

Abstract
The binding of Src-homology 2 (SH2) domains to phosphotyrosine (pY) sites is critical for the autoinhibition and substrate recognition of the eight Src family kinases (SFKs). The high sequence conservation of the 120 human SH2 domains poses a significant challenge to selectively perturb the interactions of even the SFK SH2 family against the rest of the SH2 domains. We have developed synthetic binding proteins, termed monobodies, for six of the SFK SH2 domains with nanomolar affinity. Most of these monobodies competed with pY ligand binding and showed strong selectivity for either the SrcA (Yes, Src, Fyn, Fgr) or SrcB subgroup (Lck, Lyn, Blk, Hck). Interactome analysis of intracellularly expressed monobodies revealed that they bind SFKs but no other SH2-containing proteins. Three crystal structures of monobody–SH2 complexes unveiled different and only partly overlapping binding modes, which rationalized the observed selectivity and enabled structure-based mutagenesis to modulate inhibition mode and selectivity. In line with the critical roles of SFK SH2 domains in kinase autoinhibition and T-cell receptor signaling, monobodies binding the Src and Hck SH2 domains selectively activated respective recombinant kinases, whereas an Lck SH2-binding monobody inhibited proximal signaling events downstream of the T-cell receptor complex. Our results show that SFK SH2 domains can be targeted with unprecedented potency and selectivity using monobodies. They are excellent tools for dissecting SFK functions in normal development and signaling and to interfere with aberrant SFK signaling networks in cancer cells.
Abbreviations: SH2, Src homology 2 domain; pY, phosphotyrosine; SFK, Src family kinase; ITC, isothermal titration calorimetry; TAP, tandem affinity purification; LC–MS/MS, liquid chromatography coupled to tandem mass spectrometry; TAP-MS, TAP-mass spectrometry; TEV, tobacco etch virus; PEG, polyethylene glycol; TCR, T-cell receptor; FACS, Fluorescence-activated cell sorting; TBS, Tris-buffered saline
Keywords: protein engineering, signaling, protein–protein interactions, oncogene, crystal structure
Graphical Abstract
Highlights
-
•
SFK SH2 domains are critical for signaling but difficult to target.
-
•
Monobodies bound with high affinity and competed with pY ligand binding.
-
•
Discrimination of SH2 domains of the two Src subfamilies was achieved.
-
•
Monobody–SH2 complex crystal structures reveal distinct interaction modes.
-
•
Monobodies selectively perturbed kinase regulation and downstream signaling.
Introduction
Src homology 2 domains (SH2) are small modular protein–protein interaction domains. In humans, 120 SH2 domains are found in 110 signaling proteins, including kinases, phosphatases, adaptor, and scaffold proteins, and cytoskeletal and small GTPase regulators [1], [2], [3]. SH2 domains recognize tyrosine-phosphorylated peptide sequences through two adjacent binding pockets, one binds the phosphotyrosine (pY) side chain, and a second pocket dictates selectivity by recognizing the + 3 side chain downstream of the pY residue [4].
The largest class of SH2 domain-containing proteins are the Src family kinases (SFKs), which consist of eight highly homologous kinases—Src, Yes, Fyn, Fgr, Hck, Lyn, Lck, and Blk, in which the SH2 domain is located N-terminal to the kinase domain. These SH2 domains mediate an intramolecular interaction with the tyrosine phosphorylated C-terminal tails of the kinase domain that is critical for keeping the kinase in the autoinhibited state [5], [6]. In contrast, active SFKs use their SH2 domain for intermolecular interactions to enable multisite processive phosphorylation of substrates [7]. N-terminal to their SH2 domain, all SFKs contain an SH3 domain that is also critical for autoinhibition and collaborates with the SH2 domain in recognizing SFK substrates [8], [9]. Three SFKs (Src, Yes, and Fyn) are ubiquitously expressed; the others are only expressed in particular cell types, such as Hck in myeloid cells or Lck in T- and NK-cells [10]. Mouse knockout studies have helped identify important functions of SFKs downstream of receptor tyrosine kinases, GPCRs, and immune cell receptors in bone, neural, and hematopoietic cell development and signaling [11].
Src was the first oncoprotein that was identified, and today, SFKs are important therapeutic targets in solid tumors and hematological malignancies [12], [13]. Several small molecule ATP-competitive tyrosine kinase inhibitors to SFKs are already in the clinic or in clinical trials but suffer from the rapid development of drug resistance and poor selectivity [14]. Although the SH2 domain of SFKs can be targeted with dominant-negative pY peptides, peptidomimetics, and small molecules [15] that reach up to low nanomolar affinities, these inhibitors cannot discriminate among the SFK SH2 domains and lack comprehensive selectivity data among all SH2 domains [15], [16]. Certain inhibitors originally developed toward selective inhibition are used even as “pan” SH2 domain affinity probes for proteomics experiments and enable the enrichment of a few dozen SH2 proteins, consistent with the paucity of truly selective SFK SH2-selective inhibitors [17].
To accelerate the studies of biologically important aspects of SFK SH2 domains in normal signaling and target them in aberrant oncogenic signaling, we aimed at developing high-performance synthetic binding proteins, termed monobodies. Monobodies are generated from large combinatorial libraries constructed on the molecular scaffold of a fibronectin type III domain [18], [19]. We have developed and characterized monobodies that target the SH2 domains of the leukemogenic Abl kinase [20], [21], [22] and the oncogenic Shp2 tyrosine phosphatase [23]. These monobodies are potent pY ligand antagonists and inhibited Abl or Shp2 signaling. These monobodies achieved very high selectivity [22], [23], which, however, can be rationalized by the fact that only one human paralog (Arg and Shp1, respectively) each exists for Abl and Shp2. Thus, selective targeting of SFKs presents a much greater challenge, given the larger number of highly homologous SFK SH2 domains.
Here, we describe the development and rigorous characterization of high-affinity monobodies to six of the eight SFK SH2 domains. We performed a thorough biochemical and structural analysis to understand the high selectivity that we observed in vitro and in cells and their effects on autoinhibited and active SFKs. This work provides in-depth understanding of SFK SH2 specificity and provides the foundation for the use of these high-precision tools to dissect SFK signaling in cells and in vivo.
Results
Generation of monobodies targeting six SFK SH2 domains
We recombinantly expressed and purified the SH2 domains of the eight SFKs (Yes, Src, Fyn, Fgr, Hck, Lyn, Lck, and Blk; SI Fig. 1a). The Blk SH2 domain nonspecifically bound to the beads used for monobody selection and the Fyn SH2 domain was not stable under the selection conditions. These SH2 domains were therefore excluded. We generated monobodies using phage and yeast display from the initial “loop-only” library and the recently developed “side-and-loop” library [18], [20], [24]. Monobody pools enriched with high-affinity binders were identified after two to three rounds of yeast display for all of these six SH2 domains (Fgr, Lck, Src, Yes, Hck, and Lyn). We determined the amino acid sequences of eight monobody clones for each of the SFK SH2 domain targets and selected two clones that had distinct amino acid sequences for further analysis (Fig. 1). All eight of the monobody clones for the Src SH2 domain had the same sequence, representing the single clone reported [Mb(Src_2)]. All but one clone [Mb(Hck_1)] were derived from the side-and-loop-library (Fig. 1). Based on their sequences, we observed two general types of clones. Yes, Src, and Fgr SH2-binding monobodies have a CD loop corresponding to the wild-type FN3 and a diversified FG loop that contains different sequences for all the different monobodies. In contrast, the Lyn and Lck SH2-targeting monobodies, and Mb(Hck_2), showed diversified CD and FG loops (Fig. 1).
Fig. 1.

Monobody libraries and selected clones. Amino acid sequences of monobodies generated from the “side and loop” library and “loop-only” library targeting different SFK SH2 domains. The sequence of the two different libraries is shown on top. “X” denotes a mixture of 30% Tyr, 15% Ser, 10% Gly, 5% Phe, 5% Trp, and 2.5% each of all the other amino acids except for Cys; “B”, a mixture of Gly, Ser, and Tyr; “J”, a mixture of Ser and Tyr; “O”, a mixture of Asn, Asp, His, Ile, Leu, Phe, Tyr, and Val; “U”, a mixture of His, Leu, Phe, and Tyr; “Z”, a mixture of Ala, Glu, Lys, and Thr. Monobodies are divided in SrcA binders (first five sequences) and SrcB binders (six sequences below). The left column shows dissociation constants in nM determined by the yeast binding assay. Full binding curves are shown in Supplementary Data (SI Fig. 1).
In order to determine the binding affinity of the selected monobody clones to the SH2 domain for which they were selected (“on-target SH2”) and the SH2 domains of the other SFKs (“off-target SH2”), we performed binding titrations in the yeast surface display format that allows for Kd estimations [20]. Mb(Lck_1) and Mb(Lck_3) bound to Lck, and Mb(Lyn_2) and Mb(Lyn_4) to Lyn with very high affinity (Kd = 10–20 nM), respectively (Fig. 1 and SI Fig. 1b). The monobodies to Src, Hck, Fgr, and Yes, which have the wild-type FN3 sequence of the CD loop, exhibited lower affinity with dissociation constants of 150–420 nM for their respective on-targets (Fig. 1 and SI Fig. 1b).
Next, we measured binding to the seven potential off-target SH2 domains across the SFK family in the yeast-display format at 250 nM SH2 concentration (Fig. 2 and SI Fig. 1c). Three important observations can be made from these experiments. All monobodies showed the strongest binding to their on-targets. The pairs of monobody clones for the same target show similar selectivity profiles. Monobodies selected for the SrcA family (Yes, Src, Fgr) SH2 domains show weak or no binding to the SrcB family (Hck, Lyn, Lck, and Blk), and SrcB-targeting monobodies exhibited the opposite tendency (Fig. 2). Additional binding titrations for selected SH2 domains to several monobodies revealed that the binding affinities for off-target SH2 domains were 5–10-fold lower than for the on-target (SI Fig. 1c). Taken together, these results suggest that these monobodies are highly selective to either SrcA or SrcB.
Fig. 2.

Yeast binding assay comparing mean fluorescent signals at 250 nM target concentration. Each panel shows the mean fluorescence signal of one monobody toward all tested targets at a concentration of 250 nM. For clarification, SrcA (Yes, Src, Fgr) and SrcB (Hck, Lyn, Lck, Blk) family members are subdivided by a dotted line. Each data point corresponds to the average of two repeats +/− SD.
In order to more precisely determine the thermodynamic binding parameters of the selected monobodies to their on- and off-target SFK SH2 domains, we prepared them as purified proteins and performed binding measurements using isothermal titration calorimetry (ITC). The binding affinities of selected monobody clones to their on-target SH2 domains were in the low to mid nanomolar range (Fig. 3), generally consistent with the yeast-display measurements. All measurements suggested a monobody:SH2 domain binding stoichiometry of 1:1 (Fig. 3). In all cases, the ΔH values were in the range of − 10 to − 20 kcal/mol, indicating that monobody binding is strongly enthalpically driven. Mb(Lck_1) and Mb(Lck_3) bound the Lck SH2 with similar affinities (23.5 nM and 7 nM, respectively; Fig. 3). Mb(Yes_1) was found to bind the Src SH2 domain with 38 nM affinity, which appeared much higher than observed in the yeast binding assay (Fig. 3 and SI Fig. 1).
Fig. 3.

ITC measurements of different monobodies with SH2 domains. All calorimetric titration of the monobodies with SH2 domain were performed at 25 °C. Each panel shows (at the top) the raw heat signal of an ITC experiment. The bottom panel shows the integrated calorimetric data of the area of each peak. The continuous line represents the best fit of the data based on a 1:1 binding model computed from the MicroCal software. All experiments were performed in 25 mM Hepes (pH 7.5) and 150 mM NaCl. A representative measurement is shown for each example with Kd value and stoichiometry (N) calculated from the fit. (a) Mb(Lck_1) (70 μM) titrated to Lck (7 μM), (b) Lck (115 μM) titrated to Mb(Lck_3) (11 μM), (c) Mb(Src_2) (180 μM) titrated to Src (12 μM), (d) Blk (142 μM) titrated to Mb(Lck_1) (13 μM), (e) Mb(Lck_3) (150 μM) titrated to Blk (15 μM), and (f) Mb(Yes_1) (215 μM) titrated to Lck (15 μM).
Further ITC experiments were performed to determine binding affinities to off-target SH2 domains that were identified in the yeast-display binding assay (SI Fig. 2). In particular, Mb(Lck_1) and Mb(Lck_3) showed remarkable discrimination properties. Whereas their Kd values for Lck, Blk, and Hck (SrcB family) were all between 6 nM and 40 nM (Fig. 3 and SI Fig. 2), we could not detect their binding to Yes and Src SH2 domains (SrcA family) at any tested protein concentration (SI Fig. 2). These results show that the selected monobodies bind their on-target SH2 domain with high affinity and display SFK subgroup selectivity.
SFK SH2 monobodies inhibit SH2–pY interactions
Our goal was to generate monobodies with high binding affinity but also with the ability to compete with SH2 pY ligand binding. We first determined if the selected monobodies blocked the pY peptide binding first using a peptide containing the pYEEI motif. This peptide bound with similar KD values of 310–675 nM to the eight SFK SH2 domains (SI Fig. 3). We produced the generated monobodies as purified proteins. The monobody Mb(Lyn_2) did not show a monodispersed gel-filtration profile and was therefore excluded from further analysis (data not shown). Most of the generated monobody clones robustly competed with pY peptide binding to their respective on-target SH2 domain, whereas a non-binding control monobody (HA4-Y87A [22]) showed no competition as expected (Fig. 4a and SI Fig. 4). Interestingly, Mb(Lck_1) and Mb(Lyn_4) did not compete with pYEEI peptide binding (Fig. 4a–b). We then tested Mb(Lck_1) and Mb(Lck_3) using two additional pY peptides with lower binding affinity for the Lck SH2 domain (pYIIP: Kd = 955 nM, and pYQPQ: Kd = 2200 nM). Unlike the results with the pYEEI peptide, both monobodies compete against these pY peptides (Fig. 4b), suggesting that both monobodies are inhibitors of the Lck SH2–pY peptide interaction.
Fig. 4.

Inhibition of pY peptide/SH2 interaction by monobodies. (a) The graph shows relative pYEEI peptide binding (in %) to SH2 domain in the presence of a monobody. All eight SH2 domains have been measured without and in presence of the monobody selected for the respective on-target. The pYEEI peptide in isolation and the SH2/pYEEI complex were set to 0% and 100% binding, respectively. Accordingly, the reduction in binding observed in conjunction with a monobody is expressed as a percentage. Each data point corresponds to the average of at least two repeats +/− SD. (b) Relative peptide binding (in %) of three different peptides (pYEEI, pYQPQ, pYIIP) to the Lck SH2 domain in the presence of each of the two Lck monobodies [Mb(Lck_1) or Mb(Lck_3)]. The full sequence of the peptides can be found in the Materials and Methods section. (c) The monobodies Mb(Lck_3) or HA4-Y87A or buffer alone was added to biotinylated Lck or Src SH2 domains immobilized on Streptavidin-coated beads prior to incubation with lysate from stimulated Jurkat T cells. Immunoblot analysis after pull-down using an anti-pY antibody for a 40% fraction of beads (upper blot), using an anti-Zap70 antibody and an anti-phospho-Zap70 (pY319) antibody for a 40% fraction of beads (second and third blot from top), and using an anti-His-tag antibody to detect the recombinant SH2 domains and monobodies for a 10% fraction of beads (lower blot).
We next analyzed SFK subgroup binding selectivity using the pY peptide competition assay. All selected monobodies were tested against all SFK SH2 domains. In this assay, the submicromolar affinity of the pYEEI peptide necessitated the SH2 concentration to be in a similar range, and we also needed to use an excess monobody concentration (fivefold higher concentration than the SH2 concentration) to ensure that SH2 was nearly fully saturated with a monobody. This setup ensures that even low affinity binding to off-targets shows inhibition. We observed that the SrcA family-targeting monobodies were better inhibitors of the SrcA than the SrcB family SH2 domains, whereas the opposite was observed for the SrcB family-targeting monobodies (SI Fig. 4). These results are in line with the binding selectivity data (Fig. 2, Fig. 3, and SI Fig. 1c and 2) and confirm the general selectivity of the monobodies for the SrcA or SrcB subgroups.
We next sought to study the potential of the developed monobodies as functional antagonists of SH2 domain ligand binding in a cellular proteome containing full-length proteins rather than isolated pY peptides. We therefore pulled down Lck SH2-interacting proteins from T-cell receptor-stimulated Jurkat cells with the recombinant biotinylated Lck SH2 domain. Competition with recombinant Mb(Lck_3) monobody, but not the non-binding control monobody HA4-Y87A [22], strongly reduced the overall interactions with tyrosine-phosphorylated Jurkat cell proteins (SI Fig. 5a). Importantly, the interaction with Zap70, which is phosphorylated on Y319, a known interactor of the Lck SH2 domain in activated T-cells, was efficiently disrupted by Mb(Lck_3) (Fig. 4c). As expected, Mb(Lck_3) was not able to compete with known interactors of the Src SH2 domain, as Mb(Lck_3) is not able to bind to it. In summary, we have identified several monobodies that are SrcA- or SrcB-selective competitors of SH2–pY peptide and protein–ligand interactions.
SFK monobodies bind to SFK family members with high selectivity in cells
To evaluate the specificity of selected monobodies in complex cellular proteomes, we chose the chronic myeloid leukemia cell line K562 and the T-cell acute lymphoblastic leukemia cell line Jurkat, as each expresses a different spectrum of SFKs. Members of the SrcA group (Yes, Src, and Fyn) are expressed in both cell lines, whereas SrcB members are expressed in a cell-type-specific manner in either cell line: Jurkat cells express Lck, whereas K562 cells express Lyn [25], [10], [26]. We stably expressed Mb(Src_2), Mb(Yes_1), Mb(Yes_3), Mb(Lck_1), and Mb(Lck_3) as tandem affinity purification (TAP)-tagged proteins in K562 cells, and Mb(Lck_1) and Mb(Lck_3) additionally in Jurkat cells (Fig. 5a). The monobodies were expressed at comparable levels in both cell lines, and the monobodies and their on-targets were efficiently retrieved after the second affinity purification step, as confirmed by immunoblot analysis (Fig. 5b and SI Fig. 5b–d). Captured proteins were visualized by silver staining and identified by liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS; Fig. 5c–d, Table 1, and SI Tables 1–3). In the Mb(Src_2), Mb(Yes_1), and Mb(Yes_3) TAP experiments, Yes, Src, and Fyn were identified among the most abundant proteins by LC–MS/MS, whereas Mb(Lck_1) and Mb(Lck_3) bound predominantly to Lck in Jurkat cells and to Lyn in K562 cells. These results are consistent with the dominant ~ 58-kDa bands detected by silver staining (Fig. 5c–d, Table 1, and SI Tables 1–3). In the biological replicate of the Mb(Lck_1) and Mb(Lck_3) TAP, other SFKs were identified in K562 and Jurkat cells, but at a much lower abundance (Table 1 and SI Tables 2–3). Importantly, no other SH2 domain-containing proteins were enriched in any Mb(Src_2), Mb(Lck_1), and Mb(Lck_3) samples, despite the expression of ~ 90 SH2 domains in these cells [23], [26]. For Mb(Yes_1) and Mb(Yes_3), low levels of Btk and Tec kinases, which both contain an SH2 domain, were detected (Table 1 and SI Table 1). The LC–MS/MS result did not reveal any other off-targets.
Fig. 5.

SFK SH2 monobody interactome analysis (a) Schematic representation of a TAP-tagged monobody and the purification method including two copies of the B1 domain of staphylococcal protein G (2xProt.G), TEV protease recognition site, streptavidin-binding peptide (SBP), and Myc-tag N-terminal to the monobody. (b) Immunoblot analysis of TAP of Mb(Lck_3) monobody complexes from Jurkat cells. TE, total extract; SN1, supernatant IgG beads; TEV, eluate after TEV cleavage; SN2, supernatant streptavidin beads; E1, eluate from streptavidin beads; BB, boiled streptavidin beads to control the efficiency of elution. The bait protein and the main target of Mb(Lck_3) were identified by immunoblotting using an anti-Myc or anti-Lck antibody, respectively. (c) Mb(Src_2), Mb(Yes_1), and Mb(Yes_3) monobody complexes after TAP (10% of the E1 fractions) from K562 cells, separated by SDS-PAGE and visualized with silver staining. (d) Mb(Lck_1) and Mb(Lck_3) monobody complexes after TAP (10% of the E1 fractions) from K562 and Jurkat cells, separated by SDS-PAGE and visualized with silver staining.
Table 1.
Overview of mass spectrometry results from the interactome analysis of monobodies Mb(Src_2), Mb(Yes_1), Mb(Yes_3), Mb(Lck_1), and Mb(Lck_3)
| Cell line | Bait | Total number of proteins identified⁎ |
Number of proteins meeting the selection criteria⁎⁎ |
All identified SH2 domain-containing proteins in each sample (in brackets: total spectrum counts, rank⁎⁎⁎) |
|||
|---|---|---|---|---|---|---|---|
| 1st TAP | 2nd TAP | 1st TAP | 2nd TAP | 1st TAP | 2nd TAP | ||
| K562 | Mb(Lck_1) | 68 | 252 | 16 | 50 | Lyn (112, 2); Lck (7, 34) | Lyn (1523, 1); Hck (141, 13); Lck (133, 15); Src (44, 38) |
| Mb(Lck_3) | 31 | 335 | 6 | 31 | Lyn (53, 1) | Lyn (1614, 1); Hck (172, 9); Lck (130, 15); Src (45, 32) | |
| Jurkat | Mb(Lck_1) | 33 | 285 | 3 | 23 | Lck (114, 1); Lyn (1, 22) | Lck (1811, 1); Src (210, 6); Lyn (138, 13); Hck (131, 16) |
| Mb(Lck_3) | 25 | 275 | 6 | 19 | Lck (109, 1); Lyn (1, 19) | Lck (1930, 1); Lyn (177, 6); Hck (162, 7); Src (159, 9) | |
| K562 | Mb(Src_2) | 39 | 15 | Src (35, 7); Yes1 (31, 8); Fyn (31, 8) | |||
| Mb(Yes_1) | 46 | 13 | Yes1 (101, 4); Fyn (76, 6); Src (61, 7); Lyn (12, 20); Tec (6, 27); Btk (1, 47) | ||||
| Mb(Yes_3) | 259 | 75 | Yes1 (295, 1); Fyn (139, 6); Src (107, 11); Lyn (32, 28); Btk (4, 105) | ||||
Proteins were scored as identified if at least two unique peptides were detected. The complete list of identified proteins is provided in Supplementary Tables 1–3.
Selection criteria: Proteins with a selectivity score of > 0.75 (present in < 25% of all TAP monobody experiments from our internal database), with a total spectrum count of > 1% of the most abundant protein in the sample, excluding different isoforms of keratin and the bait protein itself.
All identified proteins, including common contaminants, such as Keratin, were sorted by the number of assigned spectra from highest to lowest.
These results demonstrate the ability of the studied monobodies to efficiently bind their full-length target protein in complex cellular proteomes. They only bind detectably to certain other SFK SH2 domains, but no other off-targets. To the best of our knowledge, these are the most selective reagents targeting SFK SH2 domains reported to date.
Structural basis for binding specificity of SFK SH2 monobodies
To understand the structural basis for SH2 binding and the observed selectivity of the SFK SH2 monobodies, we determined the crystal structures of three monobody/SFK SH2 complexes: Mb(Yes_1)/Yes SH2, Mb(Lck_1)/Lck SH2, and Mb(Lck_3)/Lck SH2 at 1.95, 2.85, and 2.40 Å resolution, respectively (Table 2 and Fig. 6). All these monobodies bound to the surfaces of their targeted SH2 domain that overlap with the pY peptide-binding pockets, as expected from their ability to inhibit SH2–pY peptide interactions (Fig. 6a–d). Mb(Lck_1) and Mb(Lck_3) had a very similar orientation with respect to the SH2 domain, whereas that of Mb(Yes_1) was distinct (Fig. 6b–d).
Table 2.
Data collection and refinement statistics (molecular replacement)
| Mb(Lck_3)/Lck SH2 | Mb(Lck_1)/Lck SH2 | Mb(Yes_1)/Yes SH2 | |
|---|---|---|---|
| Data collection | |||
| Space group | P41212 | P3221 | I4122 |
| Cell dimensions | |||
| a, b, c (Å) | 81.83, 81.83, 105.96 | 91.40, 91.40, 88.78 | 101.86, 101.86, 139.62 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 120.00 | 90.00, 90.00, 90.00 |
| Resolution (Å) | 40.92–2.40 (2.49–2.40)⁎ | 45.70–2.85 (2.95–2.85)⁎ | 50.00–1.95 (1.98–1.95)⁎ |
| Rmerge | 3.0 (76.6) | 6.2 (1.0) | 0.13 (0.00) |
| I/σI | 37.91 (2.90) | 23.79 (1.63) | 24.44 (1.50) |
| Completeness (%) | 100.0 (99.00) | 100.0 (100.0) | 99.8 (99.7) |
| Redundancy | 7.2 (7.5) | 5.5 (5.7) | 19.1 (16.1) |
| Refinement | |||
| Resolution (Å) | 2.40 | 2.85 | 1.95 |
| No. of reflections | 14,544 | 10,316 | 27,104 (2649) |
| Rwork/Rfree | 0.20/0.25 | 0.23/0.27 | 0.19/0.22 |
| No. of atoms | |||
| Protein | 1555 | 1424 | 1652 |
| Ligand/ion | 2 | 15 | 33 |
| Water | 32 | – | 240 |
| B-factors | |||
| Protein | 68.40 | 79.67 | 33.18 |
| Ligand/ion | 63.77 | 115.24 | 46.73 |
| Water | 62.16 | – | 45.10 |
| rmsd | |||
| Bond lengths (Å) | 0.012 | 0.008 | 0.008 |
| Bond angles (°) | 1.16 | 1.31 | 0.84 |
One crystal for each structure.
Fig. 6.

Co-crystal structures of monobodies bound to SFK SH2 domains. (a) Structure pYEEI peptide (pink) bound to LCK-SH2 (pdb: 1LKK). The lower panel shows the atoms involved in the interaction with the epitope for the indicated peptide highlighted in pink. Epitope residues are defined as those atoms within 5 Å of an atom of the pYEEI peptide. (b) The monobody Mb(Yes_1) (blue) binding to the SH2 of Yes. (c) Mb(Lck_1) binding to SH2 domain of Lck, and (d) the binding of Mb(Lck_3) to Lck SH2. (e) Surface representation of Yes–SH2 domain showing the residues specific for SrcA or SrcB family members and the residues that are all identical in all eight SH2 domains. (f) Structural alignment of Mb(Lck_1) and Mb(Lck_3) paratope bound to Lck SH2 (upper panel). Mb(Lck_1) (cyan) and Mb(Lck_3) (bright green) residues involved in binding to Lck are shown as sticks. A pY peptide is shown in yellow. A detailed view of the CD loop residues of Mb(Lck_1) (middle panel) and of Mb(Lck_3) (lower panel) shows the arrangement of the residues compared to the + 1 Glu in the pYEEI peptide (yellow sticks). Residue numbering of monobodies is according to Fig. 1.
Mb(Yes_1) uses its long FG loop, the most extensively diversified region in the library (Fig. 1), to present an extended segment that is positioned perpendicular to the central SH2 β-beta sheet over the peptide-binding cleft, mimicking the canonical backbone conformation of a pY ligand peptide (Fig. 6a and b). Tyr83 (residue numbering is according to Fig. 1) in the FG loop is inserted into the pY pocket of the SH2 domain (SI Fig. 6a), where it makes cation–π interactions with Arg17 and Lys65 of SH2. Interestingly, a sulfate ion, possibly from the crystallization buffer, was found at the position that is occupied by the phosphate group of the pY peptide ligand in SH2–pY peptide complexes (SI Fig. 6a). Thus, the FG loop of Mb(Yes_1) mimics the backbone conformation and the pY moiety of a pY peptide ligand. This binding mode is very similar to that previously observed for the HA4 monobody bound to the Abl SH2 domain with a Cα rmsd for the complex of only 1.7 Å (SI Fig. 7) [22]. Mb(Yes_1) buries 702 Å2 of the solvent-exposed surface of the Yes SH2 domain, which substantially extends beyond the core pY peptide-binding interface (346 Å2 for the pYEEI peptide).
Fig. 7.

EF loop residues of SrcB family dictate the subgroup specificity of Mb(Lck) monobodies. (a) Co-crystal structure of Mb(Lck_3) bound to Lck SH2 domain with the zoomed region of FG loop (monobody) binding to the EF loop of Lck SH2 domain. The sticks shown in white are critical residues, which are specific for the three members of the SrcB group: Lck, Lyn, and Blk. The blue residues (also represented as sticks) are the amino acids found in Src at the same position. (b) ITC of Mb(Lck_3) to the Src SH2 domain (left panel) and Src-S78P/Q81T/N83P mutant (right panel). Each panel shows (at top) the raw heat signal of an ITC experiment. The bottom panel shows the integrated calorimetric data of the area of each peak. The continuous line represents the best fit of the data based on a 1:1 binding model computed from the MicroCal software if the data allowed the fitting of an isotherm. Both experiments were performed in 25 mM Hepes (pH 7.5) and 150 mM NaCl. A representative measurement is shown for each example with Kd value and stoichiometry (N) calculated from the fit. The titration experiments were performed at 80 μM of Mb(Lck_3) titrated to 8 μM of Src SH2 and at 95 μM of Mb(Lck_3) titrated to 9 μM of Src- S78P/Q81T/N83P.
Although only 10 of the 20 diversified positions are identical between Mb(Lck_1) and Mb(Lck_3) (Fig. 1), these monobodies bind to Lck SH2 in a very similar manner (Fig. 6c–d). Interestingly, in contrast to Mb(Yes_1) and in previously reported SH2-binding monobodies, Mb(Lck_1) and Mb(Lck_3) do not bind the pY pocket. Instead, their CD loops block the + 3 specificity pocket of Lck SH2, and the rest of the diversified positions including the FG loop recognize surfaces on the opposite side of the Lck SH2 domain from the pY pocket (Fig. 6c–d). Their FG loops bind to the EF loop of the Lck SH2 domain. Mb(Lck_1) and Mb(Lck_3) bury 798 Å2 and 693 Å2, respectively, of the solvent-exposed surface of the Lck SH2 domain, which corresponds to 14% and 11% of the total surface.
When the two structures are superimposed using the Lck SH2 domain, the similarities and differences between Mb(Lck_1) and Mb(Lck_3) became clear. 11 residues contacting the SH2 domain (defined here as those within 5 Å of an SH2 atom) that are identical between the two monobodies have nearly identical conformations (Fig. 6e). Although their CD loops are the least conserved region between them, they both would clash with the + 2 and + 3 residues of a pY ligand peptide based on the superposition of the Lck SH2-pYEEI structure (Fig. 6e). The two CD loops take on distinct conformations (Fig. 6e). Ser44 of Mb(Lck_3) is located closer to the backbone of the + 1 residue, possibly creating an additional steric clash (Fig. 6f). In contrast, the corresponding residue in Mb(Lck_1) is Pro44 that creates a kink in the backbone conformation and moves the CD loop away from the pY moiety. These observations suggest that the CD loop of Mb(Lck_3) is more effective in interfering with the binding of the pY moiety to the pY-binding pocket than the CD loop of Mb(Lck_1), although it is clear that both monobodies block the interactions of the + 1, + 2 ,and + 3 positions of a pY peptide with the Lck SH2 domain. Indeed, when we replaced the CD loop of Mb(Lck_1) with that of Mb(Lck_3), this hybrid monobody competed with the pYEEI peptide and Mb(Lck_3) and retained the SFK subgroup selectivity (SI Fig. 4). These results demonstrate that the sequence and conformation of the CD loop are critical for efficient blockade of pY ligand binding of the Lck SH2 domain by Mb(Lck_1) and Mb(Lck_3).
We next aimed at understanding the structural basis for the ability of Mb(Lck_1) and Mb(Lck_3) to discriminate SFK SH2 subgroups. Among the epitope residues (defined as Lck SH2 residues within 5 Å of a monobody atom), we identified distinct amino acids at positions in the EF loop between the SrcA and SrcB SH2 domains, which make contacts with the SYG motif of FG loop of Mb(Lck_1) and Mb(Lck_3) (Fig. 7a). Three of these residues, Pro78, Thr81, and Pro83, are identical in Lyn and Blk but are more bulky or polar residues in the SrcA family (Fig. 7a and SI Fig. 9). To test the importance of these residues for subgroup selectivity, we mutated these three positions in the EF loop of the Src SH2 domain to the respective residues of Lck (S78P/Q81T/N83P; Fig. 7a). ITC measurements showed that Mb(Lck_3) bound to the S78P/Q81T/N83P mutant of the Src SH2 domain (Kd = 64 nM), whereas Mb(Lck_3) showed no detectable binding to the wild-type Src SH2 domain (Fig. 7b). Notably, the Kd value is less than 10-fold greater than that of Mb(Lck_3) to the Lck SH2 domain. As expected, Mb(Lck_3) acted as pY peptide competitor for the Src S78P/Q81T/N83P mutant protein (data not shown). These results indicate that the ability of monobodies to bind to the pY peptide-binding site and simultaneously to recognize the EF loop sequence enables them to discriminate SrcA versus SrcB SH2 domains.
SFK-targeting monobodies activate recombinant Src and Hck kinase activity
The SH2 domain of SFKs has a dual role in regulating kinase activity and signaling. In the autoinhibited conformation of SFKs, the SH2 domain stabilizes the clamped conformation by an intramolecular interaction with a pY residue in the C-terminal tail emanating from the kinase domain [5], [27]. This C-terminal phosphorylation event is catalyzed by the Csk kinases. In contrast, in activated SFKs, the SH2 mediates intermolecular interactions that are critical to localize SFKs to particular signaling complexes and for processive multisite phosphorylation of substrates [7].
In order to test the effect of the monobodies on SFK kinase activity, we used a continuous spectrophotometric in vitro kinase assay (Fig. 8a) [28]. We chose the SrcA-selective Mb(Yes_1) monobody and the SrcB-selective Mb(Lck_3) monobody. We used recombinant Src (SrcA group) and chose Hck among the SrcB group, as Lck is more difficult to express. The SH3-SH2-kinase domain units of both SFKs were purified and assayed in the absence and presence of the inhibitory Csk kinase.
Fig. 8.

SFK monobodies activate autoinhibited recombinant Src and Hck. (a) Schematic representation of the in vitro kinase assay setup, in which recombinant Src or Hck or preincubated with the SFK negative regulatory kinase Csk and/or recombinant monobodies before assaying the phosphorylation of a SFK substrate peptide with a continuous spectrophotometric assay. (b and c) In vitro kinase activity of Src and Hck was measured in the absence or presence of Csk and set to 1.0. Relative changes in kinase activity are shown at the indicated concentrations of (b) Mb(Yes_1) or (c) Mb(Lck_3). Each data point corresponds to the average of three repeats +/− SD. Control experiments are shown in SI Fig. 10.
Mb(Yes_1) robustly activated the activity for Src in a concentration-dependent manner (Fig. 8b). The relative increase in Src kinase activity by Mb(Yes_1) was enhanced in the presence of Csk, as Csk decreases the basal Src activity. Mb(Yes_1) also activated Hck, but less potently, in line with its lower binding affinity and lower pY competition activity to Hck as compared to Src. When testing the Mb(Lck_3) monobody, we also observed a concentration-dependent increase in Hck kinase activity in the presence of Csk, whereas no activation of Src was observed in line with the lack of binding of Mb(Lck_3) to Src (Fig. 8c). A non-binding control monobody (HA4-Y87A [22]) with no affinity for SFK SH2 domains did not result in significant changes in kinase activity (SI Fig. 10a). Likewise, Mb(Yes_1) and Mb(Lck_3) had no effect on the activity of the isolated Src or Hck kinase domains in the absence or presence of Csk (SI Fig. 10b), demonstrating that the observed effects of these monobodies on kinase activation are SH2 domain dependent. These results suggest that SFK SH2 monobodies interfere with the autoinhibited conformation of specific SFKs and activate kinase activity.
Lck-targeting monobodies inhibit proximal T-cell receptor signaling
We finally sought to assess whether the SFK monobodies interfere with SFK-dependent signaling events in cells. Given the critical role of the Lck SH2 domain in the activation of the Zap70 kinase in the activated T-cell receptor signaling complex [29], we set out to study the effects of Mb(Lck_1) and Mb(Lck_3) on Zap70 phosphorylation. We stably expressed Myc-tagged Mb(Lck_1), Mb(Lck_3), and the non-binding control monobody HA4-Y87A in Jurkat T-cell acute lymphoblastic leukemia cells, stimulated these cells with an anti-T-cell receptor (TCR) antibody, and monitored the phosphorylation of the activation loop of Zap70 (Y493; Fig. 9). We observed a strong decrease of Zap70 phosphorylation upon TCR activation and a mild decrease of basal Zap70 phosphorylation in Mb(Lck_3) expressing cells, as compared with HA4-Y87A expressing cells. Mb(Lck_1) had milder effects in line with its weaker pY competition activity (Fig. 9). Interestingly, the protein expression levels of Mb(Lck_3) were much lower as compared to Mb(Lck_1) and HA4-Y87A, while the mRNA levels were similar (Fig. 9 and data not shown). This may indicate a possible negative regulatory mechanism limiting Mb(Lck_3) protein expression. Collectively, these results show that Mb(Lck_3) can interfere with Lck-mediated signaling in cells and inhibit the activation of Zap70.
Fig. 9.

Mb(Lck_3) and Mb(Lck_1) inhibit Zap70 phosphorylation by Lck in stimulated Jurkat cells. (a) Wild-type Jurkat cells or Jurkat cells stably expressing Mb(Lck_1), Mb(Lck_3), or HA4-Y87A monobodies were stimulated with an anti-TCR antibody or mock-treated. The whole cell lysates were immunoblotted against the indicated antibodies. Representative immunoblots of two independent experiments done in two technical replicates each are shown. Immunoblots against anti-myc of lysates from unstimulated cells are shown to assess monobody expression levels. (b) The Y493-phosphorylated Zap70 signal was quantified and normalized by the total Zap70 protein level. For each experiment, the pZap70:Zap70 ratio of the unstimulated HA4-Y87A control cells was set to 1.0. Average values and standard deviations from two independent experiments done in two technical replicates were used, and P values were calculated using an unpaired t-test. *P ≤ 0.05, **P ≤ 0.01.
Discussion
The identification of selective inhibitors of SH2 domains is challenged by the high structural conservation of the pY peptide binding cleft and the expression of dozens of SH2-containing proteins in any given cell. While peptidomimetics and small molecule inhibitors with high binding affinities have been developed, their selectivity was only poorly characterized, as only one or a few, in most cases, evolutionarily distant SH2 domains, were tested as potential off-targets. In contrast, the monobodies to the SFK SH2 domains that we report on here have been rigorously characterized for their binding selectivity and binding mode.
We found that 8 of the 11 monobody clones that we characterized were strong antagonists of pY ligands, even though our selection was unbiased, that is, no positive selection for a specific epitope was included. These observations underline the notion of previous studies that showed monobody binding to hotspots of protein–protein interactions in structurally diverse targets including other SH2 domains [18], [22], [23], [30], [31].
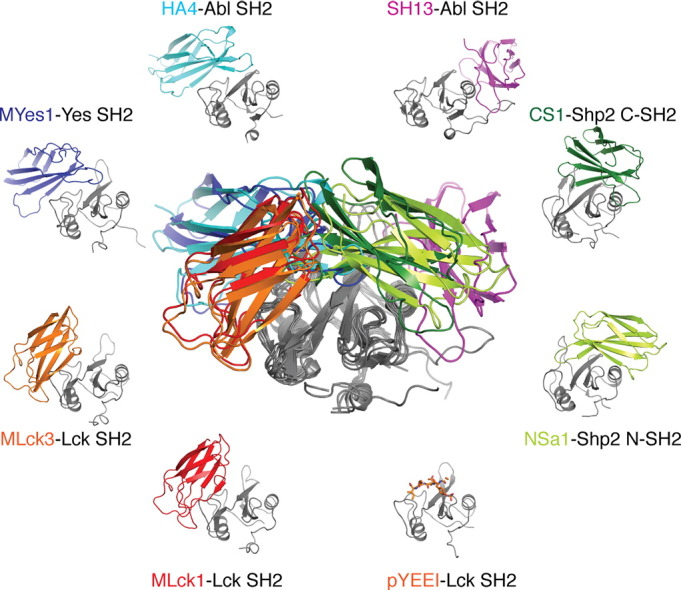
Together with the three crystal structures presented here, we now have seven crystal structures of SH2 domain–monobody complexes and thereby can distinguish at least four different binding modes that strongly differ in the way the monobody engages the SH2 domain (SI Fig. 7). In particular, the observed structural mechanisms of inhibition of pY ligand binding are amazingly different. The Mb(Yes_1) and HA4 monobodies closely mimic a pY peptide ligand by a tyrosine residue in their FG loop and an inorganic sulfate and phosphate, respectively, in the pY pocket (SI Fig. 6a) [22]. In contrast, Mb(Lck_1) and Mb(Lck_3) use their CD loops to block the + 3 specificity pocket (Fig. 6). The Shp2 monobodies NSa1 and CS1 show a pY-independent mode of interaction with the Shp2 SH2 domains, while still mimicking the bound peptide, and can bind the same epitope in two opposite orientations [23]. Lastly, the SH13 monobody binds a surface that is not overlapping with the pY ligand binding site of the Abl SH2 domain but that is involved in a critical allosteric intramolecular interaction with the Abl kinase domain [18]. Of note, more potent inhibitors of this allosteric interface could be developed using negative selection and a decoy competitor strategy [20], [21].
The lack of competition between Mb(Lck_1) and the pYEEI peptide is noteworthy but may be caused by the specific assay conditions tuned to identify pY-competing monobodies for all SFK SH2 domains and the particular binding mode of Mb(Lck_1). In contrast, it is important to note that Mb(Lck_1) inhibited the binding of two other pY peptides that have lower affinity than pYEEI (Fig. 4b), indicating that Mb(Lck_1) is a less potent inhibitor than Mb(Lck_3). In line with this view, Mb(Lck_1) inhibits T-cell activation less pronounced than Mb(Lck_3) (Fig. 9). Upon T-cell receptor activation, Mb(Lck_3) and Mb(Lck_1) will likely be able to interfere with the interaction of the Lck SH2 domain with a pY site in Zap70. This may dislodge Lck from the T-cell receptor complex and impair its proper signaling and Zap70 activation. The crystal structures of Mb(Lck_1) and Mb(Lck_3) in complex with Lck SH2 show that the pY binding pocket of the SH2 domain is less occluded by the CD loop of Mb(Lck_1) than Mb(Lck_3) (Fig. 6e), rationalizing the observed differences in both assays.
The presented work provided us with additional insight on the important issue of monobody selectivity. We observed that all monobodies can strongly discriminate between the two SFK subfamilies—SrcA and SrcB. Given that most of the surface-exposed residues within the SrcA and SrcB families are identical (Fig. 6f), we believe that we have reached a limit of binding selectivity. To obtain single SFK-selective binders, we will require the identification of new binding modes, in which less conserved surfaces on the opposite side of the pY ligand binding cleft are targeted, while still maintaining pY ligand competition, and also the extensive use of negative selection.
A TAP-mass spectrometry (TAP-MS) experiment is a rigorous method to determine monobody selectivity in the physiologically relevant cellular context and signaling pathways of the monobody target. Importantly, the monobody is expressed in the complex and competitive environment of the cellular proteome and analyzed using unbiased mass spectrometry. Still, the cell-type-specific expression of certain SFKs and their relative expression levels need to be considered to interpret selectivity among all SFKs. Therefore, it appears that a combination of TAP-MS along with binding assays (Fig. 2) focusing on important potential off-targets is a well-suited approach to comprehensively assess monobody selectivity.
In conclusion, our results provide ample evidence that the SH2 domains of SFKs can be effectively and specifically targeted with monobodies with a variety of mechanisms of action. These monobodies can now be used as excellent tools to dissect SFK signaling in normal development and signaling and to interfere with aberrant SFK signaling network in cancer cells.
Materials and Methods
Cell lines
K562 and Jurkat cells were purchased from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany).
Antibodies and reagents
Antibodies were purchased from Cell Signaling Technology [Yes (#3201), Lck (#2657), Zap70 (#2705, #2709), pZap70(Y319) (#2701), pZap70(Y493) (#2704)], Thermo Scientific [V5 Tag (#MA5-15253)], Streptavidin-Dylight650 (#84547), Millipore [T-cell receptor, clone C305 [#05-919)], Sigma Aldrich [Anti-Mouse IgG FITC antibody (#F0257)], and Rockland [Myc-tag (#600-432-381)]. Streptavidin Magnesphere Paramagnetic Particles (Promega #Z5481) were used for the recombinant SH2 pull-down and during the monobody selection.
Cloning
We have cloned the eight SFK SH2 domains (complete sequences and construct boundaries; SI Fig. 8) and all reported monobodies into a pHBT vector (modified pET vector) containing a 6xHis tag, tobacco etch virus (TEV) protease cleavage site, and an Avi-tag for biotinylation. SH2 domains and monobodies were cloned into the BamHI and XhoI restriction sites of the pHBT vector.
Biotinylation of Avi-tagged proteins
In vivo biotinylation was performed by co-transforming Escherichia coli cells with a BirACm plasmid and a pHBT plasmid containing the AviTag gene fusion. After the OD600 of the expression culture reached 0.5–0.8, cells were induced with 0.5 mM of IPTG, and a final Biotin concentration of 50 μM from a 50 mM stock in DMSO was added. Cells were further grown overnight before purification. In vitro biotinylation was performed using recombinant BirA. We added 100 μM AviTag-fused protein in 952 μL of PBS to 5 μL 1 M magnesium chloride, 20 μL 100 mM ATP, 20 μL 50 μM glutathione S-transferase-BirA, and 3 μL 50 mM D-Biotin. The samples were incubated for 1 h at 30 °C with gentle mixing on a rocking/shaking platform. Biotinylation efficacy was tested by a streptavidin-paramagnetic bead pull-down experiment.
Monobody selection
General methods for phage and yeast-display library sorting and gene shuffling have been described [18], [22]. The monobody libraries used have been reported [32]. Briefly, after three rounds of phage display, selected loops were amplified and yeast cells were transformed with enriched monobody sequences. The yeast cells were sorted in a flow cytometer Fluorescence-activated cell sorting (FACS) based on positive signal for monobody display and binding of the biotinylated target. Clones were isolated, sequenced, and cloned into pHBT plasmids.
Yeast binding assay
The yeast binding assay was performed as previously described [20]. Concentrations of biotinylated targets ranged from 10 nM to 5 μM in Tris-buffered saline (TBS) with 0.1% BSA. Target protein, yeast cells, and a mouse anti-V5 antibody were incubated for 30 min at RT. After two washing steps, streptavidin-DyLight650 and a FITC-coupled anti-mouse IgG were added and incubated for 30 min at room temperature. Samples were analyzed on a Gallios (Beckman Coulter) or BD Accuri C6 (BD Bioscience) flow cytometer. Kd values were determined from plots of the mean fluorescence intensity versus target concentration by fitting the data to a 1:1 binding model using Prism (Graphpad).
Protein expression and purification
BL21(DE3) E. coli cells were transformed with pHBT-SH2 or pHBT-monobody constructs. The overnight culture has been added to fresh LB to reach a starting culture between 0.05 and 0.1, and the cells were grown at 37 °C with 190 rpm until the OD reached 0.5–0.7. The culture was then induced with 0.5 mM IPTG final concentration and further grown at 24– 27 °C for 5 h.
Cells were pelleted at 5000g for 15 min at 4 °C. The pellets were resuspended in Buffer A [25 mM Tris (pH 7.5), 300 mM NaCl, 5% glycerol, and depending on sample, 1 mM DTT). An Avestin Emulsiflex homogenizer was used for lysis. The soluble protein was separated from cell debris by centrifugation at 45,000g for 30 min at 4 °C. The protein was purified by affinity- and size-exclusion chromatography using Ni-NTA resin and a Superdex 75 10/300 GL column, respectively. Kinases were expressed as described previously [33].
ITC
ITC measurements were performed on a MicroCal iTC200 (GE) instrument. The proteins were extensively dialyzed against 25 mM Hepes (pH 7.5) and 150 mM NaCl and degassed; concentration was determined by measuring UV absorbance at 280 nm. The protein in the syringe was titrated in 16 steps with 0.49 μL for the first and 2.49 μL for the other steps. Concentrations were adjusted based on assumed affinities and signal strength of the interaction. The MicroCal software was used to determine thermodynamic parameters.
Fluorescence polarization
We used three different FITC-labeled pY-peptides: EPQpYEEIPIYLK-(FITC) (abbreviated “pYEEI” in the figures), (FITC)-TEGQpYQPQPA (“pYQPQ”), and (FITC)-ADNDpYIIPLPD (“pYIIP”). We mixed 125–500 nM peptide with TBS [50 mM Tris (pH 7.5) and 150 mM NaCl] or 12.5 μM monobody and added it to 2.5 μM SH2 domain. The final assay volume was 50 μL, and fluorescence polarization measurements were performed at room temperature in a black 96-well plate using an M5 plate reader (Molecular Devices). The wavelength was set to 525 nm; filter at 515 nm. The raw data were normalized to the free peptide in TBS background measurement.
X-ray crystallography
Purified monobody and SH2 proteins were mixed in TBS [50 mM Tris (pH 7.5) and 150 mM NaCl] + 0.5 mM TCEP and were run on a Superdex 75 10/300 GL column to separate complex from single protein. The purified complexes were concentrated in amicon vivaspin tubes to 10–17 mg/ml. The Mb(Yes_1)/YES SH2 complex was crystallized in hanging drop plates by mixing 100 nL of protein with 100 nL of buffer conditions. Later, crystals were optimized using a 24-well format (hanging drop) by mixing 1 μL of protein with 1 μL of buffer conditions. Best resolution data were achieved when using crystals grown in 2 M ammonium sulfate, 0.2 M lithium sulfate, and 0.1 M CAPS/NaOH (pH 10.25). The Mb(Lck_1)/Lck SH2 and Mb(Lck_3)/Lck SH2 complexes were crystallized using sitting drop plates. After several days, we observed crystals in several conditions. Best diffraction data were obtained from crystals grown in 0.2 M lithium sulfate, 0.1 M Tris (pH 8.5), 10% polyethylene glycol (PEG) 8K + 10% PEG1K for Mb(Lck_1)/Lck, and 0.1 M Mes (pH 6.5) and 15% PEG 20,000 for Mb(Lck_3)/Lck. Diffraction data for Mb(Yes_1)/Yes SH2 were collected at the Advanced Photon Source (Argonne National Laboratory, Chicago, USA) beamline 19-BM, and the Mb(Lck_1)/Lck SH2 and Mb(Lck_3)/Lck SH2 crystals at Swiss Lightsource (SLS, Villigen, Switzerland). Crystal structures were determined by molecular replacement (molrep, CCP4, and PHENIX) using a monobody structure excluding loop regions (PDB 3UYO and 3K2M), Lyn SH2 structure (PDB 4TZI), and Lck SH2 structures (PDB 1LKK, 1LCK, and 1BHH). Manual model building, solvent addition, and refinement of all three structures were conducted iteratively using Coot and phenix.refine. The models were of good geometry, with 94% (Mb(Lck_1)/Lck SH2), 97% (Mb(Lck_3)/Lck SH2), and 96% (Mb(Yes_1)/Yes SH2) in favored regions of the Ramachandran plot and 1.1% (Mb(Lck_1)/Lck SH2), 0.5% (Mb(Lck_3)/Lck SH2), and 0% (Mb(Yes_1)/Yes SH2 of Ramachandran outliers.
Stable cell line generation for TAP-MS
Monobodies were cloned as an N-terminal fusion to the GS-TAP tag Gateway destination vector as previously described [22]. Jurkat and K562 cells stably expressing TAP-tagged monobodies were generated by retroviral infection and FACS sorting, as previously described [22]. Monobodies Mb(Lck_1), Mb(Lck_3), and HA4-Y87A were cloned into a hPGK-IRES-GFP Gateway destination vector [gift from Dr. J. Huelsken (EPFL)]. Jurkat cells stably expressing the monobodies were generated by lentiviral infection and FACS sorting and used for the T-cell signaling studies.
TAP purifications
We used 2–3 × 109 K562 or Jurkat cells stably expressing the SFK-targeting monobodies for TAP purifications essentially as described in Ref. [34]. Protein complexes were eluted after the two affinity purification steps using 0.1 M hydrochloric acid and immediately neutralized with 0.5 M Triethylammonium bicarbonate. The efficiency of elution was checked by boiling the beads in SDS-PAGE sample buffer. Then, 10% of the eluates were resolved by SDS-PAGE (4–20% gel; Bio-rad) and silver stained, whereas the remaining eluate were prepared for mass spectrometric analysis.
Mass spectrometric analysis
The remaining part of each TAP experiment was separated by SDS-PAGE and stained with R-250 Commassie Blue Solution. Gel lanes were cut into pieces and subjected to in-gel digestion with trypsin. Extracted peptides were separated by reversed-phase chromatography on a Dionex Ultimate 3000 RSLC nano UPLC system (Dionex) on-line connected in line with either an Orbitrap Elite or an Orbitrap Q-Exactive Mass Spectrometer (Thermo Fischer Scientific). Raw data were processed with Proteome Discoverer (v. 1.3) and searched with Mascot against a human database (UniProt release 2012_04; 86,747 sequences). Data were further processed, inspected, and visualized using the Scaffold 3 software.
In vitro kinase assays
Monobodies Mb(Yes_1), Mb(Lck_3), and HA4-Y87A at increasing concentrations (0 μM, 0.5 μM, 2.5 μM, 10 μM, 25 μM) were tested against these kinase combinations: (1) Src + Csk, (2) Src alone, (3) Src kinase domain (Src KD) alone, (4) Src KD + Csk, and (5). Hck + Csk, 6. Hck alone. All assays were performed with or without an Src optimal substrate peptide (AEEEIYGEFAKKK). Kinase activity was measured by a continuous spectrophotometric assay [28], where substrate phosphorylation and ATP regeneration are coupled to NADH oxidation via pyruvate kinase/lactate dehydrogenase. The NADH concentration was monitored by absorbance at 340 nm in a Molecular Devices (SPECTRA Max 340PC384) plate reader. Reactions were performed at 30 °C in 75 μL volume containing 100 mM Tris (pH 7.5), 10 mM MgCl2, 74 U/mL pyruvate kinase, 104 U/mL lactate dehydrogenase, 1 mM phosphoenolpyruvate, 0.282 mM NADH, 0.5 mM ATP, and 0.5 mM Src optimal substrate peptide. Reactions were started by the addition of 30 nM protein kinase with the exception of Src KD and Csk, where 16 nM and 80 nM were used, respectively. All reactions were performed in triplicates. Relative kinase activity toward substrate peptide was computed by subtracting kinase background activity from reaction containing the substrate peptide.
T-cell stimulation experiments for SH2 domain pull-down and signaling studies
Jurkat cells were stimulated with anti-TCR antibody clone C305 (Millipore) at a concentration of 100 ng/ml for 5 min at 37 °C. The cells were cooled to 4 °C immediately after stimulation by the addition of ice-cold PBS and centrifugation. Cells were lysed in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1% NP-40, 5 mM EDTA, 5 mM EGTA, and protease and phosphatase inhibitors), and the protein content was quantified using the Bradford reagent. We used 50–100 μg of total cell lysate for immunoblotting analysis. Magnetic beads coated with streptavidin were incubated with either biotinylated Lck or Src SH2 domain for 2 h at 4 °C, washed three times with TBS, and then incubated for 30 min with a threefold excess of Mb(Lck_3) or HA4-Y87A as a control. After washing three times with TBS, the beads were incubated for 2 h with 3 mg of lysate from stimulated Jurkat cells. After three washing steps, the beads were boiled for 5 min in SDS-PAGE loading buffer, and bound proteins were analyzed by SDS-PAGE, followed by silver staining or immunoblotting.
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 5MTJ (Mb(Yes_1)/Yes SH2), 5MTM (Mb(Lck_3)/Lck SH2), and 5MTN (Mb(Lck_1)/Lck SH2).
Acknowledgments
We thank S. Georgeon for expert technical assistance, S. Vallaghé for help with TAP data analysis, G. Gish for providing the pYEEI peptide, A. Reynaud for help with crystallization, and all members of the Hantschel and Koide lab for help and discussions, and the EPFL Proteomics and Flow Cytometry Core Facilities for expert support with MS analysis and cell sorting, respectively. This work was supported by the ISREC Foundation, Swiss National Centre of Competence in Research-Chemical Biology, European Research Council (Grant ERC-2016-CoG 682311-ONCOINTRABODY to O.H.), U.S. National Institutes of Health Grants R01-GM090324 (to S.K) and P30CA014599 (to The University of Chicago Comprehensive Cancer Center). E.B. is supported by a National Science Foundation Graduate Research Fellowship and the Stony Brook University Center for Inclusive Education IMSD-MERGE NIH funded fellowship (5R25GM103962_02). Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank the Swiss Light Source for support with X-ray data collection.
Edited by Sachdev Sidhu
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2017.03.023.
Contributor Information
Shohei Koide, Email: Shohei.Koide@nyumc.org.
Oliver Hantschel, Email: oliver.hantschel@epfl.ch.
Appendix A. Supplementary Data
Supplementary material
References
- 1.Sadowski I., Stone J.C., Pawson T. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol. Cell. Biol. 1986;6:4396–4408. doi: 10.1128/mcb.6.12.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 3.Liu B.A., Jablonowski K., Raina M., Arce M., Pawson T., Nash P.D. The human and mouse complement of SH2 domain proteins-establishing the boundaries of phosphotyrosine signaling. Mol. Cell. 2006;22:851–868. doi: 10.1016/j.molcel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Waksman G., Shoelson S.E., Pant N., Cowburn D., Kuriyan J. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 5.Superti-Furga G., Fumagalli S., Koegl M., Courtneidge S.A., Draetta G. Csk inhibition of c-Src activity requires both the SH2 and SH3 domains of Src. EMBO J. 1993;12:2625–2634. doi: 10.1002/j.1460-2075.1993.tb05923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sicheri F., Moarefi I., Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 7.Pellicena P., Stowell K.R., Miller W.T. Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J. Biol. Chem. 1998;273:15,325–15,328. doi: 10.1074/jbc.273.25.15325. [DOI] [PubMed] [Google Scholar]
- 8.Pawson T. SH2 and SH3 domains in signal transduction. Adv. Cancer Res. 1994;64:87–110. doi: 10.1016/s0065-230x(08)60835-0. [DOI] [PubMed] [Google Scholar]
- 9.Hantschel O., Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 10.Thomas S.M., Brugge J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 11.Lowell C.A., Soriano P. Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 1996;10:1845–1857. doi: 10.1101/gad.10.15.1845. [DOI] [PubMed] [Google Scholar]
- 12.Martin G.S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S., Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol. Sci. 2012;33:122–128. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hantschel O. Unexpected off-targets and paradoxical pathway activation by kinase inhibitors. ACS Chem. Biol. 2015;10:234–245. doi: 10.1021/cb500886n. [DOI] [PubMed] [Google Scholar]
- 15.Kraskouskaya D., Duodu E., Arpin C.C., Gunning P.T. Progress towards the development of SH2 domain inhibitors. Chem. Soc. Rev. 2013;42:3337–3370. doi: 10.1039/c3cs35449k. [DOI] [PubMed] [Google Scholar]
- 16.Quartararo J.S., Wu P., Kritzer J.A. Peptide bicycles that inhibit the Grb2 SH2 domain. Chembiochem. 2012;13:1490–1496. doi: 10.1002/cbic.201200175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofener M., Heinzlmeir S., Kuster B., Sewald N. Probing SH2-domains using inhibitor affinity purification (IAP) Proteome Sci. 2014;12:41. doi: 10.1186/1477-5956-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koide A., Wojcik J., Gilbreth R.N., Hoey R.J., Koide S. Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 2012;415:393–405. doi: 10.1016/j.jmb.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koide A., Bailey C.W., Huang X., Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 20.Wojcik J., Lamontanara A.J., Grabe G., Koide A., Akin L., Gerig B., Hantschel O., Koide S. Allosteric inhibition of Bcr-Abl kinase by high affinity monobody inhibitors directed to the Src homology 2 (SH2)-kinase Interface. J. Biol. Chem. 2016;291:8836–8847. doi: 10.1074/jbc.M115.707901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grebien F., Hantschel O., Wojcik J., Kaupe I., Kovacic B., Wyrzucki A.M., Gish G.D., Cerny-Reiterer S., Koide A., Beug H., Pawson T., Valent P., Koide S., Superti-Furga G. Targeting the SH2-kinase Interface in Bcr-Abl inhibits leukemogenesis. Cell. 2011;147:306–319. doi: 10.1016/j.cell.2011.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wojcik J., Hantschel O., Grebien F., Kaupe I., Bennett K.L., Barkinge J., Jones R.B., Koide A., Superti-Furga G., Koide S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 2010;17:519–527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sha F., Gencer E.B., Georgeon S., Koide A., Yasui N., Koide S., Hantschel O. Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proc. Natl. Acad. Sci. U. S. A. 2013;110:14,924–14,929. doi: 10.1073/pnas.1303640110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koide A., Koide S. Monobodies: antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol. Biol. 2007;352:95–109. doi: 10.1385/1-59745-187-8:95. [DOI] [PubMed] [Google Scholar]
- 25.Yamanashi Y., Mori S., Yoshida M., Kishimoto T., Inoue K., Yamamoto T., Toyoshima K. Selective expression of a protein-tyrosine kinase, p56lyn, in hematopoietic cells and association with production of human T-cell lymphotropic virus type I. Proc. Natl. Acad. Sci. U. S. A. 1989;86:6538–6542. doi: 10.1073/pnas.86.17.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hantschel O., Gstoettenbauer A., Colinge J., Kaupe I., Bilban M., Burkard T.R., Valent P., Superti-Furga G. The chemokine IL-8 and the surface activation protein CD69 are markers for Bcr-Abl activity in CML. Mol. Oncol. 2008;2:272–281. doi: 10.1016/j.molonc.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X., Brodeur S.R., Gish G., Songyang Z., Cantley L.C., Laudano A.P., Pawson T. Regulation of c-Src tyrosine kinase activity by the Src SH2 domain. Oncogene. 1993;8:1119–1126. [PubMed] [Google Scholar]
- 28.Barker S.C., Kassel D.B., Weigl D., Huang X., Luther M.A., Knight W.B. Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry. 1995;34:14843–14851. doi: 10.1021/bi00045a027. [DOI] [PubMed] [Google Scholar]
- 29.Wang H., Kadlecek T.A., Au-Yeung B.B., Goodfellow H.E., Hsu L.Y., Freedman T.S., Weiss A. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb. Perspect. Biol. 2010;2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka S.-I., Takahashi T., Koide A., Ishihara S., Koikeda S., Koide S. Monobody-mediated alteration of enzyme specificity. Nat. Chem. Biol. 2015;11:762–764. doi: 10.1038/nchembio.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spencer-Smith R., Koide A., Zhou Y., Eguchi R.R., Sha F., Gajwani P., Santana D., Gupta A., Jacobs M., Herrero-Garcia E., Cobbert J., Lavoie H., Smith M., Rajakulendran T., Dowdell E., Okur M.N., Dementieva I., Sicheri F., Therrien M., Hancock J.F., Ikura M., Koide S., O'Bryan J.P. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017;13:62–68. doi: 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilbreth R.N., Koide S. Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr. Opin. Struct. Biol. 2012;22:413–420. doi: 10.1016/j.sbi.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeliger M.A., Young M., Henderson M.N., Pellicena P., King D.S., Falick A.M., Kuriyan J. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005;14:3135–3139. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burckstummer T., Bennett K.L., Preradovic A., Schutze G., Hantschel O., Superti-Furga G., Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material