

37-kDa LRP mediates the GnRH-II induced increase in MMP-2 expression that plays a critical role in ovarian cancer cell invasion.
Abstract
GnRH-II enhances ovarian cancer cell invasion in an autocrine manner. We have now found that GnRH-II increases 37-kDa laminin receptor precursor (LRP) production in GnRH receptor (GnRHR)-positive OVCAR-3 and CaOV-3 ovarian cancer cells, while small interfering RNA (siRNA)-mediated depletion of GnRH-II or GnRHR mRNA abrogates this. The invasiveness of ovarian cancer cells is also reduced >85% by siRNA-mediated knockdown of LRP levels and >50% by pretreatment of Matrigel with a synthetic peptide that blocks interactions between laminin and the 67-kDa nonintegrin laminin receptor which comprises two LRP subunits. Conversely, overexpressing LRP in CaOV-3 cells increases their invasiveness 5-fold, while overexpressing LRP with a nonfunctional laminin-binding site does not. Depletion of LRP by siRNA treatment reduces CaOV-3 cell attachment to laminin-coated plates by ∼80% but only reduces their binding to Matrigel by ∼20%. Thus, while LRP influences CaOV-3 cell adhesion to laminin, LRP must act in other ways to enhance invasion. Matrix metalloproteinases (MMPs) are key mediators of invasion, and LRP siRNA treatment of OVCAR-3 and CaOV-3 cells inhibits MMP-2 but not MMP-9 mRNA levels. Overexpressing LRP in these cells increases MMP-2 production specifically, while a laminin-binding deficient LRP does not. Importantly, LRP siRNA treatment abolishes GnRH-II–induced MMP-2 production, and invasion in OVCAR-3 and CaOV-3 cells, which was also seen after MMP-2 siRNA treatment. These results suggest that GnRH-II–induced LRP expression increases the amount of the 67-kDa nonintegrin laminin receptor, which appears to interact with laminin in the extracellular matrix to promote MMP-2 expression and enhance ovarian cancer cell invasion.
It is increasingly apparent that GnRH-II acts as an autocrine regulator in nonpituitary tissues in addition to its role in promoting gonadotropin synthesis and steroid hormone production (1, 2), and GnRH-II is emerging as an important player in cancer cell biology (3). We have previously reported that epidermal growth factor increases GnRH-II production in ovarian cancer cells, and that this enhances their invasive potential by binding to the GnRH type I receptor (GnRHR) (4), but the downstream mechanisms responsible for this autocrine action of GnRH-II remained to be defined.
Matrix metalloproteineases (MMPs) are a family of secreted or membrane-bound enzymes that degrade extracellular matrix (ECM) molecules, thereby facilitating cancer cell migration or invasion (5). The expression of MMP-2 and MMP-9 is significantly higher in ovarian cancer, as compared with normal ovarian tissues (6–8). Moreover, in vitro studies have demonstrated that MMP-2 (9) and MMP-9 (10, 11) contribute to the invasive and metastatic potential of ovarian carcinoma, and that GnRH-I acts through the GnRHR to increase their proteolytic activities in ovarian cancer cell lines (12). Interestingly, the 37-kDa laminin receptor precursor (LRP) has also been reported to increase the activity of MMP-2 and thereby promote breast cancer cell (13) and melanoma cell (14) invasiveness.
The LRP homodimerizes to form the 67-kDa nonintegrin laminin receptor (67LR) (15, 16), and an increase in 67LR levels has been found in a variety of common cancers, as compared with their corresponding normal tissues (17, 18). In many cases, a positive correlation between LRP expression and tumor aggressiveness or metastatic potential has also been found (17). At the cell surface 67LR can interact with laminin, and this is thought to influence tumor cell-attachment (19–21), migration (22), angiogenesis (23), invasion, and metastasis (19, 20, 24). A palindromic sequence (LWMMWL) within LRP acts as the major laminin-1 binding site of 67LR (25–28), and a synthetic polypeptide (peptide G) that includes this sequence binds to laminin-1 with high affinity and can thereby block the interaction between the 67LR and laminin-1 (26). In addition, the amount of 67LR on the cell surface is increased by GnRH-I and GnRH-II in normal and cancerous T cells, and this induces their invasive and metastatic potential (29). We have therefore explored the possibility that LRP influences the way that GnRH-II acts in an autocrine manner to increase ovarian cancer invasion via altered MMP expression.
Results
GnRH-II modulates ovarian cancer cell invasion in an autocrine manner
Treatment of ovarian cancer cells with epidermal growth factor increases their GnRH-II mRNA levels, and this contributes to their invasive potential (4). We have now treated two ovarian cancer cell lines with GnRH-II small interfering RNA (siRNA) before an invasion assay, and this demonstrated that knockdown of GnRH-II mRNA levels in OVCAR-3 (Fig. 1A) and CaOV-3 (Fig. 1B) cells significantly reduces their invasive properties (Fig. 1, C and D). Importantly, this GnRH-II siRNA-mediated reduction in the invasiveness of both cell lines was reversed upon exogenous treatment of GnRH-II (Fig. 1, C and D). These results confirm and extend our previous finding that GnRH-II functions in an autocrine manner to modulate the invasive potential of ovarian cancer cells (4).
Fig. 1.

GnRH-II acts in an autocrine manner to enhance ovarian cancer cell invasion. OVCAR-3 (A) and CaOV-3 (B) cells were treated with 100 nm GnRH-II siRNA (si-GnRH-II) or 100 nm control siRNA (si-Ctrl) for 24 h. In both cases, the mRNA levels of GnRH-II and GnRHR were measured by RT-qPCR and expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with control siRNA (si-Ctrl). C and D, The cells were then seeded into Matrigel-coated transwells and cultured for 48 h in the presence of 10 nm GnRH-II. Noninvading cells were removed from the upper side of the filter, and nuclei of invading cells were stained with Hoechst 33258. Upper panels show representative photomicrographs of cells attached to the lower membrane of the transwells in the invasion assays, while lower panels show the quantitative results of these assays, with results expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with control siRNA (si-Ctrl).
GnRH-II increases LRP production in ovarian cancer cells
Overexpression of LRP influences cancer cell metastasis and invasiveness (17), and we therefore compared the LRP mRNA levels in normal ovarian surface epithelial cells and three serous ovarian carcinoma cell lines (i.e., OVCAR-3, CaOV-3, and SKOV-3 cells). The results indicate that LRP mRNA levels are much higher in CaOV-3 and SKOV-3 cells than in OVCAR-3 cells or normal ovarian surface epithelial cells (Fig. 2A, upper panel). These three ovarian cancer cell lines exhibit different invasive properties, and this is evident in our invasion assays. For instance, SKOV-3 cells were seeded at the lowest cell density but demonstrate the highest cell invasiveness, while CaOV-3 and OVCAR-3 cells were seeded at a higher cell density and show a lower cell invasive potential (Fig. 2A, lower panel). Interestingly, the LRP mRNA levels in these three ovarian cancer cell lines corresponds to their ranking in terms of invasiveness (SKOV-3 > CaOV-3 > OVCAR-3) (Fig. 2A).
Fig. 2.

GnRH-II induces the production of LRP in ovarian cancer cells. A, LRP mRNA levels in OVCAR-3, CaOV-3, and SKOV-3 cells were measured by RT-qPCR and expressed relative to the levels in normal ovarian surface epithelial (OSE) cells. *, P < 0.05 compared with OSE. In parallel experiments, OVCAR-3, CaOV-3, and SKOV-3 cells were subjected to an invasion assay for 24 h. Lower panels show representative photomicrographs of cells attached to the lower membrane of transwells in the invasion assay. Below the photomicrographs, the numbers of invasive cells were quantified in five fields and expressed as means ± sem of three independent experiments. B, OVCAR-3, CaOV-3, and SKOV-3 cells were treated with 10 nm of GnRH-II and LRP mRNA (left panel) and LRP (right panel) levels were determined after 8 h and 24 h, respectively. RT-qPCR results are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with untreated control (Ctrl). OVCAR-3 (C and E) and CaOV-3 (D and F) cells were treated with 100 nm GnRH-II siRNA (si-GnRH-II), 100 nm GnRHR siRNA (si-GnRHR), or 100 nm control siRNA (si-Ctrl) for 24 h and then treated with 10 nm GnRH-II for a further 24 h. Cells were harvested and protein extracts were subjected to Western blotting and probed for LRP, GnRHR, or β-actin as a normalization control.
To examine whether GnRH-II treatment induced LRP production in ovarian cancer cells, OVCAR-3 and CaOV-3 cells were treated with 10 nm GnRH-II for 8 h or 24 h before harvesting for measurements of LRP expression. As shown in Fig. 2B, GnRH-II increased LRP mRNA and LRP levels in both OVCAR-3 and CaOV-3 cells, which express relatively high levels of GnRHR; whereas SKOV-3 cells, a cell line that expresses limited amounts of GnRHR (4), did not respond in this way to GnRH-II treatment (Fig. 2B). This is in accordance with our previous finding that GnRH-II treatment does not affect the invasive potential of SKOV-3 cells (4). Furthermore, when SKOV-3 cells were treated with GnRHR siRNA before invasion assays, the results show that depletion of GnRHR in this cell line does not have any effects on its basal invasiveness (Supplemental Fig. 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org/).
A GnRH-II/GnRHR autocrine loop regulates LRP levels in ovarian cancer cells
When OVCAR-3 (Fig. 2C) and CaOV-3 cells (Fig. 2D) were treated with GnRH-II siRNA, a reduction in LRP levels was observed that could be reversed by treatment of the cells with exogenous GnRH-II. To verify that GnRH-II acts through the GnRHR to regulate LRP production, the cells were treated with GnRHR siRNA before treatment with GnRH-II. The results show that depletion of GnRHR in OVCAR-3 (Fig. 2E) and CaOV-3 cells (Fig. 2F) inhibits both basal and GnRH-II-induced LRP levels, without altering GnRH-II mRNA levels (Supplemental Fig. 2). These data suggest that GnRH-II acts via the GnRHR in an autocrine manner to regulate LRP production in ovarian cancer cells.
LRP promotes ovarian cancer cell invasion
To assess the role of LRP in ovarian cancer cell invasion, siRNA was used to knockdown LRP levels in CaOV-3 (Fig. 3A) and SKOV-3 cells (Fig. 3B) before invasion assays using Matrigel-coated transwells. Cells treated with LRP siRNA exhibited a significant reduction in their invasiveness, as compared with cells treated with control siRNA, suggesting that LRP up-regulates this property of ovarian cancer cells.
Fig. 3.
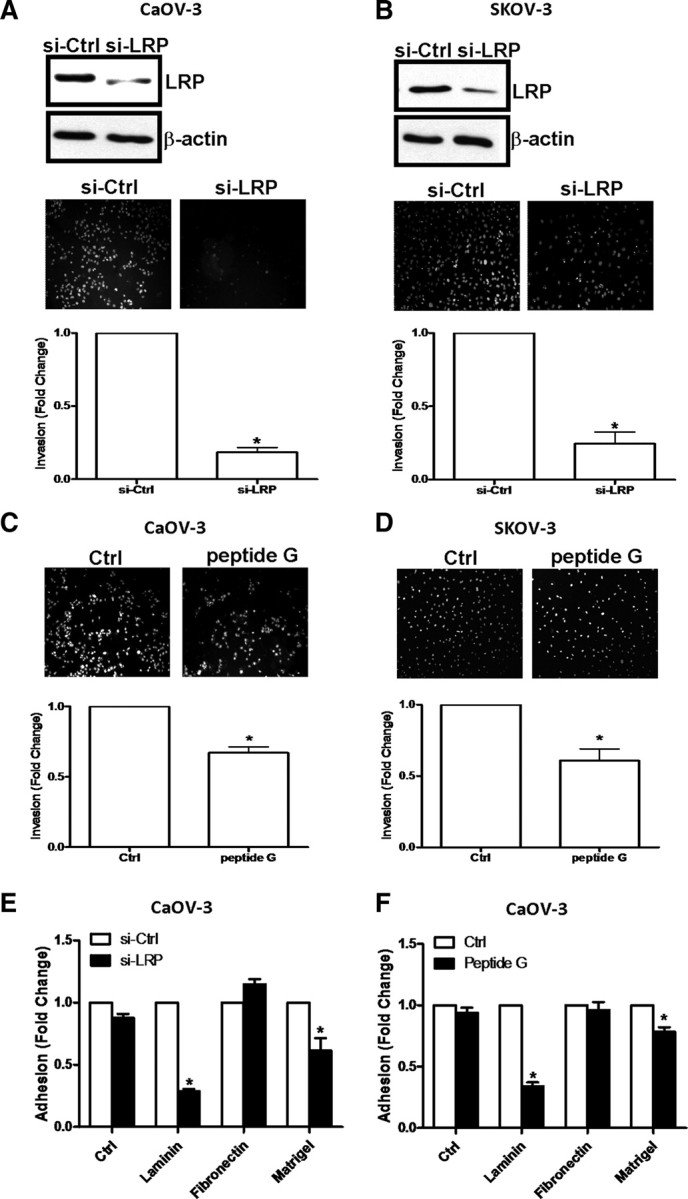
LRP mediates the invasive potential of ovarian cancer cells. CaOV-3 (A) and SKOV-3 (B) cells were treated with 75 nm LRP siRNA (si-LRP) or 75 nm control siRNA (si-Ctrl) before invasion assays for 48 h (CaOV-3 cells) or 24 h (SKOV-3 cells). In both cases, the efficiency of LRP siRNA was verified by Western blotting (upper panels). Synthetic peptide G (1 μg) was preincubated with the Matrigel-coated transwells for 30 min before CaOV-3 (C) and SKOV-3 (D) cells were subjected to invasion assays. In A, B, C, and D, representative photomicrographs of cells attached to the lower membrane of the transwells in the invasion assays are shown, and the lower panels show the quantitative results of these assays, with results expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with control siRNA (si-Ctrl) in A and B or untreated control (Ctrl) in C and D. E, CaOV-3 cells were treated with si-LRP or si-Ctrl before adhesion assays for 8 h on uncoated (Ctrl) tissue culture plates, or plates coated with laminin, fibronectin, or Matrigel. Results are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with control siRNA (si-Ctrl). F, Peptide G (1 μg) was preincubated with the uncoated (Ctrl) or coated tissue culture plates (i.e., with fibronectin, laminin, or Matrigel) for 30 min before CaOV-3 cells were tested in an adhesion assay for 8 h. Results are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with untreated control (Ctrl).
Peptide G mimics the laminin binding site of LRP and will competitively block interactions between cell surface 67LR and laminin within an ECM (30). Laminin is a major component of the synthetic ECM, Matrigel, and the basal invasiveness of CaOV-3 (Fig. 3C) and SKOV-3 cells (Fig. 3D) was reduced by ∼50% when peptide G was preincubated with Matrigel-coated transwells. We therefore also conducted adhesion assays to determine whether the effects of LRP on ovarian cancer cell invasion are simply caused by a loss of cell adhesion. In laminin-coated tissue culture plates, depletion of LRP with siRNA treatment in CaOV-3 cells resulted in an ∼75% inhibition of cell adhesiveness, while only an ∼20% inhibition of cell adhesion was observed in Matrigel-coated tissue culture plates (Fig. 3E). In addition, preincubation of laminin-coated tissue culture plates with peptide G reduced the adhesion of CaOV-3 cells by ∼60%, while peptide G treatment of Matrigel-coated plates reduced the adhesiveness of these cells to the ECM by only ∼15% (Fig. 3F). Thus, while these results suggest that the interaction between 67LR and laminin facilitates CaOV-3 cell adhesion to the ECM, it does not entirely account for the adhesive properties of ovarian cancer cells or the substantial loss in their invasiveness when LRP levels were depleted.
To further verify that LRP promotes ovarian cancer cell invasion, we first stably transfected CaOV-3 cells with a LRP expression vector and identified two independent CaOV-3 cell clones that overexpress LRP by Western blotting (Fig. 4A). Using transwell invasion assays, we found that the LRP overexpressing CaOV-3 cells were significantly more invasive than CaOV-3 cells stably transfected with a control expression vector (Fig. 4B). Furthermore, this increased invasiveness of LRP overexpressing cells was abolished when they were pretreated with LRP siRNA (Fig. 4C). Moreover, when CaOV-3 cells were engineered to overexpress a laminin-binding deficient LRP, they showed no change in invasiveness (Fig. 5A) or adhesion to Matrigel (Fig. 5B). Taken together, these results suggest that increases in LRP production enhance ovarian cancer cell invasiveness in vitro through an increase in the amounts of cell surface 67LR that can interact with laminin within Matrigel.
Fig. 4.
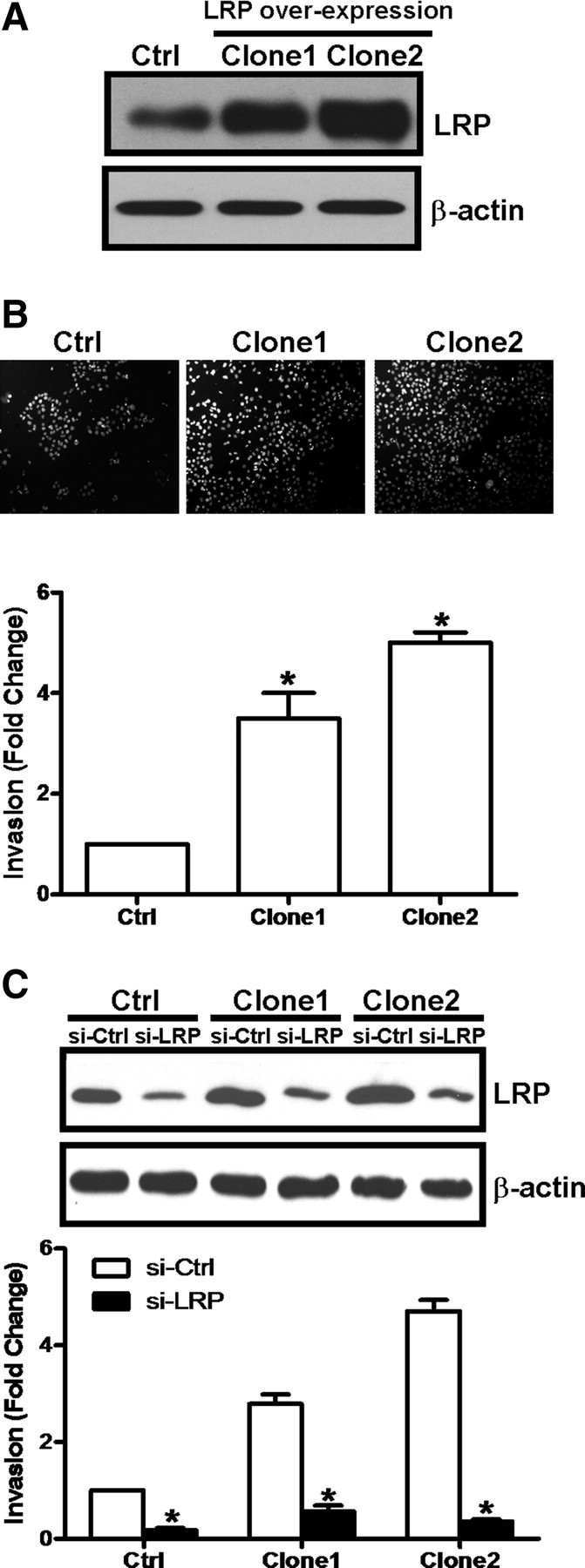
Overexpressing LRP up-regulates ovarian cancer cell invasiveness. CaOV-3 cells were stably transfected with expression vectors for wild-type LRP or eGFP as a control (Ctrl). A, Overexpression of LRP in two independent clones (i.e., Clones 1 and 2) was verified by Western blotting. B, In parallel experiments, these stably transfected CaOV-3 cells were subjected to invasion assays for 48 h. The upper panel shows representative photomicrographs of cells attached to the lower membrane of the transwells in the invasion assays, and the lower panels show the quantitative results of these assays, with results expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with control (Ctrl). C, LRP overexpressing cells (Clones 1 and 2) and control (Ctrl) cells were treated with 75 nm LRP siRNA (si-LRP) or 75 nm control siRNA (si-Ctrl) before invasion assays for 48 h. The efficiency of LRP siRNA in these LRP overexpressing clones were verified by Western blotting of LRP (upper panel). Results of invasion assays are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with the control siRNA (si-Ctrl).
Fig. 5.
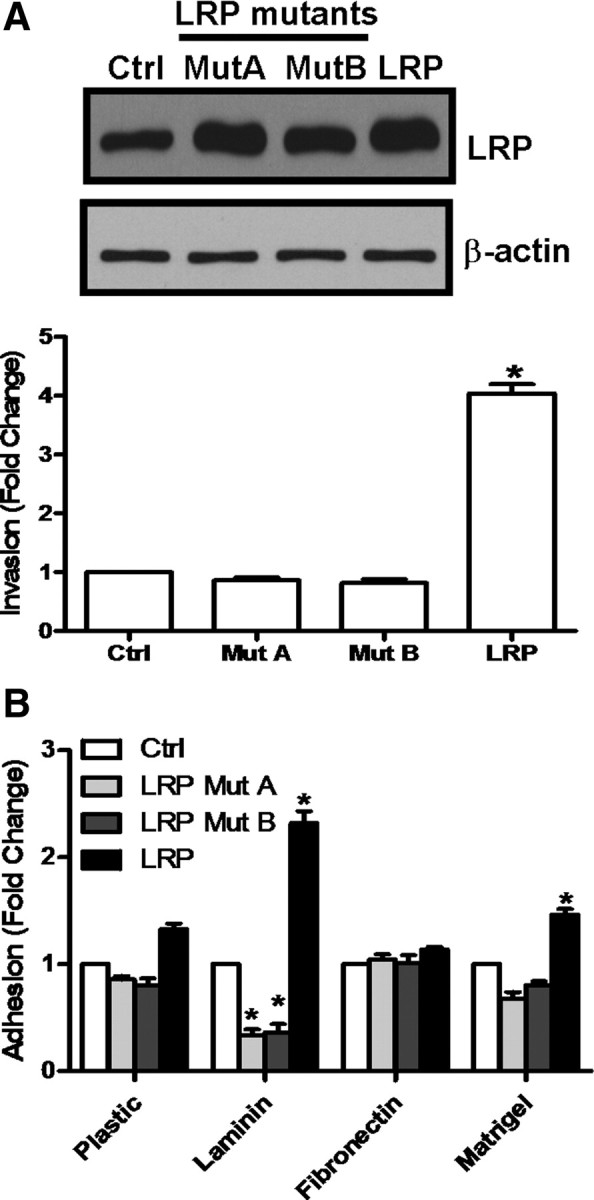
The laminin binding domain of LRP plays an important role in ovarian cancer cell invasion. A, CaOV-3 cells were stably transfected with wild-type LRP (LRP) or two different mutated LRP expression vectors (i.e., Mut A, Mut B). The relative LRP levels in these cells, and cells expressing eGFP as a control (Ctrl), were compared by Western blotting (upper panel), and invasion assays using these cells were performed for 48 h (lower panel). B, In parallel experiments, these stably transfected cells were also subjected to adhesion assays for 8 h. In both A and B, results are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with the control cells (Ctrl).
LRP is an obligate intermediate in GnRH-II–induced MMP-2 production and ovarian cancer cell invasion
Because it has been reported that LRP modulates the expression of MMP-2 in breast cancer cells (13), we examined whether altered LRP levels influences MMP-2 or MMP-9 expression in OVCAR-3 and CaOV-3 cells (Fig. 6). In these experiments, siRNA-mediated knockdown of LRP levels decreased the basal mRNA levels of MMP-2 but not MMP-9 in both cell lines (Fig. 6, A, C, D, and F). Moreover, transient overexpression of LRP in these cells specifically increased the levels of MMP-2 mRNA but not MMP-9 mRNA (Fig. 6, B, C, E, and F). In addition, no increases in MMP-2 mRNA (Fig. 6G) or MMP-2 (Fig. 6H) levels were detected in CaOV-3 cells overexpressing the laminin-binding deficient LRP mutant. These experiments indicate that an interaction between the 67LR and laminin specifically increases MMP-2 production in ovarian cancer cells.
Fig. 6.

LRP regulates MMP-2 production in ovarian cancer cells. OVCAR-3 and CaOV-3 cells were treated with (A, D) 75 nm LRP siRNA (si-LRP) or 75 nm control siRNA (si-Ctrl), or (B, E) transiently transfected with 1 μg wild-type LRP expression vector or 1 μg eGFP expression vector as a control. C and F, In both cases, the efficiency of LRP siRNA and overexpression of LRP in these cells were verified by RT-qPCR measurements of LRP mRNA levels. Total RNA from transfected cells was used to prepare cDNA for RT-qPCR analysis to evaluate the effects of LRP on MMP-2 and MMP-9 mRNA levels, which are expressed in A and D as a percentage over control siRNA (si-Ctrl) values, or in B and E as a percentage of corresponding values in the control (Ctrl) cells. G, Total RNA of CaOV-3 cells stably overexpressing mutated LRP (i.e., Mut A and Mut B), eGFP control vector (Ctrl), and wild-type LRP (LRP) was isolated and subjected to RT-qPCR to measure the MMP-2 and MMP-9 mRNA levels expressed as percentage over cells expressing the control vector (Ctrl). Results are expressed as the mean ± sem of three independent experiments. *, P < 0.05 compared with respective controls. H, In parallel experiments, protein lysates from these stably overexpressing cells were harvested and subjected to Western blotting for their relative MMP-2, MMP-9, and LRP protein levels or β-actin as a normalization control. The Western blotting data are expressed as mean ± sem of three independent experiments (right panel). *, P < 0.05 compared with the control cells (Ctrl).
In line with a previous report that GnRH-I/GnRHR signaling up-regulates MMP-2 expression in ovarian cancer cells (12), we also found that treatment of OVCAR-3 (Fig. 7A) and CaOV-3 cells (Fig. 7B) with GnRH-II increased MMP-2 mRNA levels in a time-dependent manner. In addition, depletion of GnRHR in OVCAR-3 and CaOV-3 cells abolished the GnRH-II-stimulated increases in MMP-2 levels (Fig. 7, C and D; Supplemental Fig. 3, A and B) in both cell lines and confirmed that GnRH-II/GnRHR interactions stimulate MMP-2 expression. In addition, we further verified that GnRH-II treatment did not affect MMP-2 mRNA levels in SKOV-3 cells (Supplemental Fig. 4A), which are known to have very low GnRHR levels (4). Moreover, depletion of GnRHR levels in SKOV-3 cells by GnRHR siRNA treatment does not influence MMP-2 levels in these cells (Supplemental Fig. 4B). Importantly, depletion of LRP by LRP siRNA treatment abolished the GnRH-II–induced increase in MMP-2 production in OVCAR-3 (Fig. 7E and Supplemental Fig. 3C) and CaOV-3 cells (Fig. 7F and Supplemental Fig. 3D).
Fig. 7.

LRP mediates the GnRH-II–induced production of MMP-2 by ovarian cancer cells. OVCAR-3 (A) and CaOV-3 (B) cells were treated with 10 nm GnRH-II for up to 72 h. Total RNA from treated cells was used to prepare cDNA for RT-qPCR analysis to evaluate the effect of GnRH-II on MMP-2 mRNA levels. RT-qPCR results are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with untreated control (Ctrl). C and D, OVCAR-3 and CaOV-3 cells were treated with 100 nm GnRHR siRNA (si-GnRHR) or 100 nm control siRNA (si-Ctrl) for 24 h and then treated with 10 nm GnRH-II for 48 h. E and F, OVCAR-3 and CaOV-3 cells were treated with 75 nm LRP siRNA (si-LRP) or 75 nm control siRNA (si-Ctrl) for 24 h and then treated with 10 nm GnRH-II for 48 h. In C–F, cells were harvested and protein extracts were subjected to Western blotting with antibodies against MMP-2, GnRHR, LRP, or β-actin as a normalization control.
To explore this further, CaOV-3 cells were treated with LRP siRNA or MMP-2 siRNA before invasion assays, and the results of these experiments indicate that depletion of either LRP (Fig. 8A) or MMP-2 (Fig. 8C) abolishes GnRH-II–induced invasion. Furthermore, depletion of MMP-2 levels in CaOV-3 cells did not affect GnRH-II, GnRHR, or LRP levels in this cell line (Supplementary Fig. 5). Importantly, preincubation of Matrigel-coated transwells with peptide G attenuated the proinvasive effects of GnRH-II in CaOV-3 cells (Fig. 8B). These observations suggest that LRP is a key intermediary in GnRH-II–stimulated MMP-2 production and may thereby play a pivotal role in GnRH-II-induced ovarian cancer cell invasiveness.
Fig. 8.

LRP and MMP-2 are key mediators of GnRH-II enhanced invasion in ovarian cancer cells. A, CaOV-3 cells were treated with 75 nm LRP siRNA (si-LRP) or 75 nm control siRNA (si-Ctrl) for 24 h, and with 10 nm GnRH-II for a further 48 h during an invasion assay. B, In parallel experiments, 1 μg of peptide G was preincubated with Matrigel-coated transwells for 30 min before invasion assay in the presence of 10 nm GnRH-II for 48 h. C, CaOV-3 cells were treated with 75 nm MMP-2 siRNA (si-MMP-2) or 75 nm control siRNA (si-Ctrl) for 24 h, before an invasion assay in the presence of 10 nm GnRH-II for 48 h. Results are expressed as mean ± sem of three independent experiments. *, P < 0.05 compared with control siRNA (si-Ctrl) in A and C or untreated control (Ctrl) in B. The efficiency of LRP siRNA and MMP-2 siRNA were verified by Western blot analysis of LRP (right panels in A and C). In parallel experiments, si-MMP-2–treated cells were harvested and protein extracts were subjected to Western blotting with antibodies against LRP, GnRHR, or β-actin as a normalization control.
Discussion
Both GnRH-II (31) and GnRHR (32) are more abundant in malignant ovarian tumors than in benign ovarian tissues, and this underscores the importance of understanding GnRH-II function in the context of ovarian cancer. Building on evidence that GnRH-II acts in an autocrine manner to promote ovarian cancer cell invasion (4), we have now found that this action of GnRH-II involves a GnRHR-dependent up-regulation of LRP production. This was not completely surprising because LRP has been reported to be a downstream target of GnRH-II/GnRHR signaling in T cell lymphomas and to be associated with the metastatic potential of these cancer cells (29). However, our results also show that LRP is a critical intermediary in GnRH-II actions that led to an increase in the invasive properties of OVCAR-3 and CaOV-3 cells. We believe this is important because LRP can dimerize and form the cell surface 67LR that functions as a binding protein for laminin in the ECM (17). In support of this, we used peptide G to block cell surface 67LR interactions with laminin during our ovarian cancer cell adhesion and invasion assays, and we found that this reduces the GnRH-II–enhanced invasive potential of OVCAR-3 and CaOV-3 cells. This is important because increased 67LR levels have been observed in a wide range of malignancies, including ovarian carcinomas (33, 34), and correlate with minimal differentiation, disease progression, and poor survival (35, 36). Accordingly, the invasive potential of the ovarian cancer cell lines we have used in our studies is directly related to their LRP levels, and this behavior can be manipulated by changing their LRP levels.
Interactions between cancer cells and laminin in the ECM promote tumor dissemination by several mechanisms, including increasing cell proliferation, adhesion, migration, or invasion (37–39). In our in vitro studies, altered LRP levels did not influence ovarian cancer cell proliferation (data not shown). However, while the adhesion of these cells to laminin-coated tissue culture plates was largely dependent on the expression of LRP, their adhesion to an artificial ECM (Matrigel) was only modestly affected by altering LRP expression. This suggested to us that the requirement for LRP, in relation to the GnRH-II–induced invasive properties of ovarian cancer cells, cannot simply be attributed to an alteration in adhesion to the ECM.
Although it is known that LRP is a multi-functional protein in eukaryotic cells and that 67LR also acts as an endocytotic receptor (17), our data imply that the laminin-binding domain of LRP plays a key role in the invasion of ovarian cancer cells after treatment of GnRH-II. The importance of interactions between cancer cells and ECM components, like laminin, is well documented in terms of their metastatic potential (40–43). It is also known that cancer cells interact with laminin via several cell-surface laminin-binding proteins, including dystroglycan and the integrins α1β1, α2β1, α3β1, α6β1, α6β4, and α7β1, as well as 67LR (44). Our data indicate that LRP plays a key role in GnRH-II–induced increases in the invasive properties of ovarian cancer cells and that this effect most likely relies on an interaction between the cell surface 67LR and laminin. Although the 67LR is not thought to act independently as a cell surface signaling molecule (17), it may act as an accessory with other cell surface laminin-binding proteins that function as bona fide signaling molecules upon interaction with laminin at sites other than that recognized by 67LR (17). For example, 67LR physically associates with α6β4 integrin (45), and coregulation of their membrane localization is important for tumorigenesis (35, 36). In addition, up-regulation of α6β4 integrin increases its interaction with laminin and activates mitogen-activated protein kinase signaling (46) and also appears to stimulate the production of proteases, including MMP-2, MMP-9, and MT1-MMP, that are important in ECM degradation during cell invasion (47).
Laminin induces the production of proteases, like MMPs, as one mode of promoting cancer cell invasion (48). It has previously been reported that GnRH-I treatment of ovarian cancer cells increases MMP-2 and MMP-9 production (12), and we have now shown that the autocrine actions of GnRH-II act through the GnRHR to exert the same effects. Others have shown that siRNA-mediated depletion of 67LR levels significantly reduces tumor aggressiveness with a decrease in the MMP-2 mRNA levels and proteolytic activity of melanoma cells (14). Likewise, we have found that LRP levels influence MMP-2 but not MMP-9 expression in ovarian cancer cells. Importantly, this effect was not seen when a laminin-binding deficient LRP was overexpressed in these cells, suggesting that the specific induction of MMP-2 expression is entirely dependent on the laminin-binding properties of LRP or the 67LR. Thus, our data also imply that the GnRH-mediated induction of MMP-9 expression in ovarian cancer cells must be mediated by some other pathway unrelated to the increased expression of LRP. However, we also conclude that LRP-induced MMP-2 expression must be the predominant mechanism responsible for the autocrine actions of GnRH-II in relation to its effects on the increased invasive potential of some ovarian cancer cells. It should also be noted that MMP-2 is synthesized as a latent zymogen that requires proteolytic cleavage by membrane type I matrix metalloproteinase (MT1-MMP) to achieve its full proteolytic potential (49, 50). This is of interest because we have also recently obtained evidence that MT1-MMP is a downstream target of GnRH-II/GnRHR signaling (51) and is independent of the actions of LRP (data not shown).
We and others have demonstrated previously that GnRH-I and GnRH-II have no effect on SKOV-3 cell invasiveness, and we attributed this finding to the low GnRHR levels in SKOV-3 cells as compared with OVCAR-3 and CaOV-3 cells (4). Our results also now indicate that GnRH-II treatment does not affect LRP or MMP-2 levels in SKOV-3 cells. In addition, GnRHR siRNA treatment has no effects on SKOV-3 cell invasiveness or LRP and MMP-2 levels. However, siRNA-mediated reductions in LRP levels or peptide G treatment markedly reduce the invasive potential of SKOV-3 cells. Thus, while LRP contributes to the invasive properties of SKOV-3 cells, this is not influenced by the GnRH-II/GnRHR autocrine loop as in OVCAR-3 and CaOV-3 cells, and other mechanisms may influence LRP expression in SKOV-3 cells.
Our present studies provide important insights into a novel aspect of GnRH-II function in ovarian cancer cell biology. In summary, we propose that the epidermal growth factor–stimulated increase in GnRH-II production by ovarian cancer cells (4) subsequently acts in an autocrine manner via the GnRHR to stimulate LRP production. Elevated LRP levels increase tumor cell interactions with laminin within the ECM, and this is an obligatory step in GnRH-II stimulation of MMP-2 production which plays a key role in ovarian cancer cell invasion.
Materials and Methods
Cells and cell culture
The human ovarian adenocarcinoma cell lines, OVCAR-3, CaOV-3, and SKOV-3, were obtained from American Type Culture Collection (Manassas, VA). Normal ovarian surface epithelial cells were obtained by scraping the ovarian surface at surgery or laparoscopy for nonmalignant disorders, as previously described (52). Cell cultures were maintained in M199/MCDB105 (Invitrogen Inc., Burlington, ON, Canada) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories Inc., Logan, UT, USA) at 37 C in a humidified atmosphere of 5% CO2. The cells were subcultured when they reached about 90% confluence using a trypsin/EDTA solution (0.05% trypsin, 0.5 mm EDTA).
Antibodies and reagents
GnRH-II analog (DArg6-Azagly10-GnRH-II) was purchased from Bachem (Belmont, CA). Peptide G (IPCNNKGAHSVGLMWWMLAR), corresponding to amino acids 161–180 of LRP, was synthesized at the University of British Columbia (Vancouver, BC, Canada). The monoclonal LRP antibody (MLuC5) and the polyclonal β-actin antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The monoclonal GnRHR antibody, monocolonal MMP-2 antibody, and monoclonal MMP-9 antibody were obtained from Neomarkers (Fremont, CA). Horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG were obtained from Bio-Rad Laboratories (Hercules, CA). Horseradish peroxidase-conjugated donkey anti-goat IgG was obtained from Santa Cruz Biotechnology.
Reverse transcription quantitative real-time PCR (RT-qPCR)
After treatment with 10 nm GnRH-II, medium was removed from the culture dish and RNA was extracted using Trizol (Invitrogen). Total RNA (2 μg) was reverse transcribed into cDNA using a first-strand cDNA synthesis kit (GE Healthcare Bio-Science, Piscataway NJ) following the manufacturer's procedure. The primers used for SYBR Green RT-qPCR were designed using Primer Express Software v2.0 (Applied Biosystems, Foster City, CA). The primers for GnRH-II mRNA are as follows: sense, 5′-CTGCTGACTGCCCACCTT; and antisense, 5′-GCTTTCCTCCAGGGTACCAG. The primers for GnRHR are as follows: sense, 5′-ACTGTTCCGACTTTGCTGTTGCT; and antisense, 5′-ACCGCTCCCTGGCTATCAC. The primers for LRP are as follows: sense, 5′-ATGTCCTGCAAATGAAGGAGG; and antisense, 5′-TGGAAGTCAAGATTGGTGCCA. The primers for MMP-2 are as follows: sense, 5′-TACACCAAGAACTTCCGTCTGT; and antisense, 5′-AATGTCAGGAGAGCTCCCCATA. The primers for MMP-9 are as follows: sense, 5′-GCCACTACTGTGCCTTTGAGTC; and antisense, 5′-CCCTCAGAGAATCGCCAGTACT. The primers for GAPDH are as follows: sense, 5′-GAGTCAACGGATTTGGTCGT; and antisense, 5′-GACAAGCTTCCCGTTCTCAG. The reactions were performed using SYBR Green PCR Master Mix (Applied Biosystems). All RT-qPCR experiments were run in triplicate, and a mean value was used for the determination of mRNA levels. Negative controls containing water instead of sample cDNA were used in each experiment. Relative quantification of the mRNA levels of GnRH-II, GnRHR, LRP, MMP-2, and MMP-9 was performed using the comparative Cq method with GAPDH as the reference gene and with the formula 2−ΔΔCq.
Plasmid constructs and cell transfections
The LRP and enhanced green fluorescent protein (eGFP) cDNA constructs in the same plasmid expression vector (pReciever-M02) were purchased from GeneCopoeia (Rockville, MD). The LMWWML site within coding sequence of the LRP expression plasmid was mutated to the corresponding sequence of the 37-kDa LRP orthologs found in Arabidopsis (CLFWLL) and Saccharomyces (LIWYLL) because they lack laminin-binding activity but retain other highly-conserved functions of LRP (53). Mutagenesis was performed using the Quickchange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the LRP construct as template and the following oligonucleotide primers: LRP Mut A (mutated sequence underlined encodes CLFWLL): forward, 5′-CTCAGTGGGTTGCTTGTTCTGGTTGCTGGCTCGGGAAGTTCTG and reverse, 5′-CAGAACTTCCCGAGCCAGCAACCAGAACAAGCAACCCACTGAG; LRP Mut B, (mutated sequence underlined encodes LIWYLL): forward, 5′-CTCAGTGGGTTTGATCTGGTACTTGCTGGC-TCGGGAAGTTCTG and reverse, 5′-CAGAACTTCCCGAGCCAGC-AAGTACCAGATC AAACCCACTGAG. The mutated constructs were sequenced to verify that only the desired mutations had occurred.
CaOV-3 cells overexpressing eGFP, wild-type LRP (LRP), or mutated LRP (Mut A and Mut B) were selected with 600 μg/ml geneticin (Invitrogen) and cloned by limiting dilution. The eGFP expression vector was used as a transfection control, and clones expressing eGFP were verified by fluorescence microscopy, while LRP mRNA and protein levels in clones expressing wild-type or mutant LRP were measured by RT-qPCR or Western blotting.
Transfections of expression plasmids were performed using Lipofectamine LTX and PLUS Reagent (Invitrogen), while siRNA transfections were carried out using Lipofectamine RNAiMAX Reagent (Invitrogen) following the manufacturer's protocol. Briefly, 5 × 105 cells were seeded into six-well tissue culture plates 1 d before transfection with 100 nm GnRH-II siRNA (si-GnRH-II), 100 nm GnRHR siRNA (si-GnRHR), 75 nm 37 kDa LRP siRNA (si-LRP), or a nontargeting control siRNA (si-Ctrl) (Dharmacon, Inc., Lafayette CO). After 6 h, the medium in each well was replaced with 2 ml of 0.5% FBS medium and the cells were further incubated overnight (18 h). The culture medium was then removed, and the cells were treated with 10 nm GnRH-II in 0.5% serum medium for the times indicated.
Adhesion assay
Equal amounts of laminin (2 μg/cm2), fibronectin (5 μg/cm2), and growth factor–reduced Matrigel (5 μg/cm2) were used to coat 96-well tissue culture plates (Becton Dickinson Labware, Franklin Lakes, NJ) for 2 h at 37 C. Cells were trypsinized and seeded in the coated tissue culture plates in serum-free medium and allowed to adhere at 37 C for 8 h. The plates were then washed twice with cold PBS, and the adherent cells were fixed with ice-cold methanol, stained with crystal violet, and quantified by measuring absorbance at 630 nm.
Invasion assay
Transwell cell culture inserts (8-μm pore size, 24-well, BD Biosciences) were coated with 40 μl of 1 mg/ml growth factor–reduced Matrigel (BD Biosciences). Cells in M199/MCDB105 medium supplemented with 0.5% FBS were incubated for 48 h against a gradient of 10% FBS for OVCAR-3 cells (1.5 × 106 per insert) and CaOV-3 cells (1.25 × 106 per insert) and 24 h of 5% FBS for SKOV-3 cells (1 × 106 per insert). Cells on the lower side of the membrane were fixed with ice-cold methanol, stained with Hoechst 33258 (Sigma), and the number of nuclei were counted using a Zeiss Axiophot epifluorescent microscope equipped with a digital camera and Northern Eclipse 6.0 software (Empix Imaging, Mississauga, ON). Individual experiments had duplicate inserts and five microscopic fields were counted per insert.
Data analysis
Results are shown as the mean ± sem of three independent experiments and were analyzed by one-way ANOVA followed by Tukey test using PRISM software (GraphPad Software Inc., San Diego, CA). Means were considered significantly different from each other at P < 0.05.
Acknowledgments
P.C.K.L. is the recipient of a Child and Family Research Institute Distinguished Scholar Award. G.L.H. is a Tier 1 Canada Research Chair in Reproductive Health. S.L.P. was the recipient of a Cordula and Gunter Paetzold graduate studentship.
Address all correspondence and requests for reprints to: Peter C.K. Leung, Ph.D., Department of Obstetrics and Gynecology, University of British Columbia, Room 2H-30, 4490 Oak Street, Vancouver, British Columbia, Canada, V6H 3V5. E-mail: peleung@interchange.ubc.ca.
This work was supported by an operating grant from the Canadian Institutes of Health Research to P.C.K.L.
Disclosure Summary: The authors have nothing to declare.
Footnotes
- 67LR
- 67-kDa nonintegrin laminin receptor
- ECM
- extracellular matrix
- eGFP
- enhanced green fluorescent protein
- FBS
- fetal bovine serum
- GnRHR
- GnRH receptor
- LRP
- laminin receptor precursor
- MMP
- matrix metalloproteinease
- siRNA
- small interfering RNA.
References
- 1. So WK , Cheng JC , Poon SL , Leung PC. 2008. Gonadotropin-releasing hormone and ovarian cancer: a functional and mechanistic overview. FEBS J 275:5496–5511 [DOI] [PubMed] [Google Scholar]
- 2. Cheung LW , Wong AS. 2008. Gonadotropin-releasing hormone: GnRH receptor signaling in extrapituitary tissues. FEBS J 275:5479–5495 [DOI] [PubMed] [Google Scholar]
- 3. Chen CL , Cheung LW , Lau MT , Choi JH , Auersperg N , Wang HS , Wong AS , Leung PC. 2007. Differential role of gonadotropin-releasing hormone on human ovarian epithelial cancer cell invasion. Endocrine 31:311–320 [DOI] [PubMed] [Google Scholar]
- 4. Poon SL , Hammond GT , Leung PC. 2009. Epidermal growth factor-induced GnRH-II synthesis contributes to ovarian cancer cell invasion. Mol Endocrinol 23:1646–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roy R , Yang J , Moses MA. 2009. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol 27:5287–5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hijova E. 2005. Matrix metalloproteinases: their biological functions and clinical implications. Bratisl Lek Listy 106:127–132 [PubMed] [Google Scholar]
- 7. Schmalfeldt B , Prechtel D , Harting K , Späthe K , Rutke S , Konik E , Fridman R , Berger U , Schmitt M , Kuhn W , Lengyel E. 2001. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res 7:2396–2404 [PubMed] [Google Scholar]
- 8. Murthi P , Barker G , Nowell CJ , Rice GE , Baker MS , Kalionis B , Quinn MA. 2004. Plasminogen fragmentation and increased production of extracellular matrix-degrading proteinases are associated with serous epithelial ovarian cancer progression. Gynecol Oncol 92:80–88 [DOI] [PubMed] [Google Scholar]
- 9. Kenny HA , Kaur S , Coussens LM , Lengyel E. 2008. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest 118:1367–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roomi MW , Monterrey JC , Kalinovsky T , Rath M , Niedzwiecki A. 2010. In vitro modulation of MMP-2 and MMP-9 in human cervical and ovarian cancer cell lines by cytokines, inducers and inhibitors. Oncol Rep 23:605–614 [DOI] [PubMed] [Google Scholar]
- 11. Agarwal A , Covic L , Sevigny LM , Kaneider NC , Lazarides K , Azabdaftari G , Sharifi S , Kuliopulos A. 2008. Targeting a metalloprotease-PAR1 signaling system with cell-penetrating pepducins inhibits angiogenesis, ascites, and progression of ovarian cancer. Mol Cancer Ther 7:2746–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheung LW , Leung PC , Wong AS. 2006. Gonadotropin-releasing hormone promotes ovarian cancer cell invasiveness through c-Jun NH2-terminal kinase-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9. Cancer Res 66:10902–10910 [DOI] [PubMed] [Google Scholar]
- 13. Berno V , Porrini D , Castiglioni F , Campiglio M , Casalini P , Pupa SM , Balsari A , Ménard S , Tagliabue E. 2005. The 67 kDa laminin receptor increases tumor aggressiveness by remodeling laminin-1. Endocr Relat Cancer 12:393–406 [DOI] [PubMed] [Google Scholar]
- 14. Givant-Horwitz V , Davidson B , Reich R. 2004. Laminin-induced signaling in tumor cells: the role of the M(r) 67,000 laminin receptor. Cancer Res 64:3572–3579 [DOI] [PubMed] [Google Scholar]
- 15. Romanov VI , Wrathall LS , Simmons TD , Pinto da Silva P , Sobel ME. 1995. Protein synthesis is required for laminin-induced expression of the 67-kDa laminin receptor and its 37-kDa precursor. Biochem Biophys Res Commun 208:637–643 [DOI] [PubMed] [Google Scholar]
- 16. Rao CN , Castronovo V , Schmitt MC , Wewer UM , Claysmith AP , Liotta LA , Sobel ME. 1989. Evidence for a precursor of the high-affinity metastasis-associated murine laminin receptor. Biochemistry 28:7476–7486 [DOI] [PubMed] [Google Scholar]
- 17. Nelson J , McFerran NV , Pivato G , Chambers E , Doherty C , Steele D , Timson DJ. 2008. The 67 kDa laminin receptor: structure, function and role in disease. Biosci Rep 28:33–48 [DOI] [PubMed] [Google Scholar]
- 18. Liu L , Sun L , Zhao P , Yao L , Jin H , Liang S , Wang Y , Zhang D , Pang Y , Shi Y , Chai N , Zhang H , Zhang H. 2010. Hypoxia promotes metastasis in human gastric cancer by up-regulating the 67-kDa laminin receptor. Cancer Sci 101:1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Satoh K , Narumi K , Abe T , Sakai T , Kikuchi T , Tanaka M , Shimo-Oka T , Uchida M , Tezuka F , Isemura M , Nukiwa T. 1999. Diminution of 37-kDa laminin binding protein expression reduces tumour formation of murine lung cancer cells. Br J Cancer 80:1115–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Canfield SM , Khakoo AY. 1999. The nonintegrin laminin binding protein (p67 LBP) is expressed on a subset of activated human T lymphocytes and, together with the integrin very late activation antigen-6, mediates avid cellular adherence to laminin. J Immunol 163:3430–3440 [PubMed] [Google Scholar]
- 21. Mafune K , Ravikumar TS. 1992. Anti-sense RNA of 32-kDa laminin-binding protein inhibits attachment and invasion of a human colon carcinoma cell line. J Surg Res 52:340–346 [DOI] [PubMed] [Google Scholar]
- 22. Vande Broek I , Vanderkerken K , De Greef C , Asosingh K , Straetmans N , Van Camp B , Van Riet I. 2001. Laminin-1-induced migration of multiple myeloma cells involves the high-affinity 67 kD laminin receptor. Br J Cancer 85:1387–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka M , Narumi K , Isemura M , Abe M , Sato Y , Abe T , Saijo Y , Nukiwa T , Satoh K. 2000. Expression of the 37-kDa laminin binding protein in murine lung tumor cell correlates with tumor angiogenesis. Cancer Lett 153:161–168 [DOI] [PubMed] [Google Scholar]
- 24. Givant-Horwitz V , Davidson B , Reich R. 2005. Laminin-induced signaling in tumor cells. Cancer Lett 223:1–10 [DOI] [PubMed] [Google Scholar]
- 25. Hundt C , Peyrin JM , Ha|fkk S , Gauczynski S , Leucht C , Rieger R , Riley ML , Deslys JP , Dormont D , Lasmézas CI , Weiss S. 2001. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J 20:5876–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castronovo V , Taraboletti G , Sobel ME. 1991. Functional domains of the 67-kDa laminin receptor precursor. J Biol Chem 266:20440–20446 [PubMed] [Google Scholar]
- 27. Castronovo V , Taraboletti G , Sobel ME. 1991. Laminin receptor complementary DNA-deduced synthetic peptide inhibits cancer cell attachment to endothelium. Cancer Res 51:5672–5678 [PubMed] [Google Scholar]
- 28. Magnifico A , Tagliabue E , Butó S , Ardini E , Castronovo V , Colnaghi MI , Ménard S. 1996. Peptide G, containing the binding site of the 67-kDa laminin receptor, increases and stabilizes laminin binding to cancer cells. J Biol Chem 271:31179–31184 [DOI] [PubMed] [Google Scholar]
- 29. Chen A , Ganor Y , Rahimipour S , Ben-Aroya N , Koch Y , Levite M. 2002. The neuropeptides GnRH-II and GnRH-I are produced by human T cells and trigger laminin receptor gene expression, adhesion, chemotaxis and homing to specific organs. Nat Med 8:1421–1426 [DOI] [PubMed] [Google Scholar]
- 30. Ardini E , Sporchia B , Pollegioni L , Modugno M , Ghirelli C , Castiglioni F , Tagliabue E , Ménard S. 2002. Identification of a novel function for 67-kDa laminin receptor: increase in laminin degradation rate and release of motility fragments. Cancer Res 62:1321–1325 [PubMed] [Google Scholar]
- 31. Serin IS , Tanriverdi F , Ata CD , Akalin H , Ozcelik B , Ozkul Y , Kelestimur F. 2010. GnRH-II mRNA expression in tumor tissue and peripheral blood mononuclear cells (PBMCs) in patients with malignant and benign ovarian tumors. Eur J Obstet Gynecol Reprod Biol 149:92–96 [DOI] [PubMed] [Google Scholar]
- 32. Wilkinson SJ , Kucukmetin A , Cross P , Darby S , Gnanapragasam VJ , Calvert AH , Robson CN , Edmondson RJ. 2008. Expression of gonadotrophin releasing hormone receptor I is a favorable prognostic factor in epithelial ovarian cancer. Hum Pathol 39:1197–1204 [DOI] [PubMed] [Google Scholar]
- 33. van den Brûle FA , Berchuck A , Bast RC , Liu FT , Gillet C , Sobel ME , Castronovo V. 1994. Differential expression of the 67-kD laminin receptor and 31-kD human laminin-binding protein in human ovarian carcinomas. Eur J Cancer 30A:1096–1099 [DOI] [PubMed] [Google Scholar]
- 34. van den Brûle FA , Castronovo V , Ménard S , Giavazzi R , Marzola M , Belotti D , Taraboletti G. 1996. Expression of the 67 kD laminin receptor in human ovarian carcinomas as defined by a monoclonal antibody, MLuC5. Eur J Cancer 32A:1598–1602 [DOI] [PubMed] [Google Scholar]
- 35. Ménard S , Castronovo V , Tagliabue E , Sobel ME. 1997. New insights into the metastasis-associated 67 kD laminin receptor. J Cell Biochem 67:155–165 [PubMed] [Google Scholar]
- 36. Ménard S , Tagliabue E , Colnaghi MI. 1998. The 67 kDa laminin receptor as a prognostic factor in human cancer. Breast Cancer Res Treat 52:137–145 [DOI] [PubMed] [Google Scholar]
- 37. Engbring JA , Kleinman HK. 2003. The basement membrane matrix in malignancy. J Pathol 200:465–470 [DOI] [PubMed] [Google Scholar]
- 38. Patarroyo M , Tryggvason K , Virtanen I. 2002. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin Cancer Biol 12:197–207 [DOI] [PubMed] [Google Scholar]
- 39. Ekblom P , Lonai P , Talts JF. 2003. Expression and biological role of laminin-1. Matrix Biol 22:35–47 [DOI] [PubMed] [Google Scholar]
- 40. Määttä M , Bützow R , Luostarinen J , Petäjäniemi N , Pihlajaniemi T , Salo S , Miyazaki K , Autio-Harmainen H , Virtanen I. 2005. Differential expression of laminin isoforms in ovarian epithelial carcinomas suggesting different origin and providing tools for differential diagnosis. J Histochem Cytochem 53:1293–1300 [DOI] [PubMed] [Google Scholar]
- 41. Kuwashima Y , Uehara T , Kurosumi M , Kishi K , Shiromizu K , Matsuzawa M , Takayama S. 1995. Basement membrane status in undifferentiated carcinomas of the ovary. Immunohistochemical distribution of type IV collagen and laminin. Eur J Gynaecol Oncol 16:181–186 [PubMed] [Google Scholar]
- 42. Byers LJ , Osborne JL , Carson LF , Carter JR , Haney AF , Weinberg JB , Ramakrishnan S. 1995. Increased levels of laminin in ascitic fluid of patients with ovarian cancer. Cancer Lett 88:67–72 [DOI] [PubMed] [Google Scholar]
- 43. Capo-Chichi CD , Smith ER , Yang DH , Roland IH , Vanderveer L , Cohen C , Hamilton TC , Godwin AK , Xu XX. 2002. Dynamic alterations of the extracellular environment of ovarian surface epithelial cells in premalignant transformation, tumorigenicity, and metastasis. Cancer 95:1802–1815 [DOI] [PubMed] [Google Scholar]
- 44. Sasaki T , Fässler R , Hohenester E. 2004. Laminin: the crux of basement membrane assembly. J Cell Biol 164:959–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ardini E , Tagliabue E , Magnifico A , Butò S , Castronovo V , Colnaghi MI , Ménard S. 1997. Co-regulation and physical association of the 67-kDa monomeric laminin receptor and the alpha6beta4 integrin. J Biol Chem 272:2342–2345 [DOI] [PubMed] [Google Scholar]
- 46. Miyamoto S , Teramoto H , Coso OA , Gutkind JS , Burbelo PD , Akiyama SK , Yamada KM. 1995. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol 131:791–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hood JD , Cheresh DA. 2002. Role of integrins in cell invasion and migration. Nat Rev Cancer 2:91–100 [DOI] [PubMed] [Google Scholar]
- 48. Kleinman HK , Koblinski J , Lee S , Engbring J. 2001. Role of basement membrane in tumor growth and metastasis. Surg Oncol Clin N Am 10:329–338, ix [PubMed] [Google Scholar]
- 49. Sato H , Takino T , Okada Y , Cao J , Shinagawa A , Yamamoto E , Seiki M. 1994. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370:61–65 [DOI] [PubMed] [Google Scholar]
- 50. Strongin AY , Collier I , Bannikov G , Marmer BL , Grant GA , Goldberg GI. 1995. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 270:5331–5338 [DOI] [PubMed] [Google Scholar]
- 51. Poon SL , Lau M-T , Hammond GL , Leung PCK. 2011. Gonadotropin-releasing hormone-II increases membrane type I metalloproteinase production via β-catenin signaling in ovarian cancer cells. Endocrinology 10.1210/en.2010-0942 [DOI] [PubMed]
- 52. Leung EH , Leung PC , Auersperg N. 2001. Differentiation and growth potential of human ovarian surface epithelial cells expressing temperature-sensitive SV40 T antigen. In Vitro Cell Dev Biol Anim 37:515–521 [DOI] [PubMed] [Google Scholar]
- 53. Ardini E , Pesole G , Tagliabue E , Magnifico A , Castronovo V , Sobel ME , Colnaghi MI , Ménard S. 1998. The 67-kDa laminin receptor originated from a ribosomal protein that acquired a dual function during evolution. Mol Biol Evol 15:1017–1025 [DOI] [PubMed] [Google Scholar]