

Abstract
Inherited modifications in protein structure frequently cause a loss-of-function by interfering with protein synthesis, transport, or stability. For the obesity-linked melanocortin-4 receptor (MC4R) and other G protein-coupled receptors, many mutants are intracellular retained. The biogenesis and trafficking of G protein-coupled receptors are regulated by multiple factors, including molecular chaperone networks. Here, we have investigated the ability of the cytosolic cognate 70-kDa heat-shock protein (Hsc70) chaperone system to modulate cell surface expression of MC4R. Clinically occurring MC4R mutants S58C, P78L, and D90N were demonstrated to have reduced trafficking to the plasma membrane and to be retained at the endoplasmic reticulum (ER). Analyses by fluorescence recovery after photobleaching revealed that the mobility of MC4R mutant protein at the ER was reduced, implying protein misfolding. In cells expressing MC4R, overexpression of Hsc70 resulted in increased levels of wild-type and mutant receptors at the cell surface. MC4R and Hsc70 coimmunoprecipitated, and fluorescence recovery after photobleaching analyses showed that increasing cellular levels of Hsc70 promoted the mobility of ER retained MC4R. Moreover, expression of HSJ1b, a cochaperone that enhances degradation of Hsc70 clients, reduced cellular levels of MC4R. Hsp70 and Hsp90 chaperone systems collaborate in the cellular processing of clients. For MC4R, inhibition of endogenous Hsp90 by geldanamycin reduced receptor levels. By contrast, expression of the Hsp90 cochaperone Aha1 (activator of Hsp90 ATPase) increased cellular levels of MC4R. Finally, we demonstrate that signaling of intracellular retained MC4R mutants is increased in cells overexpressing Hsc70. These data indicate that cytosolic chaperone systems can facilitate rescue of intracellular retained MC4R by improving folding. They also support proteostasis networks as a potential target for MC4R-linked obesity.
The melanocortin-4 receptor (MC4R) is a G protein-coupled receptor (GPCR) that plays an important role in maintaining energy homeostasis by influencing feeding. Normal activation of MC4R in the central nervous system decreases food intake and increases energy expenditure, leading to a reduction of body fat stores (1). MC4R controls food intake dependent on its agonist α-melanocyte stimulating hormone (α-MSH), which acts as a satiety signal, and its antagonist Agouti-related protein, which provides an orexigenic signal. Integration of α-MSH and Agouti-related protein inverse/competitive antagonist binding regulates MC4R signaling via activation of adenylate-dependent production of cAMP. Genome-wide association studies have identified common variants near MC4R as associated with obesity and insulin resistance (2). Polymorphisms associated with obesity include those that have a potential effect on the regulation of MC4R expression. Furthermore, mutations in MC4R result in severe early onset obesity that is inherited in a codominant manner (3–5). In patients with heterozygous mutations in MC4R obesity is generally thought to be due to haploinsufficiency. However, some mutants may have a dominant-negative effect as a consequence of dimerization with wild-type (WT) receptor (6). Mutations have been reported at a prevalence of 1/1000 in the general United Kingdom population and account for up to 6% of all cases of severe obesity in some studies (5, 7, 8). MC4R must traffic to the cell surface for normal function. The most common class of clinically occurring MC4R mutants causes intracellular retention of the receptor (5, 9–11).
Polytopic membrane proteins, such as GPCR, are synthesized at the endoplasmic reticulum (ER) and trafficked by the secretory pathway to sites of function. Exit from the ER is regulated by quality-control systems and is conditional on proteins achieving their native conformation (12, 13). Aberrantly folded polypeptides are retained in the ER until retrotranslocation into the cytosol and degradation by the ubiquitin-proteasome system. Protein folding, quality control, translocation, and degradation are dependent on molecular chaperone systems, with ER chaperones playing essential roles in the processing of membrane proteins. These include the lectin-like chaperones calnexin and calreticulin, the ER luminal 70-kDa heat-shock protein (Hsp70) family member BiP (HSPA5) and the glucose regulated protein Grp94, the ER paralog of Hsp90. For membrane proteins, mutations that cause conformational rearrangement may increase the ER's burden of aberrantly folded peptides, disrupting ER homeostasis and causing ER stress. The accumulation of aberrantly folded proteins at the ER triggers the unfolded protein response (UPR), which involves the up-regulation of ER chaperone expression and activation of ER-associated degradation pathways (12, 13). For example, when the archetypal GPCR rhodopsin harbors the clinically occurring P23H mutation, it is folding defective, accumulates in the ER, and stimulates the UPR (14). Moreover, there is evidence that stabilizing protein structure and promoting chaperone-mediated protein homeostasis (proteostasis) can increase functional expression of mutated ER retained proteins in some paradigms. These include MC4R, where cell surface expression of ER retained mutants has been reported to be rescued by the chemical chaperone 4-phenyl butyric acid (15).
Cytoplasmic domains of transmembrane proteins are also able to interact with molecular chaperones that reside in the cytosol. This is illustrated by the essential cognate cytosolic Hsp70 protein Hsc70 (HSPA8), which has been demonstrated to interact with cytoplasmic domains of the nonglycosylated form of the GPCR angiotensin II type 1 receptor (16). Hsc70 functions in multiple cellular processes. These include polypeptide folding, protein degradation, translocation across membranes, and protein-protein interactions. Hsc70 does not function in isolation but is dependent on cochaperones that regulate its ATP-dependent cycles of client protein binding and release. Key regulators are J-domain containing DnaJ/Hsp40 proteins that recruit Hsc70 to specific cellular locations and roles (17–19). The J-protein family incorporates members that are anchored to the cytoplasmic face of the ER by prenylation, e.g. HSJ1b (DNAJB2) (20). Hsc70 and associated J-proteins are part of a wider chaperone network functioning with cytosolic Hsp90 in folding and quality control of some clients. Hsp90 also operates in an ATP-dependent manner, is regulated by cochaperones, and has diverse cellular roles (21). These include a possible function in quality control of some polytopic membrane proteins, because the Hsp90 cochaperone Aha1 (activator of Hsp90 ATPase) has been shown to modulate trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR)ΔF508 mutant, which is aberrantly folded (22).
In this study, we identify that the functional expression of WT and intracellular retained mutant MC4R is modulated by expression of cytosolic chaperones. Specifically, we show that Hsc70 interacts with MC4R and promotes cell surface expression of intracellular retained mutants resulting in increased signaling in response to the α-MSH analogue Nle4, DPhe7-α-MSH (NDP-MSH). Moreover, the DnaJ protein HSJ1b, which acts as a shuttling factor for the sorting of Hsp70 clients to the proteasome (23), reduces cellular levels of MC4R. Inhibition of Hsp90's ATP binding also reduces cellular levels of MC4R, whereas stimulation of its ATPase activity has the converse effect. Together, these data indicate the cytosolic Hsp70/Hsp90 chaperone system has the ability to modulate GPCR processing and can facilitate functional rescue of misfolded MC4R mutants.
Results
Mutations in MC4R cause intracellular retention of the receptor at the ER
For clinically occurring MC4R mutants, intracellular retention is the most common molecular defect. To investigate MC4R misfolding, we selected three mutants, S58C, P78L, and D90N (for further details, please see Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) that have been reported to have abnormal levels of cell surface expression (9, 15, 24, 25). To confirm that these MC4R mutants did not traffic efficiently to the plasma membrane (PM), we compared their subcellular localization and levels of cell surface expression with that of WT MC4R. HEK293 cells were transfected with a vector for expression of WT or mutant hemagglutinin tagged MC4R (HA-MC4R), processed for immunofluorescence, and imaged by confocal microscopy (Fig. 1A). WT HA-MC4R partially localized to the PM, whereas S58C, P78L, and D90N showed a more reticular distribution that overlapped with an ER marker (Fig. 1 and Supplemental Fig. 1A). To quantify cell surface expression of MC4R mutants, an In-Cell Western based assay was used where only the extracellular HA tag of HA-MC4R was detected and compared with total HA-MC4R. Levels of receptor at the PM, relative to total cellular levels, were quantified for mutants and compared with cell surface expression of the WT protein (Fig. 1, B and C). HA-MC4R mutants S58C, P78L, and D90N showed an approximate 80% reduction in levels of receptor at the cell surface. To further confirm this reduction in cell surface expression, cells that had not been detergent permeabilized were processed for immunofluorescence. For WT HA-MC4R, cell surface expression was detected. This staining was greatly reduced for mutants where the extracellular HA-MC4R was at very low levels at the cell surface (Supplemental Fig. 1B). The reduction in cell surface levels of MC4R mutants correlated with a reduction in cAMP activity measured using a luciferase reporter assay (Fig. 1D).
Fig. 1.

MC4R mutants S58C, P78L, and D90N are intracellular retained. A, Confocal analyses of HEK293 cells expressing either WT HA-MC4R, HA-MC4R(S58C), HA-MC4R(P78L), or HA-MC4R(D90N). After fixation, cells were permeabilized and processed for immunofluorescent detection of HA-MC4R (red). Scale bar, 10 μm. B and C, Cell surface expression of WT HA-MC4R was compared with that of HA-MC4R mutants S58C, P78L, and D90N. Levels of WT and mutant HA-MC4R were quantified by immunofluorescent detection of the extracellular HA tag in formaldehyde-fixed HEK293 cells. Total cellular levels of the HA tag were detected after PM permeabilization with detergent (Triton-X 100), to allow entry of anti-HA and fluorescent dye-conjugated secondary antibodies into cells. This In-Cell Western assay was performed in 24-well cell culture plates 16 h after transfection. Visualization (B) and quantification (C) of the fluorescent signals was achieved using an infrared imaging system. D, Using a luciferase reporter system for cAMP activity, MC4R signaling was quantified for WT and mutant HA-MC4R. Cells were cotransfected with plasmids for expression of WT or mutant MC4R, a luciferase reporter construct for MC4R signaling, and a vector for renilla expression. Sixteen hours after transfection, cells were stimulated with 10−6 m NDP-MSH for 6 h, and luciferase activity was measured. Values were normalized to renilla activity to control for variability in transfection. Error bars represent 2× se. Con, Control.
Misfolded MC4R mutants are aggregation prone and have reduced diffusional mobility
Misfolded proteins are prone to aggregation and have altered chaperone interactions. We generated constructs for expression of MC4R as a fusion with green fluorescent protein (MC4R-GFP). In HEK293, localization of WT MC4R-GFP was predominantly PM, whereas the S58C, P78L, and D90N mutants were intracellular retained (Fig. 2A), similar to the HA-MC4R mutants. Because MC4R is expressed in neurons, in the paraventricular nucleus of the hypothalamus, and other brain regions, we also investigated subcellular localization of the MC4R-GFP fusion proteins in a neuronal cell line. SK-N-SH cells were transfected for expression of WT and mutant MC4R-GFP. Again, WT MC4R-GFP localized at the PM and was associated with intracellular membranes, whereas mutants exhibited a more perinuclear localization. Interestingly, SK-N-SH cells expressing mutant MC4R-GFP frequently had a more rounded phenotype with inclusions present in many cells (Fig. 2B). We quantified the incidence of inclusions in MC4R-GFP expressing cells, blind to experimental status, 24 h after transfection (Fig. 2C). There was a higher incidence of MC4R-GFP inclusions present in S58C (37 ± 6%), P78L (56 ± 15%), and D90N (31 ± 7%) than WT (12 ± 3%) expressing cells.
Fig. 2.

Mutant MC4R forms intracellular inclusions in a neuronal cell line. A and B, Confocal analyses of HEK293 (A) or SK-N-SH (B) cells expressing either WT MC4R-GFP, MC4R(S58C)-GFP, MC4R(P78L)-GFP, or MC4R(D90N)-GFP. Scale bar, 10 μm. C, Quantification of the incidence of inclusions in WT and mutant MC4R-GFP expressing cells. A minimum of 50 transfected cells was scored for each treatment from three separate transfections. Scoring was conducted blind to experimental status 24 h after transfection. Error bars represent 2× se. *, P < 0.05.
To further probe the characteristics of intracellular retained MC4R mutants S58C, P78L, and D90N, we used fluorescence recovery after photobleaching (FRAP). FRAP has been used extensively to study protein mobility, and indirectly protein aggregation, within the ER, for example with a temperature sensitive mutant of vesicular stomatitis virus G protein (26). For FRAP analyses, the presence of WT MC4R at the PM meant that bleaching had to be confined to the ER; therefore, regions (2 μm2) of perinuclear reticular fluorescence were selected, photobleached, and recovery monitored. For WT MC4R-GFP greater than 80%, recovery had occurred by 40 sec after photobleaching (Fig. 3). No gross changes in the architecture of the ER were observed. For MC4R mutants, we envisaged that disrupted normal folding would alter diffusional mobility in ER membranes. For all mutants investigated, there was a significant reduction in the magnitude of FRAP compared with WT MC4R-GFP in HEK293 cells (Fig. 3, A and B). We also investigated the mobility of MC4R mutant proteins in the ER of SK-N-SH cells and observed similar results (Fig. 3, C and D). FRAP measurements were not made in cells with visible inclusions. Together, these data further support misfolding of MC4R mutants S58C, P78L, and D90N at the ER.
Fig. 3.
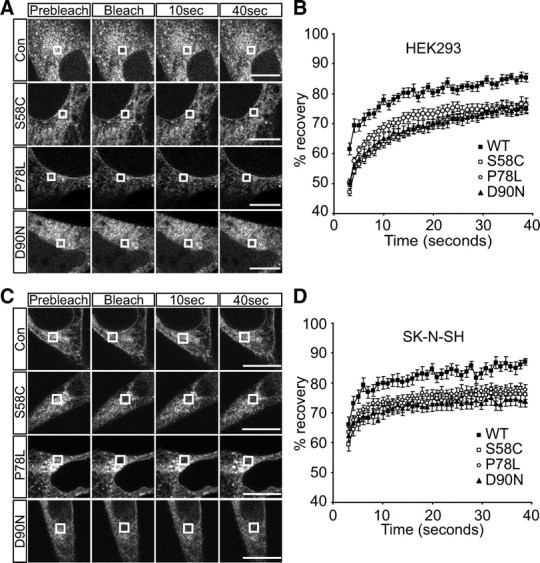
Misfolded MC4R is less mobile than WT protein at ER membranes. Representative images of live cell FRAP analysis of WT and mutant MC4R-GFP in HEK293 (A) and SK-N-SH cells (C). Graphical representation of recovery after photobleaching of WT and mutant MC4R-GFP in HEK293 (B) and SK-N-SH cells (D). A 2 × 2 μm area of the ER corresponding to the boxed areas was photobleached and the cells images every second for 40 sec. Fluorescent intensities of the 2 × 2 μm area were normalized to prebleach levels at 100%. Error bars represent se, n = 15. Con, Control.
Hsc70 modulates cell surface expression of MC4R
Polytopic membrane proteins can interact with both ER and cytosolic proteins. We hypothesized that, as well as ER chaperones, the core cytosolic Hsc70 chaperone systems would influence MC4R biogenesis and quality control. Therefore, In-Cell Western was used to compare PM expression (normalized for variations in total cellular levels) of HA-MC4R in cells cotransfected with plasmid for expression of Hsc70-V5 or empty V5 vector (Fig. 4A). A comparison was also made with two other Hsp70 family members (the ER protein BiP and the heat inducible Hsp70-3) and Hsp110 (data not shown), of these proteins Hsc70 coexpression altered cell surface levels of MC4R. Because chaperone systems that influence WT receptor biogenesis and quality control may be able to modulate trafficking of intracellular retained mutants, we next investigated the effects of Hsc70 expression on MC4R mutants. Hsc70 overexpression increased cell surface levels of transiently expressed mutant MC4R by more than 2-fold in HEK293 cells (Fig. 4A). Confocal analyses also supported increased cell surface expression of MC4R mutants S58C, P78L, and D90N in cells with exogenous GFP-Hsc70 (Fig. 4, B–E). CoIP was used to ascertain whether MC4R interacts with Hsc70. HEK293 cells were transfected for expression of epitope-tagged MC4R and Hsc70. HA-MC4R was IP, and immunoblot (IB) analyses detected bound V5-tagged Hsc70, indicating that MC4R can interact either directly or as part of a complex with Hsc70 (Fig. 4F).
Fig. 4.

Hsc70 increases cell surface levels of intracellular retained MC4R mutants and coimmunoprecipitates (coIP) with the receptor. A, HEK293 cells were transfected with vectors for expression of WT or mutant MC4R and Hsc70-V5, or V5 (empty vector). Cell surface levels of mutant MC4R are shown relative to WT protein. Error bars represent 2× se. *, P < 0.05. B–E, Confocal analysis of HEK293 cells expressing WT (B), S58C (C), P78L (D), or D90N (E) HA-MC4R in combination with GFP or Hsc70-GFP. Arrow indicates cell surface expression. Scale bar, 10 μm. F, HEK293A cells were transfected with plasmids for expression of HA-MC4R alone or in combination with Hsc70-V5. Cells were lysed, and immunoprecipitation (IP) of HA-MC4R was then performed. IP were IB with anti-HA and anti-V5. Hsc70-V5 is indicated by an arrow, and an asterisk highlights immunoglobulin.
Next, we generated clonal HEK293 cells with stable expression of WT or mutant HA-MC4R. Clones with the lowest levels of total MC4R expression that could be robustly detected were selected. Heterologous overexpression of Hsc70 in these cells resulted in an increase in both total and cell surface levels of mutant MC4R (Fig. 5, A and B). Significantly, there was a 6-fold increase in cell surface levels of MC4R(P78L). Together, these data strongly implicate a role for the Hsc70 chaperone machinery in the processing of WT and misfolded MC4R.
Fig. 5.

Hsc70 promotes cell surface expression of mutant MC4R in stable cell lines. A and B, Clonal HEK293 cell lines stably expressing WT or mutant MC4R were transfected for the expression of Hsc70–V5, or with the empty V5 vector. Total cellular levels (A) and cell surface expression (B) of MC4R were quantified by “In-Cell Western.” Error bars represent 2× se. *, P < 0.05.
Coexpression of Hsc70 reduces mutant MC4R-GFP inclusion incidence and increases ER mobility
SK-N-SH cells expressing intracellular MC4R mutants as GFP fusions frequently formed inclusions (Fig. 2, B and C). When Hsc70 was coexpressed with MC4R-GFP mutants, we observed a reduction in inclusion incidence (Fig. 6A). This was significant (P < 0.001) for P78L (18 ± 5%) and D90N (17 ± 3%). For S58C, the reduction in inclusion incidence was not significant, and for cells expressing WT MC4R-GFP, no change in inclusion incidence was observed.
Fig. 6.

Intracellular retained mutant MC4R inclusion incidence is reduced and mobility at the ER increased in cells with elevated levels of Hsc70. A, Quantification of the incidence of inclusions in WT and mutant MC4R-GFP expressing cells with and without heterologous expression of Hsc70. A minimum of 50 transfected cells were counted for each treatment from three separate transfections. Counting was blind to experimental status 24 h after transfection. Error bars represent 2× se. B–D, Graphical representation of recovery after photobleaching of MC4R-GFP mutants S58C (B), P78L (C), and D90N (D) in cells cotransfected with a plasmid for expression of Hsc70-V5 or empty V5 plasmid (Con). A 2 × 2 μm area of the ER corresponding to the boxed areas was photobleached, and the cells images every second for 40 sec. Fluorescent intensities of the 2 × 2 μm area were normalized to prebleach levels at 100%. Error bars represent se, n = 15.
To further investigate whether Hsc70 was potentially improving folding of mutant MC4R, we compared FRAP in MC4R-GFP expressing cells cotransfected with a plasmid for Hsc70 expression or empty vector. Coexpression of Hsc70 with MC4R mutants increased the degree of fluorescence recovery (Fig. 6, B–D). This increase was significant (P < 0.05) for S58C (Fig. 6B) and P78L (Fig. 6C). Hsc70 did not alter the mobility of WT MC4R (data not shown). This improved diffusional mobility of mutant MC4R in the presence of exogenous Hsc70 suggests reduced aggregation and/or altered chaperone interactions of the mutant proteins.
HSJ1b expression reduces cellular levels of MC4R
Hsc70 function is regulated by cochaperones. Of particular importance are the DnaJ cochaperones, which recruit Hsc70 to specific cellular functions and stimulate its ATPase activity. To test for involvement of DnaJ proteins in MC4R biogenesis and/or quality control, we used In-Cell Western analyses to investigate the effect of six family members on MC4R levels (Fig. 7A). We observed that coexpression of HSJ1b had a significant negative effect on MC4R cell surface levels. HSJ1b is anchored to the cytoplasmic face of the ER and promotes proteasomal degradation of Hsc70 clients. HSJ1b interaction with the ubiquitin-proteasome system is dependent on its ubiquitin interaction motif (UIM) (20, 23). To further investigate HSJ1b-mediated reduction in MC4R levels in HEK293 cells, transiently expressing WT HA-MC4R were cotransfected for expression of HSJ1b, an HSJ1b UIM mutant, or empty vector. Surprisingly, although coexpression of HSJ1b resulted in a significant reduction in total cellular levels of MC4R, this was only partly dependent on HSJ1b having a functional UIM domain (Fig. 7, B and C). CoIP indicated that HSJ1b was in a complex with MC4R (Fig. 7D). Contrastingly, DNAJB4, which was included as a negative control, because its heterologous expression did not alter MC4R levels (Fig. 7A), did not coIP (Fig. 7D).
Fig. 7.
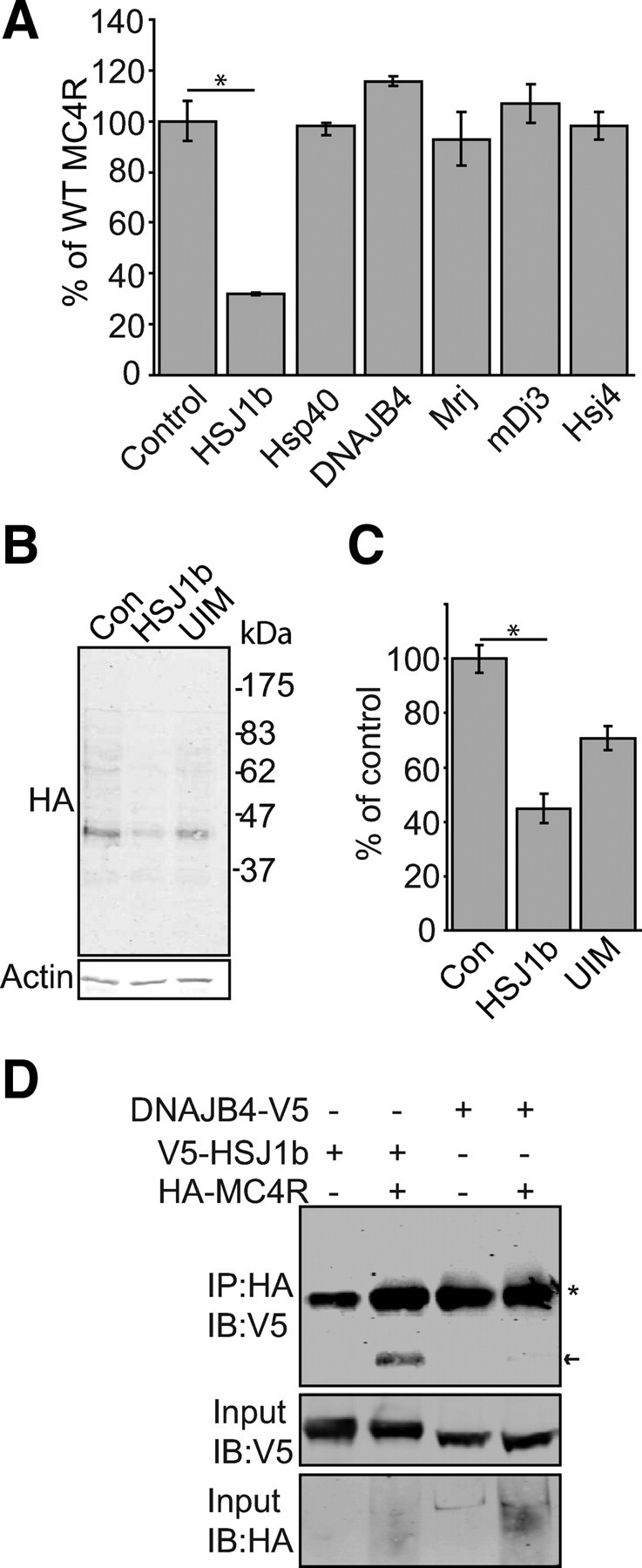
HSJ1b expression reduces cellular levels of MC4R. A, HEK293 cells were transiently cotransfected for expression of WT HA-MC4R and the DnaJ proteins HSJ1b (DNAJB2), Hsp40 (DNAJB1), Hsc40 (DNAJB4), Mrj (DNAJB6), mDj3 (DNAJA3), or Hsj4 (DNAJA4). In-Cell Western was performed to determine cell surface levels of MC4R, shown relative to a control, where pcDNA3.1 3xHA-MC4R was cotransfected with empty V5 vector. B, IB of HA-MC4R levels in HEK293 cells cotransfected with empty myc vector (Con) or vectors for expression of WT myc-HSJ1b (HSJ1b) or a UIM-deficient form of myc-HSJ1b (UIM). HA-MC4R IB detects multiple bands (these represent different glycoforms of the protein). C, Cell surface levels of HA-MC4R in cells coexpressing HSJ1b or HSJ1b(UIM) or transfected with empty vector (Con) were also analyzed by In-Cell Western 24 h after transfection. D, To investigate whether MC4R and HSJ1b are in a complex, coIP was performed. HEK293 cells were transfected with plasmids for expression of HA-MC4R alone or in combination with V5-HSJ1b or DNAJB4-V5, as a negative control. Cells were lysed, and IP of HA-MC4R was then performed. IP were IB with anti-HA and anti-V5. V5-HSJ1b is indicated by an arrow, and an asterisk highlights immunoglobulin. Error bars represent 2× se. *, P < 0.05.
Cellular levels of MC4R are altered by manipulation of the cytosolic Hsp90 chaperone system
Pharmacological induction of heat-shock proteins via the heat-shock transcription factor-1 pathway can be achieved using the Hsp90 inhibitor geldanamycin (GA). Therefore, to test whether treatment with this drug promoted MC4R trafficking, we treated cells transfected for expression of MC4R with GA. Surprisingly, after 24 h of expression, total cellular levels of WT HA-MC4R were significantly reduced, 37 ± 6% after 24 h of 1.0 μm GA treatment (Fig. 8A). Using the P78L mutant, we assessed whether this effect was observed for intracellular retained MC4R. Again, levels were reduced. This reduction in total cellular levels of WT MC4R was reflected by a reduction in levels at the cell surface (Fig. 8B). For P78L, the already low levels of cell surface HA-MCR were not significantly further reduced by GA treatment.
Fig. 8.

Manipulation of Hsp90 activity alters cellular levels of MC4R. A and B, Levels of MC4R were reduced in cells treated with the Hsp90 inhibitor GA. HEK293 cells transiently expressing HA-MC4R were treated with 0, 0.4 or 1.0 μm GA for 24 h after transfection. Total (A) and cell surface (B) levels of HA-MC4R were analyzed by In-Cell Western. C and D, Expression of the Hsp90 cochaperone Aha1 increases cellular levels of mutant MC4R. HEK293 cell lines stably expressing WT or mutant HA-MC4R were transfected with plasmid for the expression of Aha1 or empty vector, cell surface (C) and total receptor levels (D) were quantified by In-Cell Western analyses. Error bars represent 2× se. *, P < 0.05.
Hsc70 cooperates with Hsp90 in client protein folding and quality control (27). Therefore, we hypothesized that the Hsp90 chaperone system may also play a role in MC4R biogenesis. To look at the effects of increasing Hsp90 activity on MC4R biogenesis, we heterologously expressed the Hsp90 cochaperone Aha1, in cells stably expressing HA-MC4R. Aha1 stimulates ATPase activity of Hsp90, therefore accelerating cycles of client protein binding and release (28) and has been shown to play a role in quality control of another misfolded membrane protein, CFTR (22). Aha1 expression resulted in a more than 2-fold increase in cellular levels of P78L as well as an increase in total levels of the WT and S58C protein (Fig. 8C). For S58C and P78L, this corresponded to a slight increase in cell surface MC4R and, surprisingly, a decrease in cell surface levels of WT receptor (Fig. 8D). For S58C, this change was significant (P < 0.05). However, confocal analyses did not reveal a dramatic increase in cell surface HA-MC4R staining in cells cotransfected with Aha1 (data not shown). That the increase in total levels of MC4R was larger than the increase in cell surface expression, in the Aha1 transfected cells, suggests the Hsp90 chaperone system may promote stability of misfolded receptor at the ER, but a limited amount of this protein is sufficiently folded to traffic to the cell surface. We were unable to coIP Aha1 or Hsp90 with MC4R (data not shown). This may be because chaperone/client protein interactions are frequently transient.
Signaling by intracellular retained MC4R mutants increased in cells overexpressing Hsc70
We next asked whether the increase in cell surface expression of MC4R mutants, in cells with elevated levels of Hsc70, resulted in greater MC4R signaling in response to NDP-MSH. We did not investigate the effect of Aha1 expression on signaling, because we observed no increase in cell surface levels of MC4R mutants with this chaperone. Activity of cAMP was measured by firefly luciferase reporter assay and normalized against a renilla luciferase control. In cells transiently transfected for expression of MC4R and Hsc70, functional expression of WT and mutant receptor increased (Fig. 9).
Fig. 9.

Functional expression of MC4R mutants is increased in cells with increased levels of Hsc70. MC4R signaling in cells stimulated with 10−6 m NDP-MSH for 6 h was quantified by a dual luciferase reporter assay for cAMP activity. Cells with transient expression of WT or mutant HA-MC4R were cotransfected with vectors for expression of V5-Hsc70 or empty vector and luciferase reporter vectors. Error bars represent 2× se. *, P < 0.05.
Discussion
The data presented here supports the idea that intracellular retained MC4R mutants are trapped at the ER because of misfolding. The increased overlap of MC4R and ER marker staining in cells expressing intracellular retained mutants relative to WT protein indicates ER retention. The presence of inclusions of mutant MC4R-GFP in neuroblastoma-derived SK-N-SH cells and the reduced mobility of intracellular retained MC4R indicate misfolding. This is complementary to the findings of a previous study that reported ER retained MC4R variants are ubiquitinated more than WT protein and induce expression of the ER stress protein CCAAT/enhancer-binding protein homolgous protein (CHOP), indicating perturbation of normal ER homeostasis and activation of the UPR (15).
Obesity can be considered as a central nervous system disorder because of the role of MC4R and other appetite-controlling proteins in the neuroregulation of energy balance. The presence of misfolded protein intermediates disrupts proteostasis and is detrimental to neuronal function and survival (29). Indeed, neurons are particularly sensitive to protein misfolding and frequently partition aberrantly folded protein species into inclusions, as we observed for MC4R-GFP mutants in SK-N-SH cells. It is unknown whether the presence of misfolded MC4R has consequences for neurons in vivo. However, obesity generally (diet induced and genetic) is associated with ER stress, including in the hypothalamus (30).
Importantly, we have identified that Hsc70 can modulate functional expression of WT and mutant MC4R. Coexpression of Hsc70 with MC4R increased total cellular levels of the receptor and functional expression of misfolding mutants at the cell surface. The interaction of Hsc70 with MC4R, and the increased mobility of intracellular retained MC4R mutants in the presence of Hsc70, points to the chaperone stabilizing and/or promoting the folding of the receptor. This fits with the canonical protein folding function of Hsc70 and the previous report that Hsc70, with the J-protein Hdj-2 (DNAJA1), facilitates early steps in biogenesis of another polytopic membrane protein, CFTR (31). The J-protein HSJ1b reduced cellular levels of MC4R, in accordance with its function as a shuttling factor for sorting Hsc70 clients to the proteasome in neurons (23) and our previous work showing it modulates rhodopsin trafficking (20). Interestingly, Hdj-2 is farnesylated, whereas HSJ1b is geranyl-geranylated, a modification that is required for its modulation of rhodopsin processing, indicating that a J-protein localized to the cytoplasmic face of the ER may be a requirement for Hsc70 to interact with polytopic membrane proteins. The J-domain protein responsible for integrating Hsc70 activity into MC4R folding remains to be identified.
Inhibition of Hsp90 ATPase activity with GA, which binds the Hsp90 ATP binding site, reduced total levels of MC4R and thus the amount of receptor at the cell surface. Conversely, stimulating Hsp90's ATPase activity, by augmented expression of the cochaperone Aha1, increased levels of MC4R. Therefore, these data may suggest that Hsp90 is acting as a holdase for MC4R, and increased association with Hsp90 leads to degradation of the receptor. This contrasts the effects of Aha1 on CFTR(ΔF508), where knockdown of the chaperone allows mutant protein to escape ER-associated degradation pathways and couple to ER export machinery (22). Aha1 has been suggested to function by regulating the dwell time of clients with Hsp90, integrating chaperone function with client folding kinetics (32). It seems likely that the optimal chaperone environment for folding and exit from the ER is variable between different client proteins and will be in part dependent on the dynamics of Hsp90 interaction.
To our knowledge, this is the first study to show that modulation of molecular chaperone systems can rescue cell surface expression of intracellular retained MC4R mutants. Stabilization of misfolded MC4R by mechanisms other than direct manipulation of the cellular chaperone environment has previously been shown to promote functional expression. For example, the kosmotrope 4-phenyl butyric acid increased cell surface expression of WT and intracellular retained mutant MC4R (15). In addition, MC4R-selective ligands have been shown to exhibit pharmacological chaperone activities for intracellular retained mutants, with their binding promoting cell surface expression (33, 34). Importantly, these studies and data presented here suggest that for some aberrantly folded MC4R mutants, if they can traffic to the cell surface, they may retain a degree of function. This has important therapeutic implications for obesity caused by misfolding mutations in MC4R. Kosmotropes, small molecule ligands, and pharmacological inducers of chaperones may be especially useful in the restoration of misfolded GPCR trafficking if used in combination. It is also expected that the optimal manipulation of the protein-folding environment will be dependent on the conformational consequences of specific receptor residue mutations. The ability of Hsc70 to increase cell surface levels of MC4R mutants suggests that therapeutically targeting cytosolic chaperones has validity as a strategy to promote cell surface expression of ER retained membrane proteins. Finally, because ER stress is a feature of obesity, strategies to restore proteostasis are likely to be of wider relevance for the treatment of obesity.
Materials and Methods
Expression constructs and site directed mutagenesis
cDNA clones for human MC4R triple HA-tagged at the N terminus (3xHA-MC4R-pcDNA3.1) were obtained from Missouri S&T cDNA Resource Center (Rolla, MO). S58C, P78L, and D90N mutant MC4R constructs were made by site directed mutagenesis using the WT HA-MC4R construct as a template. This was also used as a template for the MC4R-GFP constructs made by cloning MC4R into pEGFP-N1 (CLONTECH, Mountain View, CA). All constructs were confirmed by sequencing. pcDNA5/FRT/TO-Hsc70 constructs and DnaJ constructs (35) were a gift from Harm Kampinga (University of Groningen, Groningen, The Netherlands). Aha1-containing F1007 vector was a gift from Paul Workman (The Institute of Cancer Research, London, United Kingdom).
Cell culture and transfections
HEK293 cells were maintained in DMEM and SK-N-SH cells in 50:50 DMEM:F12. Media were supplemented with 10% fetal calf serum and contained 50 U/ml−1 penicillin/50 μg/ml−1 streptomycin. Cells were transiently transfected using Lipofectamine Plus (Invitrogen, Carlsbad, CA). To generate HEK293 cell lines stably expressing WT or mutant HA-MC4R, cells were transfected as described above and selected with Geneticin (G418) at 1.2 mg/ml−1. Clonal isolates were selected and assayed for HA-MC4R expression by Western blot analyses.
Fluorescence based detection of MC4R cell surface expression by In-Cell Western
Cells were seeded in 24-well plates and transfected with the appropriate plasmids. Twenty-four hours after transfection, cells were washed with PBS and fixed in 3.7% formaldehyde for 10 min. Cells were permeabilized with 0.025% Triton X-100 to assay total cellular levels or left unpermeabilized to detect cell surface expression. Cells were then blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, NE) for 1 h, followed by incubation with anti-HA antibody (clone HA-7; Sigma, St. Louis, MO) in 1:10,000 dilution for 1 h. Cells were then washed with PBS. IRDye 680 secondary antibody was added at 1:1000 (Invitrogen) for 1 h. Cells were washed and fluorescent signal measured using a LI-COR Odyssey plate reader.
IB and coIP
T25 flasks of HEK293 cells were transiently cotransfected with WT or mutant HA-MC4R and with either Hsc70-V5, HSJ1b-V5, or DNAJB4-V5. Twenty-four hours after transfection, cells were lysed in 750 μl of a buffer A containing 50 mm Tris/HCl (pH7.4), 500 mm NaCl, 1 mm EDTA, and 0.5% Triton X-100, including protease and phosphatase inhibitors (P8340; Sigma) and incubated on ice for 30 min. Lysates were then centrifuged at 13,000 × g for 10 min at 4 C. The supernatant was collected and 50 μl of prewashed anti-HA agarose-conjugate beads added (clone HA-7; Sigma). After 16 h of incubation, agarose beads were collected and washed repeatedly with 1200 μl of buffer A. Bound proteins were eluted in 50 μl of SDS-PAGE sample buffer and analyzed by IB as described previously using anti-V5 antibody (clone V5–10; Sigma) at a dilution of 1:2000.
Luciferase cAMP reporter assay
HEK293 cells were plated on six-well plates and cotransfected with 1000 ng of WT or mutant MC4R, Hsc70, α-GSU-846 and 100 ng of pRL-CMV Renilla luciferase reporter plasmid constructs. Twenty-four hours after transfection, cells were stimulated with 10−6 NDP-MSH for 6 h. For Hsc70 experiments, luciferase values were normalized to a control where MC4R was not included. Cells were lysed and a Dual-Luciferase Reporter Assay performed according to the manufacturer's instructions (Promega, Madison, WI). Luciferase activity was measured using a multiplate reader (LUMIstar; BMG LABTECH GmbH, Ortenberg, Germany). For analyses, firefly luciferase activity was normalized relative to Renilla luciferase activity.
Confocal microscopy and FRAP
Cells were grown on sterile coverslips or in glass bottom culture dishes (MatTek Corp., Ashland, MA). Immunofluorescent processing and subsequent confocal microscopy were as described previously (36). FRAP analyses were performed at 37 C on the temperature-controlled stage of a Zeiss LSM510 confocal microscope (Carl Zeiss, Oberkochen, Germany). Bleaching of a defined region of an individual cell (indicated by boxes in Fig. 3A) was performed using a 488-nm laser line at 100% power and transmission over two iterations. Image acquisition was performed by scanning the entire field at less than 4% laser power. Recovery was monitored over 40 cycles of imaging with a 1-sec interval between each acquisition. Fluorescence intensity in regions of interest was quantified using the Zeiss Zen software.
Statistical analyses
Data are expressed as mean ± se. For FRAP data, area under the curve was estimated using the trapezoidal method. Significance was tested by unpaired t test or one-way ANOVA as appropriate.
Acknowledgments
We thank Professor Harm Kampinga (University of Groningen, Groningen, The Netherlands) and Dr. Jurre Hageman for pcDNA5/FRT/TO-Hsc70 and DnaJ constructs, and Professor Paul Workman (The Institute of Cancer Research, London, United Kingdom) for Aha1 reagents.
This work was supported by St Bartholomew's and the Royal London Charitable Foundation.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CFTR
- Cystic fibrosis transmembrane conductance regulator
- CoIP
- coimmunoprecipitation
- ER
- endoplasmic reticulum
- FRAP
- fluorescence recovery after photobleaching
- GFP
- green fluorescent protein
- GPCR
- G protein-coupled receptor
- HA-MC4R
- hemagglutinin tagged MC4R
- Hsc70
- cognate Hsp70 protein
- Hsp70
- 70-kDa heat-shock protein
- IB
- immunoblot
- IP
- immunoprecipitation
- MC4R
- melanocortin-4 receptor
- α-MSH
- α-melanocyte stimulating hormone
- NDP-MSH
- α-MSH analogue Nle4, DPhe7-α-MSH
- PM
- plasma membrane
- UIM
- ubiquitin interaction motif
- UPR
- unfolded protein response
- WT
- wild-type.
References
- 1. Balthasar N , Dalgaard LT , Lee CE , Yu J , Funahashi H , Williams T , Ferreira M , Tang V , McGovern RA , Kenny CD , Christiansen LM , Edelstein E , Choi B , Boss O , Aschkenasi C , Zhang CY , Mountjoy K , Kishi T , Elmquist JK , Lowell BB. 2005. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123:493–505 [DOI] [PubMed] [Google Scholar]
- 2. Loos RJ , Lindgren CM , Li S , Wheeler E , Zhao JH , Prokopenko I , Inouye M , Freathy RM , Attwood AP , Beckmann JS , Berndt SI , Jacobs KB , Chanock SJ , Hayes RB , Bergmann S , Bennett AJ , Bingham SA , Bochud M , Brown M , Cauchi S , Connell JM , Cooper C , Smith GD , Day I , Dina C , et al. 2008. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet 40:768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeo GS , Farooqi IS , Aminian S , Halsall DJ , Stanhope RG , O'Rahilly S. 1998. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 20:111–112 [DOI] [PubMed] [Google Scholar]
- 4. Vaisse C , Clement K , Guy-Grand B , Froguel P. 1998. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 20:113–114 [DOI] [PubMed] [Google Scholar]
- 5. Tan K , Pogozheva ID , Yeo GS , Hadaschik D , Keogh JM , Haskell-Leuvano C , O'Rahilly S , Mosberg HI , Farooqi IS. 2009. Functional characterization and structural modeling of obesity associated mutations in the melanocortin 4 receptor. Endocrinology 150:114–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biebermann H , Krude H , Elsner A , Chubanov V , Gudermann T , Grüters A. 2003. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes 52:2984–2988 [DOI] [PubMed] [Google Scholar]
- 7. Alharbi KK , Spanakis E , Tan K , Smith MJ , Aldahmesh MA , O'Dell SD , Sayer AA , Lawlor DA , Ebrahim S , Davey Smith G , O'Rahilly S , Farooqi S , Cooper C , Phillips DI , Day IN. 2007. Prevalence and functionality of paucimorphic and private MC4R mutations in a large, unselected European British population, scanned by meltMADGE. Hum Mutat 28:294–302 [DOI] [PubMed] [Google Scholar]
- 8. Farooqi IS , Keogh JM , Yeo GS , Lank EJ , Cheetham T , O'Rahilly S. 2003. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 348:1085–1095 [DOI] [PubMed] [Google Scholar]
- 9. Nijenhuis WA , Garner KM , van Rozen RJ , Adan RA. 2003. Poor cell surface expression of human melanocortin-4 receptor mutations associated with obesity. J Biol Chem 278:22939–22945 [DOI] [PubMed] [Google Scholar]
- 10. Yeo GS , Lank EJ , Farooqi IS , Keogh J , Challis BG , O'Rahilly S. 2003. Mutations in the human melanocortin-4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum Mol Genet 12:561–574 [DOI] [PubMed] [Google Scholar]
- 11. Lubrano-Berthelier C , Dubern B , Lacorte JM , Picard F , Shapiro A , Zhang S , Bertrais S , Hercberg S , Basdevant A , Clement K , Vaisse C. 2006. Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin Endocrinol Metab 91:1811–1818 [DOI] [PubMed] [Google Scholar]
- 12. Hirsch C , Gauss R , Horn SC , Neuber O , Sommer T. 2009. The ubiquitylation machinery of the endoplasmic reticulum. Nature 458:453–460 [DOI] [PubMed] [Google Scholar]
- 13. Vembar SS , Brodsky JL. 2008. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 9:944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin JH , Li H , Yasumura D , Cohen HR , Zhang C , Panning B , Shokat KM , Lavail MM , Walter P. 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science 318:944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Granell S , Mohammad S , Ramanagoudr-Bhojappa R , Baldini G. 2010. Obesity-linked variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol Endocrinol 24:1805–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lanctôt PM , Leclerc PC , Escher E , Guillemette G , Leduc R. 2006. Role of N-glycan-dependent quality control in the cell-surface expression of the AT1 receptor. Biochem Biophys Res Commun 340:395–402 [DOI] [PubMed] [Google Scholar]
- 17. Kampinga HH , Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11:579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheetham ME , Caplan AJ. 1998. Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperon 3:28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hageman J , van Waarde MA , Zylicz A , Walerych D , Kampinga HH. 2011. The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem J 435:127–142 [DOI] [PubMed] [Google Scholar]
- 20. Chapple JP , Cheetham ME. 2003. The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J Biol Chem 278:19087–19094 [DOI] [PubMed] [Google Scholar]
- 21. Taipale M , Jarosz DF , Lindquist S. 2010. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11:515–528 [DOI] [PubMed] [Google Scholar]
- 22. Wang X , Venable J , LaPointe P , Hutt DM , Koulov AV , Coppinger J , Gurkan C , Kellner W , Matteson J , Plutner H , Riordan JR , Kelly JW , Yates JR , Balch WE. 2006. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127:803–815 [DOI] [PubMed] [Google Scholar]
- 23. Westhoff B , Chapple JP , van der Spuy J , Höhfeld J , Cheetham ME. 2005. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr Biol 15:1058–1064 [DOI] [PubMed] [Google Scholar]
- 24. Tao YX , Segaloff DL. 2003. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology 144:4544–4551 [DOI] [PubMed] [Google Scholar]
- 25. Lubrano-Berthelier C , Durand E , Dubern B , Shapiro A , Dazin P , Weill J , Ferron C , Froguel P , Vaisse C. 2003. Intracellular retention is a common characteristic of childhood obesity-associated MC4R mutations. Hum Mol Genet 12:145–153 [DOI] [PubMed] [Google Scholar]
- 26. Nehls S , Snapp EL , Cole NB , Zaal KJ , Kenworthy AK , Roberts TH , Ellenberg J , Presley JF , Siggia E , Lippincott-Schwartz J. 2000. Dynamics and retention of misfolded proteins in native ER membranes. Nat Cell Biol 2:288–295 [DOI] [PubMed] [Google Scholar]
- 27. Höhfeld J , Cyr DM , Patterson C. 2001. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep 2:885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panaretou B , Siligardi G , Meyer P , Maloney A , Sullivan JK , Singh S , Millson SH , Clarke PA , Naaby-Hansen S , Stein R , Cramer R , Mollapour M , Workman P , Piper PW , Pearl LH , Prodromou C. 2002. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell 10:1307–1318 [DOI] [PubMed] [Google Scholar]
- 29. Muchowski PJ , Wacker JL. 2005. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22 [DOI] [PubMed] [Google Scholar]
- 30. Ozcan L , Ergin AS , Lu A , Chung J , Sarkar S , Nie D , Myers MG , Ozcan U. 2009. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9:35–51 [DOI] [PubMed] [Google Scholar]
- 31. Meacham GC , Lu Z , King S , Sorscher E , Tousson A , Cyr DM. 1999. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J 18:1492–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koulov AV , Lapointe P , Lu B , Razvi A , Coppinger J , Dong MQ , Matteson J , Laister R , Arrowsmith C , Yates JR , Balch WE. 2010. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol Biol Cell 21:871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. René P , Le Gouill C , Pogozheva ID , Lee G , Mosberg HI , Farooqi IS , Valenzano KJ , Bouvier M. 2010. Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J Pharmacol Exp Ther 335:520–532 [DOI] [PubMed] [Google Scholar]
- 34. Fan ZC , Tao YX. 2009. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J Cell Mol Med 13:3268–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hageman J , Kampinga HH. 2009. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperon 14:1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chung TT , Webb TR , Chan LF , Cooray SN , Metherell LA , King PJ , Chapple JP , Clark AJ. 2008. The majority of adrenocorticotropin receptor (melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab 93:4948–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]