

Abstract
Interstitial deletions affecting the long arm of chromosome 3 have been associated with a broad phenotype. This has included the features of blepharophimosis–ptosis–epicanthus inversus syndrome, Dandy-Walker malformation, and the rare Wisconsin syndrome. The authors report a young female patient presenting with features consistent with all 3 of these syndromes. This has occurred in the context of a de novo 3q22.3q24 microdeletion including FOXL2, ZIC1, and ZIC4. This patient provides further evidence for the role of ZIC1 and ZIC4 in Dandy-Walker malformation and is the third reported case of Dandy-Walker malformation to have associated corpus callosum thinning. This patient is also only the seventh to be reported with the rare Wisconsin syndrome phenotype.
Keywords: Dandy-Walker malformation, microdeletion, Wisconsin syndrome
Interstitial 3q deletions have been associated with a broad phenotype including dysmorphic facial features, ear and digit abnormalities, hypogonadism, intracranial malformations, cardiac and renal defects, intellectual disability, and developmental delay.1 Most reported cases have features consistent with blepharophimosis–ptosis–epicanthus inversus syndrome (OMIM: 110100). The blepharophimosis–ptosis–epicanthus inversus syndrome is a rare disorder associated with a collection of characteristic eyelid malformations—short palpebral fissures (blepharophimosis), ptosis, epicanthus inversus, and telecanthus. It is inherited in an autosomal-dominant manner with close to half of cases thought to arise from a de novo mutation.2 A strong association has been established between blepharophimosis–ptosis–epicanthus inversus syndrome and deletions in the 3q23 region causing haploinsufficiency of the FOXL2 gene (OMIM: 605597).3
Dandy-Walker malformation (OMIM: 220200) is the most common congenital defect of the cerebellum. It is defined by hypoplasia and upward rotation of the cerebellar vermis with associated dilatation of the fourth cerebral ventricle.4 Dandy-Walker malformation has been described in association with a wide variety of genetic disorders including 3q microdeletions. It is thought to arise sporadically with no defined Mendelian inheritance.5 Recent reports have proposed a link between Dandy-Walker malformation and interstitial 3q24 deletions affecting the ZIC1 (OMIM: 600470) and ZIC4 (OMIM: 608948) candidate genes.6
Wisconsin syndrome is a very rare phenotype first delineated by John M. Opitz in 1976 and later reported by Cohen.7 It was originally described as a combination of craniosynostosis, intellectual disability, upslanted palpebral fissures, small ears, and recessed fourth toes with short fourth metatarsals. The genetic basis of Wisconsin syndrome has been narrowed to the 3q24q25 region after the identification of further cases with 3q microdeletions.4
In this article, the authors describe a female patient with a phenotype encompassing the features of blepharophimosis–ptosis–epicanthus inversus syndrome, Dandy-Walker malformation, and Wisconsin syndrome. Genomic analysis revealed a large de novo microdeletion at 3q22.3q24, which includes FOXL2, ZIC1, and ZIC4. This case provides further evidence for the pathogenicity of ZIC1 and ZIC4 in Dandy-Walker malformation. This patient is also one of few cases linking the Wisconsin syndrome phenotype to 3q microdeletions.
Case Report
The patient was the first child of healthy nonconsanguineous parents. She had an uncomplicated antenatal period and was delivered at term. In the first few months of life, she was noted to be clinically microcephalic with a head circumference below the third percentile. She also exhibited several dysmorphic features. These included several eyelid abnormalities such as short and upslanted palpebral fissures, blepharophimosis, ptosis, epicanthus inversus, and telecanthus. She was also noted to have a prominent nose, coarse features, small low-set ears, and recessed fourth toes.
The patient was sent for comparative genomic hybridization (CGH) array testing at 2 months of age. Analysis was undertaken on a peripheral blood sample using the Affymetrix CytoScan 750 K platform (Affymetrix, CA, USA). This system has an effective copy number resolution of 200 kb and a long contiguous stretches of homozygosity resolution of 5 Mb. The reference sequence used for mapping was Genome Reference Consortium Human Build 37 (hg19). Software analysis was performed with the Affymetrix Chromosome Analysis Suite. Testing demonstrated a female profile with a large 12 Mb interstitial deletion within chromosome 3 from band 3q22.3 to 3q24 (chr3:136,403,035-148,341,113). This microdeletion is depicted in Figure 1 and encompasses 60 RefSeq genes. Subsequent karyotype testing of the patient’s parents demonstrated no abnormalities, confirming that her microdeletion had arisen de novo.
Figure 1.
Graphical representation of the RefSeq genes within the patient’s interstitial deletion at 3q22.3q24. Generated using the University of California Santa Cruz genome browser (https://genome.ucsc.edu).
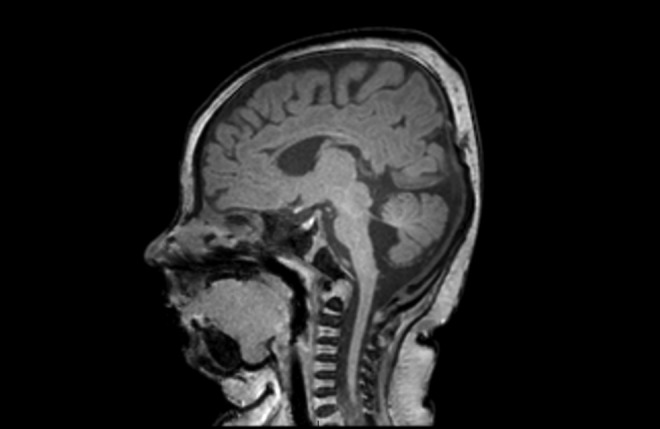
The magnetic resonance imaging of the patient’s brain was undertaken at 4 months of age as shown in Figure 2. This revealed hypoplasia of the cerebellar vermis with associated enlargement of the fourth cerebral ventricle. The patient also had a thinned corpus callosum and general prominence of her cerebrospinal fluid spaces consistent with a reduction in overall cerebral volume. By 8 months of age, the patient had also undergone renal and cardiac ultrasounds, which were normal. Ophthalmology assessment demonstrated significant anisometropia with a spherical refractive error of +1.25 in the right eye and −2.25 in the left. The patient underwent bilateral frontalis suspension surgery at 8 months of age using Visitec slings to lift her upper eyelids. She also underwent bilateral epicanthic fold reductions at 13 months of age to improve the cosmetic appearance of her eyes.
Figure 2.

T1-weighted sagittal magnetic resonance imaging (MRI) of the patient’s brain. Hypoplasia of the cerebellar vermis is shown with associated enlargement of the fourth cerebral ventricle.
At 6 months of age, the patient showed a tendency to increased extensor tone in the lower limbs with exaggerated toe grasp reflexes. She also presented with poor core stability and postural control, although her early prelingual speech and language were quite normal at this stage. By 12 months of age, the patient had a significant reduction in her tendency for excessive leg extensor tone and toe grasp. Her postural and balance control had also markedly improved. She presented overall at the lower end of normal for her developmental milestones, with gross motor and play skills equivalent to a normal 8-month-old child. Her fine motor skills and social development were progressing normally. By 18 months of age, the patient presented with excellent development of her receptive and expressive language and good progression of her motor skills.
Discussion
The authors have reported on a patient presenting with a multifaceted phenotype including microcephaly, dysmorphic facial features, digital abnormalities, intracranial malformations, and early developmental delay. Genomic analysis revealed a large de novo interstitial deletion at 3q22.3q24 including FOXL2, ZIC1, and ZIC4. The elements of this patient’s phenotype represent a combination of blepharophimosis–ptosis–epicanthus inversus syndrome, Dandy-Walker malformation, and Wisconsin syndrome. This case is one of few with Dandy-Walker malformation to have associated corpus callosum thinning. It also provides additional evidence for the role of ZIC1 and ZIC4 in this disease. This is also only the seventh reported case of a patient exhibiting the Wisconsin syndrome phenotype.
The patient’s eyelid abnormalities are consistent with the well-recognized features of blepharophimosis–ptosis–epicanthus inversus syndrome. Recent literature has established a strong association between haploinsufficiency of FOXL2 and blepharophimosis–ptosis–epicanthus inversus syndrome. The FOXL2 is a putative forkhead transcription factor gene located at 3q23 and was first described in connection with blepharophimosis–ptosis–epicanthus inversus syndrome by Crisponi et al.3 Several studies have shown that FOXL2 is present in the developing eyelids of both mice and humans, and it remains the only gene known to be associated with blepharophimosis–ptosis–epicanthus inversus syndrome.3,8-11 Most cases of blepharophimosis–ptosis–epicanthus inversus syndrome are thought to arise from intragenic mutations of FOXL2, with a smaller proportion resulting from 3q23 microdeletions.9 There remains uncertainty about the correlation between the extent of FOXL2 deletion or mutation with the severity of the blepharophimosis–ptosis–epicanthus inversus syndrome phenotype.12 Several recent reports have described patients with blepharophimosis–ptosis–epicanthus inversus syndrome with an intact FOXL2 region but nearby interstitial deletions.9-10 This suggests the presence of FOXL2 regulatory elements within 3q23. Deletion or mutation of these regulatory regions may be sufficient to cause blepharophimosis–ptosis–epicanthus inversus syndrome rather than disruption of FOXL2 itself.
Two types of blepharophimosis–ptosis–epicanthus inversus syndrome have been established after initial description by Zlotogora et al.13 Type 1 includes the characteristic eyelid features as well as female infertility, whereas type 2 involves the eyelid abnormalities alone. Several reports have been made of patients with blepharophimosis–ptosis–epicanthus inversus syndrome and associated ovarian failure.14,15 This has been reinforced by studies demonstrating the expression of FOXL2 in developing ovaries and that FOXL2 mutations can be associated with ovarian tumours.16,17 The patient will require close follow-up during adolescence to establish whether her ovarian function will be affected.
The posterior fossa pathology this patient presented with is suggestive of a mild variant of Dandy-Walker malformation. There was also associated reduction in overall cerebral volume and thinning of the corpus callosum. The genetic basis of Dandy-Walker malformation remains relatively poorly understood, and it has been reported in association with many chromosomal abnormalities. Grinberg et al6 defined the first critical region at 3q24q25.1 after comparing 8 patients with Dandy-Walker malformation and 3q deletions. Some of these patients had milder variants of Dandy-Walker malformation similar to this patient’s presentation. ZIC1 and ZIC4 were suggested as possible candidate genes as they encode zinc finger proteins that have been linked to cerebellar and spinal cord development.4 Mouse models have demonstrated that deletion of ZIC1 and ZIC4 can cause variable hypoplasia of the cerebellum and similar posterior fossa abnormalities to Dandy-Walker malformation.6,18 There have subsequently been at least 7 additional cases reported with features of Dandy-Walker malformation and associated 3q deletions affecting ZIC1 and ZIC4.4-5,19-22 A comparison of the 15 total cases is presented in Table 1. There is a wide variation in the severity of Dandy-Walker malformation between these patients. Almost all were reported to have some associated developmental delay and approximately half had concurrent blepharophimosis–ptosis–epicanthus inversus syndrome. The patient provides further evidence for the pathogenicity of ZIC1 and ZIC4 in Dandy-Walker malformation. This is also only the third reported case of Dandy-Walker malformation to have associated corpus callosum thinning.
Table 1.
Overview of DWM Patients With 3q Deletions.
| Current Patient, 2016 | Grinberg et al,6 2004 | Grinberg et al,6 2004 | Grinberg et al,6 2004 | Grinberg et al,6 2004 | Grinberg et al,6 2004 | Grinberg et al,6 2004 | Grinberg et al,6 2004 | |
|---|---|---|---|---|---|---|---|---|
| Patient details | ||||||||
| Identifier | − | LR00-225 | LR03-384 | LR01-306 | LR01-242 | LR02-027 | LR03-317 | LR01-273 |
| 3q deletion | 3q22.3q24 | 3q22.2q25.2 | 3q22q26 | 3q23q25.31 | 3q23q25.31 | 3q23q25.1 | 3q24q25.1 | 3q22.2q25.1 |
| DWM features | ||||||||
| Severity of imaging changes | + | ++ | +++ | + | + | +++ | ++ | ++ |
| Other features | ||||||||
| Corpus callosum thinning | + | NR | NR | NR | NR | NR | NR | NR |
| Microcephaly | + | NR | NR | NR | NR | NR | NR | NR |
| BPES | + | + | − | + | − | − | − | + |
| Developmental delay | + | + | + | + | + | + | + | + |
| Grinberg et al,6 2004 | Lim et al,19 2011 | Ramieri et al,20 2011 | Tohyama et al,5 2011 | Weber et al,22 2011 | D’Amours,21 2012 | Ferraris et al,4 2013 | Ferraris et al,4 2013 | |
| Patient details | ||||||||
| Identifier | LR01-325 | − | − | − | HD24 | CCM067 | CCM095 | |
| 3q deletion | 3q25.1q25.33 | 3q22.3q25.2 | 3q22.1q25.2 | 3q23q25.1 | 3q23q25.1 | 3q25.1q25.32 | 3q22.3q25.31 | 3q23q26.1 |
| DWM features | ||||||||
| Severity of imaging changes | ++ | ++ | ++ | + | ++ | + | +++ | +++ |
| Other features | ||||||||
| Corpus callosum thinning | NR | NR | NR | NR | NR | NR | + | + |
| Microcephaly | − | − | + | − | + | − | NR | NR |
| BPES | − | + | + | − | + | − | + | − |
| Developmental delay | + | + | + | + | + | NR | + | + |
Abbreviations: BPES, blepharophimosis–ptosis–epicanthus inversus syndrome; DWM, Dandy-Walker malformation; NR, not reported.
Note. + = mild, ++ = moderate, +++ = severe.
The patient’s unusual dysmorphic features were reminiscent of the rare Wisconsin syndrome phenotype. Ko et al23 reported the second case of Wisconsin syndrome in 2002 and identified an associated 3q microdeletion. Willemsen et al24 subsequently described the third and fourth cases and narrowed the critical region to 3q24q25. Ferraris et al4 then recently reported the fifth and sixth cases and proposed more accurate diagnostic criteria—coarse facies, prominent nasal tip, bushy eyebrows, high arched/upsweeping eyebrows, and a full/everted lower lip. A comparison of the 6 cases is presented in Table 2. This patient is only the seventh reported case of the Wisconsin syndrome phenotype and contributes to the association between Wisconsin syndrome and 3q deletions. This is also only the second Wisconsin syndrome case to have associated blepharophimosis–ptosis–epicanthus inversus syndrome and the third with concurrent Dandy-Walker malformation. This patient differs from the other reported cases in that her developmental delay only manifested early in life and had improved markedly by 18 months of age. The other reported patients with Wisconsin syndrome all had persisting developmental delay and intellectual disability beyond this age.
Table 2.
Overview of Patients With WS Having 3q Deletions.
| Current Patient, 2016 | Cohen,7 1986 | Ko,23 2003 | Willemsen,24 2011 | Willemsen,24 2011 | Ferraris et al,4 2013 | Ferraris et al,4 2013 | |
|---|---|---|---|---|---|---|---|
| Patient details | |||||||
| Identifier | − | − | − | 1 | 2 | CCM067 | CCM095 |
| 3q deletion | 3q22.3q24 | NR | 3q23q25 | 3q23q25.3 | 3q25.1q25.33 | 3q22.3q25.31 | 3q23q26.1 |
| Original WS features | |||||||
| Intellectual disability | − | + | + | + | + | + | + |
| Upslanted palpebral fissures | + | + | + | + | − | + | − |
| Small ears | + | + | − | − | − | − | − |
| Recessed fourth toes | + | + | + | NR | NR | + | + |
| Revised WS features | |||||||
| Coarse facies | + | + | + | + | + | + | + |
| Prominent nasal tip | + | + | + | + | + | + | + |
| Bushy eyebrows | + | + | + | + | + | + | + |
| High arched/upsweeping eyebrows | + | + | + | + | + | + | + |
| Full/everted lower lip | + | + | + | + | + | + | + |
| Other features | |||||||
| BPES | + | − | − | − | − | + | − |
| DWM | + | NR | NR | NR | NR | + | + |
Abbreviations: BPES, blepharophimosis–ptosis–epicanthus inversus syndrome; DWM, Dandy-Walker malformation; NR, not reported; WS, Wisconsin syndrome.
Note. + = present, − = absent.
Footnotes
Author Contributions: AR was involved in patient care and manuscript development. DC acted as treating consultant and was also responsible for patient care and manuscript development.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval: This study was approved by the UnitingCare Health Human Ethics Research Committee.
References
- 1. Weise A, Mrasek K, Klein E, et al. Microdeletion and microduplication syndromes. J Histochem Cytochem. 2012;60(5):346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oley C, Baraitser M. Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES syndrome). J Med Genet. 1988;25(1):47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crisponi L, Deiana M, Loi A, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27(2):159–166. [DOI] [PubMed] [Google Scholar]
- 4. Ferraris A, Bernardini L, Sabolic Avramovska V; Italian CBCD Study Group. Dandy-Walker malformation and Wisconsin syndrome: novel cases add further insight into the genotype-phenotype correlations of 3q23q25 deletions. Orphanet J Rare Dis. 2013;8:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tohyama J, Kato M, Kawasaki S, et al. Dandy-Walker malformation associated with heterozygous ZIC1 and ZIC4 deletion: report of a new patient. Am J Med Genet A. 2011;155(1):130–133. [DOI] [PubMed] [Google Scholar]
- 6. Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. 2004;36(10):1053–1055. [DOI] [PubMed] [Google Scholar]
- 7. Cohen MM. Craniosynostosis: Diagnosis, Evaluation, and Management. New York, NY: Raven Press; 1986. [Google Scholar]
- 8. Zahanova S, Meaney B, Łabieniec B, Verdin H, De Baere E, Nowaczyk MJ. Blepharophimosis-ptosis-epicanthus inversus syndrome plus: deletion 3q22.3q23 in a patient with characteristic facial features and with genital anomalies, spastic diplegia, and speech delay. Clin Dysmorphol. 2012;21(1):48–52. [DOI] [PubMed] [Google Scholar]
- 9. Beysen D, Raes J, Leroy BP, et al. Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet. 2005;77(2):205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alao MJ, Lalèyè A, Lalya F, et al. Blepharophimosis, ptosis, epicanthus inversus syndrome with translocation and deletion at chromosome 3q23 in a black African female. Eur J Med Genet. 2012;55(11):630–634. [DOI] [PubMed] [Google Scholar]
- 11. Cocquet J, Pailhoux E, Jaubert F, et al. Evolution and expression of FOXL2. J Med Genet. 2002;39(12):916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Baere E, Beysen D, Oley C, et al. FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am J Hum Genet. 2003;72(2):478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zlotogora J, Sagi M, Cohen T. The blepharophimosis, ptosis, and epicanthus inversus syndrome: delineation of two types. Am J Hum Genet. 1983;35(5):1020–1027. [PMC free article] [PubMed] [Google Scholar]
- 14. Siewert AL, Stein Q, Flanagan J, Hansen KA. Blepharophimosis-ptosis-epicanthus inversus syndrome and hypergonadotropic hypogonadism. Fertil Steril. 2008;90(5):e11–e12 [DOI] [PubMed] [Google Scholar]
- 15. Corrêa FJ, Tavares AB, Pereira RW, Abrão MS. A new FOXL2 gene mutation in a woman with premature ovarian failure and sporadic blepharophimosis-ptosis-epicanthus inversus syndrome. Fertil Steril. 2010;93(3):3–6. [DOI] [PubMed] [Google Scholar]
- 16. Laissue P, Vinci G, Veitia RA, Fellous M. Recent advances in the study of genes involved in non-syndromic premature ovarian failure. Mol Cell Endocrinol. 2008;282(1-2):101–111. [DOI] [PubMed] [Google Scholar]
- 17. Shah SP, Köbel M, Senz J, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360(26):2719–2729. [DOI] [PubMed] [Google Scholar]
- 18. Aruga J, Minowa O, Yaginuma H, et al. Mouse Zic1 is involved in cerebellar development. J Neurosci. 1998;18(1):284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim BC, Park WY, Seo EJ, Kim KJ, Hwang YS, Chae JH. De novo interstitial deletion of 3q22.3-q25.2 encompassing FOXL2, ATR, ZIC1, and ZIC4 in a patient with blepharophimosis/ptosis/epicanthus inversus syndrome, Dandy-Walker malformation, and global developmental delay. J Child Neurol. 2011;26(5):615–618. [DOI] [PubMed] [Google Scholar]
- 20. Ramieri V, Tarani L, Costantino F, et al. Microdeletion 3q syndrome. J Craniofac Surg. 2011;22(6):2124–2128. [DOI] [PubMed] [Google Scholar]
- 21. D’Amours G, Kibar Z, Mathonnet G, et al. Whole-genome array CGH identifies pathogenic copy number variations in fetuses with major malformations and a normal karyotype. Clin Genet. 2012;81(2):128–141. [DOI] [PubMed] [Google Scholar]
- 22. Weber S, Landwehr C, Renkert M, et al. Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrol Dial Transplant. 2011;26(1):136–143. [DOI] [PubMed] [Google Scholar]
- 23. Ko WT, Lam WF, Lo FM, Chan WK, Lam TS. Wisconsin syndrome in a patient with interstitial deletion of the long arm of chromosome 3: further delineation of the phenotype. Am J Med Genet A. 2003;120A(3):413–417. [DOI] [PubMed] [Google Scholar]
- 24. Willemsen MH, de Leeuw N, Mercer C, et al. Further molecular and clinical delineation of the Wisconsin syndrome phenotype associated with interstitial 3q24q25 deletions. Am J Med Genet A. 2011;155A(1):106–112. [DOI] [PubMed] [Google Scholar]