

Abstract
The effects of thyroid hormone (TH) on development and metabolism are exerted at the cellular level. Metabolism and action of TH take place intracellularly, which require transport of the hormone across the plasma membrane. This process is mediated by TH transporter proteins. Many TH transporters have been identified at the molecular level, although a few are classified as specific TH transporters, including monocarboxylate transporter (MCT)8, MCT10, and organic anion-transporting polypeptide 1C1. The importance of TH transporters for physiology has been illustrated dramatically by the causative role of MCT8 mutations in males with psychomotor retardation and abnormal serum TH concentrations. Although Mct8 knockout animals have provided insight in the mechanisms underlying parts of the endocrine phenotype, they lack obvious neurological abnormalities. Thus, the pathogenesis of the neurological abnormalities in males with MCT8 mutations is not fully understood. The prospects of identifying other transporters and transporter-based syndromes promise an exciting future in the TH transporter field.
Cellular thyroid hormone regulation requires transporters. We review the current knowledge of thyroid hormone transporters and discuss some of the major challenges in the field.
“There can be little question that the thyroid hormones must ultimately enter the tissue cells (…)” (1). This statement formulated some 50 yr ago is still a basic principle for understanding thyroid hormone (TH) action. However, the concept about the underlying mechanism for tissue uptake of TH has dramatically changed. Although Robbins and Rall (1) were convinced that TH traversed the plasma membrane via diffusion, it is currently beyond doubt that transporter proteins are required for plasma membrane transport of TH (2, 3).
The well-known effects of TH on development and metabolism are ultimately the result of changes in signaling pathways at the cellular level. Many actions of TH are mediated by changes in gene transcription, when the bioactive hormone T3 binds to its receptors [TH receptor (TRs)], which function as transcription factors (4). Deiodinating enzymes regulate intracellular concentrations of T3. The type 2 deiodinase (D2) converts the precursor T4 to T3, whereas the D3 degrades T4 to rT3 and T3 to 3,3′-diiodothyronine (T2) (5).
The prerequisite for the intracellular processes of deiodination and genomic actions of TH is the translocation of the hormone across the plasma membrane. In this review, we first describe established concepts of TH transport (the knowns). Next, we discuss several gaps in our knowledge of established TH transporters (the unknowns). Both sections will be biased toward the TH transporter monocarboxylate transporter (MCT)8. Finally, we reflect on some issues beyond the current dogmas, which may stimulate future research (the guesses). The most important knowns, unknowns, and guesses are summarized in figure 1.
Fig. 1.
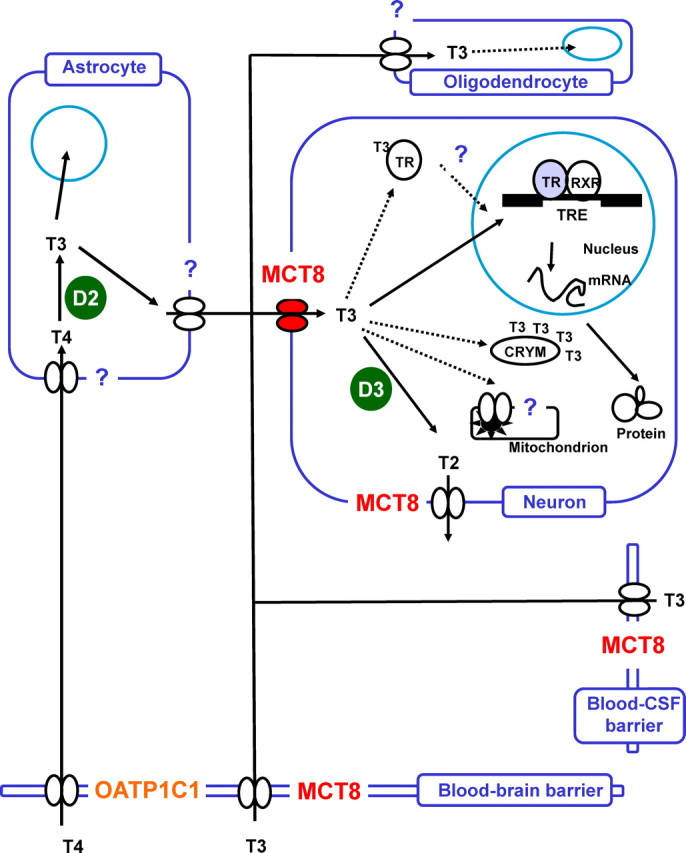
Model of TH regulation in brain. Transporters (paired ovals) are required for uptake and release of T4 and T3. D2 converts T4 to bioactive T3, whereas D3 degrades T3 to 3,3′-T2. T3 in brain is either derived from the circulation [via the blood-brain barrier or indirectly via the blood-cerebrospinal fluid (CSF) barrier] or locally produced from T4 in astrocytes by D2. Intracellular T3-binding proteins (e.g. CRYM) may store cytoplasmic T3 and function in intracellular T3 transport. Ultimately, T3 binds to its nuclear receptor (TR) and modulates gene expression of T3-target genes. T3 actions in mitochondria also require transporter proteins. All principal cells of the brain are T3 targets: neurons, astrocytes, and oligodendrocytes. The degree of certainty of this model is reflected by the color of the symbols. Filled symbols reflect established knowledge (the knowns). White symbols are surmised concepts (the unknowns). Question marks indicate possible novel players or functions in local TH regulation (the guesses). RXR, Retinoic X receptor; TRE, T3-responsive element.
The Knowns
TH transport processes
It has been known for a long time that TH accumulates in tissues. These early observations were well explained by assuming that high-affinity intracellular binding proteins determined intracellular TH accumulation (1). Based on the chemical properties of the molecule, it was rationalized that transfer of TH across the plasma membrane occurred via diffusion (6). Although it was recognized that the side chain was hydrophilic, it was assumed that the hydrophobic aromatic part of T3 and T4 interacts with the lipid bilayer, presumably constituting the first step to enter the cell. Subsequent studies were dedicated to characterize the cell surface binding properties and binding proteins for TH (for a historical overview, see Ref. 2). Important progress was made by reports showing that TH uptake was saturable, energy- and Na+-dependent in certain cell types (2). From this, it was deduced that transporter proteins were responsible for the transport of TH across the plasma membrane (2). This initiated experiments aimed to investigate kinetic properties of transport processes by in vitro and in vivo studies (2).
Only a decade ago, the first TH transporter was identified at the molecular level (7). In the following years, it was demonstrated that iodothyronine derivatives are ligands for several types of transporters, including the Na+/taurocholate cotransporting polypeptide [solute carrier family (SLC)10A1] (8, 9), multidrug resistance-associated proteins (10), the heterodimeric L-type amino acid transporters (LATs) LAT1 and LAT2 (reviewed in Ref. 11), various members of the organic anion-transporting polypeptide (OATP) family (reviewed in Refs. 12 and 13), and the MCTs MCT8 and MCT10 (reviewed in Ref. 14). Most of these transporters accept a variety of ligands, with notable exceptions for MCT8 (SLC16A2) and to a lesser extent MCT10 (SLC16A10) and OATP1C1 (SLCO1C1).
Function and expression of MCT8
Based on homology, MCT8 belongs to the MCT family. Most members of this family are orphan transporters (MCT5, MCT7, MCT9, MCT11-14), whereas MCT1-4 transport simple monocarboxylates, such as lactate and pyruvate (15). MCT6 is involved in transport of compounds with a carboxyl group, such as the diuretics bumetanide and nateglinide (16).
The MCT8 gene is located on human chromosome Xq13.2. The six exons encode a protein with 12 predicted transmembrane domains (TMDs); both N and C terminus have a cytoplasmic localization.
A major breakthrough was accomplished when rat Mct8 (rMct8) was identified as a specific transporter of TH (17). Functional characterization of rMct8 in Xenopus laevis oocytes revealed that uptake of the iodothyronines T4, T3, rT3, and 3,3′-T2 was strongly stimulated if rMct8 was expressed (17). Neither sulfated and sulfamated iodothyronines nor amino acids (Leu, Phe, Tyr, and Trp) and lactate were transported by rMct8. This specificity was substantiated by inhibition studies, demonstrating strong effects of iodothyronine derivatives on rMct8-mediated T3 and T4 uptake, in contrast with several other compounds.
Subsequent studies of human MCT8 showed that its TH transport characteristics are highly similar to rMct8 (18). Importantly, it was demonstrated that MCT8 markedly enhances intracellular deiodination of various iodothyronines. Furthermore, coexpression of MCT8 with the cytoplasmic TH binding protein μ-crystallin (CRYM) resulted in a strong accumulation of intracellular TH. These data strongly supported the concept that MCT8 facilitates transport of TH across the plasma membrane and, thus, provides intracellular compartments, including cytosol and endoplasmic reticulum with TH. In contrast to other members of the MCT family (MCT1-4), expression and function of MCT8 appears to be independent of the ancillary proteins basigin, embigin, or neuroplastin (19). MCT8 rather appears to form homodimers as evidenced by in vitro studies (19, 20).
In different species, MCT8 expression has been detected in numerous tissues, including brain, heart, liver, kidney, adrenal, and thyroid gland (21, 22). Because mutations in MCT8 result in a neurological phenotype (see below), MCT8 expression in mouse brain has been investigated in further detail. Intense staining of Mct8 was observed in the choroid plexus (23, 24, 25, 26). Prominent Mct8 expression was also noted in different cortical layers, parts of the limbic system (amygdala, hippocampus, and olfactory bulb) and hypothalamus (23, 25). Furthermore, MCT8 is expressed in microvessels of mice and humans (25, 26). This is in agreement with functional studies suggesting that Mct8 is important for transport of T3 across the blood-brain barrier (25, 26). In humans, MCT8 expression has been documented in cortex, hippocampus, and circumventricular organs (25, 26, 27). Furthermore, MCT8 is present in different parts of the human hypothalamus and folliculostellate cells of the anterior pituitary (27, 28). Taken together, these data demonstrate that MCT8 has a distinct expression pattern in brain.
Knowledge about MCT8 regulation is limited, although several studies have incidentally reported on changes in Mct8 expression under different conditions. With advancing age, Mct8 expression alters as evidenced by changes in Mct8 expression during brain development of mice, in livers of old rats and in human placenta with advancing gestation (23, 29, 30). Elevated MCT8 expression was found in placenta associated with severe intrauterine growth restriction, in femurs of hypothyroid mice, and in liver and skeletal muscle of critically ill patients and rabbits (30, 31, 32).
Down-regulation of MCT8 expression in osteoblasts and rabbits with critical illness after TH treatment suggests a compensatory mechanism (31, 32). However, thyroid state is not a modulator of MCT8 per se, because hypothalamic MCT8 staining was unremarkable in a hyperthyroid subject (27). Moreover, hypothyroidism in athyroid Pax8 knockout (KO) animals induces expression of D2 in brain, ensuing increased local T3 production, whereas Mct8 expression is not affected (23). Thus, MCT8 is regulated under different conditions, although the mechanisms and appropriateness of these responses remain to be elucidated.
Mutations in MCT8
The importance of MCT8 for human physiology was unequivocally demonstrated by the identification of MCT8 mutations in males with mental retardation and endocrine abnormalities (33, 34). The clinical features are most prominently manifested in the neurological abnormalities (for an extensive description of typical clinical characteristics, see Ref. 35). Although there is phenotypic variability (see below), all affected males display marked cognitive disabilities with intelligence quotient values mostly below 40 (35). Speech development is typically absent, and the patients communicate by nonverbal acts, such as laughing, crying, and making sounds.
Most patients are characterized by a severe axial hypotonia, of which poor head control is the most obvious feature. All patients display muscle hypoplasia. Hypotonia of the limbs in childhood progresses into spastic quadriplegia with advancing age. Involuntary and dystonic movements, which can be provoked upon stimuli, are commonly described. Although height is unremarkable, weight is below the third percentile in most patients.
Heterozygous females typically do not display a neurological phenotype. Incidentally, cognitive impairments have been reported in female carriers, but a causative link with MCT8 mutations is unproven (35). To date, an obvious neurological phenotype similar to affected males has been reported in one female with a heterozygous MCT8 mutation accompanied by unfavorable X-chromosomal inactivation (36). In the first years of life, global myelination delay may be detected by magnetic resonance imaging (37, 38).
Because the clinical phenotype of the affected males was reminiscent of the Allan-Herndon-Dudley syndrome (AHDS), originally described in 1944, Schwartz et al. (39) tested the involvement of MCT8 in this disease. They demonstrated that mutations in MCT8 are the genetic explanation for affected males in AHDS families. Recently, MCT8 mutations have also been reported in patients with Pelizaeus-Merzbacher-like disease, which display clinical features similar to AHDS and elevated T3 levels (40). Apparently, it is difficult to classify genetically distinct mental retardation syndromes on the basis of clinical features. This leaves the possibility that MCT8 is affected in other eponymous syndromes clinically resembling AHDS, of which the genetic basis is as yet unknown.
Characteristic for patients with MCT8 mutations is the remarkable combination of TH serum concentrations. The most common finding are the elevated serum T3 and reduced T4, free T4, and rT3 concentrations. TSH levels are usually within the normal range, but mean TSH is doubled compared with nonaffected individuals. Regarding the endocrine derangements, it is important to consider the following. First, abnormalities may be subtle, depending on the severity of the disease (see below). Second, values must be interpreted relative to age-specific reference ranges. This is essential for serum T3 concentrations, which are higher in infants. Third, the combination of the above-mentioned TH parameters is pathognomonic for MCT8 patients, rather than a single measurement, such as serum T3 (Fig. 2).
Fig. 2.

Thyroid parameters in serum from 17 families with MCT8 mutations. Serum levels (mean ± SE) of (A) TSH, (B) fT4, (C) T3, and (D) rT3 in affected males (Pat, squares), unaffected heterozygous females (Het, triangles), and unaffected family members (Nor, circles).
To date, MCT8 mutations have been reported in over 50 families (for a recent overview, see Ref. 41). This suggests that MCT8 mutations are not a rare cause of X-linked mental retardation, although the prevalence is currently unknown.
Many different types of mutations have been identified. Approximately 30% are deletions or mutations introducing premature stop codons. Such mutations conceivably result in a predicted complete loss of transporter function, but to deduce the impact of single amino acid substitutions, insertions, or deletions on MCT8 function is complicated. Two models have been used to study the effects of MCT8 mutations on its transport capacity: mammalian cells (transiently or stably) transfected with mutant MCT8 cDNA and fibroblasts derived from MCT8 patients.
Our lab undertook the functional analysis of MCT8 mutations by transfection studies in JEG3 cells, which express little endogenous MCT8. Cells overexpressing wild-type and mutant MCT8 cDNA alone or in combination with D2 or D3 were tested for T3 and T4 transport and metabolism. Most mutants were shown to be inactive, although some exhibited (significant) residual activity. The mutants with complete loss-of-function are mostly explained by a decreased plasma membrane expression as evidenced from a predominant cytosolic localization or by a diminished or absence of protein expression (42). Trafficking of mutant MCT8 to the plasma membrane appears cell-type dependent, probably reflecting differences in the cellular machinery, such as regulatory proteins or the capacity to form heterodimers with wild-type MCT8 (19, 43).
A second system to investigate the effects of MCT8 mutations are skin fibroblasts obtained from patients (44, 45). Although MCT8 expression was not detected by Western blot analysis or immunocytochemistry, the transporter appears functionally important, because T3 and T4 uptake were reduced by 50–60% in fibroblasts from patients as compared with controls (44, 45). The advantage of this model is that the analyses are performed in the genetic background of the patient.
Although it is too early to localize hotspots in the MCT8 gene, several mutations have been reported twice in unrelated families (41). This may be explained if particular sequences are prone to mutation and/or if not all mutations affect transport function. The latter suggestion is supported by an MCT8 polymorphism (S107P), which is not associated with clinical symptoms or TH parameters (46, 47). Even apparently drastic mutations do not necessarily impinge on MCT8 transport function. Transport was not affected in a series of experiments in which MCT8 Cys-to-Ala mutants were studied (19). Furthermore, an M402I mutation in a boy with mental retardation but not typical AHDS features did not affect T3 and T4 transport in cells overexpressing the mutant MCT8 or in the patient’s fibroblasts (our unpublished data). Thus, to address the pathogenicity of MCT8 mutations, it is required to test their effects on transport function.
Treatment is currently limited to supportive care, although the first initiatives to counteract the damaging effects of the endocrine abnormalities have been reported (see The Unknowns). Diagnosis is important for providing an etiologic explanation of the disease to the family and consequently for genetic counseling.
Relevance of Mct8 KO mice for patients
Different Mct8 KO mouse models have been created to understand the mechanisms of disease in patients with MCT8 mutations (24, 25, 48, 49). Mct8 KO animals have markedly decreased T4 and rT3 levels and increased T3 and TSH levels compared with wild-type animals, thereby perfectly representing the endocrine “fingerprint” observed in MCT8 patients (48, 49).
Despite the significant expression of Mct8 in liver, T3 uptake into the liver was not different in mutant vs. control mice, suggesting the presence of other TH transporter. This explains the increased T3 concentration in liver tissue and the elevated T3-sensitive D1 expression and activity. Increased D1 activity in liver of MCT8 patients perfectly explains the decreased serum levels of rT3. Because rT3 is the preferred substrate of D1, increased D1 activity results in an increased rT3 degradation (50). Furthermore, rT3 production is probably low because of the low T4 substrate levels.
From the inappropriately high TSH levels in MCT8 patients and in Mct8 KO mice, it was hypothesized that the hypothalamus-pituitary-thyroid axis was compromised in Mct8 KO mice. Therefore, the hypothalamus and pituitary were further investigated to address at which level the negative feedback loop was affected. At the hypothalamic level, expression of the T3-responsive TRH was increased in Mct8 KO animals (51). Only high doses of T4, but not of T3, reversed the abnormal TRH expression. This observation indicates that the hypothalamus of Mct8 null mice still responds to local T4 to T3 conversion. Presumably, an impaired Mct8-mediated T3 uptake is responsible for the aberrant TRH expression.
Expression of several T3-responsive genes is not affected in the pituitary of Mct8 KO animals, indicating adequate intracellular T3 levels. This is most likely ensured by the increased pituitary D2 activity, which compensates for the decrease in serum T4, resulting in the maintenance of local T3 production. Pituitary uptake of T3 may also be diminished in Mct8 KO mice, although Mct8 is not markedly expressed in hormone-producing cells of the mouse pituitary. In Mct8 KO mice rendered hypothyroid, only a high dose of T3 is able to restore TSH levels, whereas in wild-type animals, similar TSH levels were achieved using a low dose of T3. This may be explained in part by the lack of effect of T3 on the hypothalamus.
Thus, the studies in Mct8 KO mice indicate that the hypothalamus and, consequently, the pituitary are relatively insensitive to TH. Together with the expression of MCT8 in human hypothalamus and pituitary, the animal studies provide a plausible explanation for the inappropriately elevated TSH levels in patients with MCT8 mutations.
The most unanticipated finding in Mct8 KO mice is the absence of an overt neurological phenotype (48, 49). Recently, extensive neurological and behavioral testing revealed only subtle behavioral abnormalities in Mct8 KO mice (25). These results are surprising, because uptake of T3 in brains of Mct8 null mice is largely impaired (48, 49). Consequently, T3 content in brain is also decreased (Fig. 1).
The decreased D3 activity, which is inversely regulated by its substrate T3, probably reflects a beneficial response aimed to counteract the harmful effects of decreased T3 transport. Parallel with the decreased serum T4 levels, brains of Mct8 KO animals contain less T4 than wild-type mice. In Mct8 KO brains, D2 activity, which is inversely regulated by its substrate T4, is increased. Thus, local T3 production in brain may be relatively normal due to the decrease in T4 levels and increase in D2 activity. Brain T3 levels are nevertheless markedly decreased despite high serum T3 levels and decreased T3 degradation, due to a dramatic reduction in brain T3 uptake.
T3 uptake (measured in a 15-min period) in cultured neurons from Mct8 KO mice was diminished compared with wild-type mice (25). However, expression of the T3-sensitive gene Hairless with increasing concentrations of T3 added to cultured neurons did not differ between Mct8 KO and wild-type animals (24). Also, the effect of T3 on dendritic outgrowth in cultured neurons was similar in Mct8 KO and wild-type animals (49). This may be explained by differences in neuronal cell populations used in the different studies, where subsets of neurons may be more vulnerable to lack of functional Mct8 than others. Indeed, in contrast to other brain regions, neuronal cells in the striatum are somewhat hypothyroid, as assessed by the decrease in the T3-responsive gene RC3 (49).
The above-mentioned studies suggest that Mct8 has an important role in the transport of T3 across the blood-brain barrier in mice. In mice, T4 transport into the brain is not dependent on Mct8 or is compensated by alternative transporters. Apparently, the level of T3 in brain of Mct8 KO animals is sufficient to prevent gross neurological dysfunction in mice.
MCT10, OATP1C1, and other transporters
Monocarboxylate Transporter 10
To search for novel TH transporters, it is reasonable to investigate transporter families, which recognize ligands related to TH. Because the basic structure of iodothyronines resides in the assembly of two Tyr residues, an obvious candidate to test was a T-type amino acid transporter (TAT), which mediates uptake or efflux of aromatic amino acids. In support of this hypothesis, specific interactions between T3 and aromatic amino acids, mostly Trp, have been observed in various cell types, such as erythrocytes and hepatocytes (52, 53, 54). At the molecular level, TAT1 was identified as a transporter, which facilitates transport of the aromatic amino acids Phe, Tyr, and Trp and L-3,4-di-hydroxy-phenylalanine (55, 56). Based on its homology with other MCTs, TAT1 is also named MCT10. Among the MCT family, MCT10 is most closely related to MCT8 with an amino acid sequence identity up to 49%.
MCT10 has a gene and protein structure that is similar to MCT8. It contains six exons, which code for a 515-amino acid protein. It has a typical transporter structure of 12 TMDs, and both protein termini are located intracellularly. Based on the above considerations, it was tested whether T3 and T4 are ligands for MCT10. Therefore, cells were transfected with human MCT10, and intracellular accumulation of T3 and T4 was assayed (57). These experiments were the first showing that MCT10 facilitates TH transport with a preference for T3. This was underscored by subsequent experiments, in which MCT10 was cotransfected with different deiodinases. These studies demonstrated that MCT10 enhances intracellular metabolism of T3 to a larger extent than metabolism of T4. Under identical conditions, T3 transport mediated by MCT10 exceeds that by MCT8.
T3 uptake by MCT10 is clearly dependent on the type of incubation medium used. If cells were incubated in medium containing large amounts of nutrients (DMEM/F12), MCT10-mediated T3 uptake was lower than in “poor” medium (Dulbecco’s-PBS). This may be partially explained by the presence of aromatic amino acids in the “rich” medium, which impinge on net T3 uptake. Direct assessment of aromatic amino acids on T3 uptake revealed that Trp most strongly affected net accumulation of intracellular T3 in cells transfected with MCT10. These studies suggest that MCT10 is responsible for the previous observations of the interactions between T3 and Trp. Like MCT8, MCT10 is involved in bidirectional transport of TH across the plasma membrane.
Information about physiological function and expression is limited. MCT10 is ubiquitously expressed, among others, in intestine, kidney, liver, muscle, and placenta (54, 55, 56, 58). MCT10 appears predominantly localized in the basolateral membrane (58).
Organic Anion-Transporting Polypeptide 1C1
OATP1C1 belongs to the OATP family, which is a large family, of which many members accept a wide variety of ligands. OATP1C1 has several characteristics that distinguish this transporter from other OATPs. First, OATP1C1 displays a narrower ligand specificity toward T4, rT3, T4 sulfate, estrone-3-sulfate (E1S), estradiol-17β-glucuronide, and bromosulfophthalein (12, 59). Second, it has been reported that OATP1C1 has the highest affinity for iodothyronines compared with other OATP family members (12). Recently, it has been suggested that OATP1C1 displays atypical transport kinetics, involving two different (high and low affinity) T4 binding sites (60). Third, many OATPs demonstrate a pH-dependent transport activity, except for OATP1C1. Recently, an elegant study suggested that the pH sensitivity of OATP-mediated transport depends on a His (H) residue, which is conserved in many OATPs but not in OATP1C1 (61). Interestingly, when the corresponding amino acid in OATP1C1 was mutated to a H residue, T4 transport mediated by OATP1C1 became pH dependent.
Expression of OATP1C1 has been noted in the Leydig cells of the testis and in brain capillaries in rat and mice, leading to the suggestion that Oatp1c1 is expressed at the blood-brain barrier (62, 63, 64). In addition, OATP1C1 staining is observed in many human brain regions (65). Species-specific expression differences were underscored by the observation that Oatp1c1 is markedly enriched in rodent cerebral microvessels but only marginally in human microvessels (26). An interesting aspect of Oatp1c1 regulation is the inverse relationship with TH concentrations. In hypothyroid rats, Oatp1c1 expression in brain capillaries increases, whereas the opposite is observed in hyperthyroid animals (62).
The relevance of OATP1C1 for physiology is unknown. Genetic variation in the OATP1C1 gene is not associated with changes in serum TH parameters (59). However, polymorphisms in OATP1C1 were associated with subtle changes in clinical endpoints, such as fatigue and depression in T4-substituted hypothyroid patients (66). These observations hint at a physiological role of OATP1C1 in brain.
Other transporters
Much less information is known about the growing number of transporters that have been added to the list of proteins that mediate TH transport in recent years. Most of them are multispecific (Table 1), and the relevance in vivo is presently unclear. An exception is the liver-specific transporter OATP1B1, which facilitates uptake of, among other ligands, sulfated iodothyronines, E1S, and bilirubin (67). The well-studied polymorphism V174A is associated with higher serum T4 sulfate, E1S, and bilirubin levels in humans. In vitro experiments have shown that this variant is less active in transporting these ligands, providing the explanation for the associations found in humans.
Table 1.
Thyroid hormone transporters
| Transporter1 | Iodothyronine derivates | Specificity2 | Reference |
|---|---|---|---|
| MCT8 | T3, T4, rT3, T2 | +++ | 17 18 |
| MCT10 | T3, T4 | ++ | 57 |
| Oatp1a1 | T3, T4, rT3, T2, T4S, T3S, rT3S, T2S | + | 8 |
| OATP1A2 | T4, T3, rT3 | + | 102 |
| Oatp1a3 | T4, T3 | + | 103 |
| Oatp1a4 | T4, T3 | + | 7 |
| Oatp1a5 | T4, T3 | + | 7 |
| OATP1B1 | T4, T3, T3S, T4S, rT3S | + | 67 104 |
| Oatp1b2 | T3, T4 | + | 105 |
| OATP1B3 | rT3, T4S, T3S, rT3S | + | 13 |
| OATP1C1 | T4, rT3, T3, T4S | ++ | 59 |
| OATP2B1 | T4 | + | 61 |
| OATP3A1_v1/v2 | T4 | ++ | 88 |
| Oatp4a1 | T3, T4, rT3 | + | 102 |
| OATP4C1 | T3, T4 | + | 86 |
| Oatp6b1 | T3, T4 | + | 106 |
| Oatp6c1 | T3, T4 | + | 106 |
| LAT1 | T3, T4, rT3, T2 | + | 77 |
| LAT2 | T3, T4, rT3, T2 | + | 77 |
| NTCP | T4, T3, T4S, T3S | ++ | 8 9 |
| MDR1 | T3 | + | 10 |
The human protein symbol is presented, if TH transport has been demonstrated in different species including humans.
If a transporter only transports iodothyronine derivatives, specificity is high (+++). If fewer than five other ligands are known, specificity is moderate (++). If more than five ligands are known, the transporter is denoted as multispecific (+).
The Unknowns
The progress made in the understanding of cellular TH transport in recent years has also increased our awareness that we are just beginning to recognize the complexity of this aspect of TH homeostasis. In the following sections, we will define gaps of knowledge, which require additional studies in the near future.
Monocarboxylate Transporter 8
Even though MCT8 is the TH transporter that has gained most attention during last years, much remains to be elucidated. Starting at the structural aspects of the protein, MCT8 has an intriguing N-terminal domain enriched in Pro (P) and Glu repeats, defining three so-called PEST domains. PEST domains are hydrophilic stretches of 12 or more amino acids containing at least one P, Glu, or Asp and one Ser or Thr, flanked by Lys, Arg, or H residues (68). In fact, this was the reason why the gene was initially designated as XPCT, for X-linked PEST-containing transporter (69). Approximately 10% of the mammalian proteins contains PEST domains, and it has been demonstrated that PEST-containing proteins are subject to rapid degradation (68). However, this concept was challenged recently by whole proteome profiling of mammalian cells, which demonstrated that rapidly degraded proteins are not enriched in PEST sequences (70). Thus, the function of these PEST motifs in MCT8 is still elusive.
The human MCT8 gene contains two translation start sites (TLSs), resulting in a 613- or 539-amino acid protein, depending on which TLS is used. The first TLS is lacking in species, such as rat and mice. Most studies addressing the function of (mutated) MCT8 have used an MCT8 cDNA, which only contains the second TLS (18, 20, 42, 45, 57, 71). Although the second TLS conforms better to the consensus Kozak sequence, both TLSs have an adequate sequence. This is supported by recent in vitro studies, demonstrating that translation may also start from the first start codon (72). Proteomic analysis of membranes in mesenchymal stromal cells identified MCT8 by a sequence closely to the first TLS, which is the first evidence that the first TLS is used in vivo (73).
The long MCT8 isoform contains an additional N-terminal PEST domain, but it does not change the 12 putative TMDs. Therefore, it seems unlikely that transport function is different between both isoforms. Possibly, it may have effects on expression or subcellular localization. Apparently, lack of the long isoform is not required for normal development, because a missense mutation in the first ATG (c.1A>T), making translation from this start codon impossible, was found in a nonaffected male (36). However, it cannot be excluded that this mutated sequence may function as a TLS, because it is known that non-AUG TLSs may also be used to start translation, although at a lower efficiency (74). Thus, the relevance of the long isoform in humans remains to be clarified.
Relating to the aforementioned issues, regulation of MCT8 is poorly understood. Incidental studies have reported on MCT8 expression in different (patho)physiological states (see section MCT8 under The Knowns), although underlying mechanisms are presently unclear. Future studies may initially employ cell lines, which express endogenous MCT8, or use classical promoter analysis tools to address regulation of MCT8. Theoretically, important aspects of MCT8 regulation, such as the promoter region, distinct transcriptional regulators, or interacting proteins, may be discovered in studies in patients with the clinical and endocrine phenotype of AHDS, but without mutations in the protein-encoding parts of the MCT8 gene.
Two major unknowns concern observations in patients with MCT8 mutations. First, the origins of the abnormally high serum T3 and low serum T4 levels are unknown. Several possibilities may explain this part of the endocrine phenotype. Assuming that MCT8 provides D3-expressing neurons in brain with T3, it is conceivable that inactivation of transport results in a decreased T3 inactivation. Consequently, serum T3 levels will increase, which stimulate liver D1 activity, which results in a further conversion of T4 to T3. This scenario would explain the low T4 and rT3 levels (both consumed by D1) and high T3 levels (decreased inactivation and increased T4-T3 conversion).
The causative role of D3 in the pathogenesis of the abnormal serum thyroid parameters is challenged by observations in Mct8/Pax8 double KO mice, which lack a functional thyroid gland and are dependent on exogenous TH substitution (51). T3 treatment in these animals did not result in elevated serum T3 levels, suggesting that the increased serum T3 levels are not the consequence of decreased T3 inactivation by D3 (51).
Rather, evidence suggests that T4 (and T3) accumulates in the kidneys of Mct8 KO mice, which may cause a further increase in renal T3 production (75). Moreover, a decreased thyroidal T4 and increased T3 secretion may contribute importantly to the altered serum TH levels in Mct8 KO animals (76). It is fascinating to speculate about the possible function of MCT8 in the thyroid gland. Many steps in the TH biosynthesis are well understood, but the mechanism by which TH is secreted into the bloodstream is still elusive. It should be investigated whether MCT8 in the thyroid gland plays a role in this process, and if so, to which extent a thyroid gland devoid of functional MCT8 contributes to abnormal thyroid parameters in serum.
Taken together, there are several indications to believe that a decreased D3-mediated inactivation of T3 does not play a major role in the pathogenesis of the serum TH levels. Supportive of a prominent role of D1 in the endocrine phenotype comes from observations that rT3 levels are already low at P7 in Mct8 KO mice, whereas T4 and T3 are still normal (78). Preliminary data indicate that inactivation of Mct8 in mice that already lack Dio1 and Dio2 does not further affect serum T3 and T4 levels (78). Possibly, the combination of altered deiodinase activities and accumulation of TH in tissues may explain the endocrine phenotype. Further studies are warranted to unravel the precise mechanisms of the abnormal serum T3 and T4 levels in MCT8 patients.
The other major unknown concerns the pathogenesis of the neurological phenotype in patients with MCT8 mutations, which is far from established. This is partially related to the absence of a gross neurological phenotype in Mct8 KO mice. Several attempts have been made to explain this discrepancy. First, mouse brain may respond differently to TH deficiency compared with human brain. However, it is known for years that hypothyroidism induces structural brain abnormalities in rodents, which is why hypothyroid animals have been used as models to study the effects of TH on brain development (79). Second, MCT8 may transport alternative ligands that are important for human, but not for mouse, brain development. This is mainly based on the observation that other members of the MCT family transport monocarboxylates and amino acids. However, experimental evidence for MCT8 transport of ligands in addition to iodothyronines is limited (80).
Third, the mouse may employ compensatory mechanisms, such as the expression of alternative transporters, which are lacking in humans. This is suggested by observations that Oatp1c1 is present in mouse but absent in human brain microvessels (26). It has also been suggested that LAT2 may compensate if Mct8 is deficient in mice, mainly based on different LAT2 expression levels in mice vs. humans (25). Currently, there is little evidence to indicate which of the above mechanisms is the most likely explanation for the human-mouse difference in phenotype resulting from inactivation of MCT8.
Explanations for the neurological impairment in MCT8 patients assume that neurons suffer from an insufficient T3 supply. This scenario is based on extrapolations from other diseases, in which a lack of sufficient TH plays a role in the brain phenotype, such as untreated congenital hypothyroidism or cretinism. However, this hypothesis should be experimentally validated before any definite conclusion may be drawn. A first requirement to understand the underlying mechanisms of disease is a comprehensive knowledge of MCT8 expression in human brain in comparison with the localization of other genes important for TH regulation. The identification of target genes downstream of MCT8 is essential for our understanding of the molecular derangements if MCT8 is deficient. It is also possible that inactivation of MCT8 results in an imbalance of T3 supply to neurons that express MCT8 and neurons that express other T3 transporters.
Apart from the role of MCT8 in providing cells with T3 to initiate a T3-dependent transcriptional program, MCT8 may also be involved in mediating so-called nongenomic actions. Possibly, MCT8 may also function in providing the brain with transcriptionally inactive iodothyronines. It has been reported that T4 and rT3 regulate actin polymerization and microfilament organization, whereas T3 does not alter these processes (81). If this requires the intracellular availability of these iodothyronines, transporters are also required to for their translocation across the plasma membrane. Given the low serum T4 and rT3 concentrations in MCT8 patients, diminished nongenomic actions in brain may also contribute to the phenotype.
To date, no curative therapy is available for MCT8 deficiency. Recently, the first initiatives for possible treatment of patients with MCT8 mutations have been reported. In a 16-yr-old MCT8 patient, referred for feeding problems and very low body weight, block-and-replace therapy as applied to patients with primary hyperthyroidism was tested (82). The combination of the antithyroid drug propylthiouracil and L-T4 replacement resulted in a normalization of T4, T3, and TSH levels. During this therapy, body weight significantly increased. Unfortunately, neurological improvements were not observed and perhaps also not expected. If exposure of the developing brain to abnormally low serum T4 and high T3 levels plays an important role in the pathogenesis of AHDS, early normalization of these TH levels may well be an effective therapeutic option.
Theoretically, compounds that mimic the action of TH, but rely on other transporters than MCT8 for cellular entry, would counteract the neurological damage. This prompted the investigation of the effects of 3,5-diiodothyropropionic acid (DITPA) in comparison with T4 in hypothyroid Mct8 KO mice (83). Normal doses of T4 did not correct the elevated TSH levels and D2 activity in brain of Mct8 null mice. Treatment with supraphysiological T4 doses normalized these parameters, but also increased D1 activity in the liver, indicating peripheral hyperthyroidism. Promising effects were observed with high doses of DITPA, which produced similar beneficial effects on the brain parameters, but avoided thyrotoxicity in the liver. Importantly, in Mct8 KO mice without experimental hypothyroidism, beneficial effects of DITPA treatment on the brain remained without affecting thyroid status in the liver.
Obviously, further detailed studies need to be done to rule out possible negative effects of DITPA. Furthermore, it should be investigated to which extent DITPA replaces TH in normal brain development, e.g. by comparing the effects of DITPA vs. T4 on brain development in Pax8 KO mice. However, given the large differences in neurological phenotype of humans vs. mice and the absence of an appropriate model to investigate possible treatment regimens, it seems reasonable to explore such treatment possibilities in MCT8 patients (84).
Obviously, the earlier treatment is started the higher the potential to prevent or reverse abnormalities. This requires early detection of MCT8 mutations. Most patients with MCT8 mutations have been identified from several months onwards. However, serum TH abnormalities may already be detectable at birth. Retrospective analysis of the T4-based neonatal screening for congenital hypothyroidism revealed that most patients had low T4 levels in the blood spot (45). However, based on the current screening strategy in The Netherlands, no further action was undertaken, because TSH levels and T4/TBG ratios were not abnormal. Therefore, it should be investigated whether high T3 and low rT3 levels are already present in neonates with MCT8 mutations and whether screening protocols could be adapted to identify MCT8 patients. Taken together, the first steps toward a treatment for MCT8 patients have been made. Future research should be dedicated to explore novel therapeutic options and early detection of disease.
In vivo relevance of MCT10 and OATP1C1
Although the expression patterns of MCT10 and OATP1C1 are compatible with an important role of these transporters in TH physiology, the functional relevance in vivo of MCT10 and OATP1C1 remains to be established. Because MCT10 and OATP1C1 transport other ligands in addition to TH, fundamental questions for thyroid research are whether these transporters are primarily involved in regulating TH homeostasis or whether transport of other ligands is their principal function. Mice deficient in Mct10 and Oatp1c1 are the most straightforward models to answer these questions. Similar to the story of MCT8, it is important to realize that species-specific gene expression and compensatory mechanisms may mask certain phenotypes.
In cultured cells, endogenous expression of transporters may largely differ from the cells they represent in vivo. For example, Mct8 and Oatp1c1 were highly down-regulated in cultured microvascular endothelial cells compared with freshly isolated cells (85). Once the importance of MCT10 and OATP1C1 for TH physiology has been defined, further studies are needed to characterize the interaction between TH and other ligands, as well as the expression patterns and regulatory mechanisms. Furthermore, it should be explored to which extent TH transporters form functional pairs (e.g. basolateral and apical expressed transporters facilitating transport of the same ligand). Ultimately, novel syndromes caused by pathogenic mutations in these transporters may reveal their relevance for human physiology.
Are there novel TH transporters to be expected?
The identification of numerous TH transporters during the last decade raises the question if all TH transporters have been identified. Several basic and clinical studies suggest otherwise, implicating that other TH transporters are left in store to be discovered in the future.
From studies investigating TH transport into hepatocytes evidence emerged for the existence of a high-affinity, Na+-dependent transporter (2). It should be realized, however, that most of these experiments to investigate the Na+-dependence of transport were carried out using the Na-K-ATPase blocker ouabain. Inhibition of transport by ouabain is not necessarily explained by direct effects of Na+ on the transporter but may also be caused by indirect effects of Na+, e.g. on cellular energy status or intracellular TH binding proteins. Furthermore, ouabain is a ligand for OATPs and may thus compete with TH transport (86).
Notwithstanding these remarks, the possibility of a high-affinity, Na+-dependent liver TH transporter remains an appealing explanation for these observations. In support of this view, detailed kinetic studies in a patient with abnormal serum TH concentrations suggested a defect in iodothyronine uptake by the liver (87). Thus, the molecular identity of the putative high-affinity, Na+-dependent TH transporter in the liver remains to be clarified.
In view of the complexity of the brain, it would be surprising if MCT8 and OATP1C1 are the only TH transporters in brain. Different brain areas and different cell types may express various transporters for optimal specificity in TH regulation. An obvious candidate for further research is OATP3A1, which exhibits a relative specificity for T4 transport (88). Interestingly, there are two functionally active OATP3A1 isoforms with highly specific expression patterns in rat and human brain.
MCT8 and MCT10 are localized in the basolateral membrane of tissue cells and both facilitate uptake and efflux of TH. Other TH transporters may exist, located in the basolateral or apical membrane of polarized cells, which facilitate only uptake or efflux. This is particular relevant in the current working hypothesis of local TH control in brain, where T4 is transported into astrocytes by an as yet unknown transporter and subsequently converted to T3 by D2. It is assumed that an unidentified T3 exporter exists, which ensures the release of T3 from the astrocytes, which becomes available for adjacent neurons.
So far, most studies have been dedicated to characterize TH uptake, and little information is available about TH efflux processes (10, 18, 57, 89, 90, 91, 92). Studies of TH efflux are technically complicated, because it requires prior loading of cells through specific TH uptake transporters. Such limitations may be overcome by the use of polarized cells cultured in double-chamber dishes.
Taken together, there are firm reasons to believe that novel TH transporters are waiting to be discovered.
Intracellular transport
The intracellular availability of TH is not only determined by transporters and deiodinases but also by intracellular binding proteins. An important protein is CRYM, which has a high affinity for the iodothyronines T3, T4, and rT3 in the presence of reduced nicotinamide adenine dinucleotide phosphate and for T2 in the absence of reduced nicotinamide adenine dinucleotide phosphate (93, 94). Recently, we found that CRYM is also able to bind sulfated iodothyronines (9). In CRYM KO animals, serum T3 and T4 concentrations are decreased and T3 rapidly leaves tissues (95). Nevertheless, peripheral T3 action appears not to be changed, and thus, it remains to be established which role CRYM plays in physiology. Furthermore, it is unknown whether CRYM is involved in trafficking TH to subcellular compartments or that other proteins fulfill this function. Future research may answer the basic question how and when TH is transported in the cell to various cellular compartments.
The Guesses
As extensively illustrated by the identification and characterization of plasma membrane transporters in the previous sections, the intrinsic properties of iodothyronine molecules prevent the possibility of diffusion through the lipid bilayer. However, there seems to be no a priori reason to restrict this type of TH transport to the plasma membrane. What about cellular compartments, which are also surrounded by lipid bilayers, such as mitochondria and the nucleus?
Several transporters from the SLC family have been identified as essential for transporting molecules into mitochondria. A well-known example is the ornithine transporter SLC25A15, which results in serious symptoms in humans with loss-of-function mutations in this transporter (96). As T3 largely influences transcription of mitochondrial DNA, transport of the hormone across the mitochondrial membranes is obligatory (97). To our knowledge, TH transport across mitochondrial membranes has not been studied yet, and molecular identification of one or more mitochondrial TH transporters is awaited in the future.
It has been established that transport of macromolecules (typically >40 kDa) from the cytoplasm to the nucleoplasm is mediated by nuclear-pore complexes (NPCs). Many different NPCs, which are each an assembly of multiple proteins, have been identified, and mutations therein have been linked to diseases (98). Because NPCs facilitate translocation of transcription factors across the nuclear envelope, T3 bound to its receptor may enter the nucleus and as such mediates its effects on transcription. This scenario is consistent with the observation of rapid cytoplasmic-nuclear transport of TRs (99, 100). This nuclear uptake of TR-bound T3 may also result in an uphill cytoplasmic-nuclear gradient of free T3, a phenomenon proposed by Oppenheimer and Schwartz in 1985 (101). The importance of this supposed mechanism is unclear, because the majority of TRs is located in the nucleus. In view of the size of the pores in the nuclear membrane, it is doubtful if transporters are required to transport TH into the nucleus.
Undoubtedly, novel syndromes will be recognized, the genetic basis of which resides in TH transporters mutations. The development and application of novel genomic technologies promise that understanding of genetic mechanisms of diseases will rapidly progress in the next coming years.
Life is getting complicated in the world of local TH regulation. In addition to the intricate regulatory mechanisms of the deiodinases, the discovery of specific TH transporters, of which MCT8 is currently the most prominent, has expanded our knowledge about the players involved in TH homeostasis. It is exciting to realize that there are still numbers of unknown TH transporters and related syndromes. They just need to be discovered.
Footnotes
This work was supported by The Netherlands Organization for Scientific Research Grant 9120.6093 (to W.E.V. and E.C.H.F.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online July 21, 2010
Abbreviations: AHDS, Allan-Herndon-Dudley syndrome; CRYM, μ-crystallin; D2, type 2 deiodinase; DITPA, 3,5-diiodothyropropionic acid; E1S, estrone-3-sulfate; H, His; KO, knockout; LAT, L-type amino acid transporter; MCT, monocarboxylate transporter; NPC, nuclear-pore complex; OATP, organic anion-transporting polypeptide; P, Pro; rMct8, rat Mct8; SLC, solute carrier family; T2, diiodothyronine; TAT, T-type amino acid transporter; TH, thyroid hormone; TLS, translation start site; TMD, transmembrane domain; TR, TH receptor.
References
- 1.Robbins J, Rall JE 1960. Proteins associated with the thyroid hormones. Physiol Rev 40:415–489 [DOI] [PubMed] [Google Scholar]
- 2.Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ 2001. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev 22:451–476 [DOI] [PubMed] [Google Scholar]
- 3.Visser WE, Friesema EC, Jansen J, Visser TJ 2008. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab 19:50–56 [DOI] [PubMed] [Google Scholar]
- 4.Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U 2001. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem 276:35847–35853 [DOI] [PubMed] [Google Scholar]
- 5.Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeöld A, Bianco AC 2008. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev 29:898–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hillier AP 1970. The binding of thyroid hormones to phospholipid membranes. J Physiol 211:585–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe T, Kakyo M, Sakagami H, Tokui T, Nishio T, Tanemoto M, Nomura H, Hebert SC, Matsuno S, Kondo H, Yawo H 1998. Molecular characterization and tissue distribution of a new organic anion transporter subtype (oatp3) that transports thyroid hormones and taurocholate and comparison with oatp2. J Biol Chem 273:22395–22401 [DOI] [PubMed] [Google Scholar]
- 8.Friesema EC, Docter R, Moerings EP, Stieger B, Hagenbuch B, Meier PJ, Krenning EP, Hennemann G, Visser TJ 1999. Identification of thyroid hormone transporters. Biochem Biophys Res Commun 254:497–501 [DOI] [PubMed] [Google Scholar]
- 9.Visser WE, Wong WS, van Mullem AA, Friesema EC, Geyer J, Visser TJ 2010. Study of the transport of thyroid hormone by transporters of the SLC10 family. Mol Cell Endocrinol 315:138–145 [DOI] [PubMed] [Google Scholar]
- 10.Mitchell AM, Tom M, Mortimer RH 2005. Thyroid hormone export from cells: contribution of P-glycoprotein. J Endocrinol 185:93–98 [DOI] [PubMed] [Google Scholar]
- 11.Jansen J, Friesema EC, Milici C, Visser TJ 2005. Thyroid hormone transporters in health and disease. Thyroid 15:757–768 [DOI] [PubMed] [Google Scholar]
- 12.Hagenbuch B 2007. Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrinol Metab 21:209–221 [DOI] [PubMed] [Google Scholar]
- 13.van der Deure WM, Peeters RP, Visser TJ 2010. Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OATPs, and the effects of genetic variation in these transporters. J Mol Endocrinol 44:1–11 [DOI] [PubMed] [Google Scholar]
- 14.Visser WE, Friesema EC, Jansen J, Visser TJ 2007. Thyroid hormone transport by monocarboxylate transporters. Best Pract Res Clin Endocrinol Metab 21:223–236 [DOI] [PubMed] [Google Scholar]
- 15.Halestrap AP, Meredith D 2004. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch 447:619–628 [DOI] [PubMed] [Google Scholar]
- 16.Murakami Y, Kohyama N, Kobayashi Y, Ohbayashi M, Ohtani H, Sawada Y, Yamamoto T 2005. Functional characterization of human monocarboxylate transporter 6 (SLC16A5). Drug Metab Dispos 33:1845–1851 [DOI] [PubMed] [Google Scholar]
- 17.Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ 2003. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278:40128–40135 [DOI] [PubMed] [Google Scholar]
- 18.Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH 2006. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol 20:2761–2772 [DOI] [PubMed] [Google Scholar]
- 19.Visser WE, Philp NJ, van Dijk TB, Klootwijk W, Friesema EC, Jansen J, Beesley PW, Ianculescu AG, Visser TJ 2009. Evidence for a homodimeric structure of human monocarboxylate transporter 8. Endocrinology 150:5163–5170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A 2005. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol 153:359–366 [DOI] [PubMed] [Google Scholar]
- 21.Price NT, Jackson VN, Halestrap AP 1998. Cloning and sequencing of four new mammalian monocarboxylate transporter (MCT) homologues confirms the existence of a transporter family with an ancient past. Biochem J 329(Pt 2):321–328 [DOI] [PMC free article] [PubMed]
- 22.Nishimura M, Naito S 2008. Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet 23:22–44 [DOI] [PubMed] [Google Scholar]
- 23.Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, Bauer K 2005. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology 146:1701– 1706 [DOI] [PubMed] [Google Scholar]
- 24.Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, Morte B, Bernal J 2009. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3′-triiodo-L-thyronine. Endocrinology 150:2491–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirth EK, Roth S, Blechschmidt C, Hölter SM, Becker L, Racz I, Zimmer A, Klopstock T, Gailus-Durner V, Fuchs H, Wurst W, Naumann T, Bräuer A, de Angelis MH, Köhrle J, Grüters A, Schweizer U 2009. Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci 29:9439–9449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, Zerangue N 2008. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 149:6251–6261 [DOI] [PubMed] [Google Scholar]
- 27.Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, Wiersinga WM, Swaab DF, Visser TJ, Fliers E 2005. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab 90:4322–4334 [DOI] [PubMed] [Google Scholar]
- 28.Alkemade A, Friesema EC, Kuiper GG, Wiersinga WM, Swaab DF, Visser TJ, Fliers E 2006. Novel neuroanatomical pathways for thyroid hormone action in the human anterior pituitary. Eur J Endocrinol 154:491–500 [DOI] [PubMed] [Google Scholar]
- 29.Silvestri E, Lombardi A, de Lange P, Schiavo L, Lanni A, Goglia F, Visser TJ, Moreno M 2008. Age-related changes in renal and hepatic cellular mechanisms associated with variations in rat serum thyroid hormone levels. Am J Physiol Endocrinol Metab 294:E1160–E1168 [DOI] [PubMed]
- 30.Chan SY, Franklyn JA, Pemberton HN, Bulmer JN, Visser TJ, McCabe CJ, Kilby MD 2006. Monocarboxylate transporter 8 expression in the human placenta: the effects of severe intrauterine growth restriction. J Endocrinol 189:465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Capelo LP, Beber EH, Fonseca TL, Gouveia CH 2009. The monocarboxylate transporter 8 and L-type amino acid transporters 1 and 2 are expressed in mouse skeletons and in osteoblastic MC3T3-E1 cells. Thyroid 19:171–180 [DOI] [PubMed] [Google Scholar]
- 32.Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D'Hoore A, Darras VM, Visser TJ, Van den Berghe G 2009. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol 161:243–250 [DOI] [PubMed] [Google Scholar]
- 33.Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ 2004. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364:1435–1437 [DOI] [PubMed] [Google Scholar]
- 34.Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S 2004. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz CE, Stevenson RE 2007. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrinol Metab 21:307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frints SG, Lenzner S, Bauters M, Jensen LR, Van Esch H, des Portes V, Moog U, Macville MV, van Roozendaal K, Schrander-Stumpel CT, Tzschach A, Marynen P, Fryns JP, Hamel B, van Bokhoven H, Chelly J, Beldjord C, Turner G, Gecz J, Moraine C, Raynaud M, Ropers HH, Froyen G, Kuss AW 2008. MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet 16:1029–1037 [DOI] [PubMed] [Google Scholar]
- 37.Holden KR, Zuñiga OF, May MM, Su H, Molinero MR, Rogers RC, Schwartz CE 2005. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol 20:852–857 [DOI] [PubMed] [Google Scholar]
- 38.Sijens PE, Rödiger LA, Meiners LC, Lunsing RJ 2008. 1H magnetic resonance spectroscopy in monocarboxylate transporter 8 gene deficiency. J Clin Endocrinol Metab 93:1854–1859 [DOI] [PubMed] [Google Scholar]
- 39.Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE 2005. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaurs-Barrière C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Viñas JM, De Michele G, Dan B, Brady AF, Boespflug-Tanguy O, Touraine R 2009. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol 65:114–118 [DOI] [PubMed] [Google Scholar]
- 41.Friesema EC, Visser WE, Visser TJ 2010. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol 322:107–113 [DOI] [PubMed] [Google Scholar]
- 42.Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ 2008. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology 149:2184–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinne A, Roth S, Biebermann H, Köhrle J, Grüters A, Schweizer U 2009. Surface translocation and tri-iodothyronine uptake of mutant MCT8 proteins are cell type-dependent. J Mol Endocrinol 43:263–271 [DOI] [PubMed] [Google Scholar]
- 44.Dumitrescu AM, Liao XH, Lado-Abeal J, Moeller LC, Brockmann K, Refetoff S 2004. On the mechanism producing the unusual thyroid phenotype in defects of the MCT8 gene. Thyroid 14:761 [Google Scholar]
- 45.Visser WE, Jansen J, Friesema EC, Kester MH, Mancilla E, Lundgren J, van der Knaap MS, Lunsing RJ, Brouwer OF, Visser TJ 2009. Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat 30:29–38 [DOI] [PubMed] [Google Scholar]
- 46.Lago-Lestón R, Iglesias MJ, San-José E, Areal C, Eiras A, Araújo-Vilar D, Lado-Abeal J, Domínguez-Gerpe L 2009. Prevalence and functional analysis of the S107P polymorphism (rs6647476) of the monocarboxylate transporter 8 (SLC16A2) gene in the male population of north-west Spain (Galicia). Clin Endocrinol (Oxf) 70:636–643 [DOI] [PubMed] [Google Scholar]
- 47.van der Deure WM, Peeters RP, Visser TJ 2007. Genetic variation in thyroid hormone transporters. Best Pract Res Clin Endocrinol Metab 21:339–350 [DOI] [PubMed] [Google Scholar]
- 48.Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S 2006. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147:4036–4043 [DOI] [PubMed] [Google Scholar]
- 49.Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H 2007. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest 117:627–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.St Germain DL, Galton VA, Hernandez A 2009. Minireview: defining the roles of the iodothyronine deiodinases: current concepts and challenges. Endocrinology 150:1097–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trajkovic M, Visser TJ, Mittag J, Bauer K, Heuer H 2007. The thyroid hormone transporter MCT8 is critically involved in the uptake of T3 into the mouse CNS. Thyroid 17:1171–1172 [Google Scholar]
- 52.Zhou Y, Samson M, Osty J, Francon J, Blondeau JP 1990. Evidence for a close link between the thyroid hormone transport system and the aromatic amino acid transport system T in erythrocytes. J Biol Chem 265:17000–17004 [PubMed] [Google Scholar]
- 53.Zhou Y, Samson M, Francon J, Blondeau JP 1992. Thyroid hormone concentrative uptake in rat erythrocytes. Involvement of the tryptophan transport system T in countertransport of tri-iodothyronine and aromatic amino acids. Biochem J 281(Pt 1):81–86 [DOI] [PMC free article] [PubMed]
- 54.Ritchie JW, Taylor PM 2010. Tryptophan and iodothyronine transport interactions in HepG2 human hepatoma cells. Amino Acids 38:1361–1367 [DOI] [PubMed] [Google Scholar]
- 55.Kim DK, Kanai Y, Chairoungdua A, Matsuo H, Cha SH, Endou H 2001. Expression cloning of a Na+-independent aromatic amino acid transporter with structural similarity to H+/monocarboxylate transporters. J Biol Chem 276:17221–17228 [DOI] [PubMed] [Google Scholar]
- 56.Kim DK, Kanai Y, Matsuo H, Kim JY, Chairoungdua A, Kobayashi Y, Enomoto A, Cha SH, Goya T, Endou H 2002. The human T-type amino acid transporter-1: characterization, gene organization, and chromosomal location. Genomics 79:95–103 [DOI] [PubMed] [Google Scholar]
- 57.Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ 2008. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10 (MCT10). Mol Endocrinol 22:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramadan T, Camargo SM, Summa V, Hunziker P, Chesnov S, Pos KM, Verrey F 2006. Basolateral aromatic amino acid transporter TAT1 (Slc16a10) functions as an efflux pathway. J Cell Physiol 206:771–779 [DOI] [PubMed] [Google Scholar]
- 59.van der Deure WM, Hansen PS, Peeters RP, Kyvik KO, Friesema EC, Hegedüs L, Visser TJ 2008. Thyroid hormone transport and metabolism by OATP1C1 and consequences of genetic variation. Endocrinology 149:5307–5314 [DOI] [PubMed] [Google Scholar]
- 60.Westholm DE, Salo DR, Viken KJ, Rumbley JN, Anderson GW 2009. The blood-brain barrier thyroxine transporter organic anion-transporting polypeptide 1c1 displays atypical transport kinetics. Endocrinology 150:5153–5162 [DOI] [PubMed] [Google Scholar]
- 61.Leuthold S, Hagenbuch B, Mohebbi N, Wagner CA, Meier PJ, Stieger B 2009. Mechanisms of pH-gradient driven transport mediated by organic anion polypeptide transporters. Am J Physiol Cell Physiol 296:C570–C582 [DOI] [PubMed]
- 62.Sugiyama D, Kusuhara H, Taniguchi H, Ishikawa S, Nozaki Y, Aburatani H, Sugiyama Y 2003. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem 278:43489–43495 [DOI] [PubMed] [Google Scholar]
- 63.Tohyama K, Kusuhara H, Sugiyama Y 2004. Involvement of multispecific organic anion transporter, Oatp14 (Slc21a14), in the transport of thyroxine across the blood-brain barrier. Endocrinology 145:4384–4391 [DOI] [PubMed] [Google Scholar]
- 64.Chu C, Li JY, Boado RJ, Pardridge WM 2008. Blood-brain barrier genomics and cloning of a novel organic anion transporter. J Cereb Blood Flow Metab 28:291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ 2002. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol 16:2283–2296 [DOI] [PubMed] [Google Scholar]
- 66.van der Deure WM, Appelhof BC, Peeters RP, Wiersinga WM, Wekking EM, Huyser J, Schene AH, Tijssen JG, Hoogendijk WJ, Visser TJ, Fliers E 2008. Polymorphisms in the brain-specific thyroid hormone transporter OATP1C1 are associated with fatigue and depression in hypothyroid patients. Clin Endocrinol (Oxf) 69:804–811 [DOI] [PubMed] [Google Scholar]
- 67.van der Deure WM, Friesema EC, de Jong FJ, de Rijke YB, de Jong FH, Uitterlinden AG, Breteler MM, Peeters RP, Visser TJ 2008. OATP1B1: an important factor in hepatic thyroid hormone and estrogen transport and metabolism. Endocrinology 149:1495– 1701 [DOI] [PubMed] [Google Scholar]
- 68.Rechsteiner M, Rogers SW 1996. PEST sequences and regulation by proteolysis. Trends Biochem Sci 21:267–271 [PubMed] [Google Scholar]
- 69.Lafrenière RG, Carrel L, Willard HF 1994. A novel transmembrane transporter encoded by the XPCT gene in Xq13.2. Hum Mol Genet 3:1133–1139 [DOI] [PubMed] [Google Scholar]
- 70.Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ 2008. Global protein stability profiling in mammalian cells. Science 322:918–923 [DOI] [PubMed] [Google Scholar]
- 71.Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, Grüters A, Barrett TG, Mancilla EE, Svensson J, Wemeau JL, Busi da Silva Canalli MH, Lundgren J, McEntagart ME, Hopper N, Arts WF, Visser TJ 2007. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab 92:2378–2381 [DOI] [PubMed] [Google Scholar]
- 72.Kinne A, Roth S, Biebermann H, Köhrle J, Grüters A, Schweizer U 2009. Surface translocation and T3 uptake of mutant MCT8 proteins are cell type-dependent. J Mol Endocrinol 43:263–271 [DOI] [PubMed] [Google Scholar]
- 73.Jeong JA, Ko KM, Park HS, Lee J, Jang C, Jeon CJ, Koh GY, Kim H 2007. Membrane proteomic analysis of human mesenchymal stromal cells during adipogenesis. Proteomics 7:4181–4191 [DOI] [PubMed] [Google Scholar]
- 74.Peabody DS 1989. Translation initiation at non-AUG triplets in mammalian cells. J Biol Chem 264:5031–5035 [PubMed] [Google Scholar]
- 75.Trajkovic-Arsic M, Visser TJ, Darras VM, Friesema EC, Schlott B, Mittag J, Bauer K, Heuer H 2010. Consequences of monocarboxylate transporter 8 deficiency for renal transport and metabolism of thyroid hormones in mice. Endocrinology 151:802–809 [DOI] [PubMed] [Google Scholar]
- 76.Trajkovic-Arsic M, Visser TJ, Darras VM, Friesema EC, Schlott B, Mittag J, Bauer K, Heuer H 2010. Consequences of MCT8 deficiency on renal and thyroidal transport of thyroid hormones. Endocrinology 151:802–809 [DOI] [PubMed] [Google Scholar]
- 77.Friesema EC, Docter R, Moerings EP, Verrey F, Krenning EP, Hennemann G, Visser TJ 2001. Thyroid hormone transport by the heterodimeric human system L amino acid transporter. Endocrinology 142:4339–4348 [DOI] [PubMed] [Google Scholar]
- 78.Dumitrescu AM, Liao XH, Galton VA, St Germain DL, Weiss RE, Refetoff S 2007. High circulating T3 in Mct8 deficient mice is an age related postnatal event dependent on the ontogeny of deiodinases. Endocrinology (Abstract)
- 79.Oppenheimer JH, Schwartz HL 1997. Molecular basis of thyroid hormone-dependent brain development. Endocr Rev 18:462–475 [DOI] [PubMed] [Google Scholar]
- 80.James SR, Franklyn JA, Reaves BJ, Smith VE, Chan SY, Barrett TG, Kilby MD, McCabe CJ 2009. Monocarboxylate transporter 8 in neuronal cell growth. Endocrinology 150:1961–1969 [DOI] [PubMed] [Google Scholar]
- 81.Leonard JL 2008. Non-genomic actions of thyroid hormone in brain development. Steroids 73:1008–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wémeau JL, Pigeyre M, Proust-Lemoine E, d'Herbomez M, Gottrand F, Jansen J, Visser TJ, Ladsous M 2008. Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab 93:2084–2088 [DOI] [PubMed] [Google Scholar]
- 83.Di Cosmo C, Liao XH, Dumitrescu AM, Weiss RE, Refetoff S 2009. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology 150:4450–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hameed S, Di Cosmo C, Liao XH, Weiss RE, Verge CF, Refetoff S 3,5-Diiodothyropropionic acid (DITPA) in the treatment of patients with MCT8 deficiency: a preliminary study. Lawson Wilkins Pediatric Endocrine Society/European Society for Pediatric Endocrinology, New York, NY, 2009
- 85.Lyck R, Ruderisch N, Moll AG, Steiner O, Cohen CD, Engelhardt B, Makrides V, Verrey F 2009. Culture-induced changes in blood-brain barrier transcriptome: implications for amino-acid transporters in vivo. J Cereb Blood Flow Metab 29:1491–1502 [DOI] [PubMed] [Google Scholar]
- 86.Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, Chaki T, Masuda S, Tokui T, Eto N, Abe M, Satoh F, Unno M, Hishinuma T, Inui K, Ito S, Goto J, Abe T 2004. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci USA 101:3569–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hennemann G, Vos RA, de Jong M, Krenning EP, Docter R 1993. Decreased peripheral 3,5,3′-triiodothyronine (T3) production from thyroxine (T4): a syndrome of impaired thyroid hormone activation due to transport inhibition of T4- into T3-producing tissues. J Clin Endocrinol Metab 77:1431–1435 [DOI] [PubMed] [Google Scholar]
- 88.Huber RD, Gao B, Sidler Pfändler MA, Zhang-Fu W, Leuthold S, Hagenbuch B, Folkers G, Meier PJ, Stieger B 2007. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am J Physiol Cell Physiol 292:C795–C806 [DOI] [PubMed]
- 89.Ribeiro RC, Cavalieri RR, Lomri N, Rahmaoui CM, Baxter JD, Scharschmidt BF 1996. Thyroid hormone export regulates cellular hormone content and response. J Biol Chem 271:17147–17151 [DOI] [PubMed] [Google Scholar]
- 90.Neves FA, Cavalieri RR, Simeoni LA, Gardner DG, Baxter JD, Scharschmidt BF, Lomri N, Ribeiro RC 2002. Thyroid hormone export varies among primary cells and appears to differ from hormone uptake. Endocrinology 143:476–483 [DOI] [PubMed] [Google Scholar]
- 91.Cavalieri RR, Simeoni LA, Park SW, Baxter JD, Scharschmidt BF, Ribeiro RC, Lomri N 1999. Thyroid hormone export in rat FRTL-5 thyroid cells and mouse NIH-3T3 cells is carrier-mediated, verapamil-sensitive, and stereospecific. Endocrinology 140:4948–4954 [DOI] [PubMed] [Google Scholar]
- 92.Visser WE, Friesema EC, Visser TJ 2009. Transport of thyroxine and 3,3′,5-triiodothyronine in human umbilical vein endothelial cells. Endocrinology 150:1552–1557 [DOI] [PubMed] [Google Scholar]
- 93.Vié MP, Evrard C, Osty J, Breton-Gilet A, Blanchet P, Pomérance M, Rouget P, Francon J, Blondeau JP 1997. Purification, molecular cloning, and functional expression of the human nicodinamide-adenine dinucleotide phosphate-regulated thyroid hormone-binding protein. Mol Endocrinol 11:1728–1736 [DOI] [PubMed] [Google Scholar]
- 94.Moreno M, Silvestri E, Lombardi A, Visser TJ, Goglia F, Lanni A 2003. Identification of 3,5-diiodo-L-thyronine-binding proteins in rat liver cytosol by photoaffinity labeling. Endocrinology 144:2297–2303 [DOI] [PubMed] [Google Scholar]
- 95.Suzuki S, Suzuki N, Mori J, Oshima A, Usami S, Hashizume K 2007. Micro-crystallin as an intracellular 3,5,3′-triiodothyronine holder in vivo Mol Endocrinol 21:885–894 [DOI] [PubMed] [Google Scholar]
- 96.Camacho JA, Obie C, Biery B, Goodman BK, Hu CA, Almashanu S, Steel G, Casey R, Lambert M, Mitchell GA, Valle D 1999. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet 22:151–158 [DOI] [PubMed] [Google Scholar]
- 97.Harper ME, Seifert EL 2008. Thyroid hormone effects on mitochondrial energetics. Thyroid 18:145–156 [DOI] [PubMed] [Google Scholar]
- 98.Capelson M, Hetzer MW 2009. The role of nuclear pores in gene regulation, development and disease. EMBO Rep 10:697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baumann CT, Maruvada P, Hager GL, Yen PM 2001. Nuclear cytoplasmic shuttling by thyroid hormone receptors. multiple protein interactions are required for nuclear retention. J Biol Chem 276:11237–11245 [DOI] [PubMed] [Google Scholar]
- 100.Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X 2006. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol 246:121–127 [DOI] [PubMed] [Google Scholar]
- 101.Oppenheimer JH, Schwartz HL 1985. Stereospecific transport of triiodothyronine from plasma to cytosol and from cytosol to nucleus in rat liver, kidney, brain, and heart. J Clin Invest 75:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fujiwara K, Adachi H, Nishio T, Unno M, Tokui T, Okabe M, Onogawa T, Suzuki T, Asano N, Tanemoto M, Seki M, Shiiba K, Suzuki M, Kondo Y, Nunoki K, Shimosegawa T, Iinuma K, Ito S, Matsuno S, Abe T 2001. Identification of thyroid hormone transporters in humans: different molecules are involved in a tissue-specific manner. Endocrinology 142:2005–2012 [DOI] [PubMed] [Google Scholar]
- 103.Takeuchi A, Masuda S, Saito H, Abe T, Inui K 2001. Multispecific substrate recognition of kidney-specific organic anion transporters OAT-K1 and OAT-K2. J Pharmacol Exp Ther 299:261–267 [PubMed] [Google Scholar]
- 104.Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, Nomura H, Unno M, Suzuki M, Naitoh T, Matsuno S, Yawo H 1999. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J Biol Chem 274: 17159–17163 [DOI] [PubMed]
- 105.Cattori V, Hagenbuch B, Hagenbuch N, Stieger B, Ha R, Winterhalter KE, Meier PJ 2000. Identification of organic anion transporting polypeptide 4 (Oatp4) as a major full-length isoform of the liver-specific transporter-1 (rlst-1) in rat liver. FEBS Lett 474:242–245 [DOI] [PubMed] [Google Scholar]
- 106.Suzuki T, Onogawa T, Asano N, Mizutamari H, Mikkaichi T, Tanemoto M, Abe M, Satoh F, Unno M, Nunoki K, Suzuki M, Hishinuma T, Goto J, Shimosegawa T, Matsuno S, Ito S, Abe T 2003. Identification and characterization of novel rat and human gonad-specific organic anion transporters. Mol Endocrinol 17:1203–1215 [DOI] [PubMed] [Google Scholar]