

MIG12 influences stimulation of lipogenesis by LXR activation in the liver.
Abstract
Liver X receptor (LXR)α and LXRβ belong to the nuclear receptor superfamily and play central roles in the transcriptional control of lipid metabolism. We describe a novel LXR target, midline-1-interacting G12-like protein (MIG12), which has been recently identified as an acetyl-coenzyme A carboxylase-binding protein. The binding causes the induction of de novo fatty acid (FA) synthesis through the activation of acetyl-coenzyme A carboxylase (a rate-limiting enzyme for de novo FA synthesis). Luciferase reporter gene assays using the MIG12 gene promoter revealed the existence of a LXR-responsive element (LXRE) and carbohydrate-responsive element-binding protein (ChREBP)-responsive element named LXRE3 and carbohydrate response element 1, respectively. Deletion and mutation of LXRE3 and carbohydrate response element 1 abolished LXR and ChREBP responsiveness, respectively. Electrophoretic mobility shift assays demonstrated that the LXRα/retinoid X receptor α complex was bound to LXRE3. Treatment with high glucose concentration, which leads ChREBP activation, or LXR activator stimulated MIG12 expression in rat primary hepatocytes, and combined treatment further stimulated MIG12 expression. Furthermore, hepatic expression of MIG12 in mice was induced by refeeding. Overexpression of MIG12 stimulated and knockdown of MIG12 attenuated LXR ligand-stimulated de novo FA synthesis and triacylglycerol accumulation. These results indicate that MIG12 is a mediator for stimulation of lipogenesis by LXR activation in the liver.
Liver X receptor (LXR)α and LXRβ belong to the nuclear receptor superfamily and form heterodimers with the retinoid X receptors (RXR) to function as transcriptional regulators (1). At physiological concentrations, oxysterols (metabolites of cholesterol) activate LXR through direct binding to their ligand-binding domain (2, 3). Activated LXR bind to LXR-responsive elements (LXRE) in DNA, which comprise a direct repeat of the core sequence AGGTCA separated by four nucleotides, collectively referred to as direct repeat 4 (DR4), and regulate expression of genes involved in cholesterol, fatty acid (FA), and glucose metabolism. In addition to their role in lipid metabolism, LXR mediate immune and inflammatory responses (4). Administration of LXR activators to atherosclerosis-prone mice, low density lipoprotein receptor-deficient mice, and apolipoprotein E-deficient mice inhibits the development of atherosclerosis (5). Furthermore, LXR activation improves glucose tolerance through coordinate regulation of glucose metabolism in the liver and adipose tissue (6). Therefore, LXR are considered to be potential drug targets for cardiovascular, metabolic, and inflammatory diseases. However, LXR activation also stimulates lipogenesis in the liver, thereby causing fatty liver and hypertriglyceridemia (7). These unfavorable effects prevent the clinical use of LXR activators.
Midline-1 (MID1)-interacting G12-like protein (MIG12) was first identified as an MID1-binding protein by the use of a yeast two-hybrid system, and it was reported to cooperate with MID1 to stabilize microtubules in the central nervous system during development (8). Subsequently, it was renamed Spot 14 (S14) related, and the rat counterpart of MIG12 was named S14-like androgen-inducible protein because of its high homology to S14 at the amino acid level (9, 10). Because S14 is expressed only in lipogenic tissues, such as the liver, fat, and lactating mammary glands, it is considered to regulate lipogenesis (11). Furthermore, S14 expression is induced by lipogenesis-inducing factors, such as thyroid hormone and carbohydrate feeding, and suppressed by lipogenesis-suppressing factors, such as glucagon and catecholamine (12). Although S14 knockout mice exhibit decreased lipogenesis in lactating mammary glands, hepatic lipogenesis is not inhibited (13). Thus, MIG12, which was expressed in the liver but not in the mammary gland, was speculated to compensate for the function of S14 in the liver of S14 knockout mice (9). Aipoalani et al. (14) reported that both S14 and MIG12 are involved in hepatic lipogenesis and that the two proteins play overlapping roles in this process. However, the molecular mechanism of the MIG12-mediated increase in lipogenesis has not yet been elucidated. Recently, Kim et al. (15) showed that MIG12 is involved in hepatic lipogenesis through the activation of acetyl-coenzyme A carboxylase (ACC). MIG12 induced ACC polymerization, which in turn increased its enzymatic activity. These emerging data indicate that MIG12 is a novel regulator of de novo FA synthesis. At present, the transcriptional regulation of MIG12 gene expression remains unknown.
Lipogenesis is tightly regulated at the transcriptional level. The transcription factors LXR, sterol regulatory element-binding protein (SREBP)-1c, and carbohydrate-responsive element-binding protein (ChREBP) contribute considerably to this regulation. Because LXR stimulates SREBP-1c and ChREBP expression (16, 17), as well as FA synthase (FAS) and ACC, we considered that the detection of novel LXR target genes would lead to further understanding of the regulation of lipogenesis. In the present study, we demonstrate that LXR and ChREBP synergistically stimulated the MIG12 gene expression. Furthermore, we demonstrate that LXR-mediated induction of lipogenesis is suppressed by the knockdown of MIG12 expression even if stimulation of lipogenic gene expression, including genes encoding FAS and ACC, by LXR activator is not suppressed. These findings suggest that increased MIG12 expression is one of the factors responsible for LXR activator-mediated induction of lipogenesis.
Results
MIG12 is a novel LXR target gene
To identify new LXR target genes, we previously performed a DNA microarray analysis with MGu74v2 chips (Affymetrix, Santa Clara, CA) containing approximately 36,000 single-stranded cDNA (18). Mice were gavaged with vehicle or 50 mg/kg T0901317, a synthetic LXR agonist. After 24 h of administration, RNA was extracted from the liver of mice and used as a template to synthesize cDNA to be analyzed by DNA microarray chips. To identify novel LXR target genes, we determined that T0901317 treatment stimulated MIG12 gene expression (data not shown). To confirm this effect, we performed real-time RT-PCR. MIG12 gene expression increased approximately 4-fold after T0901317 treatment in mouse livers (Fig. 1A). Because T0901317 is a potent ligand for LXR, pregnane X receptor (PXR), and farnesoid X receptor (FXR) (19, 20), we next examined the direct involvement of FXR in the increase in MIG12 mRNA level after T0901317 treatment using FXR-null mice. Consistent with the results observed in wild-type mice (Fig. 1A), MIG12 gene expression increased in the livers of FXR-null mice (Supplemental Fig. 1A, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org), indicating that induction of gene expression by T0901317 treatment is independent of FXR activity. To confirm that ligand-activated PXR increased MIG12 mRNA level, we investigated the effect of the PXR ligand pregnenalone-16α-carbonitrile (PCN) on MIG12 expression. Although the expression of cytochrome P450 3A11, a PXR target gene, was induced by PCN administration, MIG12 gene expression was not enhanced in the livers (Supplemental Fig. 1B). These data indicate that ligand-activated PXR does not stimulate MIG12 expression in the liver. Therefore, we concluded that neither FXR nor PXR is involved in the induction of MIG12 gene expression by treatment with T0901317 in the mouse liver. To investigate the molecular mechanism of T0901317-mediated induction of the MIG12 gene, reporter assays using the promoter of the mouse MIG12 gene were performed. Because the transcription initiation site of the MIG12 gene was not known, we first analyzed the initiation site by the 5′-rapid amplification of cDNA ends method and designated it as +1 (Supplemental Fig. 2). A 2.5-kb genomic fragment of the mouse MIG12 promoter (from −2512 to +42) was used in the reporter assay. As shown in Fig. 1B, LXRα/RXRα stimulated MIG12 promoter activity, and the induction was potentiated by the addition of T0901317. The truncated MIG12 promoter pMIG12-289b-Luc retained LXR responsiveness (Fig. 1C). Inspection of the MIG12 promoter region between nucleotides −289 and −1 revealed that this region contains four putative LXR-binding elements, designated LXRE1–LXRE4. Deletion of LXRE4 resulted in inhibition of LXR responsiveness, whereas deletion of both LXRE3 and LXRE4 completely eliminated LXR responsiveness (pMIG12-198b-Luc and pMIG12-111b-Luc) (Fig. 1C). These results indicate that LXR responsiveness requires sequences between nucleotides −289 and −112 (Fig. 1C), a region that contains LXRE3 and LXRE4. The binding affinity of LXRE3 and LXRE4 was evaluated by competitive electrophoretic mobility shift assays, analyzing its ability to compete with the binding of a labeled DR4 probe to LXRα/RXRα. The DR4 sequence (Fig. 1D) was radiolabeled with [γ-32P]ATP and incubated with nuclear extracts from HEK293 cells and the indicated double-stranded unlabeled oligonucleotides (Fig. 1D) as competitors at a ratio of 50:1. Although a specific complex of retarded mobility was not observed when using nuclear extracts from mock-transfected HEK293 cells, such a complex was observed when using nuclear extracts from HEK293 cells transfected with both LXRα and RXRα expression plasmids (Fig. 1E, lanes 1 and 2), indicating that LXRα/RXRα bound to DR4 in this experiment. Unlabeled LXRE3 and DR4 oligonucleotides blocked the binding of LXRα/RXRα to DR4, whereas unlabeled LXRE4 oligonucleotides failed to block binding (Fig. 1E, lanes 3–5). Furthermore, a labeled LXRE3 probe formed a complex with LXRα/RXRα, and this complex disappeared on addition of unlabeled LXRE3 and DR4 oligonucleotides (Fig. 1E, lanes 6–9). These results indicate that LXRα/RXRα can bind to LXRE3 of the MIG12 gene promoter but not LXRE4. Furthermore, to examine whether LXRE3 is responsible for the induction of MIG12 promoter activity by LXRα/RXRα, we performed a reporter assay using a reporter construct containing the LXRE3-deleted MIG12 promoter (pMIG12-2.5kb-ΔLXRE3-Luc). As shown in Fig. 1C, deletion of LXRE3 abolished the induction of MIG12 promoter activity by LXRα/RXRα, whereas deletion of LXRE4 did not influence LXR responsiveness, indicating the involvement of LXRE3 in the LXR-mediated induction of the MIG12 gene. To determine whether LXRα binds to the MIG12 promoter region, we performed a chromatin immunoprecipitation (ChIP) assay. As shown in Fig. 1F, LXRα binds to the MIG12 gene promoter region containing LXRE3 as well as the promoter region of the SREBP-1c gene, which is known to be a LXR target gene. In contrast, LXRα did not bind to the distal region of the MIG12 gene promoter (Fig. 1F). Collectively, these results indicate that MIG12 is a novel direct target gene of LXR.
Fig. 1.
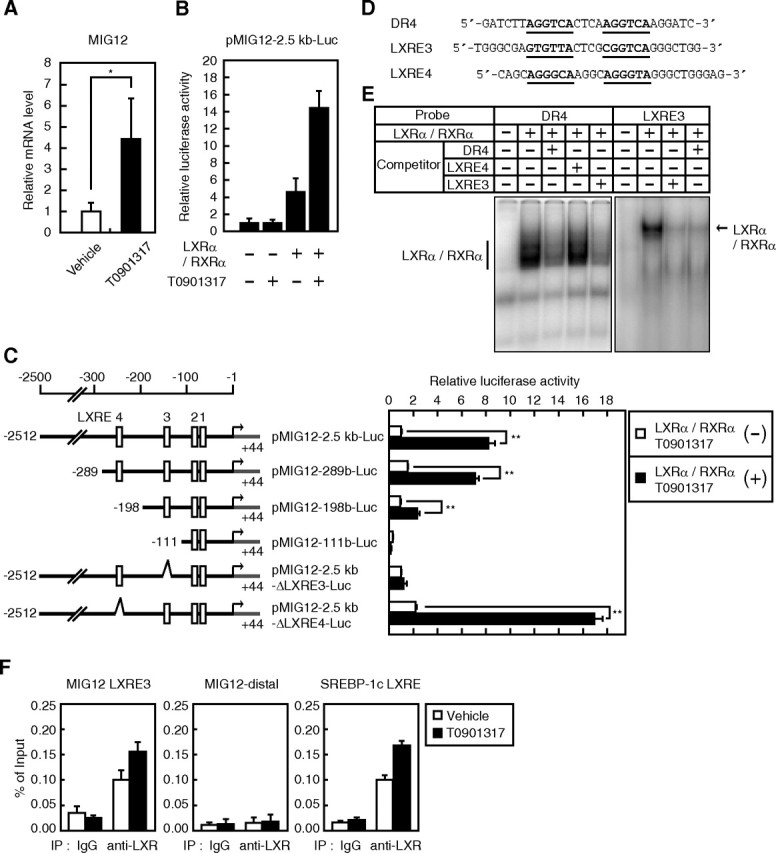
Regulation of MIG12 expression by LXR in the liver and identification of an LXRE in the MIG12 promoter. A, Mice (n = 4) were intragastrically administered the vehicle or T0901317 (50 mg/kg) and killed 24 h later. MIG12 mRNA was quantitated by real-time PCR of liver samples using 36B4 mRNA as the internal standard for normalization. B and C, HEK293 cells were transfected with 200 ng of the indicated reporter constructs together with 50 ng of pCMV-β-gal in the presence or absence of 20 ng of expression plasmids for LXRα and RXRα. Twenty-four hours after transfection, cells were placed in medium A containing the vehicle (DMSO) or 10 μm T0901317. After incubation for another 24 h, luciferase assays were performed as described in Materials and Methods. Promoter activity of pMIG12-2.5kb-Luc in the absence of LXRα and RXRα and in the presence of the vehicle is represented as 1. D, Sequences of consensus DR4, mouse MIG12 LXRE3, and LXRE4. For comparison, the MIG12 LXRE4 sequence of the bottom strand is shown. E, Competitive gel mobility-shift assays were performed as described in Materials and Methods using nuclear extracts from HEK293 cells transfected with or without LXRα and /RXRα expression plasmids, 32P-labeled DR4 (left panel) and MIG12 LXRE3 (right panel) as input probe and unlabeled oligonucleotides as competitors at 50-fold molar excess. F, Pooled livers from four mice treated with either vehicle or 50 mg/kg T0901317 were analyzed by ChIP assays using the antibody for LXRα and an isotype-matched IgG control antibody. Bound DNA was quantitated by real-time PCR and normalized to the input. All results are expressed as mean ± sd of three independent experiments. *, P < 0.05; **, P < 0.01; IP, immunoprecipitation.
LXR and ChREBP synergistically stimulate MIG12 promoter activity
Because a number of LXR target genes are known to be regulated synergistically with ChREBP and SREBP, we hypothesized that MIG12 gene expression would be regulated by these proteins. To examine this possibility, we performed reporter assays using a pMIG12-2.5kb-Luc reporter construct. Although promoter activity was not affected by the expression of the active form of SREBP-1a, ChREBP/Max-like protein X (Mlx) significantly stimulated MIG12 promoter activity (Fig. 2A) (data not shown). Serial 5′ truncations of the MIG12 gene promoter revealed that ChREBP responsiveness requires sequences between nucleotides −650 and −534 (Fig. 2A). Because this region contains a putative ChREBP-responsive sequence between −588 and −572 (CACGTGcatccGGCATG), designated as carbohydrate response element 1 (ChoRE1), we examined the effect of this sequence on ChREBP responsiveness. Introduction of the ChoRE1 mutation into the MIG12 promoter (CCCGTGcatccGGCAGG) abolished ChREBP-mediated induction of promoter activity (Fig. 2A). To determine whether ChREBP binds to the MIG12 gene promoter region, we performed the ChIP assay. As shown in Fig. 2B, ChREBP binds to the promoter region of the MIG12 gene containing ChoRE1 as well as the promoter region of liver-type pyruvate kinase (L-PK) gene, which is known to be the ChREBP target gene. In contrast, ChREBP did not bind to the distal region of the MIG12 gene promoter (Fig. 2B). These results indicate that MIG12 is a direct target gene of ChREBP. We next examined whether MIG12 promoter activity is synergistically stimulated by LXR and ChREBP. Although MIG12 promoter activity was stimulated approximately 8- and 10-fold by the individual action of LXR and ChREBP, their combination resulted in greater stimulation (approximately 60-fold), indicating that LXR and ChREBP synergistically stimulate MIG12 promoter activity (Fig. 3, top). When the reporter construct containing the LXRE3-deleted MIG12 promoter (pMIG12-2.5kb-ΔLXRE3-Luc) was used, ChREBP still stimulated promoter activity, and this induction was not influenced by LXR (Fig. 3, middle). Furthermore, LXR stimulated ChoRE1-mutated MIG12 promoter activity, and this induction was not influenced by ChREBP (Fig. 3, bottom). These results indicate that LXR and ChREBP stimulate MIG12 promoter activity, independently, only through their DNA-binding elements.
Fig. 2.

ChREBP activates the MIG12 promoter. A, HEK293 cells were transfected with 200 ng of the indicated reporter constructs together with 50 ng of pCMV-β-gal in the presence or absence of 20 ng of expression plasmids for ChREBP and Mlx. Forty-eight hours after transfection, luciferase assays were performed as described in Materials and Methods. Promoter activity of pMIG12-2.5kb-Luc in the absence of ChREBP and Mlx is represented as 1. B, Pooled livers from four mice were analyzed by ChIP assays using an antibody for ChREBP and an isotype-matched IgG control antibody. Bound DNA was quantitated by real-time PCR and normalized to the input. All results are expressed as mean ± sd of three independent experiments. *, P < 0.05; **, P < 0.01; KO, knockout; IP, immunoprecipitation.
Fig. 3.

LXR and ChREBP synergistically regulate the MIG12 promoter. HEK293 cells were transfected with 200 ng of the indicated reporter constructs together with 50 ng of pCMV-β-gal in the presence or absence of 20 ng of the indicated expression plasmids for LXRα, RXRα, ChREBP, and Mlx. Twenty-four hours after transfection, cells were placed in medium A containing the vehicle (DMSO) or 10 μm T0901317. After incubation for another 24 h, luciferase assays were performed as described in Materials and Methods. Promoter activity of pMIG12-2.5kb-Luc in the absence of LXRα, RXRα, ChREBP, and Mlx with vehicle treatment is represented as 1. Values are averages from three experiments. All results are expressed as mean ± sd. KO, Knockout.
MIG12 gene expression is synergistically stimulated by treatment with the LXR ligand and glucose
To determine whether endogenous MIG12 gene expression is also synergistically stimulated by LXR and ChREBP, we measured MIG12 mRNA levels in rat primary hepatocytes cultured with a low (5 mm) or high (25 mm) glucose concentration in the presence or absence of T0901317 for 24 h by real-time PCR. We used a high glucose concentration, because a high level of glucose is known to activate ChREBP (21). The expression of the ATP-binding cassette (ABC) A1 and L-PK genes was stimulated by treatment with T0901317 and high glucose, respectively (Fig. 4), suggesting that LXR and ChREBP are activated under each condition. MIG12 gene expression increased approximately 2-fold by treatment with T0901317 or high glucose. It was further stimulated by combined treatment, indicating synergistic stimulation of MIG12 gene expression by LXR and ChREBP.
Fig. 4.

Glucose and LXR ligand synergistically regulate MIG12 gene expression in rat primary hepatocytes. Hepatocytes were cultured for 24 h in medium C (medium 199 supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 100 nm insulin, and 100 nm dexamethasone) containing 5 mm glucose. Cells were then placed in medium C containing 5 mm glucose (L) or 25 mm glucose (H) in the presence or absence of 10 μm T0901317. After incubation for another 24 h, total RNA was extracted, and real-time PCR analysis was performed. Relative mRNA levels were obtained after normalizing to 36B4 mRNA levels. mRNA levels (MIG12, ABCA1, and L-PK) in the presence of 5 mm glucose without T0901317 are represented as 1. Values are averages from three experiments. All results are expressed as mean ± sd. Different letters above the bars indicate a significant difference between groups.
Regulation of MIG12 gene expression in livers of fasted and refed mice
Because LXR and ChREBP activities are known to be enhanced under refed conditions, we examined MIG12 gene expression under fasting and refed conditions. As expected, L-PK and phosphoenolpyruvate carboxykinase (PEPCK) gene expression increased and decreased under refed conditions and fasted conditions, respectively (Fig. 5). MIG12 gene expression in the livers of refed mice was approximately 8-fold higher than that in livers of fasted mice, indicating that MIG12 mRNA level in livers of mice is influenced by the state of feeding.
Fig. 5.

MIG12 expression in livers of fasted and refed mice. Mice in the fasted group (F) (n = 5) were fasted for 24 h, and those in the refed group (R) (n = 5) were fasted for 24 h and then refed the normal diet for 24 h. Total RNA was extracted, and real-time PCR analysis was performed. Relative mRNA levels were obtained after normalizing to 36B4 mRNA levels. The mRNA levels (MIG12, PEPCK, and L-PK) under the fasted condition are represented as 1. All results are expressed as mean ± sd. **, P < 0.01.
Overexpression of MIG12 increases and knockdown of MIG12 expression inhibits T0901317-stimulated de novo FA synthesis and intracellular triacylglycerol (TG) accumulation
Based on the finding that MIG12 stimulates de novo FA synthesis through ACC activation (15), we examined whether the induction of MIG12 expression is involved in LXR activator-mediated induction of lipogenesis. De novo FA synthesis and TG accumulation increased on treatment with T0901317, and adenovirus-meditated overexpression of MIG12 significantly potentiated these T0901317-mediated effects in rat primary hepatocytes (Fig. 6, A and B). Next, we examined whether knockdown of MIG12 expression affected de novo FA synthesis and TG accumulation. When rat primary hepatocytes were transfected with small interfering RNA (siRNA), which is designed to specifically target MIG12 mRNA, the MIG12 mRNA level decreased by 50% in the absence and by 20% in the presence of T0901317 (Fig. 6C). De novo FA synthesis and TG accumulation were significantly inhibited by the knockdown of MIG12 expression only in the presence of T0901317 (Fig. 6, D and E). These data clearly indicate that LXR ligand-induced lipogenesis is mediated, at least in part, by MIG12.
Fig. 6.

Effect of MIG12 on de novo FA synthesis and TG accumulation in rat primary hepatocytes. A and B, Hepatocytes were infected with 20 MOI of adenoviruses for the expression of β-gal (Ad-Con) or mMIG12 (Ad-MIG12) as described in Materials and Methods. At 24 (A and B) and 48 h (B) after infection, cells were placed in fresh medium B containing the vehicle (DMSO) or 10 μm T0901317. Forty-eight hours after infection, cells were treated with [14C]-acetate, and incorporation of 14C into newly synthesized FAs was determined as described in Materials and Methods (A). Seventy-two hours after infection, intracellular TG levels were determined as described in Materials and Methods (B). C–E, Hepatocytes were transfected with 150 pmol of control siRNA (siCon) or siRNA targeting MIG12 (siMIG12) as described in Materials and Methods. Twenty-four hours after transfection, cells were placed in fresh medium B containing vehicle (DMSO) or 10 μm T0901317. Seventy-two hours after transfection, total RNA was extracted, and real-time PCR analysis was performed. Relative mRNA levels were obtained after normalizing to cyclophilin mRNA levels. mRNA levels of MIG12 transfected with siCon in the absence of T0901317 are represented as 1 (C). Forty-eight hours after transfection, cells were treated with [14C]-acetate, and 14C incorporation into newly synthesized FAs was determined as described in Materials and Methods (D). Seventy-two hours after transfection, intracellular TG levels were determined as described in Materials and Methods (E). All results are expressed as mean ± sd; experiments were performed in triplicate. Similar results were obtained in three independent experiments. *, P < 0.05; **, P < 0.01.
Alteration of FA synthesis and TG accumulation by MIG12 is not mediated through the transcriptional regulation of genes involved in de novo lipogenesis
To gain insight into the mechanism by which MIG12 regulates lipogenesis, other than enzymatic activation of ACC, we analyzed the effect of MIG12 on the expression of lipogenic genes, such as FAS and ACC. Although overexpression of MIG12 did not influence FAS mRNA level, it suppressed ACC mRNA level regardless of treatment with T0901317 (Fig. 7A). Considering that overexpression of MIG12 stimulated FA synthesis and TG accumulation after treatment with T0901317 (Fig. 6, A and B), it is unlikely that reduction in ACC mRNA levels is responsible for induction of lipogenesis by overexpression of MIG12. Furthermore, although knockdown of MIG12 expression suppressed FA synthesis after the treatment with T0901317 (Fig. 6D), FAS and ACC mRNA levels were paradoxically stimulated under the same conditions (Fig. 7B). These data indicate that the regulation of lipogenesis by MIG12 does not occur through the transcriptional regulation of lipogenic genes.
Fig. 7.

Effect of MIG12 on lipogenic gene expression in rat primary hepatocytes. A, Hepatocytes were infected with 20 MOI of adenoviruses for the expression of β-gal (Ad-Con) or mMIG12 expression (Ad-MIG12) as described in Materials and Methods. At 24 and 48 h after infection, cells were placed in fresh medium B containing the vehicle (DMSO) or 10 μm T0901317. Seventy-two hours after infection, total RNA was extracted, and real-time PCR analysis was performed. Relative mRNA levels were obtained after normalizing to cyclophilin mRNA levels. mRNA levels (FAS and ACC) in cells infected with Ad-Con in the absence of T0901317 are represented as 1. B, Hepatocytes were treated and analyzed in the manner described for Fig. 6C. mRNA levels (MIG12, FAS, and ACC) of cells transfected with control siRNA (siCon) in the absence of T0901317 are represented as 1. All results are expressed as mean ± sd; experiments were performed in triplicate. Similar results were obtained in three independent experiments. *, P < 0.05; **, P < 0.01.
MIG12 does not suppress LXRα activity
Because knockdown and overexpression of MIG12 gene stimulated and suppressed T0901317-mediated ACC gene expression, respectively (Fig. 7), we examined whether MIG12 suppresses LXR activity. Although LXRα/RXRα stimulated FAS promoter activity and this induction was potentiated by T0901317 treatment, overexpression of MIG12 did not suppress LXR activity (Supplemental Fig. 3A). Expression levels of MIG12 were confirmed by immunoblotting (Supplemental Fig. 3B). These results indicate that MIG12 does not influence LXRα activity.
Discussion
Transcription factors such as LXR, SREBP-1c, and ChREBP regulate FA and TG synthesis through the induction of lipogenic gene expression. Because LXR directly activates both SREBP-1c and ChREBP gene expression, LXR is considered the master regulator of lipogenesis. In this study, we established MIG12 as a novel LXR target gene. We found that ChREBP also regulated MIG12 gene expression through ChoRE1 in the MIG12 gene promoter region, which is consistent with a previous report (22). Our results demonstrated that knockdown of MIG12 expression eliminated LXR ligand-mediated induction of lipogenesis. Furthermore, overexpression of MIG12 increased LXR ligand-stimulated FA synthesis and TG accumulation in rat primary hepatocytes. These data indicate that MIG12 is involved in the stimulation of hepatic lipogenesis by LXR ligand treatment.
It was previously reported that MIG12 mRNA expression is induced in the livers of SREBP-1c transgenic mice, and it was believed that MIG12 gene expression is directly controlled by SREBP-1 (15, 23). However, in this study, by using a reporter construct containing 2.5 kb of the MIG12 gene promoter, we demonstrate that LXR and ChREBP, but not SREBP-1, activated the MIG12 gene promoter. In addition, MIG12 mRNA expression was not induced under the cholesterol-depleted condition, which leads to activation of SREBP, in rat primary hepatocytes, indicating that SREBP-1 does not directly control MIG12 gene expression (data not shown). SREBP-1 activates glucokinase gene expression (24, 25), and increased glucose metabolism via glucokinase is necessary for both expression and function of ChREBP (21). Thus, it is possible that increased MIG12 gene expression in the livers of SREBP-1c transgenic mice is ascribable to the induction of glucokinase, which in turn leads to the activation of ChREBP. Indeed, L-PK gene expression, known to be the target gene of ChREBP, is increased in the livers of transgenic mice. Further studies are necessary to determine the molecular mechanism of this regulation.
In the present study, LXRα directly stimulated MIG12 gene expression by binding to LXRE3 in the MIG12 gene promoter, and deletion of LXRE3 completely eliminated LXR responsiveness. Furthermore, deletion of the region between nucleotides −289 and −198 in the MIG12 gene promoter, which also contains LXRE4, inhibited LXR responsiveness. However, deletion of LXRE4 did not influence LXR responsiveness (Fig. 1C). In addition, LXRα/RXRα did not bind to LXRE4 (Fig. 1D). These results indicate that sequences between nucleotides −289 and −198, but not LXRE4, are required for maximal LXR responsiveness. It has been reported that the liver receptor homolog-1 (LRH-1) is required for maximal LXR responsiveness of certain genes, including cholesterol 7α-hydroxylase, cholesteryl ester transfer protein, and FAS (26–28). Because the region between nucleotides −289 and −198 in the MIG12 gene promoter retains three putative LRH-1 binding elements (seven nucleotides matching LRH-1 binding element consisting of nine nucleotides), it can be hypothesized that LRH-1 is required for LXR responsiveness of the MIG12 gene promoter. However, overexpression of LRH-1 did not stimulate MIG12 promoter activity when using pMIG12-2.5kb-Luc (data not shown). Further studies are essential to clarify how sequences between nucleotides −289 and −198 contribute to maximal LXR responsiveness of the MIG12 gene.
It was previously reported that knockdown of MIG12 (∼20% of control) resulted in a decrease in lipogenesis in rat primary hepatocytes (14). However, our results indicate that knockdown of MIG12 (∼50% of control) does not influence FA synthesis and TG accumulation in rat primary hepatocytes in the absence of T0901317, whereas it suppresses lipogenesis in the presence of T0901317 (Fig. 6, D and E). The knockdown efficiency of MIG12 may explain this difference. Furthermore, there is a possibility that glucose concentration in the culture medium contributes to this difference. In a previous study, the effect of MIG12 knockdown on reduction of lipogenesis was examined by using a glucose concentration of 27.5 mm (14), whereas in the present study, 11 mm glucose was used. Considering that the high glucose condition caused an increase in lipogenesis and knockdown of MIG12 resulted in a decrease in lipogenesis after treatment with T0901317, which induces lipogenesis (Fig. 6, D and E), it is likely that the induced basal lipogenesis is important for observing the effects of MIG12 knockdown on lipogenesis. Furthermore, because overexpression of MIG12 stimulated lipogenesis only during T0901317 treatment (Fig. 6, A and B), it is considered that, to function as a mediator of lipogenesis, MIG12 requires induced basal lipogenic activity. This speculation is supported by the finding that MIG12 induces ACC polymerization, which in turn increases its enzymatic activity (15).
In the present study, we found that knockdown of MIG12 stimulated and overexpression of MIG12 paradoxically stimulated or suppressed T0901317-mediated ACC gene expression. We also found that MIG12 did not alter LXRα activity. At present, it is unclear how MIG12 suppresses T0901317-mediated ACC gene expression. A compensatory mechanism in response to increased lipogenesis may be involved in this regulation.
Although LXR have been considered potential drug targets for cardiovascular and inflammatory diseases, clinical application of LXR ligands has been hindered because of a severe side effect, fatty liver. In the present study, we demonstrated that LXR activation stimulated MIG12 gene expression. Furthermore, LXR ligand-stimulated lipogenesis was suppressed by the knockdown of MIG12 expression. These results suggest that MIG12 is involved in the enhancement of lipogenesis by the LXR activator. The development of selective LXR ligands that activate LXR target genes other than MIG12 or the development of small compounds that inhibit MIG12 activity may therefore allow the clinical use of LXR ligands.
In conclusion, our results identify a novel LXR target gene, MIG12, and show that induction of the expression of this gene plays an important role in LXR ligand-induced lipogenesis. MIG12 mRNA levels in the livers in mice is stimulated when mice are refed than when they are fasted, indicating that MIG12 is involved in lipogenesis that occurs after feeding. Using mouse models with loss or gain of MIG12, we should be able to elucidate the function of MIG12 in lipogenesis.
Materials and Methods
Animals and diets
Male 8- to 9-wk-old C57BL/6 mice and male 6- to 8-wk-old Sprague Dawley rats were purchased from CLEA Japan (Tokyo, Japan), and FXR-null mice (29) were acclimated for 2 wk before the experiments. Animals were housed in colony cages in a 12-h light, 12-h dark cycle. Animals were fed standard chow (Labo MR Stock; Nosan Corp. Bio Department, Yokohama, Japan) ad libitum and had free access to water. Mice were housed in pathogen-free animal facilities at the University of Tokyo.
Ligand treatment experiments
Male C57BL/6 and FXR-null mice were intragastrically administered either vehicle or 50 mg/kg T0901317 or PCN in a solution containing 5% dimethylsulfoxide (DMSO), 5% Cremophor EL, and 4.5% mannitol at 0900 h. The mice were fasted until they were killed (18). After 24 h of administration, total RNA was extracted from their livers and quantitated by real-time PCR.
Cell culture
HEK293 cells were maintained in medium A (DMEM supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum). Hepatocytes were isolated by the collagenase method (30) from male Sprague Dawley rats weighing 200–300 g that were fed ad libitum. The isolated hepatocytes were seeded at a density of 1.0 × 105 cells per six-well plate (coated with rat collagen-I) in medium B (Williams' Medium E supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μm insulin, 1 μm dexamethasone, 2 mm glutamine, and 10% fetal bovine serum). After cell attachment (4 h), cells were cultured under various conditions as described in the figures. Cells were incubated at 37 C in a 5% CO2 atmosphere.
Plasmid constructs
All luciferase reporter plasmids were constructed by cloning BglII–HindIII PCR fragments coding the 5′-untranslated region of the mouse MIG12 gene into the same restriction sites of the pGL4.10 vector (Promega, Madison, WI). pMIG12-2.5kb-Luc contains the MIG12 promoter from −2512 to +44, pMIG12-650b-Luc from −650 to +44, pMIG12-534b-Luc from −534 to +44, pMIG12-289b-Luc from −289 to +44, pMIG12-198b-Luc from −198 to +44, pMIG12-111b-Luc from −111 to +44, and pMIG12-43b-Luc from −43 to +44. pMIG12-2.5kb-ΔLXRE3-Luc, pMIG12-2.5kb-ΔLXRE4-Luc, and pMIG12-2.5kb-ChRE1 KO-Luc were generated by overlap extension PCR using synthetic oligonucleotides encoding a LXRE3 deletion (5′-GTGTTActcgCGGTCA-3′), LXRE4 deletion (5′-TACCCTgcctTGCCCT-3′), and ChoRE1 mutation (5′-CACGTGcatccGGCATG-3′ → 5′-CCCGTGcatccGGCAGG-3′, changed bases underlined), respectively (31). The promoter sequence cloned in all reporter constructs except pMIG12-2.5kb-Luc is shown in Supplemental Fig. 2. A reporter plasmid, pFAS-Luc, was constructed by inserting a 1.1-kb fragment coding the 5′-promoter region into the pGL4.10 vector. Expression plasmids for ChREBP and Mlx were kindly provided by Tamio Noguchi (Osaka Ohtani University, Osaka, Japan). Expression plasmids for LXRα and RXRα were described previously (32). An expression plasmid for 3×FLAG-MIG12 was constructed by inserting fragments coding for mouse MIG12 into p3×FLAG-CMV7.1 (Sigma, St. Louis, MO).
Antibodies
Monoclonal anti-FLAG (M2) antibody was purchased from Sigma. Peroxidase-conjugated affinity-purified goat antimouse IgG was purchased from Jackson ImmunoResearch (West Grove, PA).
ChIP assays
ChIP assays were performed essentially as previously described (33) with the following minor modifications. Male C57BL/6 mice were intragastrically administered either vehicle or 50 mg/kg T0901317 in a solution containing 5% DMSO, 5% Cremophor EL, and 4.5% mannitol at 0900 h. After 24 h of administration, livers were collected, flushed with PBS, and frozen immediately in liquid nitrogen. Samples from four mice were pulverized and pooled. Three hundred milligrams from each pool were fixed in PBS with 1% formaldehyde for 10 min and quenched with glycine for 5 min. Samples were then Dounce homogenized in hypotonic buffer [10 mm HEPES (pH 7.9), 1.5 mm MgCl2, 10 mm KCl, 0.2% Nonidet P-40, 1 mm EDTA, and 5% sucrose] and layered over cushion buffer [10 mm Tris-HCl (pH 7.5), 15 mm NaCl, 60 mm KCl, 1 mm EDTA, and 10% sucrose], followed by centrifugation at 200 × g to collect the crude nuclear pellet. Subsequent ChIP steps were performed as previously described (34). Three micrograms of protein from sonicated chromatin was used for a pull-down assay with mouse IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rabbit IgG (Santa Cruz Biotechnology, Inc.), anti-LXRα (C-19; Santa Cruz Biotechnology, Inc.), or anti-ChREBP (NB400-135; Novus Biologicals, Littleton, CO). Real-time PCR was performed with the following primers: MIG12 promoter LXRE3 region forward primer (5′-AGAAAGGGTTGGGCGAGTGTTACT-3′) and MIG12 promoter LXRE3 region reverse primer (5′-CTGCCCATTCCAGGTGCTCAATTT-3′), MIG12 distal region forward primer (5′-CGTAGTAAAAGAGCATTGCATCTG-3′) and MIG12 distal region reverse primer (5′-GAGCTCGTCCTGGTCACTGT-3′), SREBP-1c promoter LXRE region forward primer (5′-ACAGAGCTTCCGGGATCAAA-3′) and SREBP-1c promoter LXRE region reverse primer (5′-GCAACCATCCCCGAAAAG-3′), MIG12 promoter ChoRE1 region forward primer (5′-CGTGCATCCGGCATGT-3′) and MIG12 promoter ChoRE1 region reverse primer (5′-GATGACCGGAATGCTTGTGT-3′), L-PK promoter ChoRE region forward primer (5′-GGATGCCCACTACAGCCTCTGTTAAA-3′) and L-PK promoter ChoRE region reverse primer (5′-AACCAGCTAGCATCTCTCTTGCCA-3′).
Luciferase assays
HEK293 cells were plated in 12-well plates at a density of 1.0 × 105 cells/well, cultured with medium A for 20 h, and then transfected with 200 ng of one of the reporter plasmids, 20 ng of the indicated expression plasmid, and 50 ng of pCMV-β-galactosidase (β-gal) (an expression plasmid for β-gal) by the calcium phosphate method. Twenty-four hours after transfection, cells were placed in medium A containing the vehicle (DMSO) or 10 μm T0901317. After incubation for another 24 h, luciferase assays were performed. Luciferase and β-gal activities were measured as described previously (35). Normalized luciferase values were determined by dividing luciferase activity by β-gal activity.
Real-time PCR
Total RNA was extracted from mouse livers and rat primary hepatocytes using the RNeasy Mini kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions. cDNA was synthesized and amplified from 2 μg total RNA using a High-capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Real-time PCR (SYBR Green) analysis was performed using the Applied Biosystems 7000 Sequence Detection System. Expression was normalized to 36B4 or cyclophilin control. The primer sequence sets used were as follows: mouse (m) MIG12, 5′-ACTCGCTCTTTAACGCCATGAA-3′ and 5′-CATCACCGTCTGGTCCATGTT-3′; mL-PK, 5′-CTTGCTCTACCGTGAGCCTC-3′ and 5′-ACCACAATCACCAGATCACC-3′; mPEPCK, 5′-AGCCTCGACAGCCTGCCCCAGG-3′ and 5′-CCAGTTGTTGACCAAAGGCTTTT-3′; m cytochrome P450 3A11, 5′-CCGAGTGGATTTTCTTCAGC-3′ and 5′-GAGCCTCATCGATCTCATCC-3′; m36B4, 5′-CTGATCATCCAGCAGGTGTT-3′ and 5′-CCAGGAAGGCCTTGACCTTT-3′; rat (r) MIG12, 5′-GGCAGCGCCAATGGAA-3′ and 5′-GATGGACTTGAGCAGCACGTAGT-3′; rL-PK, 5′-TTTGCCTCCTTTGTGCGGAAA-3′ and 5′-TCTCCGCAGGGATCTCAATG-3′; rABCA1, 5′-CCCGGCGGAGTAGAAAGG-3′ and 5′-AGGGCGATGCAAACAAAGAC-3′; and rCyclophilin, 5′-TTGCCATTCCTGGACCCAAA-3′ and 5′-ATGGCACTGGTGGCAAGTCC-3′.
siRNA experiments
siRNA (150 pmol/six-well plate) for rat MIG12 (nucleotides 904–928 in NM_206950, CCGUGCUCUCCAAACUCACCCGCAA) and control GL2 luciferase (Bonac Co., Fukuoka, Japan) were transfected using Lipofectamine 2000 or Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) into rat primary hepatocytes according to the manufacturer's instructions.
Adenovirus vector
The cDNA of MIG12 was amplified by RT-PCR using total RNA from mouse liver and subcloned into the shuttle vector pENTR-1a at the EcoRI and XhoI sites. Then, the adenovirus vector encoding MIG12 (AdCMV-MIG12) was generated using Gateway technology.
Adenovirus infection
Adenoviruses for the expression of β-gal and mMIG12 were constructed using the ViraPower Adenoviral Expression System (Invitrogen). After cell attachment (4 h), rat primary hepatocytes were infected with 20 multiplicity of infection (MOI) of AdCMV-β-gal or AdCMV-MIG12. The medium was exchanged for fresh medium B after 24 h of infection.
Electrophoretic mobility shift assays
Nuclear extracts were prepared from HEK293 cells transfected with or without expression plasmids for LXRα and RXRα as described previously (36). The oligonucleotides used in electrophoretic mobility shift assays are listed in Fig. 1D. Double-stranded oligonucleotides were 5′-end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. Binding reactions were performed in a total volume of 34 μl consisting of 20 mm HEPES-KOH (pH 7.6), 1 mm EDTA, 10 mm (NH4)2SO4, 0.2% (wt/vol) Tween 20, 30 mm KCl, 1 mm dithiothreitol, 10 μm T0901317, 50 fmol of end-labeled probe, 1 μg of poly(dI-dC), and 10 μg of nuclear extracts from HEK293 cells, as described above. Each reaction mixture was incubated at room temperature for 20 min. Samples were then loaded on a 6% polyacrylamide gel in 0.5× Tris-borate, EDTA buffer at 200 V for 1 h. In competition assays, competing cold oligonucleotides were added to the reaction with the labeled probe at 50-fold molar excesses. After electrophoresis, the gel was dried and analyzed with a BAS2000 image analysis system (Fujifilm, Fuji, Japan).
Determination of TG accumulation
Rat primary hepatocytes were seeded at a density of 1 × 105 cells per six-well plate (coated with rat collagen-I) and cultured as described in the figure legends. Then, cells were washed with PBS, and lipids were extracted with hexane-2-propanol (3:2 vol/vol). The amount of intracellular TG accumulated was determined using the Triglyceride E-Test from Wako (Osaka, Japan) and normalized to the amount of total cellular protein determined by the BCA protein assay (Pierce, Rockford, IL), according to the manufacturer's instructions (37).
Determination of FA synthesis rates
Rat primary hepatocytes were seeded at a density of 1 × 105 cells per six-well plate (coated with rat collagen-I) and cultured as described in the figure legends. Cells were placed in fresh medium supplemented with [14C]acetate (0.5 μCi/ml) and incubated for 2 h. The cells were washed, trypsinized, and collected by centrifugation. The pelleted cells were mixed with 1 ml of 8 n KOH and 1 ml of ethanol per well. The mixture was heated at 100 C for 2 h and extracted twice with 2 ml petroleum ether. Two milliliters of the lower aqueous layer were mixed first with 1 ml of 12 n HCl and then extracted twice with 3 ml of petroleum ether. Five milliliters of the upper petroleum ether layer were dried under low heat. 14C radioactivity was determined by scintillation counting. Normalized FA synthesis rates were determined by dividing the signal of [14C]FA by the amount of total cellular protein determined by the BCA protein assay (Pierce).
Statistical analysis
All data are presented as mean ± sd. Statistical analysis was performed using StatView software. Student's t test was used to compare pairs of groups. One-way ANOVA followed by the Bonferroni/Dunn test was used to compare more than two groups.
Acknowledgments
We thank Dr. Tamio Noguchi (Osaka Ohtani University) for expression plasmids, Dr. Frank J. Gonzalez (National Institutes of Health) and Masaaki Miyata (Tohoku University) for the FXR-null mice, and Dr. Vicky Y. Lin, Dr. Steven A. Kliewer, and Dr. David J. Mangelsdorf (University of Texas Southwestern Medical Center) for the primer sequences of SREBP-1c promoter.
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages:
Nuclear Receptors: LXR-α | LXR-β | RXR-α;
Ligands: T0901317.
Footnotes
- ABC
- ATP-binding cassette
- ACC
- acetyl-coenzyme A carboxylase
- ChIP
- chromatin immunoprecipitation
- ChoRE1
- carbohydrate response element 1
- ChREBP
- carbohydrate-responsive element-binding protein
- DMSO
- dimethylsulfoxide
- DR4
- direct repeat 4
- FA
- fatty acid
- FAS
- FA synthase
- FXR
- farnesoid X receptor
- β-gal
- β-galactosidase
- L-PK
- liver-type pyruvate kinase
- LRH-1
- liver receptor homolog-1
- LXR
- liver X receptor
- LXRE
- LXR-responsive element
- m
- mouse
- MOI
- multiplicity of infection
- MID1
- midline-1
- MIG12
- MID1-interacting G12-like protein
- Mlx
- Max-like protein X
- PCN
- pregnenalone-16α-carbonitrile
- PEPCK
- phosphoenolpyruvate carboxykinase
- PXR
- pregnane X receptor
- r
- rat
- RXR
- retinoid X receptor
- siRNA
- small interfering RNA
- SREBP
- sterol regulatory element-binding protein
- TG
- triacylglycerol.
References
- 1. Willy PJ , Umesono K , Ong ES , Evans RM , Heyman RA , Mangelsdorf DJ. 1995. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9:1033–1045 [DOI] [PubMed] [Google Scholar]
- 2. Janowski BA , Willy PJ , Devi TR , Falck JR , Mangelsdorf DJ. 1996. An oxysterol signalling pathway mediated by the nuclear receptor LXR. Nature 383:728–731 [DOI] [PubMed] [Google Scholar]
- 3. Chen W , Chen G , Head DL , Mangelsdorf DJ , Russell DW. 2007. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab 5:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zelcer N , Tontonoz P. 2006. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest 116:607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Joseph SB , McKilligin E , Pei L , Watson MA , Collins AR , Laffitte BA , Chen M , Noh G , Goodman J , Hagger GN , Tran J , Tippin TK , Wang X , Lusis AJ , Hsueh WA , Law RE , Collins JL , Willson TM , Tontonoz P. 2002. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA 99:7604–7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laffitte BA , Chao LC , Li J , Walczak R , Hummasti S , Joseph SB , Castrillo A , Wilpitz DC , Mangelsdorf DJ , Collins JL , Saez E , Tontonoz P. 2003. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci USA 100:5419–5424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schultz JR , Tu H , Luk A , Repa JJ , Medina JC , Li L , Schwendner S , Wang S , Thoolen M , Mangelsdorf DJ , Lustig KD , Shan B. 2000. Role of LXRs in control of lipogenesis. Genes Dev 14:2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berti C , Fontanella B , Ferrentino R , Meroni G. 2004. Mig12, a novel Opitz syndrome gene product partner, is expressed in the embryonic ventral midline and co-operates with Mid1 to bundle and stabilize microtubules. BMC Cell Biol 5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu Q , Anderson GW , Mucha GT , Parks EJ , Metkowski JK , Mariash CN. 2005. The Spot 14 protein is required for de novo lipid synthesis in the lactating mammary gland. Endocrinology 146:3343–3350 [DOI] [PubMed] [Google Scholar]
- 10. Nishi N , Shoji H , Miyanaka H , Nakamura T. 2008. Transient up-regulation of a novel member of Spot 14 family in androgen-stimulated rat prostate. Biochim Biophys Acta 1780:1004–1009 [DOI] [PubMed] [Google Scholar]
- 11. Jump DB , Oppenheimer JH. 1985. High basal expression and 3,5,3′-triiodothyronine regulation of messenger ribonucleic acid S14 in lipogenic tissues. Endocrinology 117:2259–2266 [DOI] [PubMed] [Google Scholar]
- 12. Kinlaw WB , Schwartz HL , Towle HC , Oppenheimer JH. 1986. Opposing effects of glucagon and triiodothyronine on the hepatic levels of messenger ribonucleic acid S14 and the dependence of such effects on circadian factors. J Clin Invest 78:1091–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu Q , Mariash A , Margosian MR , Gopinath S , Fareed MT , Anderson GW , Mariash CN. 2001. Spot 14 gene deletion increases hepatic de novo lipogenesis. Endocrinology 142:4363–4370 [DOI] [PubMed] [Google Scholar]
- 14. Aipoalani DL , O'Callaghan BL , Mashek DG , Mariash CN , Towle HC. 2010. Overlapping roles of the glucose-responsive genes, S14 and S14R, in hepatic lipogenesis. Endocrinology 151:2071–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim CW , Moon YA , Park SW , Cheng D , Kwon HJ , Horton JD. 2010. Induced polymerization of mammalian acetyl-CoA carboxylase by MIG12 provides a tertiary level of regulation of fatty acid synthesis. Proc Natl Acad Sci USA 107:9626–9631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Repa JJ , Liang G , Ou J , Bashmakov Y , Lobaccaro JM , Shimomura I , Shan B , Brown MS , Goldstein JL , Mangelsdorf DJ. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev 14:2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cha JY , Repa JJ. 2007. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 282:743–751 [DOI] [PubMed] [Google Scholar]
- 18. Inoue J , Satoh S , Kita M , Nakahara M , Hachimura S , Miyata M , Nishimaki-Mogami T , Sato R. 2008. PPARα gene expression is up-regulated by LXR and PXR activators in the small intestine. Biochem Biophys Res Commun 371:675–678 [DOI] [PubMed] [Google Scholar]
- 19. Mitro N , Vargas L , Romeo R , Koder A , Saez E. 2007. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett 581:1721–1726 [DOI] [PubMed] [Google Scholar]
- 20. Houck KA , Borchert KM , Hepler CD , Thomas JS , Bramlett KS , Michael LF , Burris TP. 2004. T0901317 is a dual LXR/FXR agonist. Mol Genet Metab 83:184–187 [DOI] [PubMed] [Google Scholar]
- 21. Dentin R , Pégorier JP , Benhamed F , Foufelle F , Ferré P , Fauveau V , Magnuson MA , Girard J , Postic C. 2004. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem 279:20314–20326 [DOI] [PubMed] [Google Scholar]
- 22. Tsatsos NG , Augustin LB , Anderson GW , Towle HC , Mariash CN. 2008. Hepatic expression of the SPOT 14 (S14) paralog S14-related (Mid1 interacting protein) is regulated by dietary carbohydrate. Endocrinology 149:5155–5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horton JD , Shah NA , Warrington JA , Anderson NN , Park SW , Brown MS , Goldstein JL. 2003. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA 100:12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Foretz M , Guichard C , Ferré P , Foufelle F. 1999. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci USA 96:12737–12742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim SY , Kim HI , Kim TH , Im SS , Park SK , Lee IK , Kim KS , Ahn YH. 2004. SREBP-1c mediates the insulin-dependent hepatic glucokinase expression. J Biol Chem 279:30823–30829 [DOI] [PubMed] [Google Scholar]
- 26. Lu TT , Makishima M , Repa JJ , Schoonjans K , Kerr TA , Auwerx J , Mangelsdorf DJ. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515 [DOI] [PubMed] [Google Scholar]
- 27. Luo Y , Liang CP , Tall AR. 2001. The orphan nuclear receptor LRH-1 potentiates the sterol-mediated induction of the human CETP gene by liver X receptor. J Biol Chem 276:24767–24773 [DOI] [PubMed] [Google Scholar]
- 28. Matsukuma KE , Wang L , Bennett MK , Osborne TF. 2007. A key role for orphan nuclear receptor liver receptor homologue-1 in activation of fatty acid synthase promoter by liver X receptor. J Biol Chem 282:20164–20171 [DOI] [PubMed] [Google Scholar]
- 29. Sinal CJ , Tohkin M , Miyata M , Ward JM , Lambert G , Gonzales FJ. 2000. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744 [DOI] [PubMed] [Google Scholar]
- 30. Berry MN , Friend DS. 1969. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol 43:506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Higuchi R , Krummel B , Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16:7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arimura N , Horiba T , Imagawa M , Shimizu M , Sato R. 2004. The peroxisome proliferator-activated receptor γ regulates expression of the perilipin gene in adipocytes. J Biol Chem 279:10070–10076 [DOI] [PubMed] [Google Scholar]
- 33. Schmidt DR , Holmstrom SR , Fon Tacer K , Bookout AL , Kliewer SA , Mangelsdorf DJ. 2010. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem 285:14486–14494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanayama T , Arito M , So K , Hachimura S , Inoue J , Sato R. 2007. Interaction between sterol regulatory element-binding proteins and liver receptor homolog-1 reciprocally suppresses their transcriptional activities. J Biol Chem 282:10290–10298 [DOI] [PubMed] [Google Scholar]
- 35. Sato R , Miyamoto W , Inoue J , Terada T , Imanaka T , Maeda M. 1999. Sterol regulatory element-binding protein negatively regulates microsomal triglyceride transfer protein gene transcription. J Biol Chem 274:24714–24720 [DOI] [PubMed] [Google Scholar]
- 36. Yamada K , Tanaka T , Noguchi T. 1997. Members of the nuclear factor 1 family and hepatocyte nuclear factor 4 bind to overlapping sequences of the L-II element on the rat pyruvate kinase L gene promoter and regulate its expression. Biochem J 324:917–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takahashi Y , Ohoka N , Hayashi H , Sato R. 2008. TRB3 suppresses adipocyte differentiation by negatively regulating PPARγ transcriptional activity. J Lipid Res 49:880–892 [DOI] [PubMed] [Google Scholar]