

Abstract
Estrogens contribute to the pathogenesis of female lung cancer and function mainly through estrogen receptor-β (ERβ). However, the way in which ERβ expression is regulated in lung cancer cells remains to be explored. We have found that signal transducer and activator of transcription 3 (Stat3) activation up-regulates ERβ expression in PC14PE6/AS2 lung cancer cells in a preliminary Affymetrix oligonucleotide array study, and we sought to confirm the findings. In this study, we show that IL-6 induced ERβ mRNA and protein expression in lung cancer cells. The induction of ERβ in response to IL-6 was abolished by Janus kinase 2 inhibitor-AG490, dominant-negative mutant of Stat3, and Stat3-targeting short interfering RNA. The luciferase reporter assay and chromatin immunoprecipitation assay confirmed that IL-6-activated Stat3 binds to the ERβ promoter. Besides the Janus kinase 2/Stat3 pathway, the MEK/Erk pathway contributes to ERβ up-regulation induced by IL-6; however, the phosphoinositide 3′-kinase/Akt pathway does not. We also found that epidermal growth factor (EGF) stimulation or L858R mutation in EGF receptor (EGFR) induced Stat3 activation as well as ERβ expression in lung cancer cells. Inhibiting Stat3 activity by pharmacological or genetic approaches reduced EGF- and L858R mutant EGFR-induced ERβ expression, indicating that Stat3 activation is required for EGFR signaling-mediated ERβ up-regulation. Silencing ERβ decreased cell proliferation in lung cancer cells that overexpress L858R mutant EGFR. In conclusion, we have identified that Stat3 activation is essential for ERβ induction by IL-6, EGF, and the presence of EGFR mutation. The findings shed light on new therapeutic targets for female lung cancer, especially for those with EGFR mutations.
Lung cancer is the leading cause of cancer death throughout the world. Although cigarette smoking is considered the major risk for lung cancer, the rate of cigarette smokers among female patients with lung cancer is much lower in Asia than Western countries (1, 2). In Taiwan, the percentage of smokers in female lung cancer patients is only 10% (3). Therefore, lung cancer risk factors other than cigarette smoking should be taken into consideration. Past studies have shown that women appear to be more susceptible than men to chronic airflow obstruction and lung cancer induced by tobacco carcinogens (4, 5). Lung adenocarcinoma, which shows a weaker association with cigarette smoking than other histological types of lung cancer, is found predominantly in women (6). Also, a significant connection exists between lung cancer and postmenopausal females undergoing estrogen replacement therapy (7, 8); therefore, sex-dependent hormones, such as estrogen, may play an important role in the etiology and progression of lung cancer.
Estrogens govern many physiological functions such as cell growth, development, and differentiation through estrogen receptor (ER)-mediated signaling in a wide range of tissues (9). ER comprise two subtypes, namely ERα and ERβ, which are encoded by separate genes (10). Although ERα and ERβ have similar structures and ligand-binding patterns, the tissue distribution and relative expression levels between these two proteins are varied (11). Both ERα and ERβ are expressed in normal lung tissue and lung tumors, but ERβ gene expression appears to be more predominant than ERα (12, 13). Estrogen signaling pathways have been found to regulate normal lung development (14). In female transgenic mice models, inactivation of the ERβ gene resulted in lung abnormality and systemic hypoxia (15, 16). Recently, ERβ was shown to be the mediator of estrogen signaling in lung cancer cells and function through both genomic and nongenomic mechanisms (12, 17, 18). Together, these suggest that ERβ is the predominant ER in lung tissue and plays an important role in the physiological and pathophysiological functions of the lungs.
Signal transducers and activators of transcription 3 (Stat3), a latent cytoplasmic transcription factor, belongs to the seven-member Stat gene family of transcription factors (19). In response to growth factors and cytokines, such as epidermal growth factor (EGF) and IL-6 family of cytokines, Stat3 is activated by phosphorylation on a specific tyrosine residue 705 in the carboxy-terminal domain. Phosphorylated Stat3 forms homo- or heterodimers that translocate to the nucleus in which they bind to specific Stat3 response elements in the promoter region of target genes and activate transcription (20). Stat3 mediates expression of broadly diverse genes that control critical cellular processes such as cell growth, apoptosis, inflammation, and immune response. In our previous study, we have demonstrated that activation of Stat3 by autocrine IL-6 plays an important role in the pathogenesis of lung adenocarcinoma (21). Using Affymetrix oligonucleotide arrays (Affymetrix Inc., Santa Clara, CA), we observed an elevated expression of ERβ in Stat3-active lung adenocarcinoma cells (PC14PE6/AS2) (data not shown). For this reason, we are interested in investigating the role of activated Stat3 in the regulation of ERβ expression in lung cancer cells.
EGF receptor (EGFR) is the cell surface receptor tyrosine kinase activated by its specific ligands, such as EGF, TGF-α, amphiregulin, and others, and is essential in cell proliferation, differentiation, metabolism, and many physiological processes (22). Activated EGFR recruits and phosphorylates many cytoplasmic signaling molecules, thus initiating downstream mitogenic events, such as the MEK/Erk, the phosphoinositide 3′-kinase (PI3K)/Akt, and the Stat pathways (23). In non-small cell lung cancer (NSCLC), overexpression and kinase domain mutations in EGFR frequently occur and are linked to tumor progression. Therefore, many small molecular inhibitors, such as gefitinib and erlotinib, have been developed to target EGFR-tyrosine kinase for anticancer therapy. Rather than EGFR overexpression, EGFR mutations can predict better response to gefitinib in lung cancer (24–26), and they occur more commonly in specific subpopulations of lung cancer patients. These include patients with adenocarcinoma histology, those who have never smoked cigarettes, those of East Asian ethnicity, and females (27). Recently, it was reported that the lung cancer cell line H520, which does not express endogenous EGFR, has no expression of ERβ mRNA and protein compared with H1650 and A549 cells (28), suggesting a correlation between EGFR and ERβ expression.
Another report showed that the lung cancer cell line 273T with a Y727C EGFR point mutation expresses higher levels of ERβ than other lung cancer cell lines without EGFR mutations (29). Nose et al. (30) analyzed 447 resected primary lung adenocarcinoma specimens through an immunohistochemical assay. Although the investigators did not observe the nuclear staining of ERβ in the EGFR wild-type cohort, they found a strong nuclear expression of ERβ in 69% of patients with the EGFR mutation. They also identified that the strong expression of ERβ is associated with a favorable prognosis and better response to EGFR-tyrosine kinase inhibitor, which is likely attributable to the positive correlation between ERβ expression and EGFR mutation. Toh et al. (31) performed an immunohistochemical study on 109 East-Asian lung adenocarcinoma patients, and they found a 60% EGFR-mutation rate among ERβ-positive samples; however, ERβ positivity predicted poorer outcomes in lung cancer patients in their study. Taken together, in adenocarcinoma of the lung, ERβ expression shows a significantly positive correlation with EGFR mutations. The clinical implications, however, remain undefined. These findings suggest that ERβ may play a role in the pathogenesis of lung adenocarcinomas harboring EGFR mutations. Similar to IL-6, Stat3 is also a downstream signaling mediator of activated EGFR (32). In this study, we investigated whether Stat3 activation can up-regulate ERβ expression in lung cancer cells and whether it is essential for EGFR-induced ERβ expression.
Results
IL-6 induces Stat3 activation and ERβ expression in lung cancer cells
In a human lung adenocarcinoma cell line PC14PE6/AS2, IL-6 induced Stat3 tyrosine phosphorylation (phospho-Stat3-Y705) and ERβ expression dose dependently after 8 h of stimulation (Fig. 1A). At the dose level of 10 ng/ml, IL-6 induced Stat3 phosphorylation with peak at 0.5, 0.5, and 3 h time points, respectively, for PC14PE6/AS2, A549, and H460 cells. ERβ protein expression was induced after Stat3 activation and increased gradually thereafter, detected by Western blot analysis (Fig. 1B).
Fig. 1.

IL-6 up-regulates ERβ expression in lung cancer cells. A, PC14PE6/AS2 cells were subjected to serum starvation (0.5% FBS) overnight followed by treatment with IL-6 in different concentrations as indicated for 8 h. Tyrosine phosphorylation of Stat3 and ERβ expression were detected by Western blot analysis and normalized against total Stat3 and β-actin, respectively. Values represent the means ± sem from three separate experiments. *, P < 0.05; **, P < 0.01 vs. control. B, PC14PE6/AS2, A549, and H460 cells were serum starved (0.5% FBS) overnight followed by treatment with a fixed dosage of IL-6 (10 ng/ml) for the indicated time points. Total cell lysates were prepared and subjected to Western blotting with phospho-Stat3–Y705, general Stat3, and ERβ antibodies. Tyrosine phosphorylation of Stat3 and ERβ expression were normalized against total Stat3 and β-actin, respectively. Values represent the means ± sem from at least three separate experiments. *, P < 0.05; **, P < 0.01 vs. control. C, PC14PE6/AS2 cells were serum starved (0.5% FBS) overnight followed by treatment with IL-6 (10 ng/ml) for the indicated time points. RNA samples were collected and ERβ mRNA expression was detected by RT-PCR. The levels of ERβ mRNA were normalized by comparison of GAPDH content. The bar graph is from three different experiments, means ± sem. *, P < 0.05; **, P < 0.01 vs. control. D, PC14PE6/AS2 cells were serum starved (0.5% FBS) overnight and then pretreated with actinomycin D (Act D) (2 μg/ml) for 1 h followed by addition with or without IL-6 (10 ng/ml) for the indicated time points. Total RNA were collected and analyzed by RT-PCR. The levels of ERβ mRNA were normalized by comparison of GAPDH content and expressed as the percent change of time zero, which was set at 100%. Values represent the means ± sem of three independent experiments. *, P < 0.05; **, P < 0.01 for Act D vs. Act D + IL-6 at each time point.
IL-6 increases transcription and stability of ERβ mRNA in lung cancer cells
To investigate whether the up-regulation of ERβ protein expression is induced by IL-6 through an induction of ERβ gene transcription, we treated PC14PE6/AS2 cells with 10 ng/ml of IL-6 for 0.5–24 h. RT-PCR data show that ERβ mRNA increased gradually after IL-6 treatment in PC14PE6/AS2 cells (Fig. 1C). After exposure to Act D, an inhibitor of mRNA transcription, for 1 h, the levels of ERβ mRNA in PC14PE6/AS2 cells were analyzed at the time points of 0, 0.5, 1, 3, 5, and 8 h by the RT-PCR method. The ERβ mRNA levels declined gradually with a half-life of approximately 2 h (Fig. 1D). If IL-6 was added as a combined treatment with Act D, the half-life of ERβ mRNA was prolonged to approximately 6.5 h. Collectively the results indicated that stimulation with IL-6 increased both ERβ gene transcription and ERβ mRNA stability in lung cancer cells.
Up-regulation of ERβ by IL-6 is mediated through the Janus kinase (Jak)-2/Stat3 and the MAPK kinase (MEK)/Erk but not the PI3K/Akt pathways
IL-6 exerts its biological functions by activating three major signaling cascades, the Jak2/Stat3, MEK/Erk, and PI3K/Akt pathways (33). To determine which downstream signaling pathways of IL-6 participate in the regulation of ERβ expression, PC14PE6/AS2 cells were pretreated with a variety of pharmacological inhibitors in the presence of IL-6. As seen in Fig. 2, A and B, ERβ expression was suppressed significantly by the administration of the Jak2 inhibitor AG490 or the MEK inhibitor U0126. However, blocking PI3K/Akt activity by LY294002 did not prevent IL-6-induced ERβ expression (Fig. 2C). Next, we evaluated the benefit of cotreatment with AG490 and U0126 in PC14PE6/AS2 cells in the presence of IL-6. We found that a combined pretreatment of AG490 and U0126 greatly inhibited IL-6-induced ERβ expression at 8 h, more than that from each agent alone (Fig. 2D). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assays revealed that cell viability remained unchanged at 8 h after treatments with various agents and a combined treatment with AG490 and U0126 (data not shown).
Fig. 2.
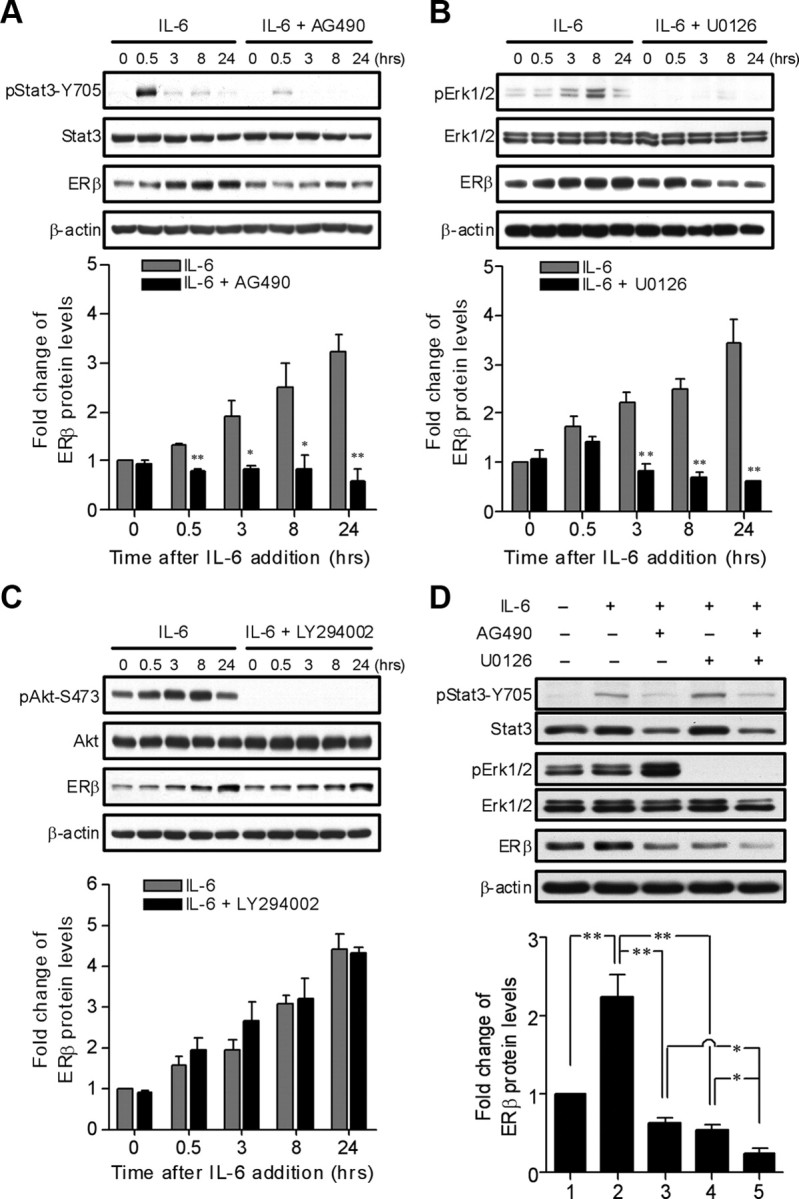
Jak2/Stat3 and MEK/ERK as downstream signaling pathways of IL-6 contribute to the regulation of ERβ expression. A–C, PC14PE6/AS2 cells were serum starved (0.5% FBS) overnight followed by pretreatment with or without the Jak2/Stat3 inhibitor (AG490, 40 μm), the PI3K/Akt inhibitor (LY294002, 20 μm), or the MEK/ERK inhibitor (U0126, 5 μm) for 1 h and then incubated with IL-6 (10 ng/ml) for the indicated time points. Whole-cell lysates were subjected to Western blotting with the indicated antibodies. β-Actin protein is shown as a loading control. The levels of ERβ were normalized against β-actin. Values represent the means ± sem of three independent experiments. *, P < 0.05; **, P < 0.01 for IL-6 vs. IL-6 + AG490, IL-6 vs. IL-6 + U0126, and IL-6 vs. IL-6 + LY294002 at each time point. D, PC14PE6/AS2 cells were serum starved (0.5% FBS) overnight followed by pretreatment with AG490 or U0126 or in combination for 1 h and then stimulated with IL-6 for 8 h. Total cell lysates were prepared and subjected to Western blotting analysis probed with the indicated antibodies. β-Actin protein is shown as a loading control. The levels of ERβ were normalized against β-actin. Values represent the means ± sem from three independent experiments. *, P < 0.05; **, P < 0.01, significant differences between groups.
Stat3 activation is directly involved in the regulation of ERβ expression
To demonstrate that Stat3 activation regulates ERβ expression, we used the PC14PE6/AS2-derived cells as follows: PC14PE6/AS2/Vec cells, which express a control empty vector; PC14PE6/AS2/S3C cells, which express S3C protein (a constitutively active mutant of Stat3 forced to form dimmers without ligand stimulation); and PC14PE6/AS2/S3D cells, which express S3D protein (a dominant negative mutant of Stat3 that fails to bind DNA) (21). PC14PE6/AS2/S3C cells express higher levels of ERβ mRNA and protein than PC14PE6/AS2/Vec cells, but lower levels of ERβ mRNA and protein were observed in PC14PE6/AS2/S3D cells (Fig. 3, A and B). Similar results were obtained in prostate cancer PC-3 cells, which do not express endogenous Stat3 (34). The expression of ERβ was up-regulated by exogenous overexpression of constitutively active mutant of Stat3 (S3C) in the cells (Supplemental Fig. 1). To further confirm that ERβ expression is regulated by Stat3, RNA interference-mediated knockdown of Stat3 was performed. Transient transfection with short interfering RNA (siRNA) targeting Stat3 greatly reduced Stat3 expression as well as phosphorylation in PC14PE6/AS2 and A549 cells in the presence or absence of IL-6. Subsequently the expression of ERβ in the cells was also suppressed (Fig. 3C). Because there is a potential Stat3 binding site (−578/−570) in the ERβ promoter, we cloned the −936/+268 region containing the putative Stat3 binding site and the −541/+268 region without the putative Stat3 binding site into the luciferase reporter vector for reporter activity assays.
Fig. 3.

ERβ expression is regulated by Stat3. A and B, The levels of ERβ mRNA and protein in the PC14PE6/AS2-derived cells stably expressing empty vector (Vec), constitutively active mutant of Stat3 (S3C), and dominant-negative mutant of Stat3 (S3D) were detected by RT-PCR and Western blotting, respectively. ERβ mRNA and protein levels were normalized with GAPDH and β-actin, respectively, and are presented as the means ± sem. *, P < 0.05; **, P < 0.01, compared with vector control cells. C, PC14PE6/AS2 and A549 cells were transfected with either scrambled control siRNA or synthetic Stat3 specific siRNA. At 48 h after the initiation of transfection, the cells were serum starved (0.5% FBS) overnight followed by treatment with or without IL-6 for 8 h. The cell extracts were collected and analyzed by Western blotting with phospho-Stat3–Y705, general Stat3, and ERβ antibodies. The levels of ERβ were normalized against β-actin. Values represent the means ± sem from three independent experiments. **, P < 0.01, significant differences between groups. D (left panel), PC14PE6/AS2 cells were cotransfected with pRL-TK plasmids in combination with either the −936/+268 (containing the putative Stat3 binding site) or the −541/+268 (with no putative Stat3 binding site) ERβ reporters. After 24 h of transfection, the cells were serum starved (0.5% FBS) overnight followed by treatment with or without IL-6 for 8 h and subjected to the dual-luciferase assay. The levels of firefly luciferase activity were normalized with Renilla luciferase activity serving as the internal control for transfection efficiency. The specific Stat3 binding sites within the human ERβ promoter were identified using TFSEARCH software (Parallel Application TRC Laboratory, RWCP, Tokyo, Japan). The results were presented as means ± sem from three independent experiments. *, P < 0.05; **, P < 0.01 vs. control. D (right panel), PC14PE6/AS2 cells stably expressing empty vector (Vec), constitutively active mutant of Stat3 (S3C), and dominant-negative mutant of Stat3 (S3D) were used to evaluate the activity of the −936/+268 or the −541/+268 ERβ reporters in a dual-luciferase reporter assay. The results were presented as means ± sem from three independent experiments. *, P < 0.05; **, P < 0.01, compared with Vec cells. E, The soluble chromatin was prepared from PC14PE6/AS2 cells exposed to IL-6 (10 ng/ml) for 0.5 h. The ChIP assay was performed using an antibody against Stat3 and an irrelevant αIgG antibody as negative control. The figure represents PCR products amplified from the final DNA extractions using pairs of primers on the ERβ promoter region as shown on the top panel. Values represent the means ± sem from three independent experiments. **, P < 0.01 vs. control.
The results show that IL-6 treatment (Fig. 3D, left panel) or ectopic expression of constitutive active Stat3 (S3C) (Fig. 3D, right panel) increased the luciferase activity of the ERβ −936/+268 reporter but not the ERβ −541/+268 reporter in PC14PE6/AS2 cells. Accordingly, the ectopic expression of the dominant-negative Stat3 (S3D) did not increase the luciferase activity in both reporters (Fig. 3D, right panel). To further determine whether activated Stat3 directly binds to the predicted region, we carried out the chromatin immunoprecipitation (ChIP) assay using two pairs of primers that cover either the predicted Stat3 binding site within the ERβ promoter or a control region upstream of the predicted Stat3 binding site. We found that, after treatment with IL-6 for 0.5 h, a PCR product from primers covering −717/−414 region but not that covering −1899/−1603 region of the ERβ promoter was detected in the immunoprecipitate captured by Stat3 antibody (Fig. 3E). Taken together, these results suggest that Stat3 enhances ERβ promoter activity through directly binding to the putative Stat3 binding site within the ERβ promoter and ERβ is a Stat3 transcriptional target.
Stat3 and ERβ interact with each other to regulate estrogen-dependent lung cancer cell growth
Although activation of Stat3 induces ERβ expression, the diarylpropionitrile (DPN, ERβ-specific agonist) stimulation did not increase the total amount of Stat3 in PC14PE6/AS2 cells and A549 cells (Fig. 4A). The transfection with ERβ siRNA into PC14PE6/AS2 cells suppressed ERβ but not Stat3 expression (Fig. 4B). Therefore, ERβ activation did not regulate Stat3 expression. Because the proliferative effects of estrogen in lung cancer cells is mediated primarily by the nongenomic action of ERβ (18) and estrogen can activate Stats through nongenomic action of ER (35), we studied the expression of phopho-Stat3 as well as Stat3 levels in lung cancer cells upon DPN stimulation. As reported previously, Stat3 is constitutively activated in PC14PE6/AS2 and A549 cells (21, 36). We found that DPN increased Stat3 phosphorylation in PC14PE6/AS2 cells but not in A549 cells (Fig. 4A). To address the physiological significance of Stat3 in modulating ERβ function, we used MTT assays to study the cell growth induced by DPN in lung cancer cells. DPN treatment induced more cell growth in lung cancer cells engineered to overexpress active form Stat3, compared with the vector control (Fig. 4C). DPN stimulation increase about 50% cell growth in PC14PE6/AS2 cells and about 25% growth in A549 cells, but the proliferative effect of DPN was suppressed when Stat3 expression was knocked down by siRNA (Fig. 4D).
Fig. 4.

The interaction between Stat3 and ERβ contributes to estrogen-dependent cell growth. A, PC14PE6/AS2 and A549 cells were depleted of estrogen by culture in phenol red-free medium containing 10% charcoal-stripped serum for 24 h followed by starvation in medium with 0.5% charcoal-stripped serum overnight. The cells were then incubated with ERβ-specific agonist DPN (10 nm) for the indicated time points. Tyrosine phosphorylation of Stat3 and total Stat3 expression were detected by Western blot analysis and normalized against total Stat3 and β-actin, respectively. Values represent the means ± sem from three separate experiments. B, PC14PE6/AS2 cells were transiently transfected with either scrambled control siRNA or ERβ-specific siRNA. After 48 h of culture, the whole-cell extracts were prepared and detected by Western blot analysis probed with Stat3 and ERβ antibodies. The levels of Stat3 protein were normalized with β-actin. Values represent the means ± sem from three separate experiments. C, PC14PE6/AS2-derived cells stably expressing empty vector (Vec) or constitutively active mutant of Stat3 (S3C) were grown in 96-well plates with phenol red-free medium containing 10% charcoal-stripped serum for 24 h, serum starved in medium with 0.5% charcoal-stripped serum for 24 h, and then incubated with or without DPN for 48 h. Cell proliferation was measured by the MTT assay. Values represent the mean ± sem of six identical wells. **, P < 0.01; ***, P < 0.01, significant differences between groups. D, PC14PE6/AS2 and A549 cells transiently transfected with either scrambled control siRNA or Stat3-specific siRNA were grown in 96-well plates with phenol red-free medium containing 10% charcoal-stripped serum for 24 h. The cells were then serum starved in medium with 0.5% charcoal-stripped serum for 24 h followed by treatment with or without DPN for 48 h under low serum (0.5% FBS) condition. Cell proliferation was measured by the MTT assay. Values represent the mean ± sem of six identical wells. NS, Not significant. *, P < 0.05; **, P < 0.01, significant differences between groups.
EGF activates Stat3 and up-regulates ERβ expression in lung cancer cells
Because Stat3 is a downstream signaling protein of normally and abnormally activated EGFR, we investigated whether Stat3 activation is required for the induction of ERβ expression by the EGFR signaling pathway. Four lung cancer cell lines (PC14PE6/AS2, A549, H1299, and H1650) were treated with EGF for 24 h, and the levels of phospho-Stat3-Y705 and ERβ were analyzed by immunoblot. EGFR activation results in autophosphorylation of five tyrosine residues (Y992, Y1068, Y1086, Y1148, and Y1173) within its intrinsic protein kinase domain (37). Among them, phosphorylated tyrosine residue 1068 and 1086 are required for Stat3 activation (38). Upon EGF stimulation, the expression of phospho-EGFR-Y1068 was augmented at about 0.5 h, and then Stat3 was activated at about 0.5–3 h, and subsequently ERβ expression was induced (Fig. 5A). The EGF-induced Stat3 activation and ERβ up-regulation was markedly prevented by either the EGFR inhibitor gefitinib or the Stat3 inhibitor S3I-201 in all the four lung cancer cell lines (Fig. 5B). To specifically address the role of Stat3 activation, siRNA against Stat3 was transiently transfected into these four cell lines. Again we found that ERβ expression was markedly suppressed when Stat3 expression had been silenced (Fig. 5C).
Fig. 5.

Stat3 activation is involved in EGF-induced ERβ expression in lung cancer cells. A, PC14PE6/AS2, A549, H1299, and H1650 cells were serum starved (0.5% FBS) overnight followed by treatment with EGF (10 ng/ml) for the indicated time points. Total cell lysates were collected and time course of ERβ expression and the phosphorylation status of both EGFR and Stat3 were analyzed by Western blotting. The levels of ERβ protein were normalized with β-actin. Values represent the means ± sem from at least three separate experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. control. B, Cells were serum starved (0.5% FBS) overnight and then pretreated with Stat3 inhibitor (S3I-201, 100 μm) or EGFR inhibitor (gefitinib, 2 μm) for 3 h followed by stimulation with EGF (10 ng/ml) for 8 h. The cell extracts were collected and analyzed by Western blotting probed with the indicated antibodies. The levels of ERβ were normalized against β-actin and presented as means ± sem from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, significant differences between groups. C, Cells were transiently transfected with either scrambled control siRNA- or Stat3-specific siRNA. After 48 h of incubation, cells were starved (0.5% FBS) overnight followed by treatment with EGF (10 ng/ml) for 8 h. The lysates of these cells were analyzed by Western blotting probed with the indicated antibodies. The levels of ERβ were normalized against β-actin and presented as means ± sem from three independent experiments. *, P < 0.05; **, P < 0.01, vs. scrambled control.
The L858R mutant EGFR up-regulates ERβ expression via Stat3 activation
The human lung adenocarcinoma cell line H1299 has a low endogenous expression of EGFR. Therefore, plasmids encoding L858R mutant EGFR were transfected into the cells to establish a stable cell line H1299/L858R (39). H1299/L858R cells strongly express phospho-EGFR-Y1068 and phospho-Stat3-Y705 as well as ERβ proteins compared with H1299/vector cells (Fig. 6A). Similarly, the up-regulation of ERβ in H1299/L858R cells was significantly reduced by the addition of S3I-201 or gefitinib (Fig. 6B) or transfection with Stat3 siRNA (Fig. 6C).
Fig. 6.

ERβ expression is up-regulated by L858R mutant EGFR via Stat3 activation. A, The levels of ERβ and phosphorylation status of both EGFR and Stat3 were compared between H1299 cells stably expressing an empty vector and L858R mutant EGFR by Western blotting. The levels of ERβ were normalized against β-actin. Values represent the means ± sem from three experiments. *, P < 0.05 vs. vector control cells. B, H1299/L858R cells were left untreated, treated with Stat3 inhibitor (S3I-201, 100 μm) or EGFR inhibitor (gefitinib, 2 μm) for 8 h. The cell extracts were collected and analyzed by Western blotting probed with the indicated antibodies. The levels of ERβ were normalized against β-actin. Values represent the means ± sem from three separate experiments. **, P < 0.01 vs. control. C, H1299/L858R cells were transiently transfected with Stat3 specific siRNA to silence Stat3 expression. Scrambled siRNA served as negative control. The cell lysates were detected by Western blot analysis probed with the indicated antibodies. The levels of ERβ were normalized against β-actin. Values represent the means ± sem from three separate experiments. **, P < 0.01 vs. scrambled control.
ERβ contributes to the cell proliferation in lung cancer cells harboring the L858R EGFR mutation
The growth rate of H1299/L858R is higher than that of H1299/vector cells (Fig. 7, A and B). We transfected ERβ siRNA into H1299/vector and H1299/L858R cells and found that the expression of ERβ was suppressed in the cells. The knockdown of ERβ in the absence of DPN stimulation decreased cell growth in H1299/L858R but not H1299/vector cells. In the presence of DPN stimulation, knockdown of ERβ diminished cell growth in both H1299/vector and H1299/L858R cells (Fig. 7A). EGF stimulation also induced cellular proliferation in H1299/vector and H1299/L858R cells, and the growth-promoting effect was blocked by transfection with ERβ siRNA in the two cell lines (Fig. 7B). The data support that ERβ contributes to EGFR-mediated growth of lung cancer cells. Finally, we tested whether DPN and EGF synergize in stimulating lung cancer cell growth. We found that DPN or EGF alone significantly stimulated cell growth of both H1299/vector and H1299/L858R cells. The combination of DPN and EGF further enhanced cell growth in H1299/vector cells but not in H1299/L858R cells (Fig. 7C). We speculate that, in H1299/L858R cells, the stimulation effect from the binding of EGF to the mutant EGFR is so strong that the addition of DPN did not induce further proliferation.
Fig. 7.
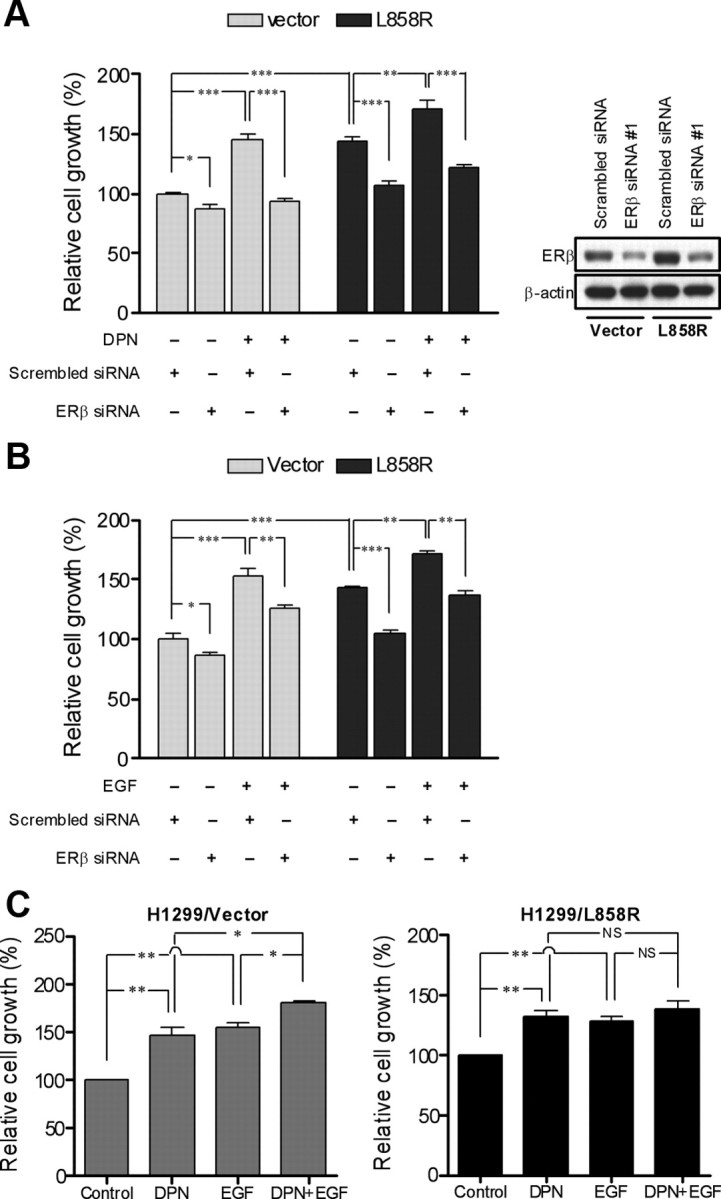
ERβ contributes to L858R mutant EGFR-induced cell growth. A, Transfection of H1299/vector and H1299/L858R cells with either scrambled control siRNA or ERβ-specific siRNA was performed, and the cells were grown in 96-well plates for 24 h. The cells were then serum starved (0.5% FBS) for 24 h followed by treatment with or without EGF for 48 h under low serum (0.5% FBS) condition. Cell proliferation was measured by MTT assay. Values represent the mean ± sem of six identical wells. *, P < 0.05; **, P < 0.01; ***, P < 0.001, significant differences between groups. siRNA knockdown of ERβ protein was confirmed by immunoblot after 48 h of transfection as shown on the right panel. B, H1299/vector and H1299/L858R cells transiently transfected with either scrambled control siRNA or ERβ-specific siRNA were grown in 96-well plates in phenol red-free medium containing 10% charcoal-stripped serum for 24 h. The cells were serum starved in medium with 0.5% charcoal-stripped serum for 24 h followed by treatment with or without DPN for 48 h under low serum (0.5% FBS) condition. Cell proliferation was measured by an MTT assay. Values represent the mean ± sem of six identical wells. *, P < 0.05; **, P < 0.01; ***, P < 0.001, significant differences between groups. C, H1299/vector and H1299/L858R cells were grown in 96-well plates in phenol red-free medium containing 10% charcoal-stripped serum for 24 h, serum starved (0.5% charcoal stripped serum) for 24 h, and then treated with DPN, EGF, or in combination for 48 h under low serum (0.5% FBS) condition. Measurement of cell proliferation was carried out by an TT assay. Values represent the mean ± sem of six identical wells. NS, Not significant. *, P < 0.05; **, P < 0.01, significant differences between groups.
Discussion
Both the Stat3 and estrogen signaling pathways are important in the pathogenesis of lung adenocarcinoma; however, the interaction between these two signaling pathways is unclear. In this study, we have offered evidence that ERβ is a downstream target gene of Stat3. We also found that activation of EGFR by EGF or L858R mutation can induce ERβ expression in lung cancer cells and ERβ up-regulation contributes to cellular proliferation. Both inhibition of Stat3 activation and knockdown of Stat3 expression suppressed activated EGFR-induced ERβ up-regulation. Therefore, Stat3 activation is essential for ERβ expression induced by EGFR activation in lung cancer cells.
In several lung cancer cell lines, we have shown that IL-6 induced Stat3 activation first and subsequently induced ERβ expression. The increase of ERβ protein expression in response to IL-6 is due to the augmentation of gene transcription because the ERβ mRNA was induced upon stimulation with IL-6. We also found that IL-6 increased the half-life of ERβ mRNA from 2 to 6.5 h in PC14PE6/AS2 cells. The report from Weigert et al. (40) showing that IL-6 up-regulates its mRNA levels through increasing its stability in human skeletal muscle cells supports our findings. When Stat3 activation had been blocked by the Jak2 inhibitor or Stat3 expression had been knocked down by Stat3 siRNA, IL-6-induced ERβ up-regulation was significantly prevented, indicating that Stat3 activation is directly involved in the regulation of ERβ expression. The assumption was supported by the luciferase reporter assay and ChIP study where activated Stat3 directly bound to the ERβ gene promoter. A study reporting that prolactin induces ERβ expression through Stat5b in rat corpus luteum and decidua of pregnancy (41) supports that Stat family members may regulate ERβ expression. Although activation of Stat3 induced ERβ expression, ERβ activation did not increase Stat3 expression in our study. However, the stimulation with DPN induced further phosphorylation of Stat3 in PC14PE6/AS2 cells. The effect might be mediated by the nongenomic actions of ERβ (35). In A549 cells, the stimulation with DPN did not induce further phosphorylation of Stat3. The discrepancy in Stat3 activation in PC14PE6/AS2 and A549 cells might be due to differential reactive oxygen species generation from DPN stimulation (Su, W. C., unpublished data). DPN treatment induced more cell growth in lung cancer cells engineered to overexpress active form Stat3, compared with the vector control. Therefore, Stat3 activation results in increased estrogen-dependent growth of lung cancer cells. Although DPN did not further increase the Stat3 activation, it induced, to a less extent than that in PC14PE6/AS2 cells, cellular proliferation in the A549 cells as well. The results indicate that the nongenomic activation of Stat3 by ERβ is contributory to but not essential for the proliferative effect from DPN.
In addition to the Jak2/Stat3, IL-6 also activates the MEK/Erk and the PI3K/Akt signaling pathways (33). Using the Jak2 inhibitor AG490, the MEK inhibitor U0126 and the PI3K inhibitor LY294002 to treat lung cancer cells, we found that the Jak2/Stat3 and the MEK/Erk pathways are involved in the regulation of ERβ expression, although the PI3K/Akt pathway is not. The up-regulation of ERα by the MAPK/Erk pathway has been reported in mouse skeletal muscle myoblasts (42). In breast cancer cells, prolonged activation of MAPK results in ERα down-regulation (43). Although there is no consensus in the results, the MEK/Erk pathway was shown to play a role in regulating ERα expression. When AG490 and U0126 were combined to treat the lung cancer cells, IL-6-induced ERβ expression was brought down further. The results highlight the possibility of a synergistic effect in treating lung cancers by the combination of the Jak2/Stat3 and the MEK/Erk pathway inhibitors.
One of the most important breakthroughs in the understanding and treatment of NSCLC is the identification of EGFR gene mutation. Approximately 90% of mutations within the EGFR gene are clustered in exon 19 (deletions) and exon 21 (point mutation at codon 858, L858R), which encode the intracellular tyrosine kinase domain (44). These activating mutations in the EGFR kinase domain are oncogenic, and their expression is necessary for tumor development and maintenance in lung tissue (45). Recently a correlation was identified between EGFR mutations and high levels of ERβ in patients with lung adenocarcinoma (30, 31, 46). Together with the finding that EGFR mutations tend to occur in female lung cancer patients, the interaction between EGFR mutations and ERβ should play a role in the tumor progression. Similar to IL-6, the EGFR signaling pathway also activates the Stat3, MEK/Erk, and PI3K/Akt signaling pathways (23). Here our findings show that EGF-induced ERβ expression requires Stat3 activation. Overexpression of L858R mutant EGFR in H1299 lung cancer cells also up-regulates ERβ expression through Stat3 activation. Silencing ERβ expression by ERβ-specific siRNA resulted in loss of L858R mutant EGFR-dependent growth of lung cancer cells. Taken together, our results suggest that the EGFR/Stat3/ERβ signaling axis has implications in lung cancer proliferation. Previously Stat3 has been reported to be required for the oncogenic effects of somatic EGFR mutation in NSCLC (47). A recent study about age- and sex-specific genomic profiles in NSCLC identified increased activation of the Stat3 pathway as a high-risk predictor (48). These studies support our findings that Stat3 participates in lung tumor progression.
Interestingly, in the absence of DPN, we observed that H1299/L858R cell growth was reduced by knockdown of ERβ (Fig. 7A). Because ER can be activated by EGF in a ligand-independent manner via the MAPK pathway (49–51), we speculated that the L858R mutant EGFR can activate ERβ without ligand stimulation. In the absence of DPN stimulation, knockdown of ERβ decreased cell growth in H1299/L858R but not H1299/vector cells. In the presence of DPN stimulation, knockdown of ERβ diminished cell growth in both H1299/L858R and H1299/vector cells. The data support our assumption.
The expression patterns of ERα and ERβ in human lung tumors remain controversial. Some studies suggested that both ERα and ERβ are present in lung cancer cells (13, 52–54); however, ERα appears to be expressed in truncated forms and mainly located in cytoplasm (52, 53). In the current study, using immunoblot analysis, we could detect ERβ expression only in the lung cancer cell lines (Supplemental Fig. 2). By RT-PCR analyses, we can detect only ERβ but not ERα mRNA in PC14PE6/AS2 and A549 lung cancer cells (data not shown). The findings are consistent with other studies that showed ERβ, but not ERα, expression in lung cancer tumor specimens and cell lines (12, 17, 18, 55, 56).
S3I-201 (NSC 74859), a Stat3 inhibitor, was identified to bind to the Stat3 Src homology 2 domain, therefore inhibiting Stat3 phosphorylation, dimerization, and DNA binding (57). In this study, S3I-210 was shown to effectively inhibit Stat3 phosphorylation, thereby suppressing the up-regulation of ERβ induced by EGF or L858R EGFR mutation in lung cancer cell lines. However, in H1299 cells, we found that EGFR phosphorylation was enhanced by a combined treatment of EGF with S3I-201 rather than with EGF treatment alone. This may be due to the nonspecific effects from S3I-201 treatment or an unknown rebound response from the H1299 cells. Further studies are required to clarify this finding.
In conclusion, we have provided the first evidence that the expression of ERβ, the predominant ER isotype expressed in lung cancer cells, is regulated by the Stat3 signaling pathway. We have also demonstrated that L858R EGFR mutation-induced constitutive activation of Stat3 strongly contributes to ERβ up-regulation and overexpression of ERβ promotes proliferation of lung cancer cells. Thus, our findings highlight Stat3 and ERβ as potential targets for improving the control of NSCLC, especially those harboring EGFR mutations.
Materials and Methods
Reagents and antibodies
The AG490, LY294002, and U0126 inhibitors were purchased from Biomol (Plymouth, PA). IL-6 and Act D were purchased from Sigma-Aldrich (St. Louis, MO). DPN was purchased from Tocris (Ellisville, MO). EGF was purchased from Cell Signaling Technology (Beverly, MA). Gefitinib was provided by AstraZeneca UK Limited (London, UK). S3I-201 was purchased from Calbiochem (San Diego, CA). Monoclonal antibodies against β-actin were purchased from Sigma-Aldrich. Polyclonal antibodies against Stat3 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Polyclonal antibodies against ERβ were purchased from Upstate Biotechnology (Lake Placid, NY). Monoclonal antibodies against ERK1/ERK2, polyclonal antibodies against phospho-ERK1 (Thr202/ Tyr204)/ERK2 (Thr185/Tyr187), Akt, phospho-Akt (Ser473), phospho-Stat3 (Tyr705), EGFR, and phospho-EGFR (Tyr1068) were purchased from Cell Signaling Technology.
Cell lines and culture conditions
The lung cancer cell lines A549, H460, and H1650 and the prostate cancer cell line PC-3 were obtained from American Type Culture Collection (Manassas, VA). The lung cancer cell line PC14PE6/AS2 and its derivatives were established previously in our laboratory (21). The lung cancer cell line H1299 and its derivatives were kindly provided by Dr. Yi-Rong Chen (National Health Research Institutes, Taiwan). NSCLC cells were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; Invitrogen). PC14PE6/AS2 and its derived cells were cultured in MEM-α (Invitrogen) supplemented with 10% FBS. PC-3 cells were cultured in Ham's F12K medium (Sigma-Aldrich) with 10% FBS. All cells were incubated at 37 C in a humidified atmosphere containing 5% CO2.
RNA preparation and RT-PCR
Total RNA was extracted from cells using the single-step TRIzol method (Invitrogen). The purity and concentrations of total RNA were determined by UV spectrophotometry (A260 and A280). One microgram of total cellular RNA was reverse transcribed for each cDNA. First-strand cDNA was synthesized using oligodeoxythymidine primer and the avian myeloma virus reverse transcriptase (Promega, Madison, WI) and used as a template for analysis of gene expression by PCR. The sequences of PCR primers were as follows: ERβ sense primer, 5′-TAG TGG TCC ATC GCC AGT TAT-3′; ERβ antisense primer, 5′-GGG AGC CAC ACT TCA CCA T-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense primer, 5′-CCA TCA CCA TCT TCC AGG AG-3′, and GAPDH antisense primer, 5′-CCT GCT TCA CCA CCT TCT TG-3′. The PCR protocol was performed as follows: enzyme activation for 1 min at 94 C, amplification with denaturation for 30 sec at 94 C, annealing for 30 sec at 58 C, elongation for 1 min at 72 C (34 cycles for ERβ and 20 cycles for GAPDH), and final extension for 7 min at 72 C. The PCR products were separated on 1% agarose gels and visualized by ethidium bromide staining. The intensity of the bands was quantified using the Gel-Pro Analyzer 4.0 software (MediaCybernetics, Bethesda, MD).
Cell lysis and Western blot analysis
The harvested cells were washed twice with PBS and lysed on ice for 30 min with whole-cell extract lysis buffer [50 mm Tris, pH 7.2–7.8; 1% Nonidet P-40; 2 mm EDTA; 100 mm NaCl; 10 mm Na3VO4; 0.1% sodium dodecyl sulfate; 10 mg/ml leupeptin; 2 mg/ml aprotinin; and 100 mm phenylmethylsulfonyl fluoride (protease inhibitors from Roche Applied Sciences, Indianapolis, IN)]. Lysates were cleared by centrifugation at 14,000 rpm for 10 min at 4 C, and protein concentration was determined by the Bradford assay (Bio-Rad Laboratories, Richmond, CA). For Western blot analysis, cell lysates were boiled for 5 min with sample buffer before being resolved in sodium dodecyl sulfate-polyacrylamide gels. The proteins were transferred to polyvinyl difluoride membrane (Millipore, Billerica, MA). The membrane was blocked with 5% nonfat dried milk in buffer of 20 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 0.1% Tween 20 for 1 h and then incubated overnight at 4 C with specific primary antibodies. Subsequently the membrane was washed with buffer of 20 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 0.1% Tween 20 and incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham Pharmacia, Uppsala, Sweden). The binding of each antibody was detected using an enhanced chemiluminescence kit (Amersham Pharmacia Biotech, Piscataway, NJ). The signals were exposed to X-ray films (Fuji, Tokyo, Japan) and analyzed by the Gel-Pro Analyzer 4.0 software (MediaCybernetics).
siRNA and transient transfection
Cells were transfected with a nonspecific random siRNA as a negative control (Invitrogen) and synthetic siRNA directed to Stat3 or ERβ (Ambion, Austin, TX) at a final concentration of 100 nm using MicroPorator MP-100 (NanoEnTek, Seoul, South Korea). Forty-eight hours after the transfection, we confirmed the knockdown of Stat3 or ERβ by Western blotting.
Reporter plasmids and luciferase assays
The reporter gene constructs bearing the different fragments of the human ERβ promoter were generated by PCR amplification of human genomic DNA. The primers for the PCR reaction were as follows: ERβ/−936, 5′-AACCCTCAAAAACCATGT-3′; ERβ/−541, 5′-AGGCCTTCCCAGTGACCT-3′ and ERβ/+268, 5′-TGCTTTTCCCGCATTAGG-3′. The 1204 bp of the 5′ flanking fragment and its deletion were then cloned into the KpnI/BglII site of firefly luciferase based pGL3-basic vector (a gift kindly provided by Dr. Ming-Derg Lai, National Cheng Kung University, Medical College, Tainan, Taiwan) and verified by sequencing. For luciferase assays, cells (5 × 104 cells/well) grown in a standard culture medium containing FBS in 24-well plates were transiently transfected with the reporter plasmids using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. To correct for transfection efficiencies, the Renilla luciferase encoding plasmid pRL-TK (0.1 μg/well) was cotransfected into the cells with the ERβ promoter-driven luciferase reporter gene constructs (0.5 μg/well). After treatment, cells were lysed, and luciferase activities were determined by a chemiluminescence assay using the dual-luciferase assay kit (Promega) and a Luminoskan Ascent luminometer (Thermo Electron Corp., Waltham, MA).
ChIP assays
Cells were serum starved overnight followed by stimulation with IL-6 (10 ng/ml) for 30 min. After cross-linking chromatin by exposure of living cells to 1% of formaldehyde, the assay was performed using the ChIP-IT ChIP kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. The protein-chromatin complexes were immunoprecipitated with polyclonal antibodies against Stat3 or nonspecific IgG as a control. The samples were then washed, and the cross-links were reversed before DNA extraction. For PCR amplification of specific region of the human ERβ promoter region, the following sets of primers were used: primers covering −717/−414 region, forward, 5′-CATTAAGCTGGGGGAACTGG-3′, and reverse, 5′-ACCAGAGAGGCTTTGGGTTT-3′; primers covering −1899/−1603 region, forward, 5′-AATGAGATGCTGTGCAGGTG-3′, and reverse, 5′-GGAGAATCGCTTGAACATGG-3′.
MTT assays
Cell proliferation was determined by the MTT assay. Briefly, cells were seeded into 96-well plates (3 × 103 cells/well). After treatment, MTT (Sigma-Aldrich) stock solution (5 mg/mL in PBS) was added to (final concentration of 0.5 mg/ml) each well and incubated for 4 h at 37 C. After centrifugation at 600 × g for 5 min, MTT solution and medium were aspirated from the wells, and dimethylsulfoxide was added to dissolve the converted dye of released MTT. The absorbance of the solutions was measured at a wavelength of 490 nm with an ELISA reader (Dynatech Technologies, Chantilly, VA).
Statistical analysis
Data were analyzed using Graph Pad Prism 4 software (Graph Pad, Inc., San Diego, CA). Differences between groups were determined using a Student's t test. Statistical significance was set at P < 0.05.
Acknowledgments
This work was supported by the National Cheng Kung University Program for Promoting Academic Excellence and Developing World Class Research Centers, Taiwan, Republic of China; Grants DOH99-TD-B-111-002 and DOH99-TD-C-111-003 from the Department of Health, Executive Yuan, Taiwan, Republic of China; and Grant NSC95-2314-B-006-083-MY3 from the National Science Council, Taiwan, Republic of China.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Act D
- Actinomycin D
- ChIP
- chromatin immunoprecipitation
- DPN
- diarylpropionitrile
- EGF
- epidermal growth factor
- EGFR
- EGF receptor
- ER
- estrogen receptor
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- Jak
- Janus kinase
- MEK
- MAPK kinase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- NSCLC
- non-small cell lung cancer
- PI3K
- phosphoinositide 3′-kinase
- siRNA
- short interfering RNA
- Stat
- signal transducer and activator of transcription.
References
- 1. Siegfried JM. 2001. Women and lung cancer: does oestrogen play a role? Lancet Oncol 2:506–513 [DOI] [PubMed] [Google Scholar]
- 2. Lam WK. 2005. Lung cancer in Asian women—the environment and genes. Respirology 10:408–417 [DOI] [PubMed] [Google Scholar]
- 3. Ger LP , Liou SH , Shen CY , Kao SJ , Chen KT. 1992. [Risk factors of lung cancer]. J Formos Med Assoc 91(Suppl 3):S222–S231 [PubMed] [Google Scholar]
- 4. Xu X , Li B , Wang L. 1994. Gender difference in smoking effects on adult pulmonary function. Eur Respir J 7:477–483 [DOI] [PubMed] [Google Scholar]
- 5. Zang EA , Wynder EL. 1996. Differences in lung cancer risk between men and women: examination of the evidence. J Natl Cancer Inst 88:183–192 [DOI] [PubMed] [Google Scholar]
- 6. Subramanian J , Govindan R. 2007. Lung cancer in never smokers: a review. J Clin Oncol 25:561–570 [DOI] [PubMed] [Google Scholar]
- 7. Taioli E , Wynder EL. 1994. Re: endocrine factors and adenocarcinoma of the lung in women. J Natl Cancer Inst 86:869–870 [DOI] [PubMed] [Google Scholar]
- 8. Liu Y , Inoue M , Sobue T , Tsugane S. 2005. Reproductive factors, hormone use and the risk of lung cancer among middle-aged never-smoking Japanese women: a large-scale population-based cohort study. Int J Cancer 117:662–666 [DOI] [PubMed] [Google Scholar]
- 9. McDonnell DP , Norris JD. 2002. Connections and regulation of the human estrogen receptor. Science 296:1642–1644 [DOI] [PubMed] [Google Scholar]
- 10. Enmark E , Pelto-Huikko M , Grandien K , Lagercrantz S , Lagercrantz J , Fried G , Nordenskjöld M , Gustafsson JA. 1997. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab 82:4258–4265 [DOI] [PubMed] [Google Scholar]
- 11. Kuiper GG , Carlsson B , Grandien K , Enmark E , Häggblad J , Nilsson S , Gustafsson JA. 1997. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology 138:863–870 [DOI] [PubMed] [Google Scholar]
- 12. Omoto Y , Kobayashi Y , Nishida K , Tsuchiya E , Eguchi H , Nakagawa K , Ishikawa Y , Yamori T , Iwase H , Fujii Y , Warner M , Gustafsson JA , Hayashi SI. 2001. Expression, function, and clinical implications of the estrogen receptor β in human lung cancers. Biochem Biophys Res Commun 285:340–347 [DOI] [PubMed] [Google Scholar]
- 13. Mollerup S , Jørgensen K , Berge G , Haugen A. 2002. Expression of estrogen receptors α and β in human lung tissue and cell lines. Lung Cancer 37:153–159 [DOI] [PubMed] [Google Scholar]
- 14. Thuresson-Klein A , Moawad AH , Hedqvist P. 1985. Estrogen stimulates formation of lamellar bodies and release of surfactant in the rat fetal lung. Am J Obstet Gynecol 151:506–514 [DOI] [PubMed] [Google Scholar]
- 15. Patrone C , Cassel TN , Pettersson K , Piao YS , Cheng G , Ciana P , Maggi A , Warner M , Gustafsson JA , Nord M. 2003. Regulation of postnatal lung development and homeostasis by estrogen receptor β. Mol Cell Biol 23:8542–8552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morani A , Barros RP , Imamov O , Hultenby K , Arner A , Warner M , Gustafsson JA. 2006. Lung dysfunction causes systemic hypoxia in estrogen receptor β knockout (ERβ−/−) mice. Proc Natl Acad Sci USA 103:7165–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hershberger PA , Vasquez AC , Kanterewicz B , Land S , Siegfried JM , Nichols M. 2005. Regulation of endogenous gene expression in human non-small cell lung cancer cells by estrogen receptor ligands. Cancer Res 65:1598–1605 [DOI] [PubMed] [Google Scholar]
- 18. Zhang G , Liu X , Farkas AM , Parwani AV , Lathrop KL , Lenzner D , Land SR , Srinivas H. 2009. Estrogen receptor β functions through nongenomic mechanisms in lung cancer cells. Mol Endocrinol 23:146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darnell JE. 1997. STATs and gene regulation. Science 277:1630–1635 [DOI] [PubMed] [Google Scholar]
- 20. Levy DE , Darnell JE. 2002. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662 [DOI] [PubMed] [Google Scholar]
- 21. Yeh HH , Lai WW , Chen HH , Liu HS , Su WC. 2006. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 25:4300–4309 [DOI] [PubMed] [Google Scholar]
- 22. Gschwind A , Fischer OM , Ullrich A. 2004. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4:361–370 [DOI] [PubMed] [Google Scholar]
- 23. Hynes NE , Lane HA. 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5:341–354 [DOI] [PubMed] [Google Scholar]
- 24. Lynch TJ , Bell DW , Sordella R , Gurubhagavatula S , Okimoto RA , Brannigan BW , Harris PL , Haserlat SM , Supko JG , Haluska FG , Louis DN , Christiani DC , Settleman J , Haber DA. 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139 [DOI] [PubMed] [Google Scholar]
- 25. Paez JG , Jänne PA , Lee JC , Tracy S , Greulich H , Gabriel S , Herman P , Kaye FJ , Lindeman N , Boggon TJ , Naoki K , Sasaki H , Fujii Y , Eck MJ , Sellers WR , Johnson BE , Meyerson M. 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500 [DOI] [PubMed] [Google Scholar]
- 26. Pao W , Miller V , Zakowski M , Doherty J , Politi K , Sarkaria I , Singh B , Heelan R , Rusch V , Fulton L , Mardis E , Kupfer D , Wilson R , Kris M , Varmus H. 2004. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 101:13306–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shigematsu H , Lin L , Takahashi T , Nomura M , Suzuki M , Wistuba II , Fong KM , Lee H , Toyooka S , Shimizu N , Fujisawa T , Feng Z , Roth JA , Herz J , Minna JD , Gazdar AF. 2005. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97:339–346 [DOI] [PubMed] [Google Scholar]
- 28. Shen H , Yuan Y , Sun J , Gao W , Shu YQ. 2010. Combined tamoxifen and gefitinib in non-small cell lung cancer shows antiproliferative effects. Biomed Pharmacother 64:88–92 [DOI] [PubMed] [Google Scholar]
- 29. Stabile LP , Lyker JS , Gubish CT , Zhang W , Grandis JR , Siegfried JM. 2005. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res 65:1459–1470 [DOI] [PubMed] [Google Scholar]
- 30. Nose N , Sugio K , Oyama T , Nozoe T , Uramoto H , Iwata T , Onitsuka T , Yasumoto K. 2009. Association between estrogen receptor-beta expression and epidermal growth factor receptor mutation in the postoperative prognosis of adenocarcinoma of the lung. J Clin Oncol 27:411–417 [DOI] [PubMed] [Google Scholar]
- 31. Toh CK , Ahmad B , Soong R , Chuah KL , Tan SH , Hee SW , Leong SS , Tan EH , Lim WT. 2010. Correlation between epidermal growth factor receptor mutations and expression of female hormone receptors in East-Asian lung adenocarcinomas. J Thorac Oncol 5:17–22 [DOI] [PubMed] [Google Scholar]
- 32. Sordella R , Bell DW , Haber DA , Settleman J. 2004. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 305:1163–1167 [DOI] [PubMed] [Google Scholar]
- 33. Hodge DR , Hurt EM , Farrar WL. 2005. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41:2502–2512 [DOI] [PubMed] [Google Scholar]
- 34. Spiotto MT , Chung TD. 2000. STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate 42:88–98 [DOI] [PubMed] [Google Scholar]
- 35. Björnström L , Sjöberg M. 2002. Signal transducers and activators of transcription as downstream targets of nongenomic estrogen receptor actions. Mol Endocrinol 16:2202–2214 [DOI] [PubMed] [Google Scholar]
- 36. Song L , Turkson J , Karras JG , Jove R , Haura EB. 2003. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 22:4150–4165 [DOI] [PubMed] [Google Scholar]
- 37. Downward J , Parker P , Waterfield MD. 1984. Autophosphorylation sites on the epidermal growth factor receptor. Nature 311:483–485 [DOI] [PubMed] [Google Scholar]
- 38. Shao H , Cheng HY , Cook RG , Tweardy DJ. 2003. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res 63:3923–3930 [PubMed] [Google Scholar]
- 39. Chen YR , Fu YN , Lin CH , Yang ST , Hu SF , Chen YT , Tsai SF , Huang SF. 2006. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene 25:1205–1215 [DOI] [PubMed] [Google Scholar]
- 40. Weigert C , Düfer M , Simon P , Debre E , Runge H , Brodbeck K , Häring HU , Schleicher ED. 2007. Upregulation of IL-6 mRNA by IL-6 in skeletal muscle cells: role of IL-6 mRNA stabilization and Ca2+-dependent mechanisms. Am J Physiol Cell Physiol 293:C1139–C1147 [DOI] [PubMed] [Google Scholar]
- 41. Frasor J , Park K , Byers M , Telleria C , Kitamura T , Yu-Lee LY , Djiane J , Park-Sarge OK , Gibori G. 2001. Differential roles for signal transducers and activators of transcription 5a and 5b in PRL stimulation of ERα and ERβ transcription. Mol Endocrinol 15:2172–2181 [DOI] [PubMed] [Google Scholar]
- 42. Hatae J , Takami N , Lin H , Honda A , Inoue R. 2009. 17β-Estradiol-induced enhancement of estrogen receptor biosynthesis via MAPK pathway in mouse skeletal muscle myoblasts. J Physiol Sci 59:181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oh AS , Lorant LA , Holloway JN , Miller DL , Kern FG , El-Ashry D. 2001. Hyperactivation of MAPK induces loss of ERα expression in breast cancer cells. Mol Endocrinol 15:1344–1359 [DOI] [PubMed] [Google Scholar]
- 44. Pao W , Miller VA. 2005. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol 23:2556–2568 [DOI] [PubMed] [Google Scholar]
- 45. Ji H , Li D , Chen L , Shimamura T , Kobayashi S , McNamara K , Mahmood U , Mitchell A , Sun Y , Al-Hashem R , Chirieac LR , Padera R , Bronson RT , Kim W , Jänne PA , Shapiro GI , Tenen D , Johnson BE , Weissleder R , Sharpless NE , Wong KK. 2006. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 9:485–495 [DOI] [PubMed] [Google Scholar]
- 46. Nose N , Uramoto H , Iwata T , Hanagiri T , Yasumoto K. 2011. Expression of estrogen receptor β predicts a clinical response and longer progression-free survival after treatment with EGFR-TKI for adenocarcinoma of the lung. Lung Cancer 71:350–355 [DOI] [PubMed] [Google Scholar]
- 47. Alvarez JV , Greulich H , Sellers WR , Meyerson M , Frank DA. 2006. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res 66:3162–3168 [DOI] [PubMed] [Google Scholar]
- 48. Mostertz W , Stevenson M , Acharya C , Chan I , Walters K , Lamlertthon W , Barry W , Crawford J , Nevins J , Potti A. 2010. Age- and sex-specific genomic profiles in non-small cell lung cancer. JAMA 303:535–543 [DOI] [PubMed] [Google Scholar]
- 49. Kato S , Endoh H , Masuhiro Y , Kitamoto T , Uchiyama S , Sasaki H , Masushige S , Gotoh Y , Nishida E , Kawashima H , Metzger D , Chambon P. 1995. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270:1491–1494 [DOI] [PubMed] [Google Scholar]
- 50. Bunone G , Briand PA , Miksicek RJ , Picard D. 1996. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 15:2174–2183 [PMC free article] [PubMed] [Google Scholar]
- 51. Tremblay A , Tremblay GB , Labrie F , Giguère V. 1999. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol Cell 3:513–519 [DOI] [PubMed] [Google Scholar]
- 52. Stabile LP , Davis AL , Gubish CT , Hopkins TM , Luketich JD , Christie N , Finkelstein S , Siegfried JM. 2002. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor α and β and show biological responses to estrogen. Cancer Res 62:2141–2150 [PubMed] [Google Scholar]
- 53. Kawai H , Ishii A , Washiya K , Konno T , Kon H , Yamaya C , Ono I , Minamiya Y , Ogawa J. 2005. Estrogen receptor α and β are prognostic factors in non-small cell lung cancer. Clin Cancer Res 11:5084–5089 [DOI] [PubMed] [Google Scholar]
- 54. Márquez-Garban DC , Chen HW , Fishbein MC , Goodglick L , Pietras RJ. 2007. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids 72:135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu CT , Chang YL , Shih JY , Lee YC. 2005. The significance of estrogen receptor β in 301 surgically treated non-small cell lung cancers. J Thorac Cardiovasc Surg 130:979–986 [DOI] [PubMed] [Google Scholar]
- 56. Schwartz AG , Prysak GM , Murphy V , Lonardo F , Pass H , Schwartz J , Brooks S. 2005. Nuclear estrogen receptor β in lung cancer: expression and survival differences by sex. Clin Cancer Res 11:7280–7287 [DOI] [PubMed] [Google Scholar]
- 57. Siddiquee K , Zhang S , Guida WC , Blaskovich MA , Greedy B , Lawrence HR , Yip ML , Jove R , McLaughlin MM , Lawrence NJ , Sebti SM , Turkson J. 2007. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA 104:7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]