

Abstract
The direct actions of transmembrane receptors within the nucleus remain enigmatic. In this report, we demonstrate that the prolactin receptor (PRLr) localizes to the nucleus where it functions as a coactivator through its interactions with the latent transcription factor signal transducer and activator of transcription 5a (Stat5a) and the high-mobility group N2 protein (HMGN2). We identify a novel transactivation domain within the PRLr that is activated by ligand-induced phosphorylation, an event coupled to HMGN2 binding. The association of the PRLr with HMGN2 enables Stat5a-responsive promoter binding, thus facilitating transcriptional activation and promoting anchorage-independent growth. We propose that HMGN2 serves as a critical regulatory factor in Stat5a-driven gene expression by facilitating the assembly of PRLr/Stat5a onto chromatin and that these events may serve to promote biological events that contribute to a tumorigenic phenotype. Our data imply that phosphorylation may be the molecular switch that activates a cell surface receptor transactivation domain, enabling it to tether chromatin-modifying factors, such as HMGN2, to target promoter regions in a sequence-specific manner.
In the mammary gland, the signaling cascades induced by the polypeptide hormone prolactin (PRL) require the presence of the cell surface PRL receptor (PRLr). Studies focusing on PRLr have described its signaling capabilities almost exclusively at the plasma membrane and have demonstrated that upon ligand binding, the PRLr-associated kinases Janus kinase 2 (Jak2), c-Src, and Tec become activated (1). These kinases in turn activate the transcription factors signal transducer and activator of transcription 5a (Stat5a) (2) and E74-like factor 5 (3), resulting in specific patterns of gene expression. These events are biologically relevant as the PRLr and its downstream signaling components have been implicated in breast cancer pathogenesis. Specifically, the PRLr is overexpressed in breast cancer tissue (4), and its increased stability and resistance to ubiquitin-mediated degradation prolongs its associated signaling events in primary breast tumors (5). Epidemiologically, high PRL serum levels are associated with a 2-fold increase in breast cancer risk (6).
The canonical pathway that is activated by PRL-PRLr engagement is Jak2/Stat5a (7). Jak2 is constitutively associated with the PRLr and is activated by ligand that induces the phosphorylation and activation of Stat5a (8). These events ultimately result in the up-regulation of PRL target genes such as CCND1 (cyclin D1) (9) and CISH (cytokine-inducible SH2-containing protein) (10), whose overexpression in breast cancers has been observed (11).
The Jak2/Stat5a pathway is widely shared with other transmembrane receptors such as the erythropoietin receptor, platelet-derived growth factor receptor, and GH receptor (GHr) (12); however, the mechanism defining the specificity of induced gene expression between these receptor-signaling pathways remains unknown. Interestingly, there have been numerous reports indicating that intact transmembrane receptors localize to the nucleus. These reports include, but are not limited to, members of the ErbB family of receptors (13–17), GHr (18, 19), and fibroblast growth factor receptor (FGFr) (16, 20–23). Additionally, a subset of these studies has demonstrated that nuclear-localized transmembrane receptors contribute to transcriptional activation. Examples include nuclear epidermal growth factor receptor (EGFr)-mediated transactivation of Aurora A, iNOS, and COX-2 genes (24–26), nuclear FGFr potentiation of c-jun (23), and nuclear GHr-mediated up-regulation of Survivin, Mybbp, and Dysadherin (19). Collectively, these observations suggest that the trafficking of a cell surface receptor into the nucleus contributes to transcriptional activation and may serve to impart specific patterns of ligand-induced gene expression. However, the specific mechanism by which intranuclear receptors serve to regulate transcription has not been fully described.
In this study, we demonstrate that PRL initiates PRLr nuclear translocation in mammary epithelial cells in vitro and breast tissue in vivo. Furthermore, we identify a novel PRLr transactivation domain (TAD) that upon phosphorylation, recruits the chromatin-modifying protein high-mobility group N2 protein (HMGN2), enabling Stat5a-mediated gene expression. This is the first report to identify a novel TAD within the PRLr and demonstrate its inducible interaction with a nucleosome-binding protein whose presumed functions are directly related to transcriptional activation.
Results
Intranuclear localization of PRLr
Preliminary observations in our laboratory and others (27, 28) led us to hypothesize that the PRLr may localize to the nucleus. Thus, cell fractionation was performed on T47D breast cancer cells, which exhibit high endogenous PRLr levels. Using an antibody specific to the PRLr extracellular domain (ECD), we detected full-length PRLr in both nonnuclear and nuclear fractions (Fig. 1A). The endoplasmic reticulum membrane protein calnexin was partitioned to the nonnuclear fraction, demonstrating that detected nuclear PRLr was not the result of contamination from newly translated endoplasmic reticulum-localized proteins. Immunoblots were also probed with anti-histone H1 to demonstrate nuclear purity and β1-integrin (Supplemental Fig. 1A, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) to demonstrate the absence of cell surface contamination in the nucleus. An antibody specific to the PRLr intracellular domain (ICD) demonstrated similar results (Fig. 1B) as did transient transfection of epitope-tagged PRLr cDNA in MCF7 breast cancer cells (Supplemental Fig. 1B) and CHO cells (Supplemental Fig. 1C). These findings were further confirmed by immunohistochemical analysis of normal and malignant breast tissue samples. In this analysis, the nuclear intensity of PRLr in tissue samples was scored from 0–3, and a score above 1.5 was counted as positive. Using this method, 12 of 16 normal breast epithelial samples and 20 of 35 malignant tissue samples showed positive nuclear staining. Representative pictures from these analyses are shown in Fig. 1C. A secondary antibody control to demonstrate antibody specificity was performed as shown in Supplemental Fig. 1D. Taken together, these results clearly demonstrate that the PRLr is present in the nucleus in vitro and in vivo.
Fig. 1.
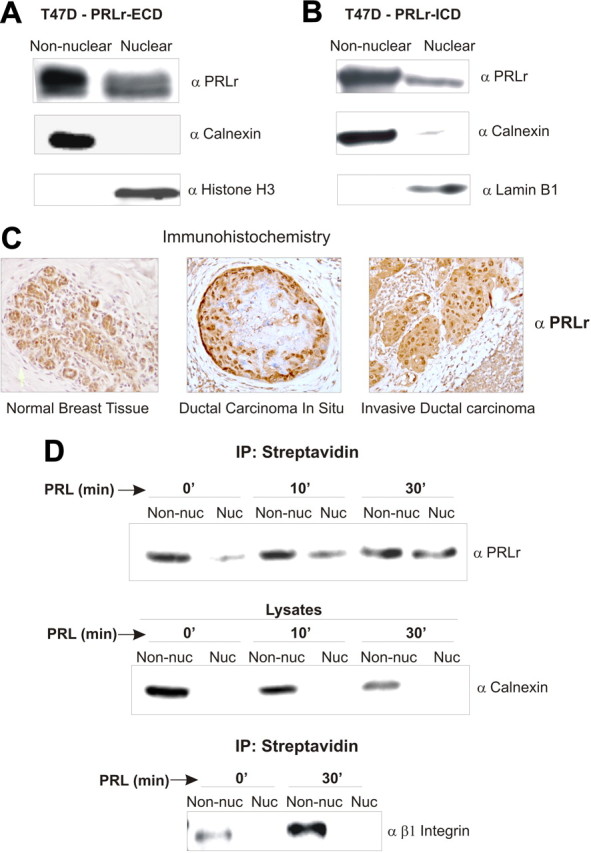
The PRLr is present in the nucleus and is translocated from the cell surface. A and B, Cell fractionation and Western blot analysis. T47D cells in normal growth conditions were partitioned into nuclear and nonnuclear fractions. An anti-PRLr antibody specific to the ECD (A) or ICD (B) was used to demonstrate the nuclear presence of full-length PRLr. To demonstrate that proteins from the endoplasmic reticulum are not contaminating nuclear fractions, nonnuclear fractions were also probed with anti-calnexin, an integral endoplasmic reticulum membrane protein. Similarly, nuclear fractions were probed with anti-histone H3 or anti-lamin B1 antibodies to demonstrate nuclear purity. C, Immunohistochemical analysis of nuclear PRLr in normal, ductal carcinoma in situ, and invasive breast tissue. Tissues were stained with an anti-PRLr ICD antibody. D, Biotinylation assay to detect cell-surface PRLr in the nucleus of PRL-stimulated T47D cells. T47D cells were serum starved for 24 h before biotinylation and PRL treatment. After cell fractionation, nuclear (Nuc) and nonnuclear (Non-nuc) fractions were immunoprecipitated with streptavidin beads. Eluted proteins were analyzed by Western blot and probed with anti-PRLr. Ten percent of nuclear and nonnuclear lysates were taken out before immunoprecipitation (IP) and analyzed by Western analysis for endoplasmic reticulum contamination in the nucleus using an anti-calnexin antibody. In a parallel experiment, T47D cells were biotinylated and PRL treated. After streptavidin immunoprecipitation, samples were analyzed by Western blot using an anti-β1-integrin antibody to demonstrate a biotinylated cell surface protein that is internalized but does not localize to the cell nucleus.
To determine whether nuclear PRLr was derived from the cell surface or from an independent cytoplasmic pool, biotinyation of T47D cells was used to track the PRLr from the cell surface. After cell fractionation and streptavidin immunoprecipitation, a significant amount of endogenous biotinylated PRLr was detected in the nucleus as a function of PRL stimulation (Fig. 1D). These results were specific to the PRLr, because the cell surface receptor, β1-integrin, despite its ability to be internalized, was not detected in the nucleus. Our results therefore suggest that a significant portion of the PRLr that accumulates in the nucleus is both derived from the cell surface and triggered by ligand stimulation.
Nuclear PRLr activates Stat5a-dependent transcription
Given that Stat5a and PRLr interact at the cell surface in a ligand-dependent manner, we postulated that PRLr/Stat5a may interact in the nucleus. PRLr nonnuclear and nuclear immunoprecipitates in T47D cells were analyzed for Stat5a binding. Indeed, a ligand-inducible interaction between the two proteins in the nucleus was observed. Comparable results were seen when using either anti-PRLr (Fig. 2A) or anti-Stat5a (Supplemental Fig. 2A) as the immunoprecipitating antibody.
Fig. 2.

Nuclear PRLr associates with Stat5a and facilitates Stat5a-driven gene expression. A, T47D cells were serum starved for 24 h before PRL treatment (10 min). Cells were then partitioned into nonnuclear (Non-nuc) and nuclear (Nuc) lysates and subjected to coimmunoprecipitation using an anti-Stat5a antibody. Eluted samples were analyzed by Western blot using an anti-PRLr antibody. Blots were stripped and reprobed for anti-Stat5a. Ten percent of nonnuclear and nuclear lysates were aliquoted before immunoprecipitation (IP) and analyzed by Western blot using an anti-calnexin antibody as a control for contamination in the nucleus. B, Top, Detail of the CISH promoter; arrows delineate primers used for ChIP. The primer sets represent the following amplicons: −-104/−68, the CISH proximal promoter, and +311/+380, the CISH downstream coding region; bottom, PRLr association at the indicated regions as assayed by ChIP. PRLr occupancy at the CISH proximal promoter or a downstream coding region was analyzed using a PRLr-ECD antibody. IgG demonstrates the negative control in this experiment, and all results are reported as fold change by normalizing to IgG conditions. The values are those of a representative experiment repeated three separate times, where sem indicates the error of three replicate measurements within one experiment. ORF, Open reading frame; C, Top, Schematic of constructs (termed PRLr-NLS or NLS-ICD) to force the nuclear localization of PRLr by the addition of an SV40 NLS; bottom, schematic of CISH luciferase reporter. D, CISH luciferase activity in MCF7 cells transfected with the indicated cDNA. Cells were transfected with cDNA for 24 h, serum starved for 24 h, and then treated with PRL for an additional 24 h. Results are presented as fold change relative to untreated empty vector control. Values are those of a representative experiment repeated three separate times where sem represents the error of three replicates performed within one experiment. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test; values for empty plus PRL vs. NLS-ICD plus PRL and PRLr plus PRL vs. PRLr-NLS plus PRL are marked in figure. Other statistical differences are as follows: empty vs. empty plus PRL, P < 0.001; empty plus PRL vs. PRLr plus PRL, P < 0.001; empty vs.NLS-ICD or PRLr-NLS, P < 0.001. ***, P < 0.001.
The finding of an interaction between the PRLr and Stat5a suggested that the two proteins exist in a functionally important nuclear complex. Previous reports have shown that Stat5a binds strongly to gamma interferon activating sequence (GAS) elements on the CISH proximal promoter after 30 min of PRL treatment (29) and were verified here (Supplemental Fig. 2B). Thus, we used chromatin immunoprecipitation (ChIP) to determine whether nuclear PRLr could also bind to the Stat5a-driven CISH proximal promoter (see schematic, Fig. 2B, top). In untreated T47D cells, an anti-PRLr-ECD antibody precipitated a small but detectable amount of DNA from the CISH proximal promoter, whereas PRL stimulation significantly enhanced this binding. PRLr promoter occupancy was observed when performing ChIP using an antibody specific to the ECD (Fig. 2B, bottom) or ICD (Supplemental Fig. 2C). As a control for nonspecific DNA binding, it was determined that the PRLr was not significantly enriched within a region downstream of the proximal promoter in the CISH open reading frame.
Based on the above findings, we hypothesized that nuclear PRLr may function as a Stat5a coactivator, where Stat5a provides the DNA-binding domain (DBD) that tethers PRLr to chromatin, and PRLr provides a TAD to promote transcription. Because knockdown or overexpression of the PRLr would alter its cell surface actions, the C-terminal addition of an simian virus 40 (SV40) large T antigen nuclear localization signal (NLS) was used to force the nuclear localization of the PRLr (termed PRLr-NLS) (see schematic, Fig. 2C; expression via Western blot, Supplemental Fig. 2D). Enhanced nuclear localization of each construct was verified by fluorescence microscopy (data not shown). To measure the effects of forced PRLr nuclear localization on Stat5a-regulated gene expression, a CISH reporter construct was used (Fig. 2C). Overexpression of wild-type PRLr potentiated reporter activation 2-fold (Fig. 2D), as has been previously reported (10). However, transfection of PRLr-NLS potentiated this effect, thus revealing a significant correlation between PRLr nuclear localization and Stat5a-induced gene expression (Fig. 2D).
To further examine this phenomenon, the PRLr ICD was fused to an SV40 NLS, and this construct was overexpressed in MCF7 cells (Supplemental Fig. 2D). Since NLS-ICD lacks a signal peptide and transmembrane domain and is therefore completely devoid of cell surface signaling capabilities, this approach enabled us to examine the nuclear-only properties of the PRLr. Indeed, forced nuclear localization of the PRLr ICD increased CISH reporter activity compared with control, further demonstrating the transcriptional function of nuclear PRLr and suggesting this function resides within the ICD (Fig. 2D).
Because the actions of the NLS-ICD construct were enhanced by ligand stimulation, this suggested that Stat5a binding to GAS elements of the CISH reporter was required for PRLr-specific transactivation. To test this possibility, the ability of NLS-ICD to activate CISH transcription in the presence of a constitutively active Stat5a (Stat5a1*6) was examined. Interestingly, the individual overexpression of Stat5a1*6, wild-type PRLr or NLS-ICD in MDA-231 cells yielded no significant effect on reporter activity. However, coexpression of NLS-ICD and Stat5a1*6 resulted in a significant increase in reporter activity. These data indicate that activated Stat5a is required for PRLr nuclear action, and in a similar vein, the nuclear presence of the PRLr-ICD is important for the transcriptional activity of Stat5a (Supplemental Fig. 2E).
A novel transactivating domain within PRLr
We hypothesized that the PRLr ICD possessed innate transactivation activity based the ability of Stat5a-mediated gene expression to be induced by the forced nuclear localization of the PRLr ICD. To identify the region that may be responsible for transactivation, the PRLr ECD or ICD were fused to the DBD of the yeast transcription activator protein, Gal4, and an upstream activating sequence (UAS)/GAL4-based reporter system was used (Fig. 3A). Although Gal4-ICD activated the Gal4-UAS reporter approximately 300-fold, Gal4-ECD yielded no significant activity (Fig. 3B). Notably, the transactivation effects observed in the presence of Gal4-ICD were 7-fold higher than Gal4-Stat5aTAD, which was previously demonstrated to be a potent transactivator as measured by the Gal4-UAS system (30).
Fig. 3.

Identification of a novel PRLr TAD. A, Top, Schematic of Gal4-UAS luciferase (Luc) reporter and Gal4-DBD PRLr fusion construct; bottom, schematic of Gal4-ECD, Gal4-ICD, Gal4-Stat5aTAD, or Gal4 control constructs. B, Gal4-UAS luciferase activity in 293T cells transfected with Gal4-ECD, Gal4-ICD, Gal4-Stat5aTAD, or Gal4 control constructs. C, Gal4-UAS luciferase assay measuring transactivation of PRLr C-terminal truncation constructs in 293T cells. D, Gal4-UAS luciferase activity of Gal4 fusion constructs in 293T cells with the indicated PRLr regions as determined by C. E, Cross-species sequence alignment of the identified PRLr TAD highlighting four conserved residues (gray). F, Gal4-UAS luciferase activity of the TAD point mutations in 293T cells as determined in F. Reported values are those of a representative experiment repeated three separate times, where sem indicates the error of quadruplicate transfections performed within one experiment. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. RLU, Relative light units. **, P < 0.01; ***, P < 0.001.
We next constructed sequential C-terminal truncations of Gal4-ICD to further fine map the PRLr TAD (Fig. 3, C and D). Using this method, we identified 44 amino acids that were necessary for PRLr transactivation (404–448). Subsequent cross-species DNA analysis highlighted a small region of homology (residues 405–411) in which four residues were highly conserved (Fig. 3E). To determine which, if any, of these residues were integral in PRLr-specific transactivation, we performed site-directed mutagenesis within the Gal4–404-448 fusion. Interestingly, Y406F or D411A mutations significantly decreased transactivation, whereas their simultaneous mutation completely ablated transactivation (Fig. 3F).
The PRLr TAD is critical for Stat5a-driven transcription
The above data led us to hypothesize that introduction of the transactivation-defective double mutation into full-length PRLr (herein referred to as PRLrYDmut), would negatively affect Stat5a-mediated transcription. Thus, stably transfected pools expressing PRLr or PRLrYDmut were generated from parental MCF10AΔp53 cells or MCF7 cells (Supplemental Fig. 3, A and B). MCF10AΔp53 cells were selected for their low level of endogenous PRLr expression and signaling and MCF7 cells for their moderate level of endogenous PRLr expression and signaling. Although overexpression of the PRLr significantly enhanced PRL-induced CISH mRNA, expression of mutant construct, PRLrYDmut significantly compromised this effect in both stable cell pools (Fig. 4A and Supplemental Fig. 3D). Expression of PRLrYDmut also significantly prevented the ability of the PRLr to enhance PRL-stimulated CISH reporter activity (Supplemental Fig. 3E).
Fig. 4.
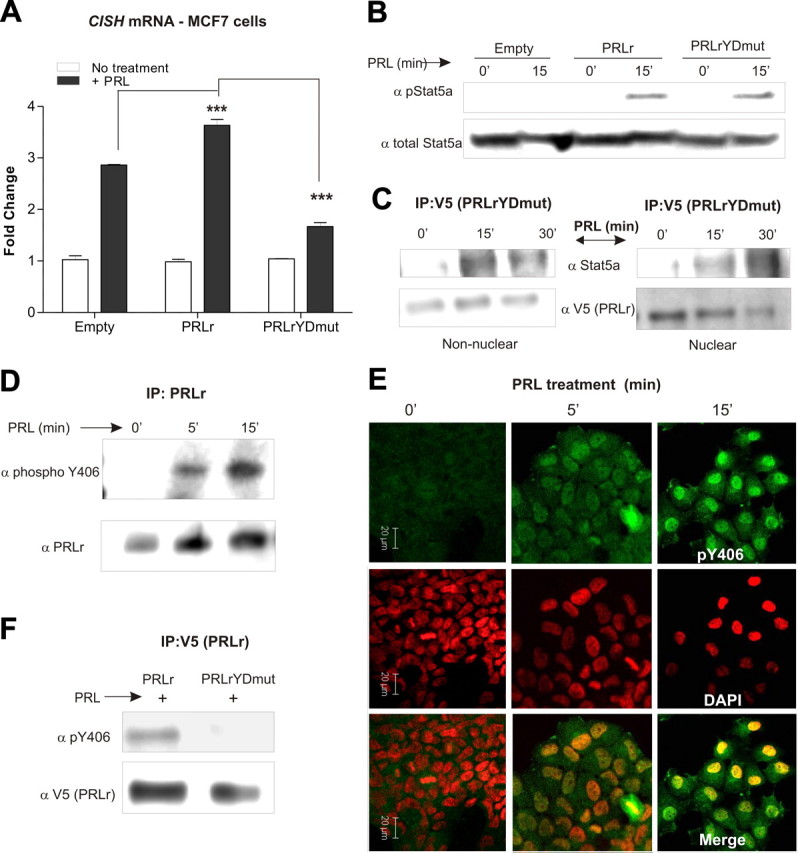
The PRLr TAD contributes to Stat5a-mediated gene expression. A, Real-time PCR performed on cDNA collected from PRL-treated MCF7 stable cell pools to determine CISH mRNA induction. MCF7 transfectants were serum starved for 24 h before 2 h PRL treatment. Values were normalized to GAPDH RNA, and fold change was calculated by comparing each value to the untreated control sample and are representative of three separate experiments with sem. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. Calculated P values of note are highlighted in the figure. Other statistical differences are as follows: E vs. E plus PRL, wild type vs. wild type plus PRL, TAD vs. TAD plus PRL, P < 0.001. B, Western blot analysis of CHO cells transiently transfected with Stat5a, Jak2, and empty vector, PRLr, or PRLrYDmut constructs. Cells were transfected and then serum starved for 24 h before PRL treatment (15 min). Blots were probed with antibodies against phospho-Stat5a (pStat5a) or Stat5a and demonstrate the ability of PRLrYDmut to activate Stat5a. C, Coimmunoprecipitation assays performed in PRL-treated T47D stable cells expressing PRLrYDmut. PRL-treated nonnuclear and nuclear lysates were subjected to coimmunoprecipitation using an anti-V5 antibody to precipitate PRLrYDmut. Bound proteins were analyzed by Western analysis using an anti-Stat5a antibody. Blots were stripped and reprobed using an anti-V5 antibody to demonstrate proper pulldown of mutant PRLr. IP, Immunoprecipitation. D, PRL-treated T47D cell lysates subjected to coimmunoprecipitation using an anti-PRLr antibody. Precipitates were analyzed by Western blot with an anti-phospho-Y406 PRLr antibody and demonstrate the phosphorylation of PRLr Y406 in a ligand-inducible manner. E, Immunofluorescence images in T47D cells using confocal microscopy. T47D cells were plated in chamber well slides and grown to 30% confluency and then serum starved for 24 h. Cells were treated with PRL for 0, 5, or 15 min and then fixed and stained with an anti-phospho-Y406 PRLr antibody. Green, PRLr phospho-Y406 PRLr; red, 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei. F, Coimmunoprecipitation analysis of PRL-treated MCF7 stable cell pools expressing wild-type PRLr or PRLrYDmut. Bound proteins were analyzed by Western analysis using an anti-phospho-Y406 PRLr antibody. ***, P < 0.001.
To ensure that the effects of PRLrYDmut were not due to deficient cell-surface signaling, the activation of downstream messengers was assessed. Western blot analysis demonstrated the ability of the transactivation-deficient mutant (PRLrYDmut) to induce Stat5a (Fig. 4B), Akt, and Erk phosphorylation (Supplemental Fig. 3E). Moreover, PRLrYDmut retained its ability to bind Stat5a both in the cytoplasm and in the nucleus (Fig. 4C). Collectively, these data indicate that mutation of the PRLr TAD does not compromise cell surface signaling relevant to the activation of downstream messengers. Therefore, the reduction in PRL-induced CISH transcription is specifically due to a defect in the transactivation capabilities of PRLrYDmut.
It was intriguing that one of the conserved residues required for PRLr transactivation was a tyrosine, since TADs are known to be regulated by a variety of posttranslational modifications including phosphorylation (31). To determine whether PRLr residue Y406 was phosphorylated, we generated a phosphospecific antibody against this residue. Indeed, Western blot analysis confirmed that Y406 was rapidly phosphorylated in response to PRL (Fig. 4D). Immunofluorescence similarly demonstrated inducible Y406 phosphorylation, and a significant portion of this phosphorylation was observed in the nucleus (Fig. 4E). Immunoprecipitation analysis also revealed the specificity of the newly generated Y406 antibody, because no detectable phosphorylation of Y406 was observed in cells expressing PRLrYDmut (Fig. 4F).
Interaction of PRLr and HMGN2
TADs are known to be responsible for stabilizing transcription factors at specific DNA loci and facilitating the recruitment of transcriptional cofactors (32). Our identification of a novel PRLr TAD led us to hypothesize that, in concert with Stat5a, nuclear PRLr may interact with other proteins involved in transcriptional regulation. A yeast two-hybrid system using the PRLr-ICD as bait was performed previously in our lab to identify potential PRLr-binding proteins (33). One of the identified proteins was HMGN2 (Gadd, S. L., and C. V. Clevenger, unpublished data). HMGN2 is known to bind to the nucleosome core particle and induce structural changes in chromatin (34). Interestingly, the intranuclear localization of HMGN2 has been shown to be associated with regions of highly active transcription (35). Based on this, we postulated that HMGN2 binds to the PRLr TAD and regulates Stat5a-mediated transcription.
To verify yeast two-hybrid results, the Gal4 system was used. HMGN2 and either wild-type or mutant Gal4-PRLr constructs were transfected into cells expressing a stably integrated Gal4-UAS reporter. Coimmunoprecipitation with anti-Gal4 followed by immunoblot analysis demonstrated the association of the PRLr TAD (404–448) and HMGN2, whereas binding was not observed between HMGN2 and the TAD mutant (Y406FD411A) (Fig. 5A) or a domain adjacent to the TAD (data not shown).
Fig. 5.

HMGN2 mediates PRLr transactivation activity. A, Coimmunoprecipitation performed on HLR cells (with stable integration of Gal4-UAS) with transient transfection of the indicated Gal4 constructs. Anti-Gal4 precipitates were subjected to Western blot analysis with an anti-HMGN2 antibody. B, Top panel, HMGN2 coimmunoprecipitation in PRL-treated (30 min) T47D cells expressing V5-tagged PRLr and PRLrYDmut. Western blots were analyzed with anti-V5 (PRLr), anti-HMGN2, anti-PRLr, or anti-phospho-PRLr Y406 antibodies. Bottom panel, The 10% input samples were probed with anti-HMGN2, anti-V5, and anti-PRLr antibodies to demonstrate equal loading (for HMGN2) and total PRLr levels (both endogenous and exogenous (V5-tagged). Note that in PRLr and PRLrYDmut input samples (lanes 4–7), anti-PRLr recognizes both endogenous PRLr and V5-tagged PRLr (which migrates ∼1 kDa higher than endogenous PRLr). C, ChIP analysis performed on PRL-treated T47D cells using an anti-HMGN2 antibody. Standard conditions (24 h serum starvation, 30 min PRL treatment) were used. D, CISH luciferase activity in MCF7 cells transfected with the indicated constructs. MCF7 cells were serum starved for 24 h and then treated for 24 h with PRL. Results are representative of three separate experiments with sem. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. Calculated P values of note are highlighted in the figure. Other statistical differences are as follows: E plus PRL vs. PRLr plus PRL, P < 0.001; empty vs. HMG2, P < 0.01; empty vs. PRLr plus HMGN2, P < 0.05. E, Real-time PCR performed on cDNA collected from T47D cells with stable expression of nonspecific shRNA (shNS) or shRNA against HMGN2 (shHMGN2). Stable T47D cells were serum starved for 24 h before 0, 1, or 4 h PRL treatment. Results are representative of three separate experiments with sem. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. Calculated P values of note are highlighted in the figure. Other statistical differences are as follows: shNS 0 PRL vs. shNS 1 h PRL/shNS 4 h PRL, P < 0.001; shHMGN2 0 PRL vs. shHMGN2 1 h PRL, P < 0.001. F, Real-time PCR performed on cDNA collected from MCF7 transfected with HMGN2 or NBDmut cDNA expression vectors. MCF7 cells were plated 24 h after transfection, and 2 μg HMGN2 or NBDmut cDNA was then transfected into cells using Fugene HD. At 24 h after transfection, cells were serum starved for an additional 24 h. Cells were then treated with PRL for 1 h. The values are those of a representative experiment repeated three separate times, where sem indicates the error of three replicate measurements within one experiment. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. Calculated P values of note are highlighted in the figure. Other statistical differences are as follows: empty vs. empty plus PRL; HMGN2 vs. HMGN2 plus PRL; NBDmut vs. NBDmut plus PRL, P < 0.001; empty plus PRL vs. NBDmut plus PRL, P < 0.05. NBDmut is the HMGN2 nucleosome-binding domain mutant harboring S24E/S28E mutations; shNS represents T47D cells expressing control shRNA. IP, Immunoprecipitation. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further confirm these results in the full-length PRLr, coimmunoprecipitation studies with stably transfected T47D cell pools were performed. PRL stimulated the association of PRLr with HMGN2, whereas mutation of the TAD (PRLrYDmut) abolished this interaction (Fig. 5B). Additionally, PRL-induced Y406 phosphorylation was detected in HMGN2 control or wild-type PRLr immunoprecipitates but not PRLrYDmut immunoprecipitates (Fig. 5B). When immunoprecipitates were analyzed for total PRLr levels, a PRL-inducible interaction was demonstrated in empty vector-, PRLr-, and PRLrYDmut-expressing cell lines. Despite this, in PRLrYDmut-expressing cells, bound PRLr failed to be Y406 phosphorylated. These data therefore suggest that PRLr/PRLrYDmut may be forming heterodimers, and the presence of PRLrYDmut in these heterodimers is sufficient to prevent phosphorylation of Y406. Coimmunoprecipitation analysis using an anti-V5 antibody further confirmed these findings (data not shown). Collectively, these results demonstrate a ligand-inducible interaction between the PRLr and HMGN2 and indicate that phosphorylation may be the molecular switch that facilitates TAD activation and subsequent HMGN2 recruitment.
We next sought to determine the extent of HMGN2 occupancy at Stat5a-target promoter regions, via ChIP. A PRL-enhanced association between HMGN2 and the CISH proximal promoter region was observed (Fig. 5C), further supporting the hypothesis that HMGN2 is a critical factor in facilitating Stat5a-mediated transcription.
The ability of HMGN2 to potentiate the Stat5a-driven transcription was next investigated. HMGN2 overexpression significantly enhanced CISH reporter activity, whereas the combination of PRLr and HMGN2 yielded an additive effect (Fig. 5D). Furthermore, short hairpin RNA (shRNA)-mediated knockdown of HMGN2 impaired PRL-induced CISH mRNA (Fig. 5E; knockdown efficiency shown in Supplemental Fig. 4, A and B). These results were confirmed using small interfering RNA (siRNA)-mediated HMGN2 depletion with a second target sequence (Supplemental Fig. 4, C and D).
High-mobility group family member HMGN1 exhibits high homology and similar function to HMGN2 (36). We therefore performed siRNA-mediated knockdown to determine whether HMGN1 also played a role in PRLr-mediated transactivation. However, as shown in Supplemental Fig. 4, E and F, HMGN1 knockdown had no effect on CISH mRNA levels. To complement siRNA data and to rule out off-target effects, a dominant-negative HMGN2 mutant was generated by replacement of two highly conserved serines within the nucleosome-binding domain (S24E/S28E, termed NBDmut). Mimicking serine phosphorylation at these sites has been previously shown to prevent HMGN2 chromatin binding (37). Interestingly, whereas wild-type HMGN2 overexpression potentiated CISH mRNA induction in MCF7 cells, NBDmut expression almost completely prevented this induction (Fig. 5F), suggesting that the HMGN2 NBD is required for its effects on Stat5a-target gene expression.
Regulation of PRLr and Stat5a promoter engagement by HMGN2
HMGN2 is thought to facilitate chromatin decompaction through its chromatin-unfolding domain (38). Based on this, we hypothesized that HMGN2 may enable Stat5a and PRLr promoter binding. To test this, ChIP on shHMGN2 or shNS cell pools was performed. As shown in Fig. 6, A and B, knockdown of HMGN2 decreased the relative amount of PRLr and Stat5a bound to the CISH promoter by approximately 80 and 40%, respectively. Taken together, these data indicate that HMGN2 may facilitate recruitment of these factors to the CISH promoter and is required for maximal PRL-induced CISH transcription.
Fig. 6.
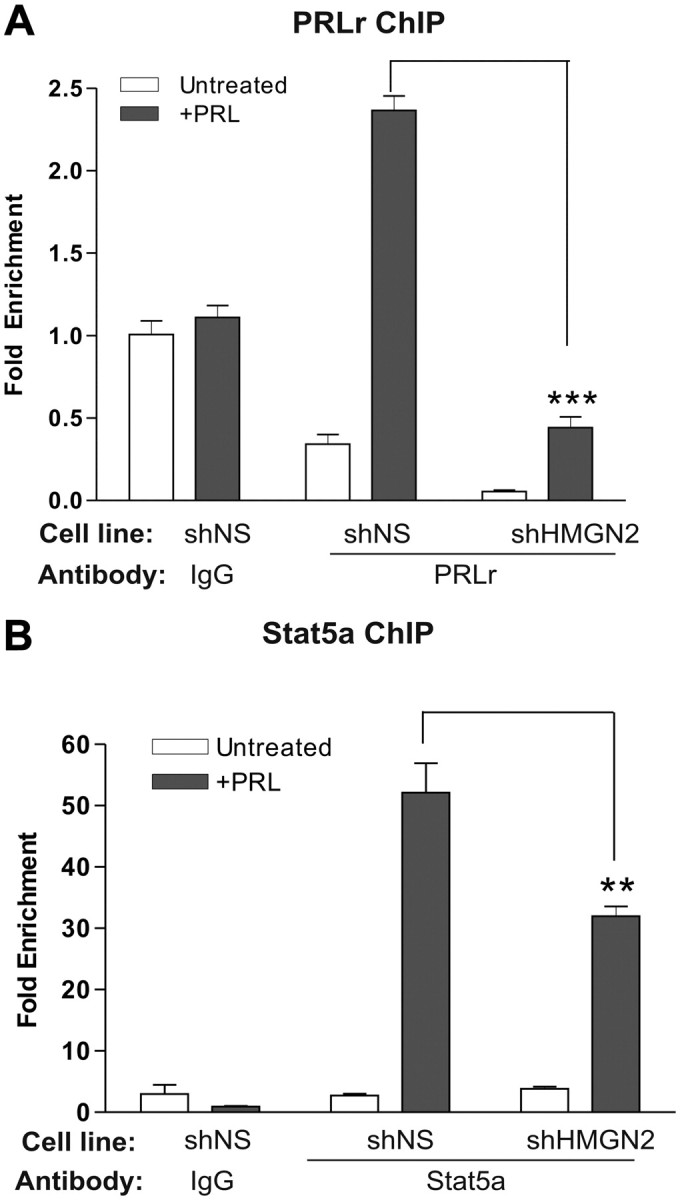
HMGN2 knockdown impairs PRLr and Stat5a promoter binding. A and B, shNS or shHMGN2 cells were serum starved for 24 h before PRL treatment. Cells were then subjected to ChIP using anti-PRLr (A) or anti-Stat5a (B) antibodies. Results were normalized to ChIP results obtained using an IgG control antibody in untreated shNS cells; values are reported as fold change. The values are those of a representative experiment repeated two separate times, where sem indicates the error of three replicate measurements within one single experiment. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. Calculated P values of note are highlighted in the figure. shNS represents T47D cells expressing control shRNA; shHMGN2 represents T47D cells expressing shRNA against HMGN2. *, P < 0.01; **, P < 0.01; ***, P < 0.001.
The PRLr TAD is necessary for a transformed phenotype in vitro
Given the association of the PRLr and its ligand in the pathogenesis of breast cancer, we sought to elucidate the functional effects of PRLr transactivation activity. Soft agar growth of cells was quantified to determine whether the transactivation function of nuclear PRLr was relevant to tumorigenic growth in vitro. Parental MCF10AΔp53 cells formed only a small number of colonies, consistent with previous reports (39). Expression of PRLr but not expression of PRLrYDmut significantly augmented colony formation (Fig. 7, A and B). These results imply that an activated PRLr TAD may be required in order for PRLr to promote a transformed phenotype.
Fig. 7.

The PRLr TAD promotes a transformed phenotype. A, MCF10AΔp53 transfectants were suspended in a 0.3% agar-medium mix supplemented with PRL (250 ng/ml) for 10 d. Each condition was photographed under phase-contrast optics. B, Quantification of the average colonies per field. Each condition was set up in triplicate wells, and error bars represent sem. All P values were calculated using one-way ANOVA with Bonferroni multiple-comparisons test. Calculated P values of note are highlighted in the figure. PRLrYDmut is transactivation-deficient PRLr harboring Y406F/D411A mutations. ***, P < 0.001.
Discussion
In this report, we provide strong evidence that in addition to its cell surface signaling functions, the PRLr possesses coactivator function; it 1) localizes to the nucleus, 2) has a defined TAD with potent activity, 3) associates with both the transcription factor Stat5a and the nucleosome-binding protein HMGN2, 4) engages with chromatin, and finally 5) aids in the activation of Stat5a-mediated gene expression. Collectively, our data imply that PRL-induced TAD phosphorylation initiates the coactivator activity of nuclear PRLr. Although potent TAD have been indentified in other transmembrane receptors (14, 40), this is the first study to identify that ligand-induced phosphorylation may be a key regulatory switch that triggers the transactivating function of a cell surface receptor.
We have performed numerous homology searches that have demonstrated the identified PRLr TAD is not shared by the larger family of cytokine receptor family members; however, this was expected for the following reasons. First, this novel domain lies outside of the membrane-proximal Box 1, X box, Box 2, V box motifs shared among all cytokine receptor family members (41). Second, a report focusing on GHr, the cytokine receptor most similar to the PRLr (42), identified a TAD within the GHr ECD, rather than the ICD (40). Thus, it is possible that other cytokine receptor family members possess unique TAD in regions unique to that specific receptor.
There are now several reports suggesting a mechanism for how transmembrane receptors localize to the nucleus. One report suggests that the protein translocon Sec61 could extract a transmembrane protein from the lipid bilayer, expelling it into the cytoplasm and leaving it accessible for nuclear import (43). Another possible mechanism is one in which a protein within the nuclear import machinery assists in extricating a receptor from the plasma membrane. This has been observed for both FGFr and the GHr ECD, where the subsequent nuclear localization of these proteins seems to be dependent on importin α/β, a component of multiple nuclear import pathways (19, 23).
The PRLr TAD is rapidly phosphorylated in response to ligand (Fig. 4D). We propose that Jak2, which is bound to the PRLr and required for PRLr signal transduction (8), may be the tyrosine kinase that is responsible for this rapid phosphorylation event. Jak2 has also been found in the nucleus, where in response to PRL, it phosphorylates the transcription factor NF1-C2 (44). However, because Jak2 activation alone is not sufficient for the full signaling properties of PRLr (45), it is possible that another kinase such as Src or Tec may be involved (46). We view these alternative kinases as less likely candidates because their kinetics of activation lag behind that of PRLr Y406 phosphorylation.
The classic model of PRL-mediated Stat5a activation involves 1) the recruitment of Stat5a to the PRLr, 2) Stat5a tyrosine phosphorylation via Jak2, 3) the formation of STAT homo- or heterodimers, and finally 4) the translocation of Stat5a homo/heterodimers into the nucleus where they bind to GAS sites in the promoters of target genes such as CISH. Although it is established that Stat5a localizes to the nucleus, it is currently unknown how this process is mediated, because Stat5 lacks the essential basic residues constituting a NLS (47). It is therefore possible that the PRLr and Stat5a remain bound and are trafficked to the nucleus together. In a previous study, it was reported that Stat3 and EGF are trafficked from the cell surface to the perinuclear region via endosomes, and inhibition of endocytosis is sufficient to prevent Stat3 nuclear entry (48). Thus, it is possible that endocytosis eventually leads to the transport of both Stat5a and PRLr into the cytoplasm, where they are then accessible to the nuclear import machinery. Because PRLr possesses no known NLS, it is possible that both Stat5a and the PRLr piggyback on another NLS-containing protein, which facilitates nuclear localization.
There are many lines of evidence that the nuclear localization of cell surface receptors may contribute to the pathology of malignant progression (13, 19, 49). In the context of these studies, we have seen that in vitro overexpression of transactivation-deficient PRLrYDmut diminishes the ability of cells to grow in soft agar. In vivo, we have observed abundant PRLr Y406 phosphorylation in malignant breast tissue as compared with normal breast epithelium with particular nuclear enhancement in metastatic tissue (Fiorillo, A. A., and C. V. Clevenger, unpublished data). Collectively, these data point to a mechanism whereby the transactivating properties of cell surface receptors may correlate with a malignant state.
We have demonstrated that the PRLr TAD serves to recruit HMGN2, which in turn enhances the binding of PRLr and Stat5a to the CISH promoter. We hypothesize that in the larger PRLr/Stat5a/HMGN2 macromolecular complex, the following events occur: 1) Stat5a provides the DBD that enables PRLr assembly onto chromatin, 2) the PRLr, through its TAD, tethers HMGN2 to the CISH promoter, thereby providing DNA sequence specificity for HMGN2, and 3) HMGN2, by modulating nucleosome phasing or posttranslational histone modifications, creates a more open chromatin conformation and enables the engagement of transcriptional elements that elevate levels of gene expression. A similar mechanism has been shown for the HMGN family member HMGN3 (50). siRNA-mediated depletion of HMGN3 prevented the binding of the transcription factor pancreatic and duodenal homeobox 1 to the Glu2 promoter and in the absence of pancreatic and duodenal homeobox 1, HMGN3 could not bind Glu2 (50). Given that HMGN family members do not possess DNA-sequence specificity, it is reasonable that an adapter molecule may be required for the binding of HMGN2 at specific loci. Therefore, HMGN2 and PRLr may depend on each other to mutually reinforce their chromatin-binding stability and assembly onto chromatin. We propose that this may be one crucial step in a series of coordinated events that regulate CISH transcription.
There are numerous and sometimes contradictory studies describing the role of HMGN2 in transcription. Evidence has suggested that HMGN2 enables chromatin modifications, specifically H3K14 acetylation (51). Using the murine ortholog of CISH, termed Cis, it has been demonstrated that in response to IL-3, H3K14 acetylation at the proximal promoter is modestly increased (52). However, this event was not sufficient for transcriptional activation, implying that Cis chromatin remodeling may also depend on a factor involved in nucleosome sliding/displacement. Because the chromatin-unfolding domain of HMGN proteins interacts with core histone tails and induces their rearrangement, leading to chromatin decompaction, (53) HMGN2 could conceivably be the missing link involved in Cis transcriptional activation. Through its interaction with core histone tails, the chromatin-unfolding domain may also serve to recruit factors that lead to posttranslational histone modifications such as H3K14 acetylation. In preliminary studies, we have observed a reduction in H3K14 acetylation at the CISH promoter as a result of HMGN2 depletion, suggesting that this may be one mechanism by which HMGN2 activates CISH transcription (data not shown).
Several lines of evidence also support a second model for HMGN whereby it counteracts chromatin compaction by linker histones. This has been shown by footprinting of HMGN bound to nucleosome core particles which indicated that HMGN binding sites overlap with the proposed binding sites for the linker histones, namely histone H1 (54). These models (i.e. HMGN2-mediated chromatin modifications or HMGN2-mediated chromatin decompaction by linked histone competition) are not mutually exclusive, and it is conceivable that HMGN could modulate the activity of both the linker histones and core histone tails within the same chromatin regions or that one mechanism would take preference over another depending on the chromatin environment.
A larger question regarding transmembrane nuclear localization relates to the physiological context of receptors functioning both at the cell surface and in the nucleus. Although it is recognized that surface receptors share many, if not most, of the same proximal signaling pathways, currently no model clearly describes how the cell distinguishes between different receptors that activate the same signaling cascades. The nuclear localization of transmembrane receptors could therefore provide a partial resolution to this conundrum of receptor/signaling pathway differentiation by contributing to specificity in output gene expression that would otherwise be compromised by the degeneracy of pathways shared among numerous receptors. We therefore propose that the ability of a cell surface receptor to recruit regulatory factors to specific promoter regions represents a general mechanism that may be applicable to the larger field of nuclear-localized cell surface receptors. In corroboration of our hypothesis and working model, two recent papers have been published, demonstrating interactions between EGFr/RNA helicase (RHA) (55) and ErbB2/progesterone receptor (56), respectively. Additionally, it has been shown that the transcription factor, coactivator activator, can interact with a TAD within the GHr ECD (40).
The concept of a nuclear transmembrane receptor functioning at both the nongenomic and genomic level can be paralleled to that of the estrogen receptor. Estrogen receptors have now been shown to function both as classic transcription factors and as signaling factors in the cytoplasm (57). This convergence of these estrogen receptor functions at multiple response elements provides an exceedingly fine degree of control for regulating gene expression. Similarly, the convergence of the cell surface and genomic actions of the PRLr may provide a unique way to confer receptor/pathway specificity at the genomic level to finely tune target-gene expression.
As shown in Fig. 8, we propose a working model in which 1) PRL stimulation induces PRLr TAD phosphorylation and PRLr nuclear localization, 2) the PRLr-mediated recruitment of HMGN2 facilitates the association of PRLr and Stat5a with the CISH proximal promoter where Stat5a provides the DBD necessary for PRLr chromatin occupancy, and 3) the coordinated assembly of HMGN2/Stat5a/PRLr onto chromatin initiates CISH transcriptional activation. We propose that this mechanism enables the cell to differentiate between multiple receptors that activate converging pathways by providing a direct connection between cell surface signaling and gene regulation in the nucleus. Given that an activated PRLr TAD is required for anchorage-independent growth and the phosphorylation of PRLr-Y406 is present in malignant breast tissue, the observations presented here may have direct pathophysiological relevance.
Fig. 8.

Model for signal-dependent PRLr transactivation. 1) Ligand induces PRLr Y406 phosphorylation and PRLr nuclear localization; 2) PRLr-mediated recruitment of HMGN2 facilitates engagement of the PRLr/Stat5a/HMGN2 complex onto the CISH promoter; 3) the coordinated assembly of this complex promotes CISH transcriptional activation. H3K14Ac, Histone 3, lysine 9 acetylation; P, phosphorylation.
Materials and Methods
PRL treatment
Human recombinant PRL was a gift from Dr. Anthony Kossiakoff, University of Chicago (Chicago, IL). PRL was added to cells to yield a final concentration of 250 ng/ml.
Cell lines and culture
T47D and MCF7 human breast cancer cell lines were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO, Grand Island, NY). The 293T/17 cells with stable integration of the SV40 large T antigen and HLR cells with stable integration of the Gal4-UAS luciferase reporter were supplied by Dr. Debu Chakravarti and were cultured in DMEM with 10% FBS and 100 μg/ml streptomycin. MCF10Adelp53 cell lines were a gift from Dr. Serge Fuchs and were cultured in DMEM/F-12 media supplemented with 10% FBS, 100 U/ml penicillin/streptomycin. All cells were incubated in a humidified 5% CO2/95% air atmosphere at 37 C.
Plasmids
Wild type PRLr in pTracer and the CISH-luciferase reporter constructs have been previously described (10). PRLr cDNA, NLS-ICD, and PRL-NLS were created using PCR that additionally added the SV40 NLS (GCCCTAAAAAGAAGCGTAAAGTCG) to each product. PCR products were ligated into pTracer EF-V5/His (Invitrogen, Carlsbad, CA) using EcoRI and NotI restriction sites. Gal4-PRLr constructs were generated by PCR to amplify specific regions of the PRLr ICD or ECD. PRLr cDNA were then cloned into Gal4-pCMX vector using EcoRI and EcoRV restriction sites. PRLrYDmut was generated using QuikChange Lightning mutagenesis (Stratagene, La Jolla, CA) using the parental PRLr pTracer construct.
pCMV-XL5 containing HMGN2 cDNA was purchased (Origene, Rockville, MD). NBDmut was constructed using QuikChange Lightning site-directed mutagenesis. All mutagenesis primer sequences are listed in Supplemental Table 1.
Cell fractionation
T47D cells were grown in 150-mm dishes until they reached 70% confluency. MCF7 or CHO cells were plated 24 h before transfection in 100-mm dishes and then transfected with 2 μg PRLr cDNA using Fugene HD (Roche, Indianapolis, IN). Cytoplasmic and nuclear extracts were subsequently obtained using NE-PER (Pierce, Rockford, IL) according to the manufacturer's protocol. The 5× Laemmli buffer was added to nonnuclear and nuclear samples. Samples were then boiled for 5 min and then subjected to SDS-PAGE using a 4–20% Tris-HCl gradient gel (Bio-Rad, Hercules, CA). Proteins were then transferred to polyvinylidene difluoride (PVDF) filters. The filters were blocked in casein blocker (Pierce) for 1 h. After blocking, the blots were incubated with the one of the following primary antibodies overnight at 4 C: anti-PRLr ECD (Invitrogen), anti-PRLr-ICD (Santa Cruz Biotechnology, Santa Cruz, CA), anti-histone H3 (Abcam, Cambridge, MA), anti-lamin B1 (Abcam), or anti-calnexin (BD Biosciences, San Jose, CA). Antibodies were diluted in casein blocker/Tris-buffered saline plus 0.05% Tween 20. Filters were washed three times with Tris-buffered saline plus 0.05% Tween 20 and then probed with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Chemiluminescence was detected with ECL Plus (Amersham, Piscataway, NJ), and images were collected on a charge-coupled device camera (Fuji, Fujifilm Life Sciences, Philadelphia, PA).
Immunohistochemistry
Immunohistochemical analysis, as previously described (4), was carried out with an anti-PRLr antibody (Santa Cruz). Sections of 5 μm from formalin-fixed paraffin-embedded tissue microarrays were deparaffinized in xylene and rehydrated in graded alcohol. After deparaffinization, heat-induced antigen retrieval by boiling slides in 10 mm citrate buffer (pH 6.0) for 20 min was carried out. The sections were blocked with a peroxidase blocking system (Dako, Carpinteria CA) for 10 min. Sequential incubations included the following: 2% normal goat serum for 20 min, anti-PRLr antibody at 1:25 dilution for 60 min at room temperature, secondary biotinylated anti-rabbit IgG for 30 min, and finally, the streptavidin-biotinylated horseradish peroxidase complex reagent (Dako) for 30 min at room temperature. After incubations, slides were washed three times in buffer for 3 min each. Sections were then exposed to the chromogen DABplus (Dako) for 5 min and counterstained in hematoxylin, dehydrated, cleared, and mounted. In this analysis, the nuclear intensity of tissue samples was scored from 0–3, and a score above 1.5 was counted as positive, as previously described (4).
Biotinylation
Biotinylation was performed using 3-sulfo-N-hydroxysuccinimide biotin (Pierce) according to the manufacturer's protocol. T47D cells in were grown in 150-mm dishes to 70% confluency and then serum starved for 24 h before biotinylation. Cells were lifted off dishes with versene, washed three times with 10 ml PBS and resuspended in 200 μl 2 mm 3-sulfo-N-hydroxysuccinimide-biotin (Pierce) for 30 min at 4 C (to prevent active internalization). Reactions were quenched for 10 min with 500 μl PBS containing 100 mm glycine. Cells were stimulated with PRL for 0, 10, and 30 min at 37 C. Samples were then separated into nonnuclear and nuclear fractions using NE-PER (Pierce) according to manufacturer's protocol, 10% of each sample was aliquoted for input. Nuclear lysates were diluted with dilution buffer (20% glycerol, 1% Triton X-100). Streptavidin-agarose beads (Pierce; 50 μl of a 50:50 slurry) were then added to 500 μl nuclear and nonnuclear fractions, and the suspension was rocked overnight at 4 C. The beads were washed five times with respective lysis buffers (cytoplasmic extraction reagent or nuclear extraction reagent). Proteins were then eluted from beads using 2× Laemmli buffer, boiled for 5 min, and then resolved by SDS-PAGE on a 4–20% gradient gel (Bio-Rad) and transferred to a PVDF membrane. Membranes were blocked as stated above (see cell fractionation) and probed with the following primary antibodies overnight at 4 C: anti-PRLr-ICD antibody and anti-β1-integrin antibody (Abcam) as a negative control. Membranes were then developed as stated above (see cell fractionation). The 5× Laemmli buffer was added to input samples, and then samples were treated as above. Membranes with input proteins were probed with an anti-calnexin primary antibody to control for nonnuclear contamination in nuclear samples.
Luciferase assay
Luciferase assays that used the CISH-luciferase reporter were conducted as previously described using MCF7 cells (10). Luciferase assays using the Gal4-UAS reporter construct were conducted as follows. 293T cells were seeded into a 24-well plate at a density of 1.5 × 105. Cells were transfected with 250 ng luciferase reporter, 2 ng renilla reporter, and 10 ng Gal4-PRLr constructs using Lipofectamine 2000. Twenty-four hours after transfection,cells were lysed through a luciferase assay using Dual Reporter Luciferase reagents (Promega, Madison, WI) and read by a Victor Microplate Reader (PerkinElmer, Norwalk, CT). All transfections were performed in triplicate or quadruplicate, and each individual sample was read in duplicate. The ratio of luciferase/renilla was calculated, and the results are reported as fold change compared with an empty vector control (Gal4-UAS reporter) or an untreated empty vector control (CISH reporter, LHRE reporter).
Cell fractionation and nuclear coimmunoprecipitation
Cells were grown in 150-mm dishes until they reached 70% confluency, followed by arrest for 24 h. Cells were then treated with PRL (250 ng/ml) for the indicated times (0, 10, or 30 min). Cytoplasmic and nuclear extracts were subsequently obtained using NE-PER (Pierce) according to the manufacturer's protocol. Nuclear lysates were diluted with dilution buffer (20% glycerol, 1% Triton X-100), and 4 μg anti-Stat5a (Invitrogen) or anti-PRLr (Santa Cruz) antibodies was added and samples rocked overnight at 4 C. Subsequently, 50 μl Dynal protein G magnetic beads (Invitrogen) were added for 1 h, and samples were collected on a magnetic particle concentrator (DynaMag-2; Invitrogen) and washed in the appropriate buffer; cytosolic samples were washed in cytoplasmic extraction reagent lysis buffer, and nuclear samples were washed in dilution buffer. Bound proteins were recovered in 2× Laemmli buffer and subjected to SDS-PAGE using a 4–20% Tris-HCl gradient gel (Bio-Rad). Proteins were then transferred to a PVDF membrane, and normal immunoblot procedures were followed as stated above.
Chromatin immunoprecipitation
ChIP was conducted as previously described (58) until DNA isolation, and then the fast ChIP was employed (59). Specifically, T47D cells were arrested in phenol-free medium for 24 h before PRL treatment (250 ng/ml) for 30 min. Anti-PRLr, anti-Stat5 (Santa Cruz), anti-HMGN2 (Millipore, Billerica, MA), and anti-H3K14 (Millipore) antibodies were used to immunoprecipitate protein-DNA complexes. Eluted DNA was subjected to real-time PCR analysis and was normalized to input. The fold change was subsequently normalized to an IgG or beads-only control. Primers for specific amplicons are listed in Supplemental Table 2.
Retroviral production
Overexpression cell lines
PRLr or PRLrYDmut in pTracer were amplified through PCR and ligated into the retroviral pBabe-GFP vector using EcoRI and SalI.
Knockdown cell lines
A predesigned HMGN2 shRNA pRFP retroviral vector was purchased from Origene, and the PRLr shRNA was designed and cloned into pRFP vector (sequence of HMGN2, GTGTCAGGCAATCTGGACTTTCCAGTGAT; PRLr, CAACTGCATAACCTTTACACT). Recombinant retroviruses for pBabe, PRLr, PRLrYDmut, scrambled shRNA, shHMGN2, and shPRLr were generated using transfection of retroviral vectors with Lipofectamine 2000 (Invitrogen) in 293T/17 cells along with pVSVG and pGalPol vectors. Viral supernatant was collected 48 h after transfection and added to the appropriate cell lines along with 8 μg/ml polybrene. Cells were spin-infected at 500 × g for 2 h at 32 C. Infected cells were enriched by fluorescence-activated cell sorting for GFP (overexpression cell lines) or puromycin selection (knockdown cell lines).
HMGN2 coimmunoprecipitation
Stably transfected T47D cells (expressing empty vector, wild-type PRLr, or PRLrYDmut) or stably transfected MCF7 cells (expressing empty vector, wild-type PRLr, or PRLrYDmut) were grown in 150-mm dishes until 70% confluency followed by arrest for 24 h before PRL treatment. Cells were washed with PBS and then pelleted at 1000 × g, and 10% of each sample was taken for input. Cell lysis and immunoprecipitation was then conducted as previously described (60). Samples were subjected to coimmunoprecipitation with 4 μg of an anti-HMNG2 antibody (Millipore), an anti-V5 antibody (Invitrogen) to recognize PRLr, or an anti-Gal4 antibody (Santa Cruz) to recognize PRLr truncation constructs. Antibodies were added to samples and incubated overnight at 4 C. Fifty microliters of Dynal protein G magnetic beads (Invitrogen) were added for 1 h, and samples were collected on a magnetic particle concentrator (DynaMag-2; Invitrogen) and washed in the appropriate buffer (as previously described, 60). Bound proteins were recovered in 2× Laemmli buffer and run on a 4–20% Tris-HCl gradient gel (Bio-Rad), followed by Western blot analysis. Membrane was blocked with casein blocker (Pierce), probed with anti-V5 or anti-HMNG2 antibodies, and visualized by chemiluminescence (GE Healthcare, Piscataway, NJ).
RT-PCR and real-time PCR
Total RNA was isolated from the following cells: MCF7 and MCF10AΔp53 stable cells expressing PRLr constructs, MCF7 cells with transient transfection of HMGN2 contructs, and T47D cells with transient or stable knockdown of HMGN2. RNA was isolated using EZ mini-prep kit (QIAGEN, Valencia, CA) as previously reported (61). cDNA was synthesized using qScript (Quanta Biosciences, Gaithersburg, MD). cDNA was diluted to 2.5 ng/μl (corresponding to RNA concentration), and 4 μl cDNA, 1 μl primers (2 μm each), and 5 μl 2× Power SYBR MasterMix was used for real-time PCR in 10 μl reaction volume performed in a 384-well plate. Real-time PCR was conducted on an ABI 7900HT thermocycler (Applied Biosystems, Foster City, CA). All real-time PCR were run in triplicate. For RT-PCR, data were normalized to GAPDH RNA or 18S rRNA. Fold change for RT-PCR is represented as 2−ΔΔCt(2−(Cttarget−Ct18SrRNA)PRL−(Cttarget−Ct18SrRNA)control) using empty vector, untreated as a baseline. Primers used are listed in Supplemental Table 2.
Immunofluorescence
T47D cells were grown on four-well chamber slides to approximately 50% confluency in normal growth medium before staining with anti-phospho-PRLrY406 (New England Biosciences, Ipswich, MA). Cells were arrested for 24 h and then treated with PRL for 0, 5, and 15 min. Cells were rinsed in PBS and then fixed in 4% paraformaldehyde/0.1 m phosphate buffer (pH 7.4) for 10 min. Cells were blocked with blocking solution (PBS, 1% BSA, 0.05% Triton X-100) for 15 min at room temperature and then incubated with a 1:25 dilution of anti-phospho-Y406 PRLr (New England Biosciences) diluted in blocking solution for 2 h at room temperature. After washing with wash solution (PBS, 0.05% Triton X-100), cells were incubated with a 1:800 dilution of secondary antibody conjugated to Alexafluor-388 (Invitrogen) for 30 min. Cells were washed twice with PBS and stained with Hoechst (1 μg/ml in PBS) for 3 min. Cells were washed three times with PBS, and then mounted using antifade mounting medium (Vector Laboratories Inc., Burlingame, CA). Images were visualized using confocal microscopy.
Western blotting
For signaling studies, the indicated cell lines (PRLr-expressing T47D, PRL-expressing MCF10AΔp53, and CHO cells transiently transfected with wild-type PRLr or PRLrYDmut) were plated in 10-mm dishes at 70% confluency. Cells were arrested and treated with 250 ng/ml PRL for the indicated time courses. Cells were lysed in RIPA buffer and subjected to Western blot as described above. Blots were probed with anti-Akt (Cell Signaling Technology, Beverly, MA), anti-phospho-Akt (Cell Signaling), anti-Erk (Cell Signaling), anti-phospho-Erk (Cell Signaling), anti-Stat5a (Santa Cruz), and anti-phospho-Stat5a (Invitrogen) antibodies.
siRNA-mediated knockdown
siRNA knockdown experiments were conducted in T47D cells as has been previously reported (62). The Accell SMARTpool siRNA against HMGN1 were purchased from Thermo Scientific (Pittsburgh, PA; catalog number E-015567-00-0005). The siRNA against HMGN2 was purchased through Origene (catalog number SR302144, sequence no. 3).
Soft agar
Soft agar was carried out as previously described (63). Colony size and number were quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Statistical analysis
Statistical analysis was performed using one-way or two-way ANOVA on GraphPad Prism version 4 (La Jolla, CA). The results are shown as the means with error bars depicting ±sem. P < 0.05 is considered as statistically significant. All experiments were performed at least three times unless otherwise indicated.
Acknowledgments
We thank Dr. Debu Chakravarti for the use of reagents for Gal4, ChIP, and retroviral experiments; Dr. Anthony Kossiakoff for providing human recombinant PRL; and Dr. Brandon Parker and Manop Buranapramest for their technical advice with ChIP and retroviral infection. We also thank Dr. Feng Fang for his guidance in cloning experimental design. Additionally, we thank Drs. Kathy Rundell, Vincent Cryns, and Debu Chakravarti for their critical review of this manuscript.
Disclosure Summary: The authors have nothing to disclose.
This work was supported by a Department of Defense BC073401 Predoctoral Traineeship Award and grants from the Avon and Lynn Sage Foundations.
Footnotes
- ChIP
- Chromatin immunoprecipitation
- DBD
- DNA-binding domain
- ECD
- extracellular domain
- EGFr
- epidermal growth factor receptor
- FBS
- fetal bovine serum
- FGFr
- fibroblast growth factor receptor
- GAS
- gamma interferon activating sequence
- GHr
- GH receptor
- HMGN2
- high-mobility group N2 protein
- ICD
- intracellular domain
- Jak2
- Janus kinase 2
- NLS
- nuclear localization signal
- PRL
- prolactin
- PRLr
- PRL receptor
- PVDF
- polyvinylidene difluoride
- shRNA
- short hairpin RNA
- siRNA
- small interfering RNA
- Stat5a
- signal transducer and activator of transcription 5a
- SV40
- simian virus 40
- TAD
- transactivation domain
- UAS
- upstream activating sequence.
References
- 1. Reynolds C , Montone KT , Powell CM , Tomaszewski JE , Clevenger CV. 1997. Expression of prolactin and its receptor in human breast carcinoma. Endocrinology 138:5555–5560 [DOI] [PubMed] [Google Scholar]
- 2. Yamashita H , Nevalainen MT , Xu J , LeBaron MJ , Wagner KU , Erwin RA , Harmon JM , Hennighausen L , Kirken RA , Rui H. 2001. Role of serine phosphorylation of Stat5a in prolactin-stimulated beta-casein gene expression. Mol Cell Endocrinol 183:151–163 [DOI] [PubMed] [Google Scholar]
- 3. Harris J , Stanford PM , Sutherland K , Oakes SR , Naylor MJ , Robertson FG , Blazek KD , Kazlauskas M , Hilton HN , Wittlin S , Alexander WS , Lindeman GJ , Visvader JE , Ormandy CJ. 2006. Socs2 and elf5 mediate prolactin-induced mammary gland development. Mol Endocrinol 20:1177–1187 [DOI] [PubMed] [Google Scholar]
- 4. McHale K , Tomaszewski JE , Puthiyaveettil R , Livolsi VA , Clevenger CV. 2008. Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma. Mod Pathol 21:565–571 [DOI] [PubMed] [Google Scholar]
- 5. Li Y , Clevenger CV , Minkovsky N , Kumar KG , Raghunath PN , Tomaszewski JE , Spiegelman VS , Fuchs SY. 2006. Stabilization of prolactin receptor in breast cancer cells. Oncogene 25:1896–1902 [DOI] [PubMed] [Google Scholar]
- 6. Hankinson SE , Willett WC , Michaud DS , Manson JE , Colditz GA , Longcope C , Rosner B , Speizer FE. 1999. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst 91:629–634 [DOI] [PubMed] [Google Scholar]
- 7. Rui H , Kirken RA , Farrar WL. 1994. Activation of receptor-associated tyrosine kinase JAK2 by prolactin. J Biol Chem 269:5364–5368 [PubMed] [Google Scholar]
- 8. Lebrun JJ , Ali S , Sofer L , Ullrich A , Kelly PA. 1994. Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the prolactin receptor and its associated tyrosine kinase JAK2. J Biol Chem 269:14021–14026 [PubMed] [Google Scholar]
- 9. Brockman JL , Schroeder MD , Schuler LA. 2002. PRL activates the cyclin D1 promoter via the Jak2/Stat pathway. Mol Endocrinol 16:774–784 [DOI] [PubMed] [Google Scholar]
- 10. Fang F , Antico G , Zheng J , Clevenger CV. 2008. Quantification of PRL/Stat5 signaling with a novel pGL4-CISH reporter. BMC Biotechnol 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raccurt M , Tam SP , Lau P , Mertani HC , Lambert A , Garcia-Caballero T , Li H , Brown RJ , McGuckin MA , Morel G , Waters MJ. 2003. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br J Cancer 89:524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clevenger CV. 2004. Roles and regulation of Stat family transcription factors in human breast cancer. Am J Pathol 165:1449–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lo HW , Xia W , Wei Y , Ali-Seyed M , Huang SF , Hung MC. 2005. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res 65:338–348 [PubMed] [Google Scholar]
- 14. Lin SY , Makino K , Xia W , Matin A , Wen Y , Kwong KY , Bourguignon L , Hung MC. 2001. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 3:802–808 [DOI] [PubMed] [Google Scholar]
- 15. Komuro A , Nagai M , Navin NE , Sudol M. 2003. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem 278:33334–33341 [DOI] [PubMed] [Google Scholar]
- 16. Ni CY , Murphy MP , Golde TE , Carpenter G. 2001. γ-Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 294:2179–2181 [DOI] [PubMed] [Google Scholar]
- 17. Offterdinger M , Schöfer C , Weipoltshammer K , Grunt TW. 2002. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol 157:929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lobie PE , Wood TJ , Chen CM , Waters MJ , Norstedt G. 1994. Nuclear translocation and anchorage of the growth hormone receptor. J Biol Chem 269:31735–31746 [PubMed] [Google Scholar]
- 19. Conway-Campbell BL , Wooh JW , Brooks AJ , Gordon D , Brown RJ , Lichanska AM , Chin HS , Barton CL , Boyle GM , Parsons PG , Jans DA , Waters MJ. 2007. Nuclear targeting of the growth hormone receptor results in dysregulation of cell proliferation and tumorigenesis. Proc Natl Acad Sci USA 104:13331–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marti U , Wells A. 2000. The nuclear accumulation of a variant epidermal growth factor receptor (EGFR) lacking the transmembrane domain requires coexpression of a full-length EGFR. Mol Cell Biol Res Commun 3:8–14 [DOI] [PubMed] [Google Scholar]
- 21. Feng S , Xu J , Wang F , Kan M , McKeehan WL. 1996. Nuclear localization of a complex of fibroblast growth factor (FGF)-1 and an NH2-terminal fragment of FGF receptor isoforms R4 and R1alpha in human liver cells. Biochim Biophys Acta 1310:67–73 [DOI] [PubMed] [Google Scholar]
- 22. Johnston CL , Cox HC , Gomm JJ , Coombes RC. 1995. Fibroblast growth factor receptors (FGFRs) localize in different cellular compartments. A splice variant of FGFR-3 localizes to the nucleus. J Biol Chem 270:30643–30650 [DOI] [PubMed] [Google Scholar]
- 23. Reilly JF , Maher PA. 2001. Importin beta-mediated nuclear import of fibroblast growth factor receptor: role in cell proliferation. J Cell Biol 152:1307–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hung LY , Tseng JT , Lee YC , Xia W , Wang YN , Wu ML , Chuang YH , Lai CH , Chang WC. 2008. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res 36:4337–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lo HW , Hung MC. 2006. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer 94:184–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lo HW , Cao X , Zhu H , Ali-Osman F. 2010. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res 8:232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rao YP , Buckley DJ , Buckley AR. 1995. The nuclear prolactin receptor: a 62-kDa chromatin-associated protein in rat Nb2 lymphoma cells. Arch Biochem Biophys 322:506–515 [DOI] [PubMed] [Google Scholar]
- 28. Rao YP , Buckley DJ , Olson MD , Buckley AR. 1995. Nuclear translocation of prolactin: collaboration of tyrosine kinase and protein kinase C activation in rat Nb2 node lymphoma cells. J Cell Physiol 163:266–276 [DOI] [PubMed] [Google Scholar]
- 29. LeBaron MJ , Xie J , Rui H. 2005. Evaluation of genome-wide chromatin library of Stat5 binding sites in human breast cancer. Mol Cancer 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Litterst CM , Kliem S , Marilley D , Pfitzner E. 2003. NCoA-1/SRC-1 is an essential coactivator of STAT5 that binds to the FDL motif in the α-helical region of the STAT5 transactivation domain. J Biol Chem 278:45340–45351 [DOI] [PubMed] [Google Scholar]
- 31. Li S , Shang Y. 2007. Regulation of SRC family coactivators by post-translational modifications. Cell Signal 19:1101–1112 [DOI] [PubMed] [Google Scholar]
- 32. Näär AM , Lemon BD , Tjian R. 2001. Transcriptional coactivator complexes. Annu Rev Biochem 70:475–501 [DOI] [PubMed] [Google Scholar]
- 33. Gadd SL , Clevenger CV. 2006. Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol Endocrinol 20:2734–2746 [DOI] [PubMed] [Google Scholar]
- 34. Crippa MP , Alfonso PJ , Bustin M. 1992. Nucleosome core binding region of chromosomal protein HMG-17 acts as an independent functional domain. J Mol Biol 228:442–449 [DOI] [PubMed] [Google Scholar]
- 35. Postnikov YV , Herrera JE , Hock R , Scheer U , Bustin M. 1997. Clusters of nucleosomes containing chromosomal protein HMG-17 in chromatin. J Mol Biol 274:454–465 [DOI] [PubMed] [Google Scholar]
- 36. Landsman D , Bustin M. 1986. Chromosomal proteins HMG-14 and HMG-17. Distinct multigene families coding for similar types of transcripts. J Biol Chem 261:16087–16091 [PubMed] [Google Scholar]
- 37. Prymakowska-Bosak M , Misteli T , Herrera JE , Shirakawa H , Birger Y , Garfield S , Bustin M. 2001. Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol Cell Biol 21:5169–5178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ueda T , Postnikov YV , Bustin M. 2006. Distinct domains in high mobility group N variants modulate specific chromatin modifications. J Biol Chem 281:10182–10187 [DOI] [PubMed] [Google Scholar]
- 39. Plotnikov A , Varghese B , Tran TH , Liu C , Rui H , Fuchs SY. 2009. Impaired turnover of prolactin receptor contributes to transformation of human breast cells. Cancer Res 69:3165–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Conway-Campbell BL , Brooks AJ , Robinson PJ , Perani M , Waters MJ. 2008. The extracellular domain of the growth hormone receptor interacts with coactivator activator to promote cell proliferation. Mol Endocrinol 22:2190–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ihle JN , Kerr IM. 1995. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 11:69–74 [DOI] [PubMed] [Google Scholar]
- 42. Boutin JM , Jolicoeur C , Okamura H , Gagnon J , Edery M , Shirota M , Banville D , Dusanter-Fourt I , Djiane J , Kelly PA. 1988. Cloning and expression of the rat prolactin receptor, a member of the growth hormone/prolactin receptor gene family. Cell 53:69–77 [DOI] [PubMed] [Google Scholar]
- 43. Liao HJ , Carpenter G. 2007. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol Biol Cell 18:1064–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nilsson J , Bjursell G , Kannius-Janson M. 2006. Nuclear Jak2 and transcription factor NF1-C2: a novel mechanism of prolactin signaling in mammary epithelial cells. Mol Cell Biol 26:5663–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lebrun JJ , Ali S , Ullrich A , Kelly PA. 1995. Proline-rich sequence-mediated Jak2 association to the prolactin receptor is required but not sufficient for signal transduction. J Biol Chem 270:10664–10670 [DOI] [PubMed] [Google Scholar]
- 46. Clevenger CV , Furth PA , Hankinson SE , Schuler LA. 2003. The role of prolactin in mammary carcinoma. Endocr Rev 24:1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams CC , Allison JG , Vidal GA , Burow ME , Beckman BS , Marrero L , Jones FE. 2004. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol 167:469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bild AH , Turkson J , Jove R. 2002. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J 21:3255–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lincoln DT , Sinowatz F , Temmim-Baker L , Baker HI , Kölle S , Waters MJ. 1998. Growth hormone receptor expression in the nucleus and cytoplasm of normal and neoplastic cells. Histochem Cell Biol 109:141–159 [DOI] [PubMed] [Google Scholar]
- 50. Ueda T , Furusawa T , Kurahashi T , Tessarollo L , Bustin M. 2009. The nucleosome binding protein HMGN3 modulates the transcription profile of pancreatic β-cells and affects insulin secretion. Mol Cell Biol 29:5264–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herrera JE , Sakaguchi K , Bergel M , Trieschmann L , Nakatani Y , Bustin M. 1999. Specific acetylation of chromosomal protein HMG-17 by PCAF alters its interaction with nucleosomes. Mol Cell Biol 19:3466–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rascle A , Lees E. 2003. Chromatin acetylation and remodeling at the Cis promoter during STAT5-induced transcription. Nucleic Acids Res 31:6882–6890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Trieschmann L , Martin B , Bustin M. 1998. The chromatin unfolding domain of chromosomal protein HMG-14 targets the N-terminal tail of histone H3 in nucleosomes. Proc Natl Acad Sci USA 95:5468–5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alfonso PJ , Crippa MP , Hayes JJ , Bustin M. 1994. The footprint of chromosomal proteins HMG-14 and HMG-17 on chromatin subunits. J Mol Biol 236:189–198 [DOI] [PubMed] [Google Scholar]
- 55. Huo L , Wang YN , Xia W , Hsu SC , Lai CC , Li LY , Chang WC , Wang Y , Hsu MC , Yu YL , Huang TH , Ding Q , Chen CH , Tsai CH , Hung MC. 2010. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc Natl Acad Sci USA 107:16125–16130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Béguelin W , Díaz Flaqué MC , Proietti CJ , Cayrol F , Rivas MA , Tkach M , Rosemblit C , Tocci JM , Charreau EH , Schillaci R , Elizalde PV. 2010. Progesterone receptor induces ErbB-2 nuclear translocation to promote breast cancer growth via a novel transcriptional effect: ErbB-2 function as a coactivator of Stat3. Mol Cell Biol 30:5456–5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Razandi M , Pedram A , Greene GL , Levin ER. 1999. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol 13:307–319 [DOI] [PubMed] [Google Scholar]
- 58. Lee TI , Johnstone SE , Young RA. 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc 1:729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nelson JD , Denisenko O , Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1:179–185 [DOI] [PubMed] [Google Scholar]
- 60. Zhu N , Hansen U. 2007. HMGN1 modulates estrogen-mediated transcriptional activation through interactions with specific DNA-binding transcription factors. Mol Cell Biol 27:8859–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fang F , Rycyzyn MA , Clevenger CV. 2009. Role of c-Myb during prolactin-induced signal transducer and activator of transcription 5a signaling in breast cancer cells. Endocrinology 150:1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fang F , Zheng J , Galbaugh TL , Fiorillo AA , Hjort EE , Zeng X , Clevenger CV. 2010. Cyclophilin B as a co-regulator of prolactin-induced gene expression and function in breast cancer cells. J Mol Endocrinol 44:319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Galbaugh T , Feeney YB , Clevenger CV. 2010. Prolactin receptor-integrin crosstalk mediated by SIRPα in breast cancer cells. Mol Cancer Res 8:1413–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]