

Abstract
GnRH is a pivotal hypothalamic neurohormone governing reproduction and sexual development. Because transcriptional regulation is crucial for the spatial and temporal expression of the GnRH gene, a region approximately 3.0 kb upstream of the mammalian GnRH promoter has been extensive studied. In the present study, we demonstrate a transcription-enhancer located in the first intron (intron A) region of the GnRH gene. This transcriptional enhancer harbors putative sex-determining region Y-related high-mobility-group box (SOX) family transcription factor-binding sites, which are well conserved across many mammalian species. The class-C SOX member proteins (SOX-C) (SOX4 and SOX11) specifically augment this transcriptional activation by binding to these SOX-binding sites. In accordance, SOX11 is highly enriched in immortalized GnRH-producing GT1-1 cells, and suppression of its expression significantly decreases GnRH gene expression as well as GnRH secretion. Chromatin immunoprecipitation shows that endogenous SOX-C factors recognize and bind to the intronic enhancer in GT1-1 cells and the hypothalamus. Accompanying immunohistochemical analysis demonstrates that SOX4 or SOX11 are highly expressed in the majority of hypothalamic GnRH neurons in adult mice. Taken together, these findings demonstrate that SOX-C transcription factors function as important transcriptional regulators of cell type-specific GnRH gene expression by acting on the intronic transcriptional enhancer.
GnRH, a pivotal hypothalamic neurohormone, plays a crucial role in both reproduction and sexual development of vertebrates. Mammalian GnRH-secreting neurons are located mainly in the preoptic area (POA) of the hypothalamus and project efferent fibers to the median eminence. GnRH is released into the hypophysial portal circulation in a pulsatile manner, controlling the biosynthesis and secretion of pituitary gonadotropins. The mammalian GnRH gene is composed of four short exons and three intervening introns. Exon 2 encodes a signal peptide sequence followed by the GnRH decapeptide, whereas the remaining exons 2, 3, and 4 encode the GnRH-associated peptide. Notably, GnRH mRNA expression is highly restricted to a subset of hypothalamic neurons and is tightly regulated by multiple humoral and neural signals (1, 2).
Transcriptional control is believed to be crucial for spatiotemporal regulation of GnRH gene expression. Therefore, extensive studies have been undertaken to characterize GnRH neuron-specific transcriptional enhancers in distal promoter regions of the mammalian GnRH genes since immortalized GnRH neuronal cell lines, such as GT1 and Gn11, became available (3, 4). In this context, a number of transcription factors, including GATA-binding protein, octamer-binding transcription factor 1, and orthodenticle homolog 2, have been proposed to function as trans-acting factors on the approximately 3.0 kb of the mammalian GnRH promoter (GnRHp) (5–8). These transcription factors, however, still cannot fully account for the GnRH gene expression taking place in a highly restricted population of hypothalamic neurons, because they are ubiquitously expressed in many tissues. The unique combination of these ubiquitous transcription factors has thus been suggested to contribute to the specificity of GnRH neurons (9). In addition, few of these trans-acting factors and their binding cis-elements on the GnRHp are well conserved throughout mammalian species (10), suggesting that as yet undiscovered transcription factors and their evoked mechanisms may underlie the cell type specificity of GnRH gene expression.
Considering the structure of the GnRH gene, it should be noted that there is a gap of over 1 kb that exists between the transcription and translation start sites, exon 1 (Ex1) and the first intron (intron A), in all mammalian species examined, suggesting that this common gene structure may, at least in part, contribute to the regulation of GnRH gene expression. Indeed, our previous studies demonstrated that the excision of GnRH intron A is rate limiting in the GnRH neuron-specific splicing, which produces mature mRNA (11–14). In this study, we tested the hypothesis that a novel transcriptional regulatory mechanism acting through a putative transcriptional enhancer in the intronic region may underlie GnRH neuron-specific GnRH gene transcription.
Results
The GnRH intron A region possesses transcription-enhancing activity
Because downstream enhancer elements within the 5′-untranslated region (UTR) often contribute to the regulation of gene transcription (15–17), we hypothesized that the 5′ noncoding region of the GnRH gene contains a putative transcriptional regulatory cis-element. The sequence alignment of the total GnRH gene structure revealed that the first exon (Ex1) and intron (intron A) are relatively conserved across mammalian species (Fig. 1A). Interestingly, the conservation pattern of intron A is distinct from the intron A and B of GnRH genes; besides the regions on the 5′ and 3′ ends of intron A, which are recognized by basic splicing machinery, internal regions of intron A (Fig. 1A, black boxes) show higher conservation scores relative to those of other introns. The region extended from 5′ end and the middle of intron A ranged from approximately 60 to 90% homologies among the five examined mammalian species. Because the GnRH Ex1 region did not exert apparent transcription-enhancing activity in our previous study (12), the present study focused on the GnRH intron A region. To examine the enhancer activity of intron A, we constructed reporter vectors containing the full-length mouse intron A sequence (1003 bp) in either the forward or reversed direction followed by the simian vacuolating virus 40 (SV40) minimal promoter (SV40p) driving luciferase expression. These constructs were transfected into GnRH-expressing GT1-1 cells, and luciferase activity was then measured. Cells harboring the vectors with the full-length intron A exhibited luciferase activity significantly increased by 2- to 3-fold compared with those harboring only the SV40p (the “empty vector” in this study) (Fig. 1B). The rat and human versions of GnRH intron A also exhibited similar enhancing activities (Fig. 1C). These findings strongly suggest that GnRH intron A contains a putative transcriptional enhancer that is likely involved in GnRH gene regulation. In an ensuing experiment, serially deleted and truncated reporter constructs revealed that 315 nucleotides (nt) of GnRH intron A, located proximal to Ex1, were responsible for its transcription enhancing activity; the enhancing activity of this region was comparable with that of the full-length intron A (Fig. 2).
Fig. 1.

Transcription-enhancing activity of GnRH intron A. A, Schematic diagram of mouse GnRH gene structure and conservation map of GnRH genomic sequences among the various mammalian species using the UCSC genome browser (http://genome.ucsc.edu). Nucleotide locations are based on the previous studies (45, 46). Threshold of conservation by PhyloP set as 0–0.3 in the conservation map. The well-conserved regions of GnRH intron A are indicated by boxes. B, Schematic representation of the reporter constructs (top) and enhancing activities (bottom) of mouse GnRH intron A on the SV40p in the forward- or reverse-oriented forms. Data are shown as mean ± se in arbitrary units (A.U.), where the mean activity of the empty vector was set at 1 (n = 3–4; **, P < 0.01 vs. empty vector). C, Schematic diagram of the reporter constructs (top) and enhancing effects (bottom) of mouse, rat, and human GnRH intron A fragments on the SV40p in GT1-1 cells. Data are shown as mean ± se in arbitrary units, where the mean activity of the empty vector was set at 1 (n = 4–6; **, P < 0.01 vs. empty vector). Data were statistically evaluated by one-way ANOVA followed by Newman-Keuls post hoc comparison.
Fig. 2.

Ex1-proximal 315-bp region of GnRH intron A exerts transcription-enhancing activity. Schematic diagram of the reporter constructs and relative luciferase activities is shown. To determine the enhancer-containing region of GnRH intron A, serially deleted and truncated fragments of mouse intron A were fused upstream of the SV40p (the beginning base of intron A was designated as 1). Luciferase activities from deletion constructs were determined in GT1-1 cells. Data are shown as mean ± se in arbitrary units (A.U.), where the mean activity of the empty reporter was set at 1 (n = 3; **, P < 0.01 vs. empty vector; one-way ANOVA followed by Newman-Keuls post hoc comparison).
Intron A located downstream of the GnRHp elicits transcription-enhancing activity
For further identification of the intronic enhancer, we used an enhancer test system resembling the native GnRH gene structure. It should be noted that GnRH intron A cannot be easily excised due to its suboptimal 3′-splicing acceptor site (11). Therefore, we developed a transcription/splicing-coupled reporter containing a minigene construct (designated as Ex1IA-315), comprising GnRH Ex1 and the centrally truncated intron A (1–315 and 897-1003 nt of the mouse intron A) with the native splicing donor and optimally mutated acceptor, as shown in Fig. 3A. The Ex1IA-315 minigene is followed by a luciferase coding sequence, which mimics the open reading frame of the GnRH gene. Then, Ex1IA-315-Luc or Ex1-Luc, its intron A-lacking control, was fused downstream to either the SV40p or mouse 3.4-kb GnRHp. Ex1IA-315 elicited similar enhancing effects on both the SV40p and GnRHp (Fig. 3B), indicating that the intronic enhancer can act on the GnRHp in its native conformation. To examine whether the intronic enhancer activity is exerted in a cell type-specific fashion, we compared the enhancing activities in additional mouse hypothalamic GnRH-expressing cell lines (N4 and N44) and mouse fibroblast NIH3T3 cells as a nonneuronal control. N4 and N44 cells express GnRH mRNA at lower but significant levels compared with GT1-1 cells (Fig. 3C, top). Relative GnRH mRNA levels in N4 and N44 cells quantified by real-time quantitative PCR were 6.53 ± 0.59 and 10.45 ± 0.80% of that observed in GT1 cells (100.00 ± 2.30% in mean ± se), respectively (n =3–4 for each cell line). Interestingly, the transcription-enhancing activity of Ex1IA-315 exhibited cell type specificity; the SV40p-Ex1IA-315-Luc reporter exerted significantly higher activities in GnRH-expressing cell lines such as GT1, N4, and N44 than non-GnRH-producing fibroblasts (Fig. 3C, bottom).
Fig. 3.
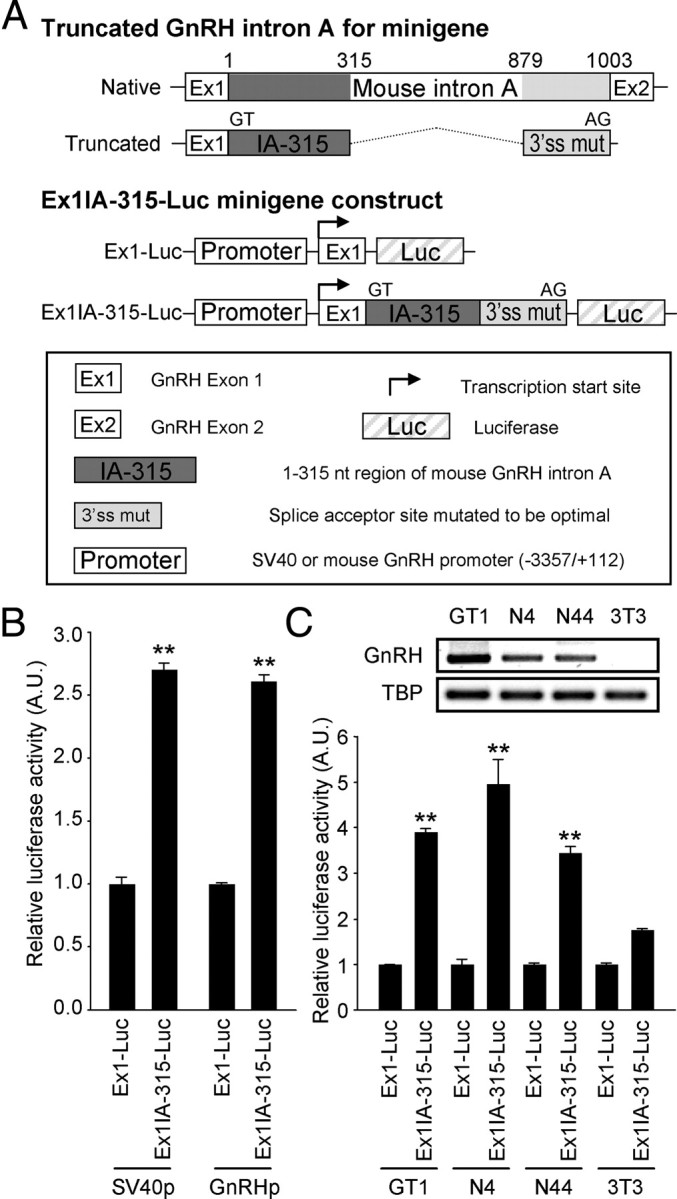
Cell type-specific transcription enhancement of the intron A located downstream of the promoter. A, Schematic diagram of the transcription/splicing-coupled Ex1IA-315-Luc reporter constructs. To mimic the native GnRH gene structure, Ex1 and the centrally truncated GnRH intron A with the restored 3′-splicing acceptor site to the consensus one were inserted between promoters and luciferase open reading frame. B, The Ex1-proximal fragment of mouse intron A augmented both the SV40 and mouse GnRHp-driven luciferase activities. Data are shown as mean ± se in arbitrary units (A.U.), where the mean activity of each Ex1-Luc reporter was set at 1 (n = 4; **, P < 0.01 vs. Ex1-Luc vector by Student's t test). C, Expression profiles of GnRH mature mRNA (top) and enhancing activities of the Ex1IA-315 fragment (bottom) in several GnRH-expressing mouse cell lines and NIH3T3 fibroblasts. TATA box-binding protein (TBP) was used as a positive control in the RT-PCR. Data are presented as mean ± se in A.U., where the mean activity of each Ex1-Luc reporter was set at 1 (n = 3; **, P < 0.01 vs. Ex1IA-315-Luc activity in NIH3T3 cell by one-way ANOVA followed by Newman-Keuls post hoc comparison).
In the ensuing experiments, serial deletion of the Ex1-proximal intron A sequence was performed to more precisely define the region containing the cis-elements. Schematic diagrams of the reporter constructs and their relative luciferase activities in comparison with those of SV40p-Ex1-Luc and SV40p-Ex1IA-315-Luc are shown in Fig. 4. The transcription-enhancing activity was seen even in the presence of the Ex1-proximal 25 nt but disappeared when only 12 nt of the intron A remained. Because our in vivo splicing assay confirmed that the Ex1-proximal 12 nt is sufficient for an efficient excision of the intronic sequence from the minigene mRNA (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org), the decreased luciferase activity in SV40p-Ex1IA-12 is unlikely to be due to a possible failure in the excision of the intronic sequence. Thus, these results strongly suggest that the region between 12 and 25 nt in the mouse GnRH intron A harbors a key cis-element for the enhancer function.
Fig. 4.

Determination of the cis-element-containing region. Schematic diagrams and relative luciferase activities of SV40p-Ex1IA-315-Luc and its serially deleted derivatives. To dissect the enhancer element containing region, Ex1IA-315 fragment was serially deleted from the 3′ end and placed between the SV40p and 3′-splicing acceptor site as indicated. Data are shown as mean ± se in arbitrary units (A.U.), where the mean activity of SV40-Luc reporter was set at 1. Statistically distinct groups are marked with different letters (n = 7; one-way ANOVA followed by Newman-Keuls post hoc comparison).
Class-C sex-determining region Y-related high-mobility-group box (SOX) (SOX-C) factors control GnRHp activity through their binding sites in intron A
To identify the exact cis-element(s) and the trans-acting partners of the intronic enhancer, we analyzed putative transcription factor-binding sites in the mouse intron A region. Using the MatInspector program (Genomatix, Munich, Germany), we found that two putative binding sites for SOX transcription factors (designated as sites 1 and 2 from the Ex1-proximal) reside within the Ex1-proximal 50 nt of mouse intron A. It is of interest to note that the SOX-binding sequences are conserved in the Ex1-proximal intron A sequence of many mammalian species, including the rat, cow, monkey, and even human (Fig. 5A). Thus far, more than 20 members of the mammalian SOX, which can be categorized into eight classes, have been identified. These SOX factors interact with the consensus DNA sequence of (A/T)(A/T)CAA(A/T)G (18). Therefore, we examined which members or classes of SOX exert a transcription enhancing effect through the intronic cis-element. Out of seven SOX factors from five different classes that were tested, SOX4 and SOX11, belonging to the SOX-C factors, dramatically augmented SV40p-driven Ex1IA-315 reporter activities (Fig. 5B). Their enhancing effects were also exerted on the GnRHp; overexpression of SOX4 or SOX11 caused a nearly 2-fold increase over empty vector control (Fig. 5C). The 3.0-kb GnRHp activity was not influenced by overexpression of either SOX4 or SOX11 in the absence of the intronic enhancer (Supplemental Fig. 2). Because all of the IA-SV40p-Luc reporters based on mouse, rat, and human GnRH gene were induced by coexpression of SOX-C factors in GT1-1 cells (Supplemental Fig. 3), it can be postulated that the activational effect of SOX-C factors on GnRH gene transcription through the enhancer elements within intron A appears to be conserved in these three species.
Fig. 5.

Involvement of SOX factors in intron A-evoked transcriptional activation of the GnRHp. A, Putative SOX-binding sites on GnRH intron A in various mammalian species. Two SOX-binding sites in mouse intron A are designated as sites 1 and 2. The 1- to 50-nt region of GnRH intron A in various mammalian species (rat, human, chimpanzee, rhesus monkey, and cow) were compared by the ClustalW program (http://www.ebi.ac.uk/clustalw/). The black characters indicate the major conserved nucleotides, whereas gray indicates the less conserved nucleotides. Putative SOX-binding sites are enclosed in boxes. B, Effects of various SOX factors on the enhancer activity of mouse GnRH intron A. The class of each SOX factor is shown in parentheses. Data are presented as fold inductions in luciferase activities of SV40p-Ex1IA-315-Luc relative to Ex1-Luc reporter (n = 3; **, P < 0.01 vs. other groups; one-way ANOVA followed by Newman-Keuls post hoc comparison). C, Effects of SOX-C on GnRHp-Ex1IA-315-Luc reporter (n = 3; **, P < 0.01 vs. pcDNA3-transfected group; one-way ANOVA).
To examine whether the SOX-C-evoked transcriptional activation is mediated through the intronic elements, we then abrogated either or both of the SOX-binding sites of mouse intron A in the Ex1IA-315-Luc construct (Fig. 6A). When both SOX-binding sites were removed by either truncation or microdeletion of core binding sequences, the effects of SOX4 and SOX11 overexpression were drastically attenuated, whereas with the removal of one of them resulted in a partial effect, suggesting that two SOX-binding sites in the mouse GnRH intron A may work additively (Fig. 6, B and C). In accordance with the result in the reporter assay, EMSA revealed that in vitro translated SOX4 and SOX11 proteins can recognize both SOX-binding sites in mouse intron A; the binding was completely abrogated when both sites were mutated but partially diminished in the probes with a deletion of one of them (Fig. 7). Taken together, these findings indicate that both of the SOX-binding sites in mouse intron A are functional and play an important role in mediating SOX-C factor-evoked transcriptional regulation.
Fig. 6.

Role of putative SOX-binding sites in the enhancing activities of intron A. A, Schematic diagram for the constructs bearing mutations at SOX-binding sites. Putative SOX-binding sites were removed by truncation or microdeletion from the IA-315 region as indicated. B and C, Enhancing activities of the mutated intron A fragment in the absence or presence of SOX-C factors. Data are shown as mean ± se in arbitrary units (A.U.), where the mean activity of SV40p-Ex1-Luc in pcDNA3-expressed cells was set at 1 (B) or presented as fold inductions for each reporter in response to coexpressed SOX-C factors (C). Statistically distinct groups are marked with different letters (n = 3–7; one-way ANOVA followed by Newman-Keuls post hoc comparison).
Fig. 7.

SOX-C factors directly bind to each SOX-binding site of mouse GnRH intron A. EMSA was performed using in vitro translated SOX-C proteins. A, Probe sequences for EMSA. SOX-binding sites are indicated by boxes, and mutated nucleotides are shown as bold characters. B, In vitro translated SOX4 and SOX11 proteins (Myc-epitope-tagged forms) were detected by immunoblotting (IB) with an anti-Myc antibody (asterisks). Rabbit reticulocyte lysate (RRL) was used as a negative control. In vitro translated SOX4 (C) or SOX11 (D) proteins were incubated with 32P-labeled wild type (WT) or various mutant probes as indicated at the bottom. Binding specificities of SOX-C factors to each probe were confirmed by the addition of 200-fold molar excess self-unlabeled probes as a cold competitor (Comp.).
SOX-C factors regulate endogenous GnRH gene expression
To determine whether SOX-C factors control endogenous GnRH gene expression, we examined the protein expression levels of SOX-C factors in GnRH-producing GT1-1 cells and the adult POA-anterior hypothalamus (POA-AH), where GnRH neurons are abundantly located. Although both SOX4 and SOX11 were expressed in the hypothalamic extracts, only SOX11 was detectable by immunoblotting in GT1-1 cells (Fig. 8A). Then, we designed short hairpin RNA (shRNA)-expressing vectors to specifically knockdown endogenous SOX4 or SOX11 (Supplemental Fig. 4) and determined whether suppression of endogenous SOX-C impaired GnRH gene transcription in GT1-1 cells. For this purpose, we measured both GnRH mRNA and primary transcript levels with a real-time RT-PCR method in the presence of either of the shSOX-C vectors or the control pU6 empty vector. Overexpression of shSOX11 but not shSOX4 suppressed endogenous SOX11 protein expression (Fig. 8B) and, accordingly, significantly decreased GnRH gene expression in GT1-1 cells. Both the mRNA and primary transcript levels of the GnRH gene transcripts were reduced by approximately half of those in the control group, indicating a down-regulation at the level of transcription (Fig. 8C). Consistent with the results of gene transcription, knockdown of the endogenous SOX11 expression significantly reduced GnRH secretion from GT1-1 cells, showing that SOX-C-mediated GnRH gene regulation is required for proper production of the neurohormone (Fig. 8D).
Fig. 8.

SOX-C factors regulate endogenous GnRH gene expression. A, Western blot analyses of SOX-C protein expression in whole-cell lysates from GT1-1 cells and mouse POA-AH. B, Knockdown of endogenous SOX-C expression in GT1-1 cells by transfection with a shRNA-expressing plasmid against the indicated SOX-C factor. Knockdown efficacy of endogenous SOX11 was validated by immunoblotting. C, Effect of endogenous SOX-C knockdown on GnRH gene expression in GT1-1 cells. Quantitative RT-PCR analyses were carried out to measure the GnRH mRNA (top) and primary transcript (bottom) levels. Data are shown as mean ± se in arbitrary units, where the mean level of the empty vector control is set at 1 (n = 4–5; **, P < 0.01 vs. control by Student's t test). D, GnRH secretion from cells transfected with control or shSOX-C-expressing plasmid were measured by RIA (n = 6; *, P < 0.05 vs. empty vector control by Student's t test).
We then examined whether endogenous SOX-C factors bind to the cis-elements identified in the intron A in vivo. Chromatin immunoprecipitation (ChIP) assay revealed that SOX11 binds to the Ex1-proximal enhancer region of intron A, and overexpression of SOX11 increases this binding in GT1-1 cells (Fig. 9A). By contrast, SOX11 did not bind to the promoter and 3′-UTR of the GnRH gene, which lack SOX-binding sites, thereby confirming the specificity of our binding assay. Histone H3 was used as a positive control, and thus all the target regions were detected when ChIP assays were carried out with an antihistone H3 antibody. We also tested SOX-C binding to their target sites on mouse GnRH intron A. Interestingly, ChIP assays using hypothalamic lysates from immature [postnatal d (P)10] and adult mice (P70) demonstrated that the intronic enhancer fragment containing two SOX-binding sites was coimmunoprecipitated with both SOX4 and SOX11 antibodies in an age-independent manner (Fig. 9B), raising the possibility that both SOX-C factors are functional in mouse GnRH neurons, in contrast to GT1-1 cells.
Fig. 9.

SOX-C factors bind to the enhancer region of intron A in vivo. A, ChIP assays were performed on pcDNA3 or SOX11-transfected GT1-1 cells using an anti-SOX11 antibody. B, ChIP assays with hypothalamic lysate from immature (P10) and adult (P70) mice were performed using SOX4 and SOX11 antibodies. An antihistone H3 (α-H3) antibody and preimmune normal rabbit serum (NRS) were used as positive and negative controls for immunoprecipitation (IP), respectively.
SOX-C factors are enriched in adult hypothalamic GnRH neurons
Although SOX-C proteins were detectable in mouse POA-AH extracts, as shown in Fig. 8A, this does not provide direct evidence of SOX-C expression in hypothalamic GnRH neurons. Therefore, we examined endogenous SOX-C expression in randomly chosen GnRH neurons located in the POA region of the hypothalamus by immunohistochemical analyses in comparison with neuronal nuclear antigen (NeuN)-positive pan-neuronal populations (Fig. 10 and Table 1). By simultaneous staining with anti-SOX4, SOX11, and GnRH antibodies, we were able to detect either SOX4 or SOX11 signals in approximately 80% of GnRH-immunoreactive (ir) neurons (81.1 ± 1.4%) (Fig. 10A). By contrast, the coexpression of SOX-C factors with NeuN-positive cells was significantly lower than GnRH-ir cells (34.8 ± 3.5%) (Fig. 10B).
Fig. 10.

SOX-C factors are enriched in adult hypothalamic GnRH neurons. Immunohistochemical analysis of the adult mouse POA region (scale bar, 20 μm). SOX4 (green) and SOX11 (blue) were simultaneously labeled with (A) GnRH or (B) NeuN (red). Arrows depict dual-ir cells for SOX4 and SOX11, whereas arrowheads indicate cells expressing either SOX4 or SOX11. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI).
Table 1.
Enriched expression of SOX-C factors in GnRH neurons in the POA
| SOX-C-ir | GnRH-ir |
NeuN-ir |
||||
|---|---|---|---|---|---|---|
| SOX4/11-ir double + | SOX4-ir single + | SOX11-ir single + | SOX4/11-ir double + | SOX4-ir single + | SOX11-ir single + | |
| % Colocalization | 29.4 ± 3.6 | 10.8 ± 1.0 | 40.9 ± 4.3 | 4.3 ± 1.3 | 9.8 ± 1.6 | 20.8 ± 0.8 |
Colocalization of SOX-C with GnRH-ir or NeuN-ir cells was quantified and is presented as the mean ± se % (n = 3; total 100 cells for GnRH-ir and 1897 cells for NeuN-ir from three different mice).
It should be noted that the expression of SOX-C proteins in GnRH-ir neurons exhibit heterogeneity; GnRH-ir neurons ir for either one or both of them were found throughout the hypothalamic region. As shown in Fig. 10 and Table 1, approximately one third of the GnRH-ir neurons (29.4 ± 3.6%) expressed both SOX4 and SOX11 simultaneously, and approximately 40% of the GnRH-ir neurons were immunoreactive for only SOX11 (40.9 ± 4.3%). These proportions are quite a bit higher than those observed in NeuN-ir neurons (4.3 ± 1.3% for SOX4/SOX11-ir double positive and 20.8 ± 0.8% for SOX11-ir single positive). On the other hand, the proportions of single SOX4-ir cells are similar in both groups (10.8 ± 1.0% for GnRH-ir vs. 9.8 ± 1.6% for NeuN-ir cells). Taken together, it appears that SOX-C factors are more enriched in GnRH neurons than other types of neurons and are expressed with heterogeneous profiles among the GnRH-producing cells.
Discussion
In the present study, we clearly demonstrate that the SOX-C transcription factors regulate GnRH gene expression through interaction with the cis-elements residing on the Ex1-proximal region of intron A. Furthermore, the SOX4 and SOX11 members of the SOX-C are abundantly expressed in hypothalamic GnRH neurons, and directly bind to the enhancer region in vivo. Numerous studies have been undertaken to elucidate enhancer elements that confer the GnRH neuron-specific gene expression and their trans-acting factors on the upstream GnRHp region (10). However, strong evidence is here presented for the first time on a novel molecular mechanism underlying GnRH gene regulation by the intronic enhancer element and the interacting SOX-C transcription factors.
It was demonstrated that the SOX-binding sites in the Ex1-proximal region of GnRH intron A serve as a downstream transcription enhancer of GnRH gene expression (Figs. 1–4). Downstream enhancer elements within the first intronic region regulate the transcription of many genes, in particular, those containing a long 5′ noncoding region. Ig (19), cystic fibrosis transmembrane conductance regulator (CFTR) (15), aquaporin 5 (16), and ubiquitin C (17) are well-known examples. In addition, recent studies reported that EGFR and ERBB2 genes are transcriptionally regulated by SOX4 through the enhancer elements within the first intron in prostate cancer cells (20). Notably, previous studies using transgenic mice bearing an approximately 3.0-kb GnRHp-driven reporter transgene have strongly suggested the requirement of additional enhancer elements outside this upstream promoter region for the highly cell type-restricted GnRH gene transcription. GnRHp-driven reporter activities have been widely reported in non-GnRH neuronal populations in extrahypothalamic regions (21, 22). Thus, these observations imply that this promoter region appears insufficient to account for the GnRH neuron-specific transcriptional control, although the upstream promoter has been successfully used to immortalize GnRH neurons to establish several cell lines (3, 4). Subsequent characterization of the exact cis-elements and the expression profiles of SOX-C indicated that the transcriptional activation mechanism of the intronic enhancer does play an important role in at least strengthening the GnRH neuron specificity of the transcription of this neurohormone gene. This notion is also supported by the fact that the transcription-enhancing activity exerted by the intron A is stronger in GnRH-expressing cell lines (Fig. 3C). The putative SOX-binding site(s) in intron A is notably well conserved in many mammalian species, from rodents to human, despite a variation in the number of the binding sites (Fig. 5A). Based on such evolutionary conservation, it can be postulated that this regulatory mechanism is crucial for the normal expression of the neurohormone.
SOX transcription factors were originally reported to regulate a variety of developmental processes throughout the entire body. Among them, the SOX-C factors consist of three members (SOX4, SOX11, and SOX12), which are characterized by a high-mobility-group box DNA binding domain and transactivation domain (18, 23). From knockout studies, loss of SOX4 or SOX11 induces embryonic lethality due to circulatory failure, whereas SOX12-deficient mice do not display any evidently defective phenotype (24–26). Hence, SOX4 and SOX11 are thought to be dominant factors in normal development. However, SOX12 has redundant functions and seems to be relatively dispensable. SOX4 and SOX11 are found to be highly expressed in the developing nervous system (23, 26) and to regulate pan-neuronal gene expression in the central nervous system (27), as well as the development of the sympathetic nervous system (28). SOX-C expression in brain tissues increases by embryonic d (E)16.5 and then gradually returns to a marginal level by E18.5 (26). However, little is known about their roles in the adult nervous system, presumably due to the embryonic lethality of these SOX-C-deficient mice. Here, we show a novel function of SOX-C in the adult central nervous system, transcriptional regulation of hypothalamic GnRH gene expression. The fact that abrogation of SOX-C expression significantly impairs GnRH gene expression as well as neurohormone secretion in GT1 cells clearly supports this notion. It is of interest to note that sex-determining region Y and SOX family members often have an affect on pre-mRNA splicing processes (29, 30). Considering that the excision of GnRH intron A is crucial for producing translatable mature mRNA, it is worth investigating whether SOX-C factors are involved in the GnRH neuron-specific excision of intron A. Suppression of SOX11 expression in GT1-1 cells barely altered the excision rate (Supplemental Fig. 5), indicating that the impact of SOX-C factors on the regulation of GnRH gene expression is primarily achieved at the level of transcription.
In contrast to the limited numbers of neuronal cells expressing either SOX4 and/or SOX11, a larger population of GnRH neurons was immunopositive for them in the adult hypothalamus (Fig. 10 and Table 1). Furthermore, SOX-C factors exhibit binding activities to the intronic enhancer region of the GnRH gene regardless of the state of sexual maturation (P10 and P70) (Fig. 9B). Such expression profiles, along with the binding activities, raise several important issues with regard to their function. First, during the gradual decrease of these factors in brain tissues that occurs after having promoted massive neuronal differentiation, SOX-C factors continue to be expressed in a subset of neuronal populations, including hypothalamic GnRH neurons, where they consistently play a certain role, even in adulthood. In this regard, it is worth noting that SOX4 is specifically maintained in a subset of islet cells of the mouse pancreas despite the overall decrease that takes place in this organ during postnatal development and then regulates insulin secretion and glucagon expression (31–33). Second, SOX-C factors tend to recognize their acting sites on the GnRH gene as early as P10 of age, suggesting that SOX-C factors may have a priming or facilitatory role in the regulation of GnRH gene expression. However, it cannot be excluded that the SOX-C binding to the intronic enhancer can be transiently regulated during certain phases of postnatal development, such as the pubertal onset period. This possibility seems worth being further investigated. Interestingly, GT1-1 cells exclusively express SOX11, and knockdown of SOX4 in these cells barely resulted in any change in GnRH gene expression or secretion, whereas hypothalamic GnRH neurons express either of the SOX-C factors. Recently, SOX-C heterogeneous expression was also reported in sympathetic ganglia neurons (28). In addition, GnRH neurons are already known to consist of heterogeneous populations in several aspects (2). Together, these observations provide a plausible explanation for the discrepancy between GT1-1 cells and hypothalamic GnRH neurons in SOX-C expression. Because the GT1-1 cell line was established by clonal selection and propagation, a single positive for SOX11 shown in the GT1-1 cells is most likely due to their origin. Nevertheless, the effects of SOX4 and SOX11 on GnRH gene transcription can be regarded as being equivalent in terms of DNA-binding (Figs. 7 and 9) and transactivational activities (Figs. 5 and 6).
Although we demonstrated the binding of SOX-C factors to their cis-elements in the GnRH gene both in vitro and in vivo, and thereby the activation of GnRH mRNA expression, the findings still raise an important question related to selectivity. Figure 5 shows that both SOX4 and SOX11 specifically augment the enhancing activity of intron A, whereas other classes of SOX factors fail to do so and often decrease the reporter activity regardless of their trans-acting properties, i.e. transcriptional activation or repression. SOX-C members are well-known transcriptional activators that contain 33 highly conserved C-terminal residues as a transactivation domain (23). However, all SOX family transcription factors are known to recognize the common consensus binding sites; thus, the selective actions of SOX-C are likely to need a further mechanism to confer DNA-binding specificity. Previous studies proposed that each SOX factor can cooperate with other protein partners to acquire its target gene-specific functions. The Pit-Oct-Unc (POU) family transcription factors are well-known binding partners of SOX factors (34, 35), and several neuron-enriched genes are reported to be regulated by such SOX-POU complexes in their highly conserved noncoding DNA regions (36). For instance, brain-1 and brain-2 of the POU family were reported as putative binding partners of SOX11 in the regulation of Fgf4 and Nestin gene expression (37, 38). Therefore, a specific binding partner belonging to other families of transcription factors contribute to the GnRH gene-specific action of SOX-C members.
In conclusion, the present study reveals a novel regulatory mechanism underlying transcriptional control of GnRH gene expression and thus provides new insight into the molecular regulation of the neurohormone. Based on these findings, it may be speculated that this novel mechanism greatly contributes to the specific activation of GnRH gene transcription in a limited population of hypothalamic neuroendocrine neurons. However, it should also be appreciated that the temporal regulation of GnRH gene expression by various neural and humoral factors is of great importance in mammalian reproduction (2, 10, 39). In this regard, it is noteworthy that SOX11 expression was recently reported to be directly regulated by depolarization in certain neurons (40). Therefore, the SOX-C and intronic enhancer-mediated regulatory mechanisms have a role in controlling GnRH gene expression, and thereby biosynthesis of the neurohormone, by integrating diverse extracellular stimuli and cell type-specific regulation. This issue needs to be addressed in future investigation.
Materials and Methods
Reagents and antibodies
Materials for cell culture were obtained from Invitrogen (Carlsbad, CA). Other chemicals, if not specified, were purchased from Sigma (St. Louis, MO). The sources for the antibodies are as follows: anti-Myc antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); goat anti-SOX4 antibody (Santa Cruz Biotechnology, Inc.); rabbit anti-SOX11 antibody (Santa Cruz Biotechnology, Inc.); anti-NeuN antibody (Millipore, Billerica, MA); antiactin antibody (Santa Cruz Biotechnology, Inc.); polyclonal (for immunohistochemistry: Abcam, Cambridge, MA; for RIA: Sigma) and monoclonal (Santa Cruz Biotechnology, Inc.) anti-GnRH antibodies; antihistone H3 antibody (Millipore); horseradish peroxidase-conjugated donkey antimouse, antirabbit, and antigoat antibodies (Jackson ImmunoResearch, West Grove, PA); Alexa Fluor 488-conjugated antigoat antibody (Invitrogen); Cy3-conjugated antimouse antibody; and Cy5-conjugated antirabbit antibody (Jackson ImmunoResearch).
Plasmids
To prepare reporter plasmids, the Ex1 and/or intron A fragment of mouse, human, and rat GnRH gene were amplified by PCR using previously described constructs as templates (12). All PCR products were cloned into the pGEM-T Easy vector (Promega, Madison, WI). Luciferase reporter plasmids were prepared by inserting each fragment into the pGL3-Promoter vector (Promega) using KpnI/BglII sites directly upstream of the SV40p or HindIII/NcoI sites downstream of the SV40p. For the series of enhancer/splicing-coupled Ex1IA-Luc constructs, PCR-amplified Ex1IA derivatives were inserted between the promoter and the luciferase coding sequence using HindIII/NcoI sites. The primer sequences for the reporter constructs are described in Supplemental Table 1. To construct mouse GnRHp-driven reporters, the 3.4-kb mouse GnRHp was excised from mouse GnRH-3446 vector (kindly provided by Helen H. Kim; University of Chicago, Chicago, Illinois) and then inserted into the pGL3-Basic vector. The various SOX expression constructs were kindly provided by the following researchers: SOX4, SOX11, and SOX21 were from Jonas Muhr (Karolinska Institute, Stockholm, Sweden); SOX5, SOX6, SOX9, and SOX10 were from Michael Wegner (Universität Erlangen, Erlangen, Germany); and SOX2 was from Shinya Yamanaka (Kyoto University, Kyoto, Japan). For the T7 promoter-based in vitro transcription/translation of SOX4 and SOX11, we subcloned Myc-tagged SOX4 and SOX11 fragments into the pcDNA3 vector (Invitrogen) using HindIII/BamHI sites. To prepare shRNA-expressing plasmids, oligonucleotides of the target sequences were annealed and cloned into the pSilencer 1.0-U6 small interfering RNA expression vector (Ambion, Austin, TX). The selected target sequences for each SOX gene are as follows: shSox4, 5′-AAC CCC AGC TCA AAC TTT GAG-3′ and shSox11, 5′-AAG AAC ATC ACC AAG CAG CAG-3′.
Cell culture and transfection
The GT1-1 cell line was kindly provided by Pamela L. Mellon (University of California, San Diego, CA). N4 and N44 cells were purchased from CELLutions Biosystems (Burlington, Ontario, Canada). N4 and N44 cell lines were established by a massive immortalization of embryonic hypothalamic cells (E15, E17, and E18) followed by clonal selection (please see Ref. 41 and manufacturer's data). After retroviral infection of SV40 large T-antigen to the primary cultured cells, mixed populations were subcloned and classified by the expression profiles of neuropeptides and receptors. Thus, their cellular origins and characteristics can be distinct from those of GT1-1 cells in spite of GnRH mRNA expression. Cells were cultured in DMEM supplemented with 4 mm glutamine, 1 mm sodium pyruvate, 100 U/ml of penicillin-streptomycin, and 10% fetal bovine serum under a humidified atmosphere containing 5% CO2 at 37 C. For the luciferase reporter assay, cells were seeded on 12-well culture plates. Each luciferase-reporter construct and/or expression vectors for the SOX genes were cotransfected with 100 ng of the thymidine kinase promoter-driven Renilla luciferase construct (pRL-TK) by using Lipofectamine PLUS reagent (Invitrogen). Thirty nanograms of reporter DNA and 500 ng of each SOX gene were used. After 36 h of transfection, luciferase activities were measured with a commercial enzyme assay kit according to the manufacturer's instructions (Promega). In the case of knockdown or ChIP assay experiments, 3 × 105 GT1-1 cells were transfected with 1.5 μg of SOX expression or shRNA-expressing plasmids using the Neon system (two pulses of 10 msec duration at 1350 V; Invitrogen). Under this condition, a greater than 80% transfection efficiency was achieved based on visual inspection of the cotransfected green fluorescent protein signals.
RNA isolation and RT-PCR
RNA expression analyses were performed as described previously with slight modifications (42). Cells were briefly washed with ice-cold PBS and stored at −70 C until use. Total RNA was isolated by the single-step acid guanidinium thiocyanate-phenol-chloroform method. One microgram of each RNA sample was reverse-transcribed with Moloney murine leukemia virus reverse transcriptase (Promega). Then, aliquots of the cDNA were subjected to conventional or quantitative real-time PCR in the presence of SYBR green I (Sigma). For quantitative real-time PCR, gene expression levels were normalized with glyceraldehyde-3-phosphate dehydrogenase. The sequences and locations (nucleotide locations for GnRH gene; for other genes, if not mentioned, the numerals were expressed based on the mRNA sequences) of primer sets are as follows: GnRH mature mRNA up, 5′-GGA AGA CAT CAG TGT CCC AGA-3′ and down, 5′-GAA GTG CTG GGG TTC TGC-3′ (+19/+2651); GnRH primary transcript up, 5′-AGC ACT GGT CCT ATG GGT TG-3′ and down, 5′-TTG CAC TAT CTG GCT CAT GC-3′ (+1217/+1354); glyceraldehyde-3-phosphate dehydrogenase up, 5′-AAC TTT GGC ATT GTG GAA GG-3′ and down, 5′-GGA TGC AGG GAT GAT GTT CT-3′ (543/674); and TATA box-binding protein up, 5′-GGG AGA ATC ATG GAC CAG AA-3′ and down, 5′-CCG TAA GGC ATC ATT GGA CT-3′ (239/351).
Western blot analysis
For immunoblotting, proteins were resolved on 8% sodium dodecyl sulfate-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore). After blocking with 10% nonfat milk in Tris-buffered saline [10 mm Tris (pH 7.6) and 150 mm NaCl] with 0.3% Tween 20 (TBS-T), the primary antibody was applied to the membrane and incubated for 1 h at room temperature: anti-Myc, 1:1000; anti-SOX4, 1:1000; anti-SOX11, and 1:1000; antiactin, 1:5000. Thereafter, the blots were washed three times with TBS-T and incubated with the horseradish peroxidase-conjugated secondary antibody for 1 h. After three washes with TBS-T, the ir bands were visualized using enhanced chemiluminescence reagent (Thermo Fisher Scientific, Rockford, IL).
Radioimmunoassay
RIA of GnRH was performed as previously described, with slight modifications (43). Briefly, GT1-1 cells (5 × 105), transfected with indicated shSOX expression plasmid, were cultured in six-well tissue culture plates. After 36 h, cells were washed twice in Dulbecco's modified PBS, and the medium was changed to 1 ml of fresh serum-free DMEM. After 2 h, the medium was collected and centrifuged for 5 min at 10,000 × g, to prevent contamination of cells, and then subjected to RIA.
Electrophoretic mobility shift assay
EMSA was carried out as previously described (44), with certain modifications. The SOX4 and SOX11 proteins were produced by a coupled in vitro transcription/translation reaction with rabbit reticulocyte lysates according to the manufacturer's instructions (TNT Quick Coupled Transcription/Translation System; Promega). DNA-protein binding reaction was performed in a 20 μl reaction mixture containing 20 mm HEPES (pH 7.9), 40 mm KCl, 0.5 mm EDTA, 0.5 mm DTT, 1 mm PMSF, 10% glycerol, 2 μg poly(dI-dC), a 32P-labeled DNA probe, and the indicated SOX protein for 30 min at 30 C. In competition experiments, a 200-fold molar excess of a cold competitor was added. After incubation, 1 μl of loading dye (15% Ficoll and 0.4% Orange G) was added to each reaction mixture, which was then immediately separated on 5% nondenaturing polyacrylamide gels. Thereafter, the gel was dried and exposed to x-ray film (Fujifilm, Greenwood, SC) at −70 C for 1 d.
ChIP assay
ChIP assays were performed using a commercial ChIP assay kit (Millipore). Sheared and precleared chromatin from GT1-1 cells or mouse hypothalamus was immunoprecipitated for 2 h at 4 C by agitating with 2 μg of the anti-SOX4, anti-SOX11, or antihistone H3 antibody. As a negative control, the supernatant was incubated with 5 μl of preimmune normal rabbit serum. Immune complexes were collected by incubation with Protein-G Sepharose beads. The sequences and binding sites of primer sets used are as follows: IA up, 5′-GTA CAG TTC TTT GTT GTT CTA GC-3′ and down, 5′-CAC AAC GTG TGT GAT GAC TTG A-3′ (+143/+244); GnRHp up, 5′-GAG GCA GCA TCT GCT AAA GG-3′ and down, 5′-GCC TGC CTC TGA AAC TTT TG-3′ (-1220/-1016); and GnRH 3′-UTR up, 5′-AAG CCA GGC AGA AGA AGA TG-3′ and down, 5′-TGG GAC TTG ATC CAC AAC AA-3′ (+4076/+4253).
Immunohistochemistry
Male mice (CD-1 strain, 10 weeks of age) were perfused with 4% paraformaldehyde in PBS, and the brains were postfixed in the same solution for 24 h. The brains were then cryoprotected in 30% sucrose and sectioned (20 μm); subsequent immunostaining was performed by a free-floating method. Brain sections containing POA were washed with PBS and blocked with 3% BSA and 0.1% Triton X-100 in PBS for 30 min. The following primary antibodies were applied overnight at 4 C: anti-GnRH, 1:500; anti-SOX4, 1:50; anti-SOX11, 1:50; and anti-NeuN, 1:1000. After three washes with PBS, the appropriate secondary antibodies conjugated with fluorescent dyes were applied for 30 min. Subsequently, the sections were washed, mounted, and observed under confocal microscopy (Nikon, Melville, NY).
Statistical analysis
All data were statistically analyzed with Student's t test and one- or two-way ANOVA using PRISM version 4.00 software (GraphPad Software, La Jolla, CA). Newman-Keuls multiple comparison was used for post hoc comparison for ANOVA. Statistical significance was set at P < 0.05.
Acknowledgments
We thank Dr. Jonas Muhr (Karolinska Institute, Stockholm, Sweden) for providing expression vectors of SOX4, SOX11, and Sox21; Dr. Michael Wegner (Universität Erlangen, Erlangen, Germany) for the expression vectors of SOX5, SOX6, SOX9, and SOX10; Dr. Shinya Yamanaka (Kyoto University, Kyoto, Japan) for the SOX2 expression vector (Addgene plasmid no. 13459); and Dr. Helen H. Kim for providing the mouse GnRHp.
This work was supported by grants from the Korea Ministry of Education, Science, and Technology (MEST) through the Brain Research Center for the 21st Century Frontier R&D Program in Neuroscience. H.-D.K., H.K.C., S.C., and M.K. were supported by a Brain Korea 21 Research Fellowships from MEST.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP
- Chromatin immunoprecipitation
- E
- embryonic day
- Ex1
- exon 1
- GnRHp
- GnRH promoter
- ir
- immunoreactive
- NeuN
- neuronal nuclear antigen
- nt
- nucleotide
- P
- postnatal day
- POA
- preoptic area
- POA-AH
- POA-anterior hypothalamus
- POU
- Pit-Oct-Unc
- shRNA
- short hairpin RNA
- SOX
- sex-determining region Y-related high-mobility-group box
- SOX-C
- class-C SOX
- SV40
- simian vacuolating virus 40
- SV40p
- SV40 minimal promoter
- TBS-T
- Tris-buffered saline with 0.3% Tween 20
- UTR
- untranslated region.
References
- 1. Son GH , Park E , Jung H , Han J , Lee KH , Seong JY , Kim K. 2005. GnRH pre-mRNA splicing: solving the mystery of a nature's knockout, hpg mouse. Biochem Biophys Res Commun 326:261–267 [DOI] [PubMed] [Google Scholar]
- 2. Herbison AE. 2006. Physiology of the gonadotropin-releasing hormone neuronal network. In: , Neill JD ed. Knobil and Neill's physiology of reproduction. 3rd ed. San Diego: Academic Press; 1415–1482 [Google Scholar]
- 3. Mellon PL , Windle JJ , Goldsmith PC , Padula CA , Roberts JL , Weiner RI. 1990. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5:1–10 [DOI] [PubMed] [Google Scholar]
- 4. Radovick S , Wray S , Lee E , Nicols DK , Nakayama Y , Weintraub BD , Westphal H , Cutler GB , Wondisford FE. 1991. Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proc Natl Acad Sci USA 88:3402–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clark ME , Mellon PL. 1995. The POU homeodomain transcription factor Oct-1 is essential for activity of the gonadotropin-releasing hormone neuron-specific enhancer. Mol Cell Biol 15:6169–6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lawson MA , Whyte DB , Mellon PL. 1996. GATA factors are essential for activity of the neuron-specific enhancer of the gonadotropin-releasing hormone gene. Mol Cell Biol 16:3596–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eraly SA , Nelson SB , Huang KM , Mellon PL. 1998. Oct-1 binds promoter elements required for transcription of the GnRH gene. Mol Endocrinol 12:469–481 [DOI] [PubMed] [Google Scholar]
- 8. Kelley CG , Lavorgna G , Clark ME , Boncinelli E , Mellon PL. 2000. The Otx2 homeoprotein regulates expression from the gonadotropin-releasing hormone proximal promoter. Mol Endocrinol 14:1246–1256 [DOI] [PubMed] [Google Scholar]
- 9. Nelson SB , Lawson MA , Kelley CG , Mellon PL. 2000. Neuron-specific expression of the rat gonadotropin-releasing hormone gene is conferred by interactions of a defined promoter element with the enhancer in GT1–7 cells. Mol Endocrinol 14:1509–1522 [DOI] [PubMed] [Google Scholar]
- 10. Lee VH , Lee LT , Chow BK. 2008. Gonadotropin-releasing hormone: regulation of the GnRH gene. FEBS J 275:5458–5478 [DOI] [PubMed] [Google Scholar]
- 11. Seong JY , Han J , Park S , Wuttke W , Jarry H , Kim K. 2002. Exonic splicing enhancer-dependent splicing of the gonadotropin-releasing hormone premessenger ribonucleic acid is mediated by tra2α, a 40-kilodalton serine/arginine-rich protein. Mol Endocrinol 16:2426–2438 [DOI] [PubMed] [Google Scholar]
- 12. Son GH , Jung H , Seong JY , Choe Y , Geum D , Kim K. 2003. Excision of the first intron from the gonadotropin-releasing hormone (GnRH) transcript serves as a key regulatory step for GnRH biosynthesis. J Biol Chem 278:18037–18044 [DOI] [PubMed] [Google Scholar]
- 13. Park E , Han J , Son GH , Lee MS , Chung S , Park SH , Park K , Lee KH , Choi S , Seong JY , Kim K. 2006. Cooperative actions of Tra2α with 9G8 and SRp30c in the RNA splicing of the gonadotropin-releasing hormone gene transcript. J Biol Chem 281:401–409 [DOI] [PubMed] [Google Scholar]
- 14. Park E , Lee MS , Baik SM , Cho EB , Son GH , Seong JY , Lee KH , Kim K. 2009. Nova-1 mediates glucocorticoid-induced inhibition of pre-mRNA splicing of gonadotropin-releasing hormone transcripts. J Biol Chem 284:12792–12800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rowntree RK , Vassaux G , McDowell TL , Howe S , McGuigan A , Phylactides M , Huxley C , Harris A. 2001. An element in intron 1 of the CFTR gene augments intestinal expression in vivo. Hum Mol Genet 10:1455–1464 [DOI] [PubMed] [Google Scholar]
- 16. Flodby P , Zhou B , Ann DK , Kim KJ , Minoo P , Crandall ED , Borok Z. 2007. Conserved elements within first intron of aquaporin-5 (Aqp5) function as transcriptional enhancers. Biochem Biophys Res Commun 356:26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bianchi M , Crinelli R , Giacomini E , Carloni E , Magnani M. 2009. A potent enhancer element in the 5′-UTR intron is crucial for transcriptional regulation of the human ubiquitin C gene. Gene 448:88–101 [DOI] [PubMed] [Google Scholar]
- 18. Bowles J , Schepers G , Koopman P. 2000. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol 227:239–255 [DOI] [PubMed] [Google Scholar]
- 19. Banerji J , Olson L , Schaffner W. 1983. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33:729–740 [DOI] [PubMed] [Google Scholar]
- 20. Scharer CD , McCabe CD , Ali-Seyed M , Berger MF , Bulyk ML , Moreno CS. 2009. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res 69:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim HH , Wolfe A , Smith GR , Tobet SA , Radovick S. 2002. Promoter sequences targeting tissue-specific gene expression of hypothalamic and ovarian gonadotropin-releasing hormone in vivo. J Biol Chem 277:5194–5202 [DOI] [PubMed] [Google Scholar]
- 22. Lawson MA , Macconell LA , Kim J , Powl BT , Nelson SB , Mellon PL. 2002. Neuron-specific expression in vivo by defined transcription regulatory elements of the GnRH gene. Endocrinology 143:1404–1412 [DOI] [PubMed] [Google Scholar]
- 23. Dy P , Penzo-Méndez A , Wang H , Pedraza CE , Macklin WB , Lefebvre V. 2008. The three SoxC proteins-Sox4, Sox11 and Sox12-exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res 36:3101–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schilham MW , Oosterwegel MA , Moerer P , Ya J , de Boer PA , van de Wetering M , Verbeek S , Lamers WH , Kruisbeek AM , Cumano A , Clevers H. 1996. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature 380:711–714 [DOI] [PubMed] [Google Scholar]
- 25. Sock E , Rettig SD , Enderich J , Bösl MR , Tamm ER , Wegner M. 2004. Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol Cell Biol 24:6635–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoser M , Potzner MR , Koch JM , Bösl MR , Wegner M , Sock E. 2008. Sox12 deletion in the mouse reveals nonreciprocal redundancy with the related Sox4 and Sox11 transcription factors. Mol Cell Biol 28:4675–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bergsland M , Werme M , Malewicz M , Perlmann T , Muhr J. 2006. The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev 20:3475–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Potzner MR , Tsarovina K , Binder E , Penzo-Méndez A , Lefebvre V , Rohrer H , Wegner M , Sock E. 2010. Sequential requirement of Sox4 and Sox11 during development of the sympathetic nervous system. Development 137:775–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohe K , Lalli E , Sassone-Corsi P. 2002. A direct role of SRY and SOX proteins in pre-mRNA splicing. Proc Natl Acad Sci USA 99:1146–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohe K , Tamai KT , Parvinen M , Sassone-Corsi P. 2009. DAX-1 and SOX6 molecular interplay results in an antagonistic effect in pre-mRNA splicing. Dev Dyn 238:1595–1604 [DOI] [PubMed] [Google Scholar]
- 31. Lioubinski O , Müller M , Wegner M , Sander M. 2003. Expression of Sox transcription factors in the developing mouse pancreas. Dev Dyn 227:402–408 [DOI] [PubMed] [Google Scholar]
- 32. Wilson ME , Yang KY , Kalousova A , Lau J , Kosaka Y , Lynn FC , Wang J , Mrejen C , Episkopou V , Clevers HC , German MS. 2005. The HMG box transcription factor Sox4 contributes to the development of the endocrine pancreas. Diabetes 54:3402–3409 [DOI] [PubMed] [Google Scholar]
- 33. Goldsworthy M , Hugill A , Freeman H , Horner E , Shimomura K , Bogani D , Pieles G , Mijat V , Arkell R , Bhattacharya S , Ashcroft FM , Cox RD. 2008. Role of the transcription factor Sox4 in insulin secretion and impaired glucose tolerance. Diabetes 57:2234–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kamachi Y , Uchikawa M , Kondoh H. 2000. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 16:182–187 [DOI] [PubMed] [Google Scholar]
- 35. Wilson M , Koopman P. 2002. Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12:441–446 [DOI] [PubMed] [Google Scholar]
- 36. Bailey PJ , Klos JM , Andersson E , Karlén M , Källström M , Ponjavic J , Muhr J , Lenhard B , Sandelin A , Ericson J. 2006. A global genomic transcriptional code associated with CNS-expressed genes. Exp Cell Res 312:3108–3119 [DOI] [PubMed] [Google Scholar]
- 37. Kuhlbrodt K , Herbarth B , Sock E , Enderich J , Hermans-Borgmeyer I , Wegner M. 1998. Cooperative function of POU proteins and SOX proteins in glial cells. J Biol Chem 273:16050–16057 [DOI] [PubMed] [Google Scholar]
- 38. Tanaka S , Kamachi Y , Tanouchi A , Hamada H , Jing N , Kondoh H. 2004. Interplay of SOX and POU factors in regulation of the Nestin gene in neural primordial cells. Mol Cell Biol 24:8834–8846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gore AC , Roberts JL. 1997. Regulation of gonadotropin-releasing hormone gene expression in vivo and in vitro. Front Neuroendocrinol 18:209–245 [DOI] [PubMed] [Google Scholar]
- 40. Sun W , Park KW , Choe J , Rhyu IJ , Kim IH , Park SK , Choi B , Choi SH , Park SH , Kim H. 2005. Identification of novel electroconvulsive shock-induced and activity-dependent genes in the rat brain. Biochem Biophys Res Commun 327:848–856 [DOI] [PubMed] [Google Scholar]
- 41. Belsham DD , Cai F , Cui H , Smukler SR , Salapatek AM , Shkreta L. 2004. Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology 145:393–400 [DOI] [PubMed] [Google Scholar]
- 42. Son GH , Chung S , Choe HK , Kim HD , Baik SM , Lee H , Lee HW , Choi S , Sun W , Kim H , Cho S , Lee KH , Kim K. 2008. Adrenal peripheral clock controls the autonomous circadian rhythm of glucocorticoid by causing rhythmic steroid production. Proc Natl Acad Sci USA 105:20970–20975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cho S , Cho H , Geum D , Kim K. 1998. Retinoic acid regulates gonadotropin-releasing hormone (GnRH) release and gene expression in the rat hypothalamic fragments and GT1-1 neuronal cells in vitro. Mol Brain Res 54:74–84 [DOI] [PubMed] [Google Scholar]
- 44. Son GH , Geum D , Chung S , Park E , Lee KH , Choi S , Kim K. 2005. A protective role of 27-kDa heat shock protein in glucocorticoid-evoked apoptotic cell death of hippocampal progenitor cells. Biochem Biophys Res Commun 338:1751–1758 [DOI] [PubMed] [Google Scholar]
- 45. Mason AJ , Hayflick JS , Zoeller RT , Young WS , Phillips HS , Nikolics K , Seeburg PH. 1986. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science 234:1366–1371 [DOI] [PubMed] [Google Scholar]
- 46. Han J , Seong JY , Kim K , Wuttke W , Jarry H. 2001. Analysis of exonic splicing enhancers in the mouse gonadotropin-releasing hormone (GnRH) gene. Mol Cell Endocrinol 173:157–166 [DOI] [PubMed] [Google Scholar]