

Epigenetic changes induced by liganded thyroid hormone receptor (TR) on pituitary genes demonstrate the role of Aurora Kinase B during transcriptional activation.
Abstract
Covalent histone modifications clearly play an essential role in ligand-dependent transcriptional regulation by nuclear receptors. One of the predominant mechanisms used by nuclear receptors to activate or repress target-gene transcription is the recruitment of coregulatory factors capable of covalently modify the amino terminal ends of histones. Here we show that the thyroid hormone (T3) produces a rapid increase in histone H3Ser10 phosphorylation (H3Ser10ph) concomitant to the rapid displacement of the heterochromatin protein 1β (HP1β) to the nuclear periphery. Moreover, we found that T3-mediated pituitary gene transcription is associated with an increase in H3Ser10ph. Interestingly, the Aurora kinase B inhibitor ZM443979 abolishes the effect of T3 on H3Ser10ph, blocks HP1β delocalization, and significantly reduces ligand-dependent transactivation. Similar effects were shown when Aurora kinase B expression was abrogated in small interfering RNA assays. In an effort to understand the underlying mechanism by which T3 increases H3Ser10ph, we demonstrate that liganded thyroid hormone receptor directly interacts with Aurora kinase B, increasing its kinase activity. Moreover, using chromatin immunoprecipitation assays, we have shown that Aurora kinase B participates of a mechanism that displaces HP1β from promoter region, thus preparing the chromatin for the transcriptional activation of T3 regulated genes. Our findings reveal a novel role for Aurora kinase B during transcriptional initiation in GO/G1, apart from its well-known mitotic activity.
In eukaryotic cells, genes are complexed with core histones and other chromosomal proteins to form either active euchromatin or silent heterochromatin (1–3). The tails of core histones are subjected to covalent posttranslational modifications which allow the recruitment of proteins such as heterochromatin protein (HP1) that modulate chromatin structure (4). In mammalian cells, HP1β is present both in pericentric heterochromatin and in euchromatin where it represses gene expression (5), while HP1α and HP1γ are mainly associated with either constitutive heterochromatin or euchromatin, respectively. In histones H3 and H4, both lysine acetylation and serine phosphorylation are associated with transcriptional activation, while lysine methylation at specific residues is linked to either transcriptional repression or activation. Lysine residues in histone tails can be mono-, di- or trimethylated by the action of histone methyltransferases. Methylation of the lysine 9 residue in H3 (H3K9me), particularly in the di- and trimethylated state, serves as a scaffold for HP1 proteins binding to the chromatin. It has been demonstrated that HP1β bound to methylated H3K9 is released from chromatin by H3Ser10 phosphorylation. This “methyl-phos switch” mechanism occurs at the onset of mitosis and is regulated by Aurora kinase-B (6, 7). The role of Aurora kinase B during mitosis is well established as chromosome passenger protein kinase that regulates centrosome separation, chromosome segregation, and cytokinesis. However, an additional report shows that Aurora kinase B also mediates the displacement of HP1β from facultative heterochromatin by Ser10 phosphorylation in terminally differentiated post-mitotic plasma cells suggesting that the “methyl-phos” switch mechanism could occur in other cell-cycle stages apart from mitosis.
Nuclear receptors constitute a family of ligand-dependent transcription factors that fulfill important roles in growth, development, and metabolism. Thyroid nuclear receptors (TRs) belong to this family and are activated upon the binding of thyroid hormone (T3). In the absence of ligand, TRs recruit corepressor complexes that contain histone deacetylases (8, 9). Ligand binding allows the release of corepressors and the recruitment of coactivators, some of which contain histone acetyltransferases. Gene expression results from the ability of transcription activators and repressors to access chromatin at specific promoter. Interestingly Aurora kinase B activity and histone posttranslational modifications have been previously related with the action of nuclear receptors. Thus, estrogens regulate H3Ser10ph in both ovarian follicle and mammary epithelial tumor cells by induction of Aurora kinase B activity (10). Other ligands such as T3 activate transcription with an increase in the dual modification H3Ser10pH/H3K14 although the underlying mechanism has yet to be elucidated.
T3 is an important regulator of pituitary genes such as GH and prolactine (PRL) genes. Both are expressed in GH4C1 cells, a pituitary somatolactotrope cell line, which is derived from a rat pituitary tumor that has been widely used as a model to study the mechanism of T3 action (11). In this system, here we show that T3 causes a rapid increase in the total cellular levels of H3Ser10 phosphorylation accompanied by HP1β delocalization to the nuclear periphery. Inhibition of Aurora B blocks this effect and inhibits T3-dependent gene expression, demonstrating a novel mechanism by which this enzyme plays an important role in the initiation of hormone-dependent gene activation. In addition to its functions during mitosis, the Aurora kinase B has an important role in the initiation of gene transcription by interaction with TR in a ligand dependent manner. Our results also suggest that thyroid receptors are important regulators of the uncoupling of Aurora kinase B regulation from the cell cycle.
Results
T3 induces HP1β redistribution to the nuclear periphery and increases global cellular levels of H3Ser10ph
To determine whether T3 controls HP1-mediated gene regulation, we first analyzed HP1β localization by immunofluorescence microscopy in pituitary GH4C1 cells which express high levels of TR. As expected (12–15), endogenous HP1β localized as condensed region into the nucleus of untreated cells grown for 48 h in hormone-depleted serum. In contrast, exposure to T3 for only 15 min caused the relocation of HP1β to the periphery of the nucleus (Fig. 1A). To demonstrate that TR was implicated in this effect we overexpressed both TR and HP1β in HEK293T cells, in which TR levels are almost undetectable. After T3 treatment, HP1β only relocated to the nuclear envelope in those cells cotransfected with both plasmids, indicating that this effect was indeed mediated by the receptor (Fig. 1B).
Fig. 1.

HP1β redistribution and levels of H3Ser10ph after T3 treatment. A, Endogenous Hp1β is relocalized in GH4C1 cells treated with T3 (5 nm). B, Simultaneous expression of pSG5-TRα with hemaglutinin (HA)-tagged Hp1β expression in HEK293T cells leads to Hp1β redistribution after incubation with T3 (100 nm) for 15 min. C, Total cellular levels of H3Ser10ph in T3-treated GH4C1 cells. H3 was used as a loading control. D, Immunofluorescence of endogenous H3Ser10ph in GH4C1 cells treated with T3 for 15 min. E, Incubation of GH4C1 cells with 100 nm estradiol for 5 or 15 min increases total cellular levels of H3Ser10 phosphorylation.
Because the H3 phosphorylation at Ser10 has been shown to prevent HP1 binding to di- and trimethylated H3K9 (16), we next analyzed the effect of T3 on H3Ser10 phosphorylation. As shown by Western blot assays, T3 caused a rapid increase of H3Ser10ph in GH4C1 cells (Fig. 1C). To confirm this data we performed immunofluorescence assays after 15 min of T3 treatment (Fig. 1D). As expected, these assays also showed an increase in the intensity of staining H3Ser10ph and demonstrated that its dispersed nuclear distribution was not altered in hormone-treated cells. Because estrogens increase H3Ser10ph in other models and GH4C1 cells contain endogenous estrogens receptor levels, we also checked whether the observed effects were nuclear receptor specific. For that reason we analyzed the effect of 5- and 15-min estrogens treatment on H3Ser10ph levels in GH4C1 cells (Fig. 1E). In agreement with the effect observed after T3 incubation, estrogens also produced a rapid increase on H3Ser10ph in this cell line.
T3 increases H3Ser10ph levels at T3-target promoters
Given the fact that we observed a global increase in H3Ser10ph levels after 15-min T3 treatment, we tested the possibility that this posttranslational modification could be found at target genes upon hormonal induction. Thus, we performed chromatin immunoprecipitation (ChIP) assays with the promoter region of the GH gene which contains thyroid hormone response elements (TREs). In agreement with previous reports (17), a small amount of TR was bound to GH promoter in the absence of hormone, while short-term exposure to T3 (15 min) provoked a significant recruitment of TR to the promoter (Fig. 2A). In parallel to the TR binding, a significant increase in H3Ser10ph marker was observed after T3 treatment, indicating an increase of this modification during hormonal activation of transcription.
Fig. 2.

T3-regulated genes showed increased levels of H3Ser10ph after 15-min treatment with 5 nm T3. A, Representative ChIP assay in GH4C1 cells using the indicated antibodies and primers flanking the TRE region of the GH promoter (left). Real-time quantitative PCR (QRT-PCR) of several ChIP assays (right). Bars represent mean ± sd (n > 3). ChIP assays were performed in GH4C1 cells maintained in the presence and absence of T3 (5 nm, 15min). B and C, Immunoprecipitated chromatin was quantified by real-time PCR using primers flanking the TRE element in the Prolactin gene and GAPDH gene primers that was used as negative control. Bars represent mean ± sd (n =3).
PRL gene is also a target of T3 activation, so we also checked the levels of H3Ser10ph after T3 induction in the PRL promoter containing TREs. As it can be observed in Fig. 2B, T3 treatment also produced an increase in the recruitment of TR to the PRL promoter together with an important increase in H3Ser10ph levels. In contrast, when we analyzed the levels of this epigenetic mark in GAPDH promoter, which does not contain TREs, we did not observe any change upon T3 treatment (Fig. 2C).
Aurora kinase B is required for T3-induced HP1β displacement and H3Ser10ph increase
The effect of T3 on H3 phosphorylation could be attributable to the activation of several kinases such as mitogen- and stress-associated protein kinase 1 (MSK1), ribosomal S6 kinases (RSK)1-2, and even to the effect controlled by Aurora kinase B where H3Ser10 phosphorylation at the onset of mitosis interferes with the interaction of HP1 with trimethylated H3K9 (6, 7). In addition, it has been reported that phosphorylation of Ser10 also markedly decreased the in vitro interaction of HP1β with both K9me3 and K9me2 peptides (16). For these reasons, we next analyzed the possible role of Aurora kinase B in the effect of T3 on both H3Ser10ph and displacement of HP1β. We found that T3-induced phosphorylation on H3Ser10 was blocked either by incubation in the presence of the specific Aurora kinase B inhibitor, ZM447439 (Fig. 3A), or knocking-down Aurora kinase B expression with specific small interfering RNA (siRNA) (Fig. 3B). Moreover, exposure to ZM447439 also inhibited the T3-induced displacement of HP1β to the periphery of the nucleus (Fig. 3C). These data indicate that Aurora kinase B mediates the effect of T3 on HP1β localization.
Fig. 3.

Inhibition of Aurora-B abolishes both the delocalization of Hp1β and H3Ser10ph induced by T3. A, H3Ser10ph levels in GH4C1 cells incubated with or without T3 and ZM447439. B, Aurora kinase B and H3Ser10ph protein expression in GH4C1 cells transfected with siRNA targeting Aurora kinase B or a control siRNA. C, Nuclear localization of HP1β in HEK293T after ectopic expression of pSG5-TRα together with hemaglutinin (HA)-tagged HP1β. Cells were treated with ZM447439 in absence or presence of T3. D, H3Ser10ph levels in GH4C1 cells incubated in the presence or absence of T3 and DMSO, SB203580 or U0126.
We wanted to check whether MSK1 or RSK1-2 was also involved in T3-mediated H3Ser10ph increase. H89 as well as Ro318220 are used to inhibit both activities, respectively, but still inhibit two or more protein kinases with similar potency. However, MSK1 respond to both mitogen- and stress-activated protein kinases (ERK and p38MAPK), while RSK1-2 is ERK target. In cell-based assays, it has been shown that these MAPK are specifically inhibited by U0126 and SB203580, respectively (18). Thus, we incubated GH4C1 cells in the absence or presence of both specific inhibitors. Figure 3D shows that none of them were able to avoid H3ser10ph increase induced by thyroid hormone. These data demonstrate that T3 effect is specifically mediated by Aurora kinase B in our cells.
A particularly striking aspect of these results is the fact that T3 effects occurred in cells that do not proliferate. Cell cycle analysis showed that 80–90% of GH4C1 cells incubated in hormone-depleted medium for 24 h remained in G0/G1 phase, and incubation with T3 for up to 8 h did not induce the entry of the cells in the G2/M phase (Fig. 4). This result implies a novel role for Aurora Kinase B at G0/G1, apart from its well-known mitotic function.
Fig. 4.
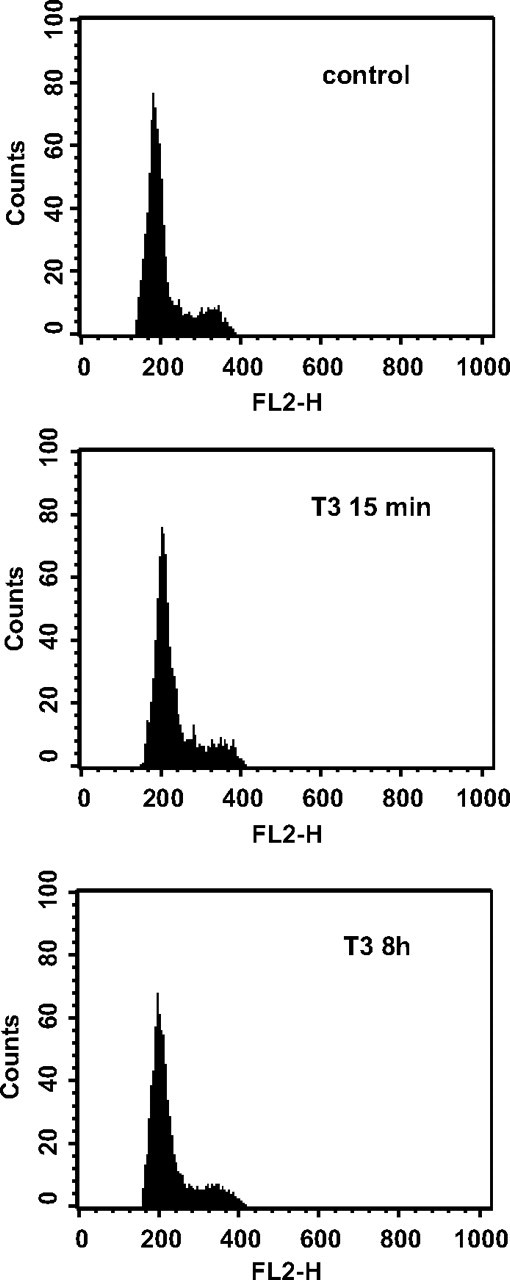
Cell-cycle profiles from GH4C1 cells treated without or with T3 for 15 min and 8 h.
TR interacts with Aurora kinase B and increases its kinase activity in a T3-dependent manner
To further study the mechanism underlying the effect of T3 on H3Ser10ph and HP1β delocalization, we analyzed the interaction of TR with Aurora kinase B. In vitro pull-down assays showed that Aurora kinase B interacted with TR both in the absence or presence of T3 (Fig. 5A). Despite the T3-induced increase in H3Ser10ph, the interaction between TR and Aurora kinase B was not modified by exposure to the hormone. The interaction between both proteins was confirmed by coimmunoprecipitation assays in GH4C1 cells (Fig. 5B). Throughout the same experiment, we tested the kinase activity of Aurora kinase B in the immunoprecipitates from control and T3-treated cells (Fig. 5B). T3 augmented significantly the kinase activity of Aurora kinase B, as reflected by the Ser10 phosphorylation of exogenous H3 used as a substrate.
Fig. 5.

A, GST-TRα or GST-0 fusion proteins were incubated with S35-labeled Aurora kinase B, and pull-down assays were performed as described (32). Where indicated, T3 (50 nm) was added to the binding assay. B, Endogenous Aurora kinase B was immunoprecipitated from GH4C1 cells in the presence or absence of 5 nm T3 and incubated with histone H3 in kinase buffer with unlabeled ATP in the presence or absence of T3. Kinase activity was evaluated by measuring H3Ser10ph levels by Western blotting with a specific antibody. Coimmunoprecipitated Aurora kinase B and TR were also detected in Western blot assays. The quantification from three different experiments is shown in the graph.
Aurora B is required for transcriptional activation of T3-responsive genes
Our results indicate that the effect of T3 on the association of HP1β with chromatin appears to be regulated by a novel mechanism involving Aurora kinase B (Figs. 3 and 5). To test whether Aurora kinase B modulated TR transcriptional activity, a TRE-containing luciferase construct (DR4-luc) was transiently transfected into GH4C1 cells. As expected, T3 strongly induced the DR4-luc activity in control cells. However, the incubation of the cells with ZM447439 significantly reduced the T3-mediated transactivation of this reporter (Fig. 6A). Similarly, this inhibitor severely impaired T3-stimulated transcription of the endogenous GH gene (Fig. 6B). In addition, ectopic expression of Aurora kinase B increased in a dose-dependent manner T3–dependent transactivation in HEK293T cells (Fig. 6C), whereas a “kinase-dead” Aurora kinase B construct (K109R) had no effect on the reporter gene (Fig. 6D).
Fig. 6.
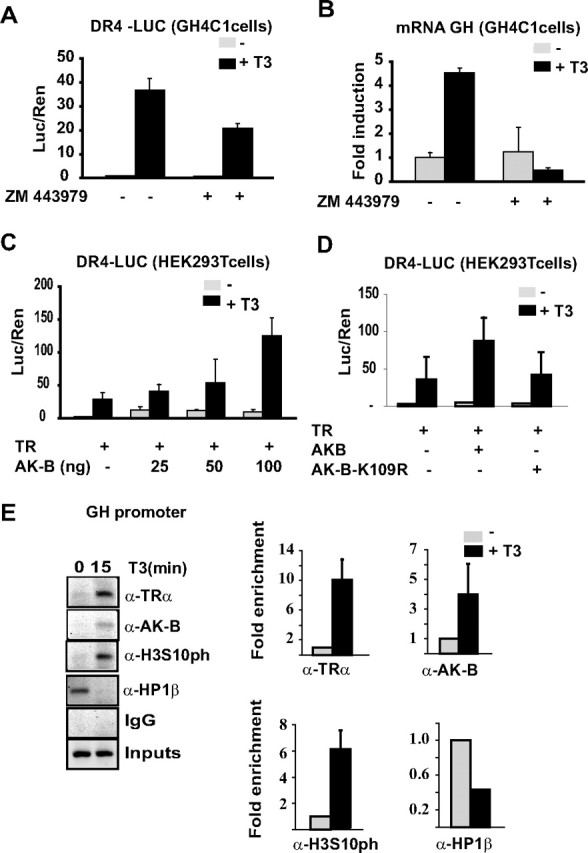
Aurora kinase B controls T3-induced transcriptional activity. A, GH4C1 cells were transiently transfected with the TRE-containing DR4-TKLUC reporter plasmid. ZM447439 treatment inhibits T3-induced transactivation. B, GH mRNA expression in GH4C1 cells, analyzed by QRT-PCR 8 h after T3 treatment in the presence or absence of ZM447439. C, A reporter plasmid containing a consensus TRE (DR4-TKLUC) was cotransfected in HEK293T cells together with expression plasmids for TRα and increased amounts of Aurora kinase B. D, Expression vectors for Aurora kinase B (50 ng) or a kinase-dead mutant (K109R, 50ng) were cotransfected with the reporter plasmid in HEK293T cells. Reporter activity was determined after 36 h in the presence or absence of T3. E, ChIP assays with the antibodies indicated in GH4C1 cells maintained for 15 min in the presence or absence of T3 (left). Quantitative PCR was performed with primers flanking the TRE sequence in the GH promoter. Bars represent the mean ± sd (n > 3, right).
To determine whether Aurora kinase B and TR associate in a ligand-dependent manner with chromatin in vivo, we next performed ChIP assays on T3-treated GH4C1 cells (Fig. 6E). In these cells, both TR and Aurora kinase B were rapidly recruited to the GH promoter in a hormone-dependent manner. The data suggest that both the T3-mediated association of Aurora kinase B with the GH promoter and the increase in its kinase activity causes the enrichment observed in H3Ser10ph after T3 treatment (Fig. 6E). Interestingly, the association of Hp1β with the promoter strongly diminished after exposure to T3. Thus, T3 induces phosphorylation of the Ser10 residue in H3 and causes displacement of HP1β from the promoter in a mechanism regulated by Aurora kinase B.
Discussion
Thyroid hormone regulates HP1β dissociation through histone covalent modifications
The results of this study point at the important role of thyroid hormone in the regulation of chromatin structure, secondary to the modulation of specific epigenetic changes in certain histone residues. Histones are subject to numerous posttranslational modifications that correlate with the state of higher-order chromatin structure and gene expression. At the moment, it is not clear whether these epigenetic marks are causative regulatory factors in chromatin structure changes, although it has been evidenced that modulation of H3Ser10 phosphorylation at interphase can function as a causative regulator of higher-order chromatin structure in Drosophila in vivo (19). Here we report that thyroid hormone receptor, in the presence of its ligand, produces the delocalization of HP1β protein to the nuclear periphery, concomitant with a significant and rapid increase in H3Ser10 phosphorylation. Our hypothesis is that removal of HP1β protein to the nuclear periphery might be an important step for rapid gene induction and efficient initiation of T3-mediated transcription. In this regard, it has been suggested that the redistribution of HP1β would be in accordance with the radial arrangement of the genome in the nucleus, in which transcriptional silent chromatin is located near the nuclear periphery and active loci are positioned more interior (20, 21, 22). However, to confirm this hypothesis, further experiments, such as FISH assays, will be necessary to determine the nuclear localization of T3 activated and repressed genes.
Previous report have demonstrated that transcriptional regulation by TR involves, in addition to the expected changes in acetylation, methylation of H3-K4, H3-K9, and H3-R17, and the dual modification in H3-K14/H3-Ser10 in xenopus oocytes (23). Moreover these authors demonstrated that TR interacts with the methyltransferase Suv39H1 that facilitates repression by unliganded TR.
Our experiments show a significant increase in total amount of H3Ser10ph in response to T3 mediated by TR receptor. Global histone changes have also been observed by other authors (20, 24–26), however it is logical to think that each gene may file different levels of such changes. In fact we have observed that despite an overall change in H3Ser10 phosphorylation of those genes that are regulated by T3 such GH or PRL, the non–T3-regulated gene, GAPDH, does not show any changes in the levels of H3Ser10ph even after T3 induction.
Two additional reports have analyzed the mechanisms by which steroid hormone–induced histone phosphorylation promotes transcriptional activation. Thus, progesterone appears to induce H3Ser10 phosphorylation promoting release of HP1γ from the target promoter (27), whereas androgens would induce H3T11 and H3T6 phosphorylation enhancing H3K9 demethylation or inhibiting H3K4 demethylation, respectively (24, 28). Interestingly, our data show that in T3-regulated genes the hormone induces both H3Ser10 phosphorylation and HP1β release. The enzymes that regulate nuclear receptor–mediated histone phosphorylation differ from one ligand to the other. For example, progesterone induced H3Ser10 phosphorylation through activation of MSK1, while androgens mediate H3T11 and H3T6 phosphorylation through either the protein kinase C–related kinase 1 or protein kinase C beta I (PKCβ1) respectively. In our study, we have analyzed the involvement of different kinases, such as MSK1, RSK2, and Aurora kinase B using specific inhibitors, and only the Aurora kinase B specific inhibitor ZM443979 was able to inhibit T3 effects, indicating that this kinase mediates T3-induced H3Ser10 phosphorylation. Strikingly, not only T3 but also estrogens are able to induce Aurora kinase B activity (10), opening the possibility that other nuclear receptors ligands could regulate the activity of this kinase in a similar manner. All these data demonstrate that different nuclear receptors show selectivity for specific kinases that modulate the transcription of hormone target genes by regulating epigenetic marks such as histone phosphorylation at different residues.
A novel function for the Aurora kinase B during transcriptional activation
The role of Aurora kinase B during mitosis is well established because this enzyme regulates cytokinesis at G2/M. Aurora kinase B is part of the chromosomal passenger complex that maintains the integrity of spindle assembly checkpoint during cell cycle. In dividing cells, Aurora kinase B is responsible for H3Ser10 phosphorylation, which in turn leads to the release of HP1β and to chromatin changes necessary for chromosome segregation (6, 7). Our data show a novel transcriptional role for Aurora kinase B, because activation of this kinase is essential for TR-dependent GH gene expression. We demonstrated in vivo and in vitro the interaction between TR and Aurora kinase B in a ligand-independent manner as well as the recruitment of Aurora kinase B to the GH promoter region upon hormone induction. A possible explanation for the induction of Aurora kinase B activity in response to T3 is that the phosphorylated form of this kinase is required for its promoter recruitment. Alternatively, factors such as nuclear receptor coactivators could be also mediate the interaction of Aurora kinase B with promoter regions.
This transcriptional effect occurs in nondividing cells at G0/G1, a cell-cycle stage where several genes are reexpressed. In noncycling differentiated plasma cells, high levels of H3Ser10 phosphorylation, generated by Aurora kinase B, have been shown to lead the delocalization of HP1β away from heterochromatin (16). These data together with our results, provide evidence that the role of Aurora kinase B is not only restricted to mitosis. Our studies have been performed in pituitary cells, but it could be interesting to check the transcriptional role of Aurora kinase B in other systems because overexpression of this kinase has been observed in hepatocellular carcinoma (29), biliary tract cancer (30), and acute myeloid leukemia among others (31). In fact, Aurora kinase B inhibitors are being evaluated in clinical trials for the treatment of cancer, because they induce apoptosis in tumor cells. Aurora kinase B could be an important regulator of genes implicated in pathological situations such as cancer. In this regard, it will be also necessary to perform microarray analyses in T3-treated cell in the presence or absence of Aurora B inhibitor to analyze which genes are transcriptionally controlled by Aurora B.
Taken together, our data demonstrate a novel mechanism by which T3-regulated gene transcription is controlled by the Aurora kinase B. Aurora kinase B mediates the T3-induced enhancement of histone H3Ser10 phosphorylation and the dissociation of HP1β from chromatin, thereby facilitating efficient transcriptional initiation of T3-target genes. The regulation of chromatin packaging with such a model would provide a particularly flexible mechanism to regulate genes that respond to hormone stimulus and are characterized by periods of induction followed by periods of silencing.
Materials and Methods
Cell culture and transfections
HEK293T and GH4C1 cells were cultured in DMEM supplemented with 10% fetal calf serum, and they were transfected by the calcium phosphate method or electroporation, respectively. The following amounts of plasmid were used per well (60 mm): 5 μg DR4TK-luc, 0.6 μg pRL-TK (renilla), 0.5 μg pSG5-TRα, 25–100 ng pcDNA-FLAG-AKB, and pcDNA–FLAG-AKB-K/R (K109R; kinase-negative). After transfection, the cells were preincubated for 48 h in DMEM supplemented with 10% AG1-X8 resin and charcoal-stripped newborn calf serum, and then they were exposed to 5 nm T3 (GH4C1) or 100 nm T3 (HEK293T) for different times where appropriate. When indicated, 2 μm ZM447439 (Tocris), 5 μm U0126 (Calbiochem), or 1 μm SB203580 (Calbiochem) were also added 30 min before the T3. These products were diluted in DMSO. Luciferase and renilla activity were assayed with a dual luciferase assay system (Promega). All experiments were repeated at least three times in duplicate, and all data are presented as the mean ± sd.
Immunoprecipitation (IP) and Western blot analyses
For Western blot assays, cells extracts were prepared in lysis buffer [50 mm Tris (pH 7.5), 150 mm NaCl, 0.5% SDS, 30 mm PPi, 0.5 m NaF, and protease inhibitors] and 5–10 μg was loaded onto 10% or 15% SDS gels. After the proteins were transferred to membranes, they were probed with the following antibodies: anti-phosphoSer10H3, anti-H3 (Upstate Biotechnology), anti-HP1β (Chemicon), anti-TRα (Santa Cruz) and anti-Aurora B (Abcam) and anti-HA (Sigma). For IP, GH4C1 cells extracts in buffer B [50 mm HEPES (pH 8), 600 mm KCL, 0.5% N-P40, 1 mm Na3VO4, 1 mm DTT, protease inhibitors]. These cell extracts (2 mg for each immunoprecipitation) were precleared for 2 h with 40 μl protein A agarose beads and incubated overnight with 60 μl protein A agarose beads previously coupled with 2 μg of the specific antibody or rabbit IgG. The immunoprecipitated proteins and 20 μg of total extract as the input were analyzed by Western blot using the antibodies indicated.
GST pull-down assays
Recombinant proteins were synthesized, purified on glutathione–Sepharose resin, and analyzed by SDS–PAGE. 35S-labeled Aurora B was generated with the TNT T7 Quick coupled in vitro transcription and translation kit and used in pull-down assays with 1 μg of GST or GST-fused proteins as described previously (32).
ChIP
ChIP experiments were essentially performed as described in the Chromatin Immunoprecipitation Assay kit (Upstate, Cat. 17-295) Sonication was performed using a Bioruptor UCD-200TM (Diagenode) following the manufacturer's instructions. GH4C1 cells were treated for 15 min in the presence or absence of T3 (5 nm), and IP was performed with the specific antibodies indicated. Quantification of ChIP was performed by real-time PCR, and the Ct values of the target sequences in the immunoprecipitate were calculated in relation to the input (INP) fractions by the comparative Ct method using the equation 2-[Ct(IP)-Ct(INP)]. Each of these values was referred to as the relative abundance with respect to untreated cells. GAPDH promoter was used in all experiments as negative control.
RNA interference experiments
Aurora-B siRNAs (2 μm, Dharmacon) were transfected with siPORT electroporation siRNA buffer (Ambion). GH4C1 cells were transfected by electroporation, as previously described (33). Briefly, 2–3 × 106 cells were mixed with reporter plasmids and exposed to a high-voltage pulse (170 to 200 V, 960 μF) in a Bio-Rad electroporator with a capacitor extender (Bio-Rad Laboratories, Richmond, CA). After 48 h, the medium was replaced by fresh medium with hormone-depleted serum, and after a further 24 h the cells were incubated with T3 for 15 min before performing Western blot analysis or for 8 h before carrying out RT-PCR assays. The decrease in the levels of Aurora-B was confirmed by Western blot assays.
Quantitative RT-PCR (QRT-PCR)
DNAse-treated RNA (2 μg) isolated using TRI Reagent (Sigma) was reverse transcribed with the SuperScript First Strand Synthesis System (Invitrogen Life Technologies) according the manufacturer's instructions. The resulting cDNA template (2 μl) was amplified with specific primers using the 2× Brilliant SYBR Green QPCR Kit (Stratagene). Product formation was detected by the incorporation of SYBR green I using ROX as a passive reference. Each value was normalized against the GAPDH gene and expressed as relative RNA abundance over time zero. Experiments were repeated at least three times and the primers used were as follows: GAPDH, forward (5′-ACACTGCATGCCATCACTGCC-3′), reverse (5′-GCCTGCTTCACCACCTTCTTG-3′); GH, forward (5′-CTGGCTGCTGACACCTACAA-3′), reverse (5′-AAGCGAAGCAATTCCATGTC-3′).
Kinase assay
GH4C1 cells were incubated for 15 min with T3 (5 nm). Aurora-B was then immunoprecipitated and incubated with H3 histone (10 μg, Roche) for 30 min at 30 C in kinase assay buffer [20 mm HEPES (pH 7.4), 150 mm KCl, 5 mm MnCl2, 5 mm NaF, 1 mm DTT, 140 μm cold ATP] in the presence or absence of T3 (50 nm). H3Ser10ph levels were detected by Western blotting with the specific antibody.
Immunofluorescence
GH4C1 cells were fixed with 4% formaldehyde for 10 min at room temperature and permeabilized with PBS containing 0.5% Triton for 10 min at room temperature. Slides were incubated overnight in antibody dilution buffer (1× phosphate-buffered saline, 1% BSA, 0.05% Triton X-100) containing the primary antibodies diluted 1:100 (anti-H3Ser10ph), 1:100 (anti-HP1), or 1:500 (anti-HA). After incubation, the slides were washed three times in antibody dilution buffer and incubated with Alexa Fluor 488- and 546-conjugated antibodies (1:500, Molecular Probes). The slides were washed three times in PBS, stained with DAPI (0.5 μg/ml), and mounted in vectashield antifade mounting medium (Vector Laboratories) to be analyzed by confocal microscopy. Confocal images were taken using a ×63/1.4–0.6 OIL on Leica microscope with the TCS SP2 confocal system. The projection images were produced using the Leica LCS software.
Cell-cycle analysis
GH4C1 cells were grown for 48 h in DMEM with hormone-depleted serum and incubated in the presence or absence of T3 for 15 min. Cells were collected and fixed in 70% ethanol for 15 min, incubated with RNase (1 mg/ml) at 37 C for 30 min, and labeled with 400 μl propidium iodide (50 μg/ml) for at least 15 min at room temperature. Cell-cycle profiles were determined by flow-cytometry on a FACScan flow cytometer, and the proportion of cells in the different phases was calculated from the DNA histograms.
Acknowledgments
We thank Dr. Tatsuka for Aurora kinase B expression vectors, Drs. M. Steller and E. Ballestar for their input at early stages of this work, and Dr. M. Vallejo, B. Cebolla, and M. Lasa for critical reading of this manuscript.
This work was supported by Grants BFU2006-13497 and BFU2009-11071 from the Ministerio de Educación y Ciencia and from the Fundación Médica Mutua Madrileña.
Present address for M.T.: Centro Nacional de Biotecnología, C/Darwin 3, Campus de la Universidad Autónoma de Madrid, 28049 Madrid. Present address for E.G.-G.: Centro de Biología Molecular Severo Ochoa, C/Nicolás Cabrera 1, Campus de la Universidad Autónoma de Madrid, 28049 Madrid.
Disclosure Summary: The authors have nothing to declare.
NURSA Molecule Pages:
Nuclear Receptors: TR-α;
Ligands: Thyroid hormone.
Footnotes
- ChIP
- Chromatin immunoprecipitation
- IP
- immunoprecipitation
- MSK1
- mitogen- and stress-associated protein kinase 1
- PRL
- prolactine
- RSK
- ribosomal S6 kinases
- siRNA
- small interfering RNA
- T3
- thyroid hormone
- TR
- thyroid nuclear receptor
- TRE
- thyroid hormone response element.
References
- 1. Elgin SC , Grewal SI. 2003. Heterochromatin: silence is golden. Curr Biol 13:R895–R898 [DOI] [PubMed] [Google Scholar]
- 2. Henikoff S. 2000. Heterochromatin function in complex genomes. Biochimica et biophysica acta 1470:O1–O8 [DOI] [PubMed] [Google Scholar]
- 3. Richards EJ , Elgin SC. 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108:489–500 [DOI] [PubMed] [Google Scholar]
- 4. Jenuwein T , Allis CD. 2001. Translating the histone code. Science 293:1074–1080 [DOI] [PubMed] [Google Scholar]
- 5. Li Y , Kirschmann DA , Wallrath LL. 2002. Does heterochromatin protein 1 always follow code? Proc Natl Acad Sci USA 99(Suppl 4):16462–16469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fischle W , Tseng BS , Dormann HL , Ueberheide BM , Garcia BA , Shabanowitz J , Hunt DF , Funabiki H , Allis CD. 2005. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438:1116–1122 [DOI] [PubMed] [Google Scholar]
- 7. Hirota T , Lipp JJ , Toh BH , Peters JM. 2005. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438:1176–1180 [DOI] [PubMed] [Google Scholar]
- 8. Chen JD , Evans RM. 1995. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377:454–457 [DOI] [PubMed] [Google Scholar]
- 9. Horner MA , Chen T , Thummel CS. 1995. Ecdysteroid regulation and DNA binding properties of Drosophila nuclear hormone receptor superfamily members. Dev Biol 168:490–502 [DOI] [PubMed] [Google Scholar]
- 10. Ruiz-Cortes ZT , Kimmins S , Monaco L , Burns KH , Sassone-Corsi P , Murphy BD. 2005. Estrogen mediates phosphorylation of histone H3 in ovarian follicle and mammary epithelial tumor cells via the mitotic kinase, Aurora B. Mol Endocrinol 19:2991–3000 [DOI] [PubMed] [Google Scholar]
- 11. Tashjian AH. 1979. Clonal strains of hormone-producing pituitary cells. Methods Enzymol 58:527–535 [DOI] [PubMed] [Google Scholar]
- 12. Maison C , Bailly D , Peters AH , Quivy JP , Roche D , Taddei A , Lachner M , Jenuwein T , Almouzni G. 2002. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30:329–334 [DOI] [PubMed] [Google Scholar]
- 13. Mateescu B , England P , Halgand F , Yaniv M , Muchardt C. 2004. Tethering of HP1 proteins to chromatin is relieved by phosphoacetylation of histone H3. EMBO Reports 5:490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minc E , Allory Y , Worman HJ , Courvalin JC , Buendia B. 1999. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 108:220–234 [DOI] [PubMed] [Google Scholar]
- 15. Peters AH , Kubicek S , Mechtler K , O'Sullivan RJ , Derijck AA , Perez-Burgos L , Kohlmaier A , Opravil S , Tachibana M , Shinkai Y , Martens JH , Jenuwein T. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12:1577–1589 [DOI] [PubMed] [Google Scholar]
- 16. Sabbattini P , Canzonetta C , Sjoberg M , Nikic S , Georgiou A , Kemball-Cook G , Auner HW , Dillon N. 2007. A novel role for the Aurora B kinase in epigenetic marking of silent chromatin in differentiated postmitotic cells. EMBO J 26:4657–4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y , Xia X , Fondell JD , Yen PM. 2006. Thyroid hormone-regulated target genes have distinct patterns of coactivator recruitment and histone acetylation. Mol Endocrinol 20:483–490 [DOI] [PubMed] [Google Scholar]
- 18. Davies SP , Reddy H , Caivano M , Cohen P. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johansen KM , Johansen J. 2006. Regulation of chromatin structure by histone H3S10 phosphorylation. Chromosome Res 14:393–404 [DOI] [PubMed] [Google Scholar]
- 20. Bartova E , Kozubek S , Jirsova P , Kozubek M , Gajova H , Lukasova E , Skalnikova M , Ganova A , Koutna I , Hausmann M. 2002. Nuclear structure and gene activity in human differentiated cells. J Struct Biol 139:76–89 [DOI] [PubMed] [Google Scholar]
- 21. Sadoni N , Langer S , Fauth C , Bernardi G , Cremer T , Turner BM , Zink D. 1999. Nuclear organization of mammalian genomes. Polar chromosome territories build up functionally distinct higher order compartments. J Cell Biol 146:1211–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skalnikova M , Kozubek S , Lukasova E , Bartova E , Jirsova P , Cafourkova A , Koutna I , Kozubek M. 2000. Spatial arrangement of genes, centromeres and chromosomes in human blood cell nuclei and its changes during the cell cycle, differentiation and after irradiation. Chromosome Res 8:487–499 [DOI] [PubMed] [Google Scholar]
- 23. Li J , Lin Q , Yoon HG , Huang ZQ , Strahl BD , Allis CD , Wong J. 2002. Involvement of histone methylation and phosphorylation in regulation of transcription by thyroid hormone receptor. Mol Cell Biol 22:5688–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Metzger E , Yin N , Wissmann M , Kunowska N , Fischer K , Friedrichs N , Patnaik D , Higgins JM , Potier N , Scheidtmann KH , Buettner R , Schule R. 2008. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol 10:53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fraga MF , Ballestar E , Villar-Garea A , Boix-Chornet M , Espada J , Schotta G , Bonaldi T , Haydon C , Ropero S , Petrie K , Iyer NG , Perez-Rosado A , Calvo E , Lopez JA , Cano A , Calasanz MJ , Colomer D , Piris MA , Ahn N , Imhof A , Caldas C , Jenuwein T , Esteller M. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Gent 37:391–400 [DOI] [PubMed] [Google Scholar]
- 26. Duong FH , Christen V , Lin S , Heim MH. Hepatitis C virus-induced up-regulation of protein phosphatase 2A inhibits histone modification and DNA damage repair. Hepatology 51:741–751 [DOI] [PubMed] [Google Scholar]
- 27. Vicent GP , Ballare C , Nacht AS , Clausell J , Subtil-Rodriguez A , Quiles I , Jordan A , Beato M. 2006. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell 24:367–381 [DOI] [PubMed] [Google Scholar]
- 28. Metzger E , Imhof A , Patel D , Kahl P , Hoffmeyer K , Friedrichs N , Muller JM , Greschik H , Kirfel J , Ji S , Kunowska N , Beisenherz-Huss C , Gunther T , Buettner R , Schule R. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature 464:792–796 [DOI] [PubMed] [Google Scholar]
- 29. Lin ZZ , Jeng YM , Hu FC , Pan HW , Tsao HW , Lai PL , Lee PH , Cheng AL , Hsu HC. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer 10:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen YC , Hu FC , Jeng YM , Chang YT , Lin ZZ , Chang MC , Hsu C , Cheng AL. 2009. Nuclear overexpression of mitotic regulatory proteins in biliary tract cancer: correlation with clinicopathologic features and patient survival. Cancer Epidemiol Biomarkers Prev 18:417–423 [DOI] [PubMed] [Google Scholar]
- 31. Oliveira FM , Lucena-Araujo AR , Leite-Cueva SD , Santos GA , Rego EM , Falcao RP. Segmental amplification of MLL gene associated with high expression of AURKA and AURKB genes in a case of acute monoblastic leukemia with complex karyotype. Cancer Genet Cytogenet 198:62–65 [DOI] [PubMed] [Google Scholar]
- 32. Mendez-Pertuz M , Sanchez-Pacheco A , Aranda A. 2003. The thyroid hormone receptor antagonizes CREB-mediated transcription. EMBO J 22:3102–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flug F , Copp RP , Casanova J , Horowitz ZD , Janocko L , Plotnick M , Samuels HH. 1987. cis-acting elements of the rat growth hormone gene which mediate basal and regulated expression by thyroid hormone. J Biol Chem 262:6373–6382 [PubMed] [Google Scholar]