ligands
| GPCR | GLP-1R residue and equivalent residue in other GPCRs |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y152 TM1 | R190 TM2 | K197 TM2 | D198 TM2 | Q234 TM3 | Y235 TM3 | W284 EC2 | R310 TM5 | H363 TM6 | E364 TM6 | E387 TM7 | T391 TM7 | |
| CALCR | H | N | H | L | * | * | H | H | Q | F | M | I |
| CALCRL | H | N | H | L | L | * | H | H | E | F | M | M |
| CRF1 | H | * | F | V | N | * | * | Q | T | Y | N | E |
| CRF2 | H | * | F | L | N | * | * | Q | T | Y | N | Q |
| GlucagonR | * | K | I | * | * | * | * | * | * | * | D | S |
| GHRHR | H | K | * | * | H | F | * | K | * | Y | * | G |
| GIP-R | * | * | R | * | * | * | * | * | * | * | * | S |
| GLP-2R | * | * | * | * | H | * | * | * | * | * | Q | S |
| PAC1 | * | * | * | * | H | * | * | K | * | Y | * | G |
| PTH1 | * | * | * | * | L | * | * | Q | * | Y | * | N |
| PTH2 | * | * | * | * | I | * | * | Q | * | Y | * | N |
| SecretinR | * | * | * | * | * | * | * | * | * | Y | * | G |
| VPAC1 | * | * | * | * | * | * | * | K | * | Y | * | G |
| VPAC2 | * | * | * | * | * | * | * | * | * | Y | * | G |
Sequences of the human family B GPCRs were aligned using the multiple-sequence alignment tool in ClustalW (http://www.clustal.org). Residues of the GLP-1R are indicated along with their likely location. Where residues are identical between the GLP-1R and the comparator family B GPCR, this is designated by an asterisk. Where residues show conservative differences, these are shown in normal text. Where residues are not conserved, these are shown in underlined italics. Conservation or lack thereof is based on the BLOSUM62 substitution matrix (54) with residues being considered conserved with a score of more than or equal to 0. Note that of the residues mutated in the GLP-1R, least conservation is shown in CALCR, CALCRL, CRF1 and CRF2, while some residues, particularly E364 and T391, show the least conservation across the receptors. These residues shown are entirely conserved in the GLP-1R across mammalian species including human, chimpanzee, sheep, dog, rat, mouse, and rhesus monkey with the exception of a conserved arginine substitution at K197 in dog and a nonconserved asparagine substitution at Y152 in rhesus monkey. CALCR, Calcitonin receptor; CALCRL, calcitonin receptor-like receptor; CRF1, corticotropin-releasing factor receptor 1; CRF2, corticotropin-releasing factor receptor 2; glucagonR, glucagon receptor; GHRHR, GH-releasing hormone receptor; GIP-R, gastric inhibitory polypeptide receptor; GLP-2R, glucagon-like peptide-2 receptor; PAC1, pituitary adenylate cyclase activating polypeptide 1 receptor type I; PTH1, PTH receptor 1; PTH2, PTH receptor 2; secretinR, secretin receptor.
Fig. 2.

Schematic representation of the transmembrane domain and connecting loops of the hGLP-1R. The linear sequence was obtained from the NCBI database (rs1042044; var105098 as used in the present study). Residues mutated in the present study are shown by white text in black circles. All of these residues are fully conserved across the cloned mammalian GLP-1Rs (chimpanzee, dog, human, mouse, rat, rhesus monkey, sheep) with the exceptions of K197, which has a conservative substitution of arginine in the dog sequence and Y152 which is replaced by serine in the rhesus monkey sequence. Dashed lines indicate missing residues. This representation is based on our final model of the GLP-1R and differs slightly from the transmembrane helices identified in the Swiss-Prot entry (P43220). Note that although W284 was selected for mutation based on its location in TM4, as suggested in Swiss-Prot, our model suggests that this residue is at the proximal end of EC2, immediately adjacent to TM4. Figure was based on one generated using the residue-based diagram editor RbDe (55).
Results
Binding of GLP-1 7–36 amide and exendin 9–39 to the wild-type (WT) human (h)GLP-1R and mutated receptors
The human (h)GLP-1R was transiently transfected into HEK-293 cells. Subsequent assays in which the binding of 0.1 nm [125I]exendin 9–39 to the receptor was competed with either exendin 9–39 (homologous) or GLP-1 7–36 amide (heterologous) revealed concentration-dependent inhibition of [125I]exendin 9–39 binding (Fig. 3). Analysis of these data revealed a distribution constant (Kd) for exendin 9–39 of −9.15 ± 0.10 (n = 5, log10 M) and an inhibition constant (KI) for GLP-1 7–36 amide of −8.22 ± 0.03 (n = 5, log10 M) (Table 2).
Fig. 3.

Binding of exendin 9–39 and GLP-1 7–36 amide to the WT hGLP-1R. Homologous and heterologous competition binding assays were carried out on membranes prepared from HEK-293 cells transiently transfected with the WT hGLP-1R using [125I]exendin 9–39. A homologous binding curve was fitted to the exendin 9–39 data and a sigmoidal curve to the GLP-1 7–36 amide data. Data show total binding and are expressed as mean ± sem, n = 5.
Table 2.
KI of GLP-1 7–36 amide (agonist) and Kd of exendin 9–39 (antagonist) for the WT and mutated hGLP-1Rs
| Receptor | Location of mutation | KI (Log10 M) (GLP-1 7–36 amide) | Kd (Log10 M) (exendin 9–39) | Receptor levels (pmol mg−1 protein) |
|---|---|---|---|---|
| WT | −8.22 ± 0.03 | −9.15 ± 0.10 | 27.50 ± 3.44 | |
| Y152A | TM1 | −6.72 ± 0.37 a | −8.13 ± 0.13 a | 1.84 ± 0.56 a |
| R190A | TM2 | −6.78 ± 0.12 a | −8.36 ± 0.12 a | 1.98 ± 0.43 a |
| K197A | TM2 | −6.86 ± 0.04 a | −8.67 ± 0.09 b | 15.77 ± 2.77 b |
| D198A | TM2 | −6.59 ± 0.04 a | −8.74 ± 0.20 | 18.18 ± 5.94 |
| Q234A | TM3 | −7.12 ± 0.04 a | −8.63 ± 0.08 b | 7.32 ± 1.45 a |
| Y235A | TM3 | −6.84 ± 0.15 a | −7.88 ± 0.12 a | 3.29 ± 0.84 a |
| W284A | EC2 | −6.77 ± 0.41 a | −8.49 ± 0.22 a | 9.33 ± 4.74 a |
| R310A | TM5 | −7.22 ± 0.13 a | −8.81 ± 0.14 | 4.73 ± 1.36 a |
| H363A | TM6 | −6.23 ± 0.16 a | −7.53 ± 0.08 a | 6.65 ± 0.53 a |
| E364A | TM6 | −6.46 ± 0.10 a | −7.28 ± 0.03 a | 11.59 ± 0.36 a |
| E387A | TM7 | −8.09 ± 0.08 | −8.56 ± 0.01 b | 28.24 ± 1.82 |
| T391A | TM7 | −7.77 ± 0.09 | −8.70 ± 0.06 | 17.75 ± 3.17 |
Using [125I]exendin 9–39 as the radiolabel, homologous and heterologous competition binding assays were carried out on membranes of HEK-293 cells transiently expressing either the WT hGLP-1R or hGLP-1Rs with single-alanine substitutions in their transmembrane domain. Homologous binding curves were fitted to determine the Kd for the antagonist exendin 9–39 at each of the receptors and KI values calculated using the Cheng-Prusoff correction on IC50 values generated from sigmoidal displacement curves using the agonist GLP-1 7–36 amide as the competing ligand. The expression levels of the receptors in each assay were calculated and are expressed as pmol mg−1 protein.
, P < 0.01;
, P < 0.05; compared with WT hGLP-1R. Data are mean ± sem with n = 5 for the WT receptor and n = 3 for each of the mutants. Single underlined are those mutations in which either agonist but not antagonist affinity was reduced or in which the reduction in agonist affinity was greater than the reduction in antagonist affinity, whereas a double underline (E387A only) indicates that the reduction in antagonist affinity was greater than the reduction in agonist affinity. These were determined by calculating the change in affinity between the WT hGLP-1R and each mutant for both the agonist and antagonist (i.e. WT KI − mutant KI and WT Kd − mutant Kd). For each mutant, the ratio of the differences in KI and Kd values was then calculated. A ratio of more than 2 was taken to indicate that binding affinity of the agonist, GLP-1 7–36 amide, was more severely affected than the binding affinity of the antagonist, exendin 9–39, whereas a ratio of less than 0.5 was taken to indicate that binding affinity of exendin 9–39 was more severely affected than the binding affinity of GLP-1 7–36 amide.
A series of hGLP-1R constructs were generated in which a single residue within a transmembrane helix (TM) or at the extracellular boundary of such a helix was replaced with alanine. In a transient expression system, all of the mutated receptors bound [125I]exendin 9–39, indicating both synthesis and expression of the constructs (Table 2 and Fig. 4, A and B). Although there was some variability in receptor expression levels between experiments, there were a number of mutations in which expression was significantly reduced (Table 2). In particular Y152A (TM1), R190A (TM2), Y235A (TM3), R310A (TM5), and H363A (TM6) expression was less than 25% of the WT receptor.
Fig. 4.
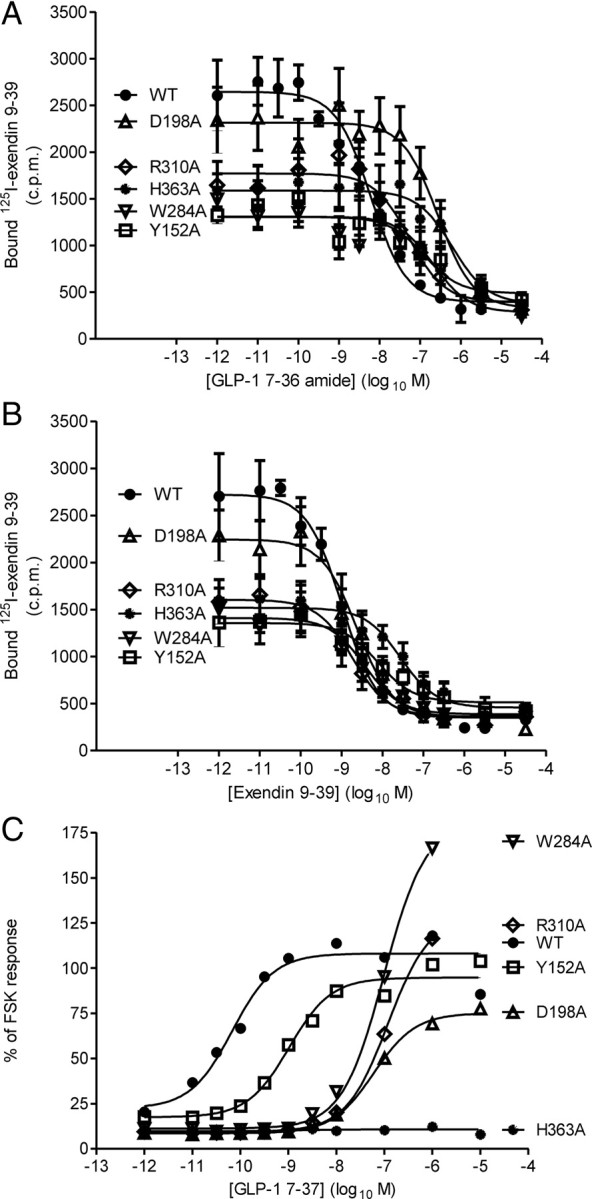
Ligand binding and cAMP generation by mutated hGLP-1Rs. A and B, Homologous and heterologous competition binding assays were carried out on membranes prepared from HEK-293 cells transiently transfected with the WT and mutated GLP-1Rs using [125I]exendin 9–39. Homologous binding curves were fitted to the exendin 9–39 data and a sigmoidal curve was fitted to the GLP-1 7–36 amide data. Data are expressed as mean ± sem with n = 5 for the WT receptor and n = 3 for each of the mutated receptors. C, Transiently transfected cells were stimulated, in the presence of 1 mm IBMX, for 1 h with varying concentrations of either GLP-1 7–37 or FSK (50 μm) at 37 C. The cAMP was extracted and measured and expressed as a proportion of the response to FSK. Sigmoidal concentration-response curves were fitted. Curves represent the means of n = 7 for the WT receptor and n = 3 for the mutated receptors (error bars omitted for clarity). In each of the panels, data from the WT hGLP-1R and the Y152A (TM1), D198A (TM2), W284A (EC2), R310A (TM5), and H363A (TM6) mutations have been shown to demonstrate the range of alterations observed. The binding affinities for GLP-1 7–36 amide and exendin 9–39 (KI and Kd values, respectively) and receptor expression levels derived from experiments on all receptor constructs are given in Table 2. Similarly, potency estimates and Emax values for cAMP generation derived from experiments on all receptor constructs are given in Table 3.
With the exception of T391A (TM7), which had no effect, all other alanine substitutions influenced the binding of one or both ligands (GLP-1 7–36 amide/exendin 9–39) (Fig. 4, A and B, and Table 2). For Y152A (TM1), R190A (TM2), Y235A (TM3), H363A (TM6), and E364A (TM6), the affinities of both GLP-1 7–36 amide and exendin 9–39 were similarly reduced. In contrast, alanine substitutions at K197 (TM2), Q234 (TM3), and W284 (EC2) preferentially reduced agonist (GLP-1 7–36 amide) affinity, whereas D198A (TM2) and R310A (TM5) reduced only agonist affinity. Note that although Swiss-Prot predicted W284 to be at the top of TM4, our model suggests that this residue may be at the proximal end of EC2, immediately adjacent to TM4 (e.g see Fig. 2), and we have therefore used this as the location throughout the text. The greatest reductions in ligand affinity resulted from mutation at either H363 or E364 (both TM6). The E387A (TM7) mutant was the only construct in which the affinity of exendin 9–39 but not GLP-1 7–36 amide was reduced, although this was a very modest reduction (∼½ log unit).
Effects of single alanine substitutions on agonist potency
Challenge of HEK-293 cells transiently expressing the WT hGLP-1R with GLP-1 7–37 resulted in a concentration-dependent increase in cAMP levels with an EC50 value of −10.16 ± 0.22 (n = 7, log10 M) (Table 3 and Fig. 4C). The Emax of the WT receptor was 114 ± 34% (n = 7) of the response to challenge with 50 μm forskolin (FSK). Neither E387A nor T391A (both TM7) influenced agonist affinity (see above), and this was consistent with a lack of effect on agonist potency (Table 3). In contrast, agonist potency was reduced for all the other constructs (Table 3 and Fig. 4C), consistent with reductions in agonist affinity (see above). Of the 12 mutants, only H363A (TM6) was essentially uncoupled from cAMP generation (Fig. 4C and Table 3) although E364A (TM6) also had a reduced Emax (44 ± 25%, n =3 of the FSK response) (Table 3). In addition to H363A, which was essentially uncoupled despite agonist binding, the mutations K197A (TM2), W284A (EC2), and R310A (TM5) resulted in much greater reductions in agonist potency than affinity (Tables 2 and 3). A similar profile of potency differences between the WT receptor and the mutated receptors was observed irrespective of whether GLP-1 7–37 or GLP-1 7–36 amide was used as the agonist in the functional assays (data not shown).
Table 3.
Agonist potency for cAMP generation by WT and mutated hGLP-1Rs
| Receptor | Location of mutation | EC50 (Log10 M) | Emax (% FSK response) |
|---|---|---|---|
| WT | −10.16 ± 0.22 | 114 ± 34 | |
| Y152A | TM1 | −8.92 ± 0.08 a | 96 ± 17 |
| R190A | TM2 | −7.93 ± 0.09 a | 112 ± 16 |
| K197A | TM2 | −7.36 ± 0.04 a | 85 ± 7 |
| D198A | TM2 | −7.17 ± 0.09 a | 77 ± 9 |
| Q234A | TM3 | −8.51 ± 0.22 a | 179 ± 96 |
| Y235A | TM3 | −8.82 ± 0.12 a | 80 ± 27 |
| W284A | EC2 | −7.03 ± 0.03 a | 183 ± 102 |
| R310A | TM5 | −7.06 ± 0.13 a | 130 ± 74 |
| H363A | TM6 | Not detected | |
| E364A | TM6 | −8.99 ± 0.03 a | 44 ± 25 |
| E387A | TM7 | −9.77 ± 0.06 | 172 ± 97 |
| T391A | TM7 | −10.21 ± 0.15 | 93 ± 9 |
HEK-293 cells transiently transfected with either the WT hGLP-1R or hGLP-1Rs with single-alanine substitutions in their transmembrane domain were stimulated, in the presence of 1 mm IBMX, for 1 h with varying concentrations of GLP-1 7–37 or FSK (50 μm) at 37 C. The cAMP was extracted and measured and expressed as a proportion of the response to FSK. Sigmoidal concentration-response curves were fitted to allow determination of EC50 and Emax values. Data are mean ± sem with n = 7 for the WT receptor and n = 3 for each of the mutants.
, P < 0.01 compared to the WT receptor. Underlined are those mutations in which cAMP responses were either not detectable or in which the reduction in EC50 was greater than the reduction in agonist affinity, KI (see Table 2). In constructs in which potency was measurable and significantly reduced, for both the EC50 and KI values, the change between the mutant and the WT hGLP-1R was determined, and the ratio of the differences in EC50 and KI values was then calculated. A ratio of more than 2 was taken to indicate that the potency (EC50) of the agonist GLP-1 was more severely affected than its binding affinity (KI).
Although expression levels did vary among the receptor constructs, there was little evidence to suggest this had a major impact on agonist potencies. For example, despite the expression of Y152A being substantially lower than the WT receptor, agonist potency was reduced in line with affinity. Indeed, there were no instances in which potency, but not affinity, was reduced. In many cases, reductions in agonist potency and affinity were similar, suggesting that reduced potency resulted from reduced agonist affinity. However, in a number of mutants (K197A, W284A, and R310A) potency was reduced more than agonist affinity, whereas H363A was uncoupled from cAMP generation. These constructs did not show the lowest levels of expression.
Three-dimensional model and helical wheel projection of the hGLP-1R
Development of the receptor model was an iterative process in which a number of models were generated, selected, and remodeled to account for incompatibilities between the model and both data within the literature and data arising from the analysis of our receptor mutants. For example, in an intermediate model (without helix remodeling) some incompatibility was observed between the model and the mutation data. As an illustration of this, Y152 was predicted to be on the outer face of TM1, orientated toward the membrane, and it was difficult to formulate a clear idea about how mutation could account for the observed reductions in ligand affinity and potency. However, this intermediate model used the structure of active opsin as a template for the transmembrane domain and did not account for misaligned secondary-structure features within the helices, such as proline residues. On remodeling of the helices to account for such features (Table 4), the orientation of this residue changed, placing it in an aromatic pocket between TM1 and TM2 [π-stacking with F195 (TM2) and hydrophobic interaction with Y148 (TM1)]. This is entirely compatible with the mutation data in which Y152A showed a significant reduction in affinity for both agonist and antagonist binding (predicted from the model as result of this pocket collapsing around the much smaller alanine residue). An additional conservative Y152F mutation showed no significant change in agonist or antagonist affinities nor a change in potency (Y152F KI, −7.98 ± 0.07; Kd, −9.02 ± 0.10; EC50, −9.93 ± 0.06 vs. WT hGLP-1R KI, −8.22 ± 0.03; Kd, −9.15 ± 0.10; EC50, −10.16 ± 0.22, all data are log10 M, mean ± sem, n = 3 for Y152F and n = 5 for WT hGLP-1R). This is compatible with the space-filling and π-stacking interactions observed with both tyrosine and phenylalanine residues. Note that in the vasoactive intestinal peptide receptor 1 (VPAC1) and vasoactive intestinal peptide receptor 2 (VPAC2) the equivalent residues (Y150 and Y134) have been argued to be important in stabilizing the active receptor conformation (20).
Table 4.
Comparison of the sequences of opsin and the GLP-1R

Sequence alignment of human GLP-1R (S136-R421) with bovine opsin, determined by the method described by Bissantz (52). As the position of proline residues (outlined) in the transmembrane helices differ between template and target, alternative templates (in italics) were used to model the shape of helices where these misalignments occur. The templates used were from opsin and were used to create the correct secondary structure at the location of these proline residues and to remove the effect of a template proline where none existed in the target. * indicates conserved residues, # indicates the location of the important C226-C296 bridge (45).
Two recent studies using photolabile probes of GLP-1 have identified spatial approximations between F12 of the ligand (L:F12) and Y145 of the receptor (21), between L:A24 and E133 and between L:G35 and E125 (22), providing potential constraints for any model of the ligand-bound GLP-1R. In our early models, L:A24 and E133 were separated by approximately 47Å. However, E133 (N-terminal domain) is within a highly flexible loop, and this was remodeled, reducing the intermolecular distance to around 30Å. Remodeling to reduce this distance further was not compatible with other constraints on the model. In our model, L:G35 and E125 (N-terminal domain) are 23Å apart. The modeling of this region is based on, and therefore consistent with, the crystal structure of the N-terminal domain of the GLP-1R with GLP-1 bound (23) and we, therefore, did not remodel this region. Our model indicates a distance of approximately 15Å between L:F12 and Y145 (top of TM1), which is acceptable, and this region was not therefore remodeled to accommodate potential interaction over a shorter distance. It is important to note that photoaffinity labeling has been used to identify spatial approximations and not to define interacting residues (21). Indeed, mutation of E125, E133, or Y145 to alanine had no impact on either GLP-1 binding affinity or function (21, 22), suggesting if any interactions did occur, they are not critical for receptor structure or function. Further, it is not clear whether such spatial approximations result from intra- or intermolecular proximity.
The three-dimensional structure of the receptor was examined to investigate the possible interactions of specific residues and the possible consequences of their mutation (Fig. 5). The approximate location and orientation of each residue is indicated in the helical wheel model (see Fig. 6).
Fig. 5.
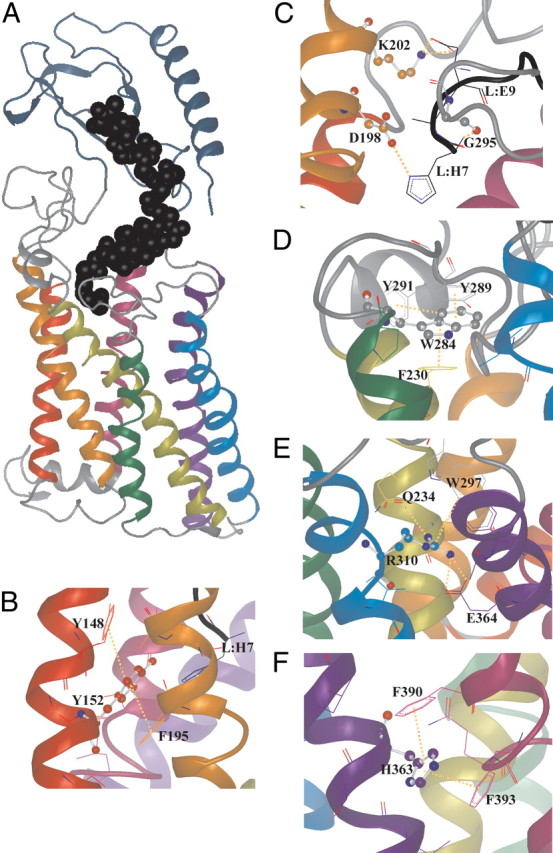
The three-dimensional (3D) model of the GLP-1R and example close-up images to highlight specific structural features and interactions. A, The 3D model showing the hGLP-1R with GLP-1 bound. GLP-1 is shown as black spheres (backbone atoms only). In all images the transmembrane helices are rainbow colored: TM1, red; TM2, orange; TM3, yellow; TM4 green; TM5, blue; TM6, indigo; TM7, violet. The N-terminal domain is gray-blue. Intracellular and extracellular loops are gray, and the ligand (GLP-1) is black. Within those amino acid residues in which some structure is shown, the colors of the helices are used to indicate carbon atoms whereas nitrogen is blue, oxygen is red, and sulfur is yellow. Nonbonded interactions are shown as dotted orange lines. B, The region surrounding Y152 (TM1) showing that this residue exists in a hydrophobic pocket interacting with some aromatic residues, providing a structured region. Mutation to alanine (Y152A) would be expected to allow conformational collapse, possibly affecting the surrounding structures including EC1. C, The region surrounding D198 (TM2) showing the interaction of this residue with H7 at the N terminus of GLP-1 (L:H7). G295 (TM3) is also predicted to interact with L:H7, and K202 is predicted to interact with L:E9. D, The region surrounding W284 (EC2) is shown to illustrate its role as a space-filling residue displaying an aromatic stacking interaction with Y289 (EC2) and F230 (TM3) that provides conformational support, particularly to EC2. E, The region surrounding R310 (TM5) showing a strong salt bridge with E364 (TM6). F, The region surrounding H363 (TM6) showing its position in an aromatic pocket formed by F390 and F393.
Fig. 6.

Helical wheel model of the transmembrane domain of the hGLP-1R. Only the upper half of the transmembrane domain is shown with the helices labeled I–VII. The N terminus of GLP-1 is also shown inserted between the transmembrane domain. The diagram represents the hydrophilic residues as circles, hydrophobic residues as diamonds, potentially negatively charged residues as triangles, and potentially positively charged residues as pentagons. Hydrophobicity is color coded: the most hydrophobic residues are green with the intensity of the green decreasing in relation to the loss of hydrophobicity. Zero hydrophobicity is coded as yellow. Hydrophilic residues are coded red with pure red being the most hydrophilic (uncharged) residue and the intensity of red decreasing through orange with loss of hydrophilicity. Residues that are potentially charged are light purple. The interaction of D198 (TM2) with residue H7 of GLP-1 (L:H7) and the interaction of K202 (TM2) with L:E9 are shown. Residues mutated in the current study are circled in red.
Interactions of the mutated residues with other amino acids in the GLP-1R structure as defined by the final model are summarized in Table 5. Each of the residues investigated interacts directly with at least one other residue, and several have the potential to interact with others through water-mediated interactions. Interestingly, although mutation of either D198 (TM2) or R310 (TM5) to alanine reduced receptor affinity for GLP-1 7–36 amide but not exendin 9–39, only D198 interacts directly with the ligand in our model. However, R310 interacts with W297 (via a π-stacking interaction) and E364 (via an electrostatic interaction). This provides a point of direct contact between TM5 and TM6 near the top of the helices, which likely stabilizes the local loop structure of EC2. W297 itself is only approximately 4Å from the ligand (F12). Our model also highlights interactions with TM7 by a number of the residues mutated in the current study. There is particularly close contact between R190 (TM2) and G395 (TM7) and between E387 (EC3) and R376 (TM7). The mutation of T391 (TM7) to alanine had no effect on either the binding or function of the GLP-1R. This is consistent with the model, in which T391 is predicted not to make any significant interactions (only a weak interaction with the aromatic ring of W297 is predicted). The relative lack of importance of T391 in family B GPCR structure is perhaps reflected in the relative lack of conservation compared with the majority of residues mutated in the present study. Given that the H363A (TM6) mutation abolished cAMP response to GLP-1 7–37 and that the model indicates that it makes direct contact with two residues in TM7 (F390 and F393), this suggests that this link between TM6 and TM7 is critical for GLP-1R function. In addition to interactions between the transmembrane helices, interactions between transmembrane helices and EC regions have been highlighted in Table 5 (e.g. Q234 of TM3 interacts directly with W297 of EC2 and F230 of TM3 interacts with W284 of EC2 via π-stacking). Interactions between the receptor and the ligand indicated by the model are highlighted in Table 6.
Table 5.
Interactions between mutated residues and the surrounding GPCR structure
| Residue | Location | Orientation | Directly interacts with |
|---|---|---|---|
| Y152 | TM1 | Inner | Y148 (TM1, HP) |
| F195 (TM2, AR) | |||
| R190 | TM2 | Inner | F187 (TM1, AR) |
| N240 (TM3, ES) | |||
| K197 | TM2 | Outer | S225 (TM3, ES) |
| D198 | TM2 | Inner | H7 (L, ES) |
| Q234 | TM3 | Inner | W297 (EC2, ES) |
| R310 (TM5, ES) | |||
| Y235 | TM3 | Outer | L189 (TM2, HP) |
| S193 (TM2, HP) | |||
| P277 (TM4, HP) | |||
| L278 (TM4, HP) | |||
| W284 | EC2 | Inner | F230 (TM3, AR) |
| Y289 (TM5, AR) | |||
| Y291 (EC2, AR) | |||
| R310 | TM5 | Inner | Q234 (TM3, ES) |
| W297 (EC2, AR) | |||
| E364 (TM6, ES) | |||
| H363 | TM6 | Inner | L359 (TM6, ES) |
| F390 (TM7, AR) | |||
| F393 (TM7, AR) | |||
| E364 | TM6 | Inner | Y241 (TM3, ES) |
| R310 (TM5, ES) | |||
| E387 | TM7 | Inner | R376 (EC3, ES) |
| T391 | TM7 | Inner | W297 (EC2, AR) |
Based on the final model generated, the interactions of each of the mutated residues with other amino acids are presented, including electrostatic interactions (hydrogen-bonds and charge attraction), aromatic π-interactions, and hydrophobic interactions (Van der Waals forces). Indicated in parentheses are the location of these amino acids within the GLP-1R structure and the type of interaction between the residues. ES, Electrostatic; AR, aromatic; HP, hydrophobic.
Table 6.
Sites of interaction between GLP-1 and the GLP-1R
| Receptor residue | Location | Directly interacts with ligand residue |
|---|---|---|
| D198 | TM2 | H7 (ES) |
| K202 | TM2 | E9 (ES) |
| D293 | EC2 | H7 (ES) |
| T11 (ES) | ||
| F12 (AR) | ||
| D15 (ES) | ||
| E294 | EC2 | T11 (ES) |
| G295 | EC2 | H7 (ES) |
| W297 | EC2 | F12 (HP) |
| N300 | EC2 | Y19 (ES) |
Based on the final model generated, the interactions of residues within the receptor transmembrane domain and residues of the ligand are presented, including electrostatic interactions (hydrogen-bonds and charge attraction), aromatic π-interactions and hydrophobic interactions (Van der Waals forces). Indicated in parentheses is the type of interaction between the residues. ES, Electrostatic; AR, aromatic; HP, hydrophobic.
Discussion
Heterologous and homologous competition binding assays demonstrated low nanomolar affinities of GLP-1 7–36 amide and exendin 9–39 at the hGLP-1R, consistent with previous reports (24–27). GLP-1 binding likely occurs through an initial, relatively low-affinity interaction between the ligand C terminus and receptor N terminus, followed by an interaction of the N terminus of GLP-1 with the receptor core to establish high affinity (13, 15, 16). This implies that the receptor N-terminal domain constrains the ligand to facilitate interaction with the transmembrane domain and that a rigid connection exists between these domains in at least one conformation. In contrast, the receptor N-terminal domain is predominantly involved in high-affinity binding of the agonist, exendin-4 (16, 28). Such different requirements for high-affinity binding may result from a stable α-helical structure within exendin-4, with GLP-1 paying an entropic penalty to form the bioactive conformation before binding (29). Although removal of the first two N-terminal amino acids of exendin-4 abolishes agonism, truncation by up to eight amino acids has no effect on affinity (16), and peptides including exendin 9–39 are high-affinity antagonists.
Recently a model of the rat GLP-1R (90.9% identity, 95.9% similarity to the human receptor) bound to GLP-1 has been presented (30). This model was based on the N-terminal domain of the corticotropin-releasing hormone receptor 2 (34.5% identity, 53.4% similarity to rat GLP-1R) and the transmembrane domain of inactive rhodopsin. However, this model could not adequately explain the consequences of a number of our mutations. For example, this literature model (kindly provided by the authors) shows Y152 (TM1), W284 (TM4 although EC2 in our model), and E364 (TM6) all orientated out of the transmembrane domain, into the membrane. However, our alanine mutations of these residues demonstrate a significant effect on both agonist and antagonist binding, indicating that these residues serve a role either interacting with the ligand or structurally in supporting the receptor conformation. Further, the crystal structure of either the exendin 9–39-bound (29) or GLP-1-bound (23) N-terminal domain of the human GLP-1R, and a model of opsin in its G protein-interacting conformation (31) are now available and we have, therefore, developed an alternative model with the most recently available and most appropriate data. The model is intended to predict the biologically relevant agonist-bound conformation. Unlike the previous model (30) our model excludes the signal peptide sequence because we have shown that this does not form part of the mature, signaling receptor (32). Sequence differences between the rat and human GLP-1R are not expected to have significant impact on the overall model.
Alanine substitutions at Y152 (TM1), R190 (TM2), Y235 (TM3), H363 (TM6), and E364 (TM6) reduced agonist and antagonist affinities to similar extents. As there is little evidence for interaction between exendin 9–39 and the transmembrane domain, this indicates general structural functions of these residues. In contrast, alanine substitutions at K197 (TM2), Q234 (TM3), and W284 (EC2) reduced agonist affinity preferentially, whereas in D198A (TM2) and R310A (TM5), only agonist affinity was reduced. These data are consistent with interaction of GLP-1, but not exendin 9–39, with transmembrane domain residues and the requirement for such residues to either directly interact with agonist or provide structure within the binding regions. The polarity of D198 is important for GLP-1 binding (19), and our model has been constrained to show the reported direct ionic interaction with H7 of GLP-1 (30). D198 and adjacent residues are also critical for ligand binding in other family B GPCRs (33–36). Although D198 does not make direct contact with other residues, the model suggests it is approximately 9Å from T149 (TM1), which may allow an interaction via a water molecule. Interestingly, T149M has been identified in a patient with type 2 diabetes (37), and the mutation has been shown to reduce agonist, but not antagonist, affinity (38). A link between T149 and D198 could explain the consequence of this mutation.
Our model suggests that residues immediately adjacent to D198 do not contact the ligand, although they may be critical in maintaining aspects of receptor structure. For example, our model suggests an interaction of K197 (TM2) with S225 (TM3), which are part of a hydrogen-bonding network that includes Q221 (EC1). This region is adjacent to the ligand-binding domain, and the observed conformation of EC1 is critical to GLP-1 binding. Substitution of this critical lysine [K197 (TM2)] will weaken the hydrogen-bonding network and result in a change in the conformation of EC1, with likely consequences for ligand binding. This marks an improvement over previous models (our unpublished data and Ref. 30) in that K197 now has a clear structural role, fitting with the observed reduction in ligand binding affinity shown here and previously (15). Such structural requirements may be critical in other family B receptors. For example, in VPAC1 and VPAC2 receptors, the equivalent residues (K195 and K179, respectively) also influence binding of the N terminus of the agonist (35, 36). A previous study has highlighted the importance of residues within EC1 to GLP-1 binding by the GLP-1R. Thus, the combined alanine substitution of residues M204 and Y205, but not the single mutants, markedly reduce GLP-1 affinity, most likely through a reduction in the combined size and hydrophobicity of the side chains (39), consistent with a change in the local conformation. The model suggests these residues are at the boundary of TM2 and EC1 and are not orientated toward the ligand. The model is consistent with the double mutant causing changes to the local conformation: especially to EC1 and thus the interactions of the N-terminal domain and the EC loops.
GLP-1 makes a number of interactions with the receptor (Table 6). The model shows the ligand to sit in a shallow groove on the top of the transmembrane domain, from where it makes various interactions with the receptor. Thus, the ligand conformation allows D293 of EC2 to interact with H7, T11, F12, and D15 of the ligand through hydrogen-bonding interactions. A previous study demonstrated that mutation of these residues to alanine reduces GLP-1 affinity by 10- to 100-fold although H7, G10, F12, T13, and D15 of GLP-1 were suggested as key for direct interactions with the receptor based on binding and functional studies (40). Clearly, interpretation of such structure-function studies is difficult in the context of predicting the sites of interaction, and other analyses are required to refine our understanding. Interactions of the ligand with EC2 may be important to binding and the structure of this region is therefore critical. Interestingly, although the polar residue R310 (TM5) shows little conservation among family B GPCRs, our model suggests a salt bridge with E364 (TM6), providing the only interaction between TM5 and TM6 in the upper half of the transmembrane domain. It is possible that mutation of R310 results in a general loss of rigidity and structure that particularly affects EC2, and this may cause the observed loss of agonist affinity. In family A GPCRs the proposed activation mechanism involves a crucial rotation of TM6, bringing the top half of the helix toward TM3 (41, 42). Because R310A (TM5) had a more profound impact on agonist potency than affinity, this suggests that the relative positioning and/or movement of TM6 and/or TM5 may also be important in the activation of family B receptors. Comparison of the active and inactive conformations of opsin [Protein Data Bank (PDB) codes 3DQB and 1U19, respectively] show that the major movement on activation is a shift in the position and tilt of TM6, along with an increase in the kink angle (31). Our model, being based on the active opsin structure, replicates this shift, although the position of the helix kink is different owing to the change in the proline position, and it is interesting to note that there are significant differences around the TM5/6 region between our model and the previous literature model (30), both in terms of conformation and alignment. In the previous model, EC2 contains a helix not present in this model, and R310 was critical in maintaining secondary structure between this helix and TM5. Our new model is more in line with the proposed model for family A GPCR activation, which is consistent with recent evidence from the family B PTH receptor 1 (43).
Our model suggests that within TM3, Q234 interacts via hydrogen bonding with W297 in EC2. This tryptophan was recently identified as being a point of approximation for L20 of GLP-1 using photoaffinity labeling, and its importance in GLP-1 binding was confirmed by mutational work, which showed that a tryptophan to alanine substitution at this position resulted in no saturable GLP-1 binding (44). These residues are approximately 20Å apart in our model. EC2 is long and inherently flexible and therefore fixed points such as this interaction and the cysteine bridge (C226 and C296) observed in family B GPCRs (45) are crucial in providing some rigidity to this loop and allowing it to adopt a conformation suitable for ligand binding, including the placement of D293 and E294. The Q234A mutation removes this structural constraint and allows the loop to be more flexible, which does not favor GLP-1 binding. Although the affinities of both ligands are affected by the mutation, the predominant effect is on GLP-1 binding. This hypothesis supports the ligand-binding mode of the current structural model and is inconsistent with the alternative ligand conformation (a single helix). Other connections that may support EC2 include an aromatic-stacking interaction between W284 of EC2 and F230 on TM3, and hydrogen bonds between K288 of EC2 and P283 of TM4 and between Y289 of EC2 and Y305 of TM5. Mutation of W284 to alanine is also likely to change EC2 conformation and the preferential effect on GLP-1 affinity again implicates this extracellular loop in GLP-1 binding, consistent with other recent work implicating this loop in both binding and receptor activation (46). Further, mutation of K288 to alanine reduces GLP-1 affinity (18), and our model indicates that this may result from the importance of this residue in the conformation of EC2 rather than the result of a direct interaction with the ligand.
As previously suggested (30), we show L:F12 sits in a hydrophobic pocket, and that L:Y19 forms an electrostatic interaction with the receptor, although our model suggests involvement of N300 rather than R227 and K288, as was highlighted previously (30). Additional interactions between E294 and L:T11, between G295 and L:H7, and between K202 (TM2) and L:E9 are also highlighted in our model (Table 6). This latter interaction may explain the reported reduction in GLP-1 affinity in a K202A receptor mutant (15).
H363 (TM6) is highly conserved in family B GPCRs, and its importance is further emphasized because mutation to alanine not only reduced both GLP-1 and exendin 9–39 affinity but also abolished functional responses. Initial modeling indicated that this residue lay close to other residues but had limited contact. Given the profound effects of the H363A mutation, the orientation of this residue was manually selected from a residue conformation library and refined using Prime (Schrödinger, Inc., Portland, OR) with side chains in the vicinity of 7.5Å unfrozen during forcefield minimization. This refinement indicates that H363 sits in an aromatic pocket and interacts with F390 and F393 on the adjacent helix (TM7). It is reasonable to argue that it serves a structural role, the π-stacking providing a sort of anchor. Indeed, this residue is highly conserved in family B GPCRs, and mutation of the equivalent residue in the VPAC1 receptor (N229) markedly reduces cAMP and abolishes Ca2+ responses (47). Interestingly, mutation of H363 to glutamine (H363Q) in the GLP-1R had no effect on either ligand affinity or agonist potency (data not shown). However, this glutamine substitution may allow for π-stacking and provide similar functionality to histidine.
Mutation of the residue adjacent to H363 [E364A (TM6)] reduced GLP-1 and exendin 9–39 affinities and agonist potency to similar extents, suggesting a more general role of this residue in receptor structure. E364 within TM6 is in a region critical for peptide binding in other family B GPCRs including the secretin (48) and PTH (49, 50) receptors. Our model suggests a direct interaction of this residue with R310 (TM5) and through this hydrogen-bonding network onto Q234 (TM3), providing key structural constraints between transmembrane helices. This is in contrast to the previous literature model (30) in which E364 (TM6) is orientated toward the membrane and mutation would be predicted therefore to have little effect on ligand affinity.
R190A results in similar reductions in ligand affinities (agonist and antagonist) and agonist potency. In the family B, VPAC1, VPAC2, and secretin receptors, the equivalent residues to R190 of the GLP-1R (R188, R172, and R166, respectively) have been argued to interact with, or come into close proximity to, an aspartic acid residue at position 3 of the endogenous agonists (33, 35, 36). In our model R190 is in close proximity (∼5Å) with the ligand (H7) and although not predicted to interact directly, a water-mediated interaction is possible. Agonists of the GLP-1R have a comparable glutamic acid residue at the same position (L:E9), and a different residue (K202) does interact directly with L:E9 (Table 6) and may serve the same structural role in the GLP-1R.
Among the mutations, only E387A at the extracellular face of TM7 reduced antagonist but not agonist affinity, albeit to a small extent. The model indicates a salt bridge interaction between E387 and R376. Because R376 is part of EC3, mutation of E387 would be expected to have an effect on EC3 conformation, which forms part of the groove in which the ligand resides. However, the small effect on antagonist affinity and data indicating that high-affinity binding of exendin 9–39 requires only the receptor N terminus (13, 16, 28), suggests that EC3 has little or no contribution to binding. The equivalent mutation in the rat secretin receptor (E351A) does, however, markedly affect binding of secretin and receptor function, possibly due to loss of a charge-charge interaction between the N terminus of secretin and the receptor (51). This is entirely consistent with our model in which the N-terminal domain is located in space very close to EC3 (closest approach is ∼5Å); however, the model is not sufficiently well tuned to discern individual residue interactions between N-terminal and transmembrane domain. The previous literature model (30) did not show a significant role for E387.
In our model, Y235 is located at the outer face of TM3, in a hydrophobic pocket between TM2 and TM4. It makes a hydrogen-bonding interaction with G273 in TM4. The role of Y235 is largely steric, and mutation to alanine would result in a collapse of the local structure, thereby accounting for the generally negative effects on ligand binding and agonist potency. Indeed, the packing effect of this bulky residue is likely to be critical because mutation to phenylalanine had no effect on the measured parameters (data not shown), which is consistent with phenylalanine substitution at the equivalent residue in the VPAC1 receptor (47). This is a clear improvement over the previous literature model (30) in which Y235 is shown to be a structural constraint restricting the flexibility of EC2 through a hydrogen bond to C226. This model would predict the same effect for both alanine and phenylalanine, whereas the reality shows quite different effects.
The current data demonstrate the importance for ligand binding and receptor activation of residues within the GLP-1R transmembrane domain. Comparison of agonist potency and ligand affinities also highlighted roles for residues conserved across mammalian GLP-1Rs or among family B GPCRs. The GLP-1R model has also facilitated understanding of the likely mechanisms through which mutations influence ligand binding and receptor activation.
Materials and Methods
Cell culture
HEK-FlpIn cells were routinely maintained in DMEM supplemented with 10% fetal bovine serum in 160-cm2 tissue culture flasks at 37 C in a 5% CO2 humidified atmosphere.
Generation of GLP-1R cDNA and mutated GLP-1Rs
WT hGLP-1R was amplified by PCR from a vector containing the hGLP-1R and the product inserted into a PCR-Script vector. It was then subcloned into the pcDNA5-FRT expression plasmid to generate pcDNA5-FRT-GLP-1R. Point mutations (see Fig. 2 and Table 1) were generated using the Quikchange II Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) using the pcDNA5-FRT-GLP1R as the starting template. Mutations at the sites indicated were all initially by alanine substitution although, as indicated in text, a number of other substitutions were performed at several sites. Mutagenic primers were designed using guidelines from the Quikchange II Site-Directed Mutagenesis kit, and coding sequences for the mutated receptors contained a start codon, stop codon, and an appropriately positioned Kozak sequence. All sequences were confirmed by automated sequence analysis. Further details of the cloning strategy are available on request.
Homologous and heterologous competition-binding assays
To explore the structure-function relationships for the binding of ligands thought to interact with either the N-terminal and transmembrane domain or predominantly the N-terminal domain, heterologous (GLP-1 7–36 amide) and homologous (exendin 9–39) competition-binding assays were performed, respectively, using [125I]exendin 9–39 as the radioligand as described below.
Transfection of cells
The growth media of cells in 160-cm2 flask at approximately 80% confluency was replaced with OptiMEM media, and the cells were transfected using a 1:4 ratio of DNA to Lipofectamine 2000 reagent (e.g. 20 μg DNA and 80 μl Lipofectamine 2000) following the manufacturer's instructions. Cells were then cultured for 6 h before the media was replaced with normal growth media. After a further 42 h, cells were used as described below.
Membrane preparation
Adherent cells were removed from tissue culture flasks using Accutase and washed in Dulbecco's PBS before being pelleted by centrifugation (600 × g, 10 min, 4 C). The pellets were then resuspended in ice-cold membrane buffer (20 mm HEPES; 1 mm EDTA; 1 mm EGTA, pH 7.4) containing protease inhibitor cocktail (one tablet 50 ml−1) and homogenized using 50 strokes of a Dounce glass homogenizer (Fisher Scientific, Loughborough, UK). The homogenate was then centrifuged at 600 × g for 10 min at 4 C. The resulting supernatant was then centrifuged at 48,000 × g for 1.5 h at 4 C, and the resulting pellet was resuspended in ice-cold membrane buffer. Samples were stored at −80 C until use.
Assay
Membrane-based binding assays were carried out in round-bottomed 96-well plates in a total volume of 200 μl with component parts being diluted in assay buffer [Hank's Balanced Salt Solution (HBSS); 1.26 mm CaCl2, 0.493 mm MgCl2, 20 mm HEPES, 0.1% (wt/vol) BSA, pH 7.4]. Initial titration experiments were performed to determine the concentration of each membrane giving a maximal total binding of approximately 1500–2000 cpm and ligand depletion of less than 10%. For the assay itself, membranes, [125I]exendin 9–39 (final concentration 0.1 nm) and either exendin 9–39 (homologous competition assays) or GLP-1 7–36 amide (heterologous competition assays) at a range of concentrations was added and binding allowed to proceed to equilibrium at room temperature for 4 h. Using a Brandel Cell Harvester that had been washed with 2% (wt/vol) BSA, membranes were collected on Whatman GF/C glass fiber filters presoaked in 0.5% polyethyleneimine. Membranes were then washed with ice-cold buffer (composition: 25 mm HEPES; 1.5 mm CaCl2; 1 mm MgSO4; 100 mm NaCl, pH 7.4) and filters were allowed to dry. Bound radioactivity was determined using a TopCount-NXT liquid scintillation counter (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Data analysis
For each receptor construct, homologous competition binding curves were constructed assuming one class of binding site. The Kd and Bmax values were determined using standard analysis of homologous competition binding data (GraphPad Software Inc., San Diego, CA). Heterologous competition binding curves were similarly constructed, and the Cheng-Prusoff equation was applied to calculate the KI values for GLP-1 7–36 amide using an affinity estimate determined by standard analysis of homologous competition binding data (GraphPad Software, Inc.) and also the Kd of exendin 9–39 determined from the homologous competition-binding assays. Heterologous competition binding data were compared using a one-site and two-site fit (GraphPad Software, Inc.) but in all cases were best fit by the one-site model (P > 0.05, data not shown), which has therefore been presented. Data are expressed as mean ± sem unless otherwise stated. Statistical analysis was by one-way ANOVA and, where P < 0.05, followed by Dunnett's post hoc test against the WT hGLP-1R.
Generation and measurement of cAMP
Transfection of cells
HEK-293 cells were seeded into six-well tissue culture plates (1 × 106 cells per well−1) in culture media and allowed to adhere overnight. The medium was then changed to OptiMEM and cells were transfected with 2 μg DNA and 8 μl Lipofectamine 2000 per well following the manufacturer's instructions. Cells were cultured for 6 h before the media was replaced with cell culture media. After a further 42 h, cells were used as described below.
Assay
Media was removed, after which the cells were collected using Accutase and pelleted by centrifugation (170 × g, 5 min). Cells were washed in assay buffer [HBSS; 1.26 mm CaCl2, 0.493 mm MgCl2, 20 mm HEPES, 0.1% (wt/vol) BSA, pH 7.4] collected by centrifugation and resuspended at a density of 0.526 × 106 cells ml−1 in assay buffer containing 1 mm 3-isobutyl-1-methylxanthine (IBMX). After 15 min, cells were added to round-bottomed 96 well plates (95,000 cells well−1) containing either GLP-1 7–37 at a range of concentrations or alternatively 50 μm FSK to directly activate adenylyl cyclase. Plates were incubated at 37 C for 1 h before stimulations were terminated with 100 μl of lysis buffer (cAMP Biotrak Enzymeimmunoassay System kit). cAMP levels were then determined following the manufacturer's instructions.
Data analysis
A standard curve was constructed and interpolated to determine the concentration of cAMP. Data are expressed as a percentage of the cAMP produced in response to 50 μm FSK. Concentration-response curves were fitted (GraphPad Software Inc.,) and EC50 values were determined. These are expressed as mean ± sem. Statistical analysis was by one-way ANOVA and, where P < 0.05, followed by Dunnett's post hoc test against the WT hGLP-1R.
Three-dimensional model and helical wheel projection of the hGLP-1R
A three-dimensional model of the human hGLP-1R (T29-L422) in complex with GLP-1 was constructed through a process of comparative modeling. Initial modeling was performed using MOE (Chemical Computing Group, Montreal, Quebec, Canada), whereas subsequent refinement and optimization were performed using Prime (Schrödinger, Inc., Portland, OR). To model the receptor in its active state, the structure of active opsin (31) (PDB code: 3DQB) was used as a template for the transmembrane domain. Initial alignment of these two sequences followed the principles laid out by Bissantz (52) in aligning family A and family B GPCRs. The alignment of the second extracellular loop was set so as to conserve the important cross-link between Cys226 and Cys296, observed in family B GPCRs (45). The structure of the human β2-adrenergic receptor has just been reported, stabilized in an agonist-bound active state by a camelid antibody fragment (nanobody) that mimics Gαs binding (42). The structural changes on activation are very similar to those of the active state of opsin, used for homology modeling in the present study, and consequently the use of the active state β2-adrenergic receptor structure as a template would not be expected to enhance the model further.
For each modeling run, 10 different models were constructed, based on alternative amino acid conformations, employing the AMBER99 force field. The best model, according to the scoring function, was selected for further refinement. The extracellular loops EC1 and EC2 were rebuilt (part of EC2 was retained to maintain the Cys cross-link) to create space to accommodate the GLP-1 ligand. The ligand was manually docked into the cavity, and its position was refined using the AMBER99 force field. The bound ligand conformation is modeled as two α-helical regions with a flexible region connecting the two (53). Because the precise conformation of the ligand where it interacts with the transmembrane domain is unknown, this region (H7-F12) was modeled with the various conformations of the solution nuclear magnetic resonance structure of exendin-4 (1JRJ) as the template. The best five conformations were manually selected, and the local structure was allowed to relax within the force field. The best fitting of these was then chosen by manual inspection. The N-terminal domain structure is taken from the x-ray crystal structure of this isolated unit (23) (PDB code: 3IOL). Although the relative orientation of the N-terminal domain and transmembrane domain is uncertain, some interactions between the ligand and the transmembrane domain (19) and between the ligand and the N-terminal domain (from the crystal structure) are understood. Therefore, in constructing the model, we were guided by the placement of the ligand with respect to both domains. The connecting loop between these two domains was built from residue structure libraries and optimized with the AMBER99 force field.
One of the difficulties in modeling family B GPCRs is the differing positions of proline residues between the model sequence and the template. Proline, being unable to take part in hydrogen bonding necessary for helix formation, results in a pronounced kink in the helix. Failure to account for this misalignment would mean a kink in the model where no kink is due and no kink where a proline residue is located. In an effort to account for this, the helices in which proline misalignment was identified were modeled based on alternative templates. A search of the PDB revealed no template structures with high similarity to the sequences of the individual helices. Therefore, the alternative templates used were the other transmembrane helices of the opsin structure, aligned in place to override the original template as indicated in Table 4. These override sections were specifically aligned to match the proline position and the best of the available templates chosen by , root mean squared deviation to the original template at either end of the helix.
For illustration, a helical wheel projection was constructed from the final model (using a program by Armstrong and Zidovetzki, available from http://rzlab.ucr.edu/scripts/wheel) to best represent the orientation of the residues in the upper (extracellular face) sections of the transmembrane domain. Some simplifications have been made; for example, kinks are not represented and the relative position of each helix is set by the location close to the ligand-binding site. Some helices, particularly TM3, are not perpendicular to the membrane and so are less well represented by the helical wheel model, toward the intracellular side of the membrane.
Materials
All tissue culture plastics were purchased from Nunc (VWR International, Lutterworth, UK). DMEM, OptiMEM, fetal bovine serum, HBSS, Lipofectamine 2000, One Shot TOP10 competent cells, pcDNA5/FRT, and HEK-FlpIn cells were purchased from Invitrogen (Paisley, UK). Accutase was obtained from Innovative Cell Technologies (San Diego, CA). GLP-1 7–36 amide, GLP-1 7–37, and exendin 9–39 were purchased from Bachem (Weil am Rhein, Germany). The cAMP Biotrak Enzymeimmunoassay kit was obtained from Amersham Biosciences (GE Healthcare UK Ltd, Little Chalfont, UK). Whatman GF/C glass filters and [125I]exendin 9–39 (specific activity 2200Ci mmol−1) were obtained from PerkinElmer (Waltham, MA). Complete protease inhibitor cocktail tablets were purchased from Roche Diagnostics (Basel, Switzerland). All primers for mutagenesis and sequencing were obtained from Eurogentec (Southampton, UK). Pfu Turbo Hotstart PCR master mix, PCR-Script, and the QuikChange (II and XL) Site-Directed mutagenesis kits were purchased from Stratagene (La Jolla, CA). QIAquick Gel Extraction Kit, QIAquick PCR purification kit, QIAprep Spin Miniprep Kit, and Qiagen HiSpeed Plasmid Maxi-prep kit were all obtained from QIAGEN (Crawley, UK). Restriction enzymes were from New England Biolabs (Ipswich, MA). All other chemicals and reagents were purchased from Sigma-Aldrich (Gillingham, UK).
Acknowledgments
Disclosure Summary: R.W., G.R., A.J.H.B., G.F.W. and D.T. are employees of AstraZeneca, (Alderely Park, Macclesfield UK). K.C. and G.B.W. received financial support from AstraZeneca for this study.
Footnotes
- EC
- Extracellular loop
- FSK
- forskolin
- GLP-1
- glucagon-like peptide-1
- GLP-1R
- GLP-1 receptor
- GPCR
- G-protein-coupled receptor
- h
- human
- HBSS
- Hank's Balanced Salt Solution
- HEK
- human embryonic kidney
- hGLP-1R
- human GLP-1R
- TM
- transmembrane helix
- VPAC1
- vasoactive intestinal peptide receptor 1
- VPAC2
- vasoactive intestinal peptide receptor 2
- WT
- wild type.
References
- 1. Orskov C , Rabenhøj L , Wettergren A , Kofod H , Holst JJ. 1994. Tissue and plasma concentrations of amidated and glycine- extended glucagon-like peptide I in humans. Diabetes 43:535–539 [DOI] [PubMed] [Google Scholar]
- 2. Bavec A , Hällbrink M , Langel U , Zorko M. 2003. Different role of intracellular loops of glucagon-like peptide-1 receptor in G-protein coupling. Regul Pept 111:137–144 [DOI] [PubMed] [Google Scholar]
- 3. Hällbrink M , Holmqvist T , Olsson M , Ostenson CG , Efendic S , Langel U. 2001. Different domains in the third intracellular loop of the GLP-1 receptor are responsible for Gαs and Gαi/Gαo activation. Biochim Biophys Acta 1546:79–86 [DOI] [PubMed] [Google Scholar]
- 4. Montrose-Rafizadeh C , Avdonin P , Garant MJ , Rodgers BD , Kole S , Yang H , Levine MA , Schwindinger W , Bernier M. 1999. Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology 140:1132–1140 [DOI] [PubMed] [Google Scholar]
- 5. Baggio LL , Drucker DJ. 2007. Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2157 [DOI] [PubMed] [Google Scholar]
- 6. Doyle ME , Egan JM. 2007. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther 113:546–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kieffer TJ , McIntosh CH , Pederson RA. 1995. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136:3585–3596 [DOI] [PubMed] [Google Scholar]
- 8. Mentlein R , Gallwitz B , Schmidt WE. 1993. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 214:829–835 [DOI] [PubMed] [Google Scholar]
- 9. Eng J , Kleinman WA , Singh L , Singh G , Raufman JP. 1992. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem 267:7402–7405 [PubMed] [Google Scholar]
- 10. Gallwitz B. 2006. Exenatide in type 2 diabetes: treatment effects in clinical studies and animal study data. Int J Clin Pract 60:1654–1661 [DOI] [PubMed] [Google Scholar]
- 11. Hoare SRJ. 2005. Mechanisms of peptide and nonpeptide ligand binding to class B G-protein coupled receptors. Drug Discov Today 10:417–427 [DOI] [PubMed] [Google Scholar]
- 12. Parthier C , Reedtz-Runge S , Rudolph R , Stubbs MT. 2009. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci 34:303–310 [DOI] [PubMed] [Google Scholar]
- 13. López de Maturana R , Willshaw A , Kuntzsch A , Rudolph R , Donnelly D. 2003. The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J Biol Chem 278:10195–10200 [DOI] [PubMed] [Google Scholar]
- 14. Wilmen A , Göke B , Göke R. 1996. The isolated N-terminal extracellular domain of the glucagon-like peptide-1 (GLP)-1 receptor has intrinsic binding activity. FEBS Lett 398:43–47 [DOI] [PubMed] [Google Scholar]
- 15. Xiao Q , Jeng W , Wheeler MB. 2000. Characterization of glucagon-like peptide-1 receptor-binding determinants. J Mol Endocrinol 25:321–335 [DOI] [PubMed] [Google Scholar]
- 16. Al-Sabah S , Donnelly D. 2003. A model for receptor-peptide binding at the glucagon-like peptide-1 (GLP-1) receptor through the analysis of truncated ligands and receptors. Br J Pharmacol 140:339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Montrose-Rafizadeh C , Yang H , Rodgers BD , Beday A , Pritchette LA , Eng J. 1997. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J Biol Chem 272:21201–21206 [DOI] [PubMed] [Google Scholar]
- 18. Al-Sabah S , Donnelly D. 2003. The positive charge at Lys-288 of the glucagon-like peptide-1 (GLP-1) receptor is important for binding the N-terminus of peptide agonists. FEBS Lett 553:342–346 [DOI] [PubMed] [Google Scholar]
- 19. López de Maturana R , Donnelly D. 2002. The glucagon-like peptide-1 receptor binding site for the N-terminus of GLP-1 requires polarity at Asp198 rather than negative charge. FEBS Lett 530:244–248 [DOI] [PubMed] [Google Scholar]
- 20. Perret J , Vertongen P , Solano RM , Langer I , Cnudde J , Robberecht P , Waelbroeck M. 2002. Two tyrosine residues in the first transmembrane helix of the human vasoactive intestinal peptide receptors play a role in supporting the active conformation. Br J Pharmacol 136:1042–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Q , Pinon DI , Miller LJ , Dong M. 2010. Spatial approximations between residues 6 and 12 in the amino-terminal region of glucagon-like peptide 1 and its receptor. A region critical for biological activity. J Biol Chem 285:24508–24518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Q , Pinon DI , Miller LJ , Dong M. 2009. Molecular basis of glucagon-like peptide 1 docking to its intact receptor studied with carboxyl-terminal photolabile probes. J Biol Chem 284:34135–34144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Underwood CR , Garibay P , Knudsen LB , Hastrup S , Peters GH , Rudolph R , Reedtz-Runge S. 2010. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem 285:723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dillon JS , Tanizawa Y , Wheeler MB , Leng XH , Ligon BB , Rabin DU , Yoo-Warren H , Permutt MA , Boyd AE. 1993. Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology 133:1907–1910 [DOI] [PubMed] [Google Scholar]
- 25. Kieffer TJ , Heller RS , Unson CG , Weir GC , Habener JF. 1996. Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology 137:5119–5125 [DOI] [PubMed] [Google Scholar]
- 26. Thorens B. 1992. Expression cloning of the pancreatic β cell receptor for the gluco-incretin hormone glucagon-like peptide-1. Proc Natl Acad Sci USA 89:8641–8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thorens B , Porret A , Bühler L , Deng SP , Morel P , Widmann C. 1993. Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9–39) an antagonist of the receptor. Diabetes 42:1678–1682 [DOI] [PubMed] [Google Scholar]
- 28. Runge S , Schimmer S , Oschmann J , Schiodt CB , Knudsen SM , Jeppesen CB , Madsen K , Lau J , Thøgersen H , Rudolph R. 2007. Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry 46:5830–5840 [DOI] [PubMed] [Google Scholar]
- 29. Runge S , Thøgersen H , Madsen K , Lau J , Rudolph R. 2008. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem 283:13440–11347 [DOI] [PubMed] [Google Scholar]
- 30. Lin F , Wang R. 2009. Molecular modeling of the three-dimensional structure of GLP-1R and its interactions with several agonists. J Mol Model 15:53–65 [DOI] [PubMed] [Google Scholar]
- 31. Scheerer P , Park JH , Hildebrand PW , Kim YJ , Krauss N , Choe HW , Hofmann KP , Ernst OP. 2008. Crystal structure of opsin in its G-protein-interacting conformation. Nature 455:497–502 [DOI] [PubMed] [Google Scholar]
- 32. Huang Y , Wilkinson GF , Willars GB. 2010. Role of the signal peptide in the synthesis and processing of the glucagon-like peptide-1 receptor. Br J Pharmacol 159:237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Di Paolo E , De Neef P , Moguilevsky N , Petry H , Bollen A , Waelbroeck M , Robberecht P. 1998. Contribution of the second transmembrane helix of the secretin receptor to the positioning of secretin. FEBS Lett 424:207–210 [DOI] [PubMed] [Google Scholar]
- 34. Du K , Nicole P , Couvineau A , Laburthe M. 1997. Aspartate 196 in the first extracellular loop of the human VIP1 receptor is essential for VIP binding and VIP-stimulated cAMP production. Biochem Biophys Res Commun 230:289–292 [DOI] [PubMed] [Google Scholar]
- 35. Solano RM , Langer I , Perret J , Vertongen P , Juarranz MG , Robberecht P , Waelbroeck M. 2001. Two basic residues of the h-VPAC1 receptor second transmembrane helix are essential for ligand binding and signal transduction. J Biol Chem 276:1084–1088 [DOI] [PubMed] [Google Scholar]
- 36. Vertongen P , Solano RM , Perret J , Langer I , Robberecht P , Waelbroeck M. 2001. Mutational analysis of the human vasoactive intestinal peptide receptor subtype VPAC2: role of basic residues in the second transmembrane helix. Br J Pharmacol 133:1249–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tokuyama Y , Matsui K , Egashira T , Nozaki O , Ishizuka T , Kanatsuka A. 2004. Five missense mutations in glucagon-like peptide 1 receptor gene in Japanese population. Diabetes Res Clin Pract 66:63–69 [DOI] [PubMed] [Google Scholar]
- 38. Beinborn M , Worrall CI , McBride EW , Kopin AS. 2005. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept 130:1–6 [DOI] [PubMed] [Google Scholar]
- 39. López de Maturana R , Treece-Birch J , Abidi F , Findlay JBC , Donnelly D. 2004. Met-204 and Tyr-205 are together important for binding GLP-1 receptor agonists but not their N-terminally truncated analogues. Protein Pept Lett 11:15–22 [DOI] [PubMed] [Google Scholar]
- 40. Adelhorst K , Hedegaard BB , Knudsen LB , Kirk O. 1994. Structure-activity studies of glucagon-like peptide-1. J Biol Chem 269:6275–6278 [PubMed] [Google Scholar]
- 41. Crocker E , Eilers M , Ahuja S , Hornak V , Hirshfeld A , Sheves M , Smith SO. 2006. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J Mol Biol 357:163–172 [DOI] [PubMed] [Google Scholar]
- 42. Rasmussen SG , Choi HJ , Fung JJ , Pardon E , Casarosa P , Chae PS , Devree BT , Rosenbaum DM , Thian FS , Kobilka TS , Schnapp A , Konetzki I , Sunahara RK , Gellman SH , Pautsch A , Steyaert J , Weis WI , Kobilka BK. 2011. Structure of a nanobody-stabilized active state of the b2 adrenoceptor. Nature 469:175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas BE , Sharma S , Mierke DF , Rosenblatt M. 2009. PTH and PTH antagonist induce different conformational changes in the PTHR1 receptor. J Bone Miner Res 24:925–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller LJ , Chen Q , Lam PC , Pinon DI , Sexton PM , Abagyan R , Dong M. 2011. Refinement of glucagon-like peptide 1 docking to its intact receptor using mid-region photolabile probes and molecular modelling. J Biol Chem 286:15895–15907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Unson CG. 2002. Molecular determinants of glucagon receptor signaling. Biopolymers 66:218–235 [DOI] [PubMed] [Google Scholar]
- 46. Mann RJ , Al-Sabah S , de Maturana RL , Sinfield JK , Donnelly D. 2010. Functional coupling of Cys-226 and Cys -296 in the glucagon-like peptide-1 (GLP-1) receptor indicates a disulfide bond that is close to the activation pocket. Peptides 31:2289–2293 [DOI] [PubMed] [Google Scholar]
- 47. Nachtergael I , Gaspard N , Langlet C , Robberecht P , Langer I. 2006. Asn229 in the third helix of VPAC1 receptor is essential for receptor activation but not for receptor phosphorylation and internalization: comparison with Asn216 in VPAC2 receptor. Cell Signalling 18:2121–2130 [DOI] [PubMed] [Google Scholar]
- 48. Dong M , Li Z , Pinon DI , Lybrand TP , Miller LJ. 2004. Spatial approximation between the amino terminus of a peptide agonist and the top of the sixth transmembrane segment of the secretin receptor. J Biol Chem 279:2894–2903 [DOI] [PubMed] [Google Scholar]
- 49. Bisello A , Adams AE , Mierke DF , Pellegrini M , Rosenblatt M , Suva LJ , Chorev M. 1998. Parathyroid hormone-receptor interactions identified directly by photocross-linking and molecular modeling studies. J Biol Chem 273:22498–22505 [DOI] [PubMed] [Google Scholar]
- 50. Monaghan P , Thomas BE , Woznica I , Wittelsberger A , Mierke DF , Rosenblatt M. 2008. Mapping peptide hormone-receptor interactions using a disulfide-trapping approach. Biochemistry 47:5889–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dong M , Pinon DI , Miller LJ. 2005. Insights into the structure and molecular basis of ligand docking to the G protein-coupled secretin receptor using charge-modified amino-terminal agonist probes. Mol Endocrinol 19:1821–1836 [DOI] [PubMed] [Google Scholar]
- 52. Bissantz C , Logean A , Rognan D. 2004. High-throughput modeling of human G-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J Chem Inf Comput Sci 44:1162–1176 [DOI] [PubMed] [Google Scholar]
- 53. Murage EN , Schroeder JC , Beinborn M , Ahn JM. 2008. Search for α-helical propensity in the receptor-bound conformation of glucagon-like peptide-1. Bioorg Med Chem 16:10106–10112 [DOI] [PubMed] [Google Scholar]
- 54. Henikoff S , Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 89:10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skrabanek L , Campagne F , Weinstein H. 2003. Building protein diagrams on the web with the residue-based diagram editor RbDe. Nucleic Acids Res 31:3856–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]