

Abstract
The ability of cancer cells to move and invade the surrounding environment is the basis of local and distant metastasis. Cancer cell movement requires dynamic remodeling of the cytoskeleton and cell membrane and is controlled by multiprotein complexes including focal adhesion kinase (FAK) or the Neural Wiskott-Aldrich Syndrome Protein (N-WASP). We show that 17β-estradiol induces phosphorylation of FAK and its translocation toward membrane sites where focal adhesion complexes are assembled. This process is triggered via a Gα/Gβ protein-dependent, rapid extranuclear signaling of estrogen receptor α interacts in a multiprotein complex with c-Src, phosphatidylinositol 3-OH kinase, and FAK. Within this complex FAK autophosphorylation ensues, and activated FAK recruits the small GTPase cdc42, which, in turn, triggers N-WASP phosphorylation. This results in the translocation of Arp2/3 complexes at sites where membrane structures related to cell movement are formed. Recruitment of FAK and N-WASP is necessary for cell migration and invasion induced by 17β-estradiol in breast cancer cells. Our findings identify an original mechanism through which estrogen promotes breast cancer cell motility and invasion. This information helps to understand the effects of estrogen on breast cancer metastasis and may provide new targets for therapeutic interventions.
ERα activates the FAK/cdc42/N-WASP/Arp-2/3 cascade and cytoskeleton remodeling in breast cancer cells, leading to enhanced motility. This may be important for estrogen-dependent breast cancer metastasis.
One of eight women develops breast cancer at some stage throughout life (1). Breast cancer is the major cause of cancer-related death in women, and its treatment is particularly difficult when metastasis occurs (2). Despite the recent improvements in survival rates, many patients relapse, and the majority of these patients die for disseminated metastatic disease, which supports the need for new therapeutic strategies.
The sex steroid estrogen plays a major role in the development and progression of breast cancer. Prolonged exposure to estrogen, i.e. early menarche, late menopause, or postmenopausal hormone replacement therapy, is associated with a greater risk of developing breast cancer (3, 4, 5). Estrogen promotes breast cancer proliferation through a number of established pathways (6). However, the effects of E2 on breast tumor cell motility or invasion are poorly understood. Indications that estrogens facilitate the progression of breast cancer mostly come from clinical studies in which antiestrogens or aromatase inhibitors have been used. In this setting patients with breast cancer treated with these drugs have a lower rate of local and distant relapse (7, 8). However, the mechanistic basis of these actions are unclear, and specifically, it is not known whether estrogens simply increase the proliferation of ER+ cells (9) or if they also provide these cells with specific abilities that help them spread locally or at distant sites (10).
Cell migration is required for cancer spread, invasion, and metastasis, and it is achieved through a dynamic remodeling of filamentous actin and of focal adhesion sites (11). This structural modification is required for the protrusion of the leading edge of the cell, for the formation of adhesion complexes, for myosin/actin-mediated cell contraction, and for the release of adhesions at the rear of the cell (12), all of which are required for cell movement (13). Recent findings indicate that sex steroid hormones are fundamental regulators of cell morphology and motility in diverse cellular types, including breast cancer cells (2, 14, 15, 16, 17) and that many of these actions are played via rapid signaling to the actin cytoskeleton achieved via the recruitment of actin-binding proteins, including the ezrin/radixin/moesin family protein, moesin and the WASP (Wiskott-Aldrich syndrome protein) family protein, WAVE-1. In these cells, estrogen exposure leads to rapid modifications of the interaction with the extracellular matrix and nearby cells (2, 14, 16, 17).
Between the many regulators of the actin cytoskeleton is focal adhesion kinase (FAK). FAK is a nonreceptor tyrosine kinase that controls a number of cellular signaling pathways, including cell motility and survival (18). FAK is particularly involved in the formation and turnover of focal adhesion sites (19, 20). FAK is also important for cancer development and progression, being overexpressed in many tumors (21, 22, 23, 24), often in the early stages of tumorigenesis (21, 25). Moreover, FAK activity is higher in migrating breast cancer cells (26), and its expression is higher in metastases as compared with primary tumors (27).
N-WASP is a scaffold that links upstream signals to the activation of the Arp2/3 complex, leading to a burst of actin polymerization. Actin nucleation by the Arp2/3-complex appears to be critical for the rapid formation of an actin network at the leading edge of the cell (28, 29) that provides the protrusive force required for the extension of filopodia and lamellipodia during cell movement (30).
In this paper we wished to identify whether the regulatory actions of estrogen on breast cancer cell motility may require actin remodeling via FAK and N-WASP and to characterize the intracellular cascades recruited during this signaling.
Results
Estrogen rapidly induces FAK phosphorylation and the formation of focal adhesion complexes
Treatment with E2 (10 nm) of T47-D breast cancer cells resulted in a rapid increase of Tyr397 and Tyr576-phosphorylation of FAK, which corresponds to activation (20) (Fig. 1A). This phenomenon was time dependent and transient, being maximal after 15–20 min and reversing to baseline after 60 min (Fig. 1A). Total immunoreactive FAK did not change during this time frame (Fig. 1A). FAK phosphorylation was found throughout a range of estrogen concentrations that fall within the physiological range (Fig. 1B). Similar responses to E2 were found in MCF-7 cells (Supplemental Figs. A and B published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org).
Fig. 1.
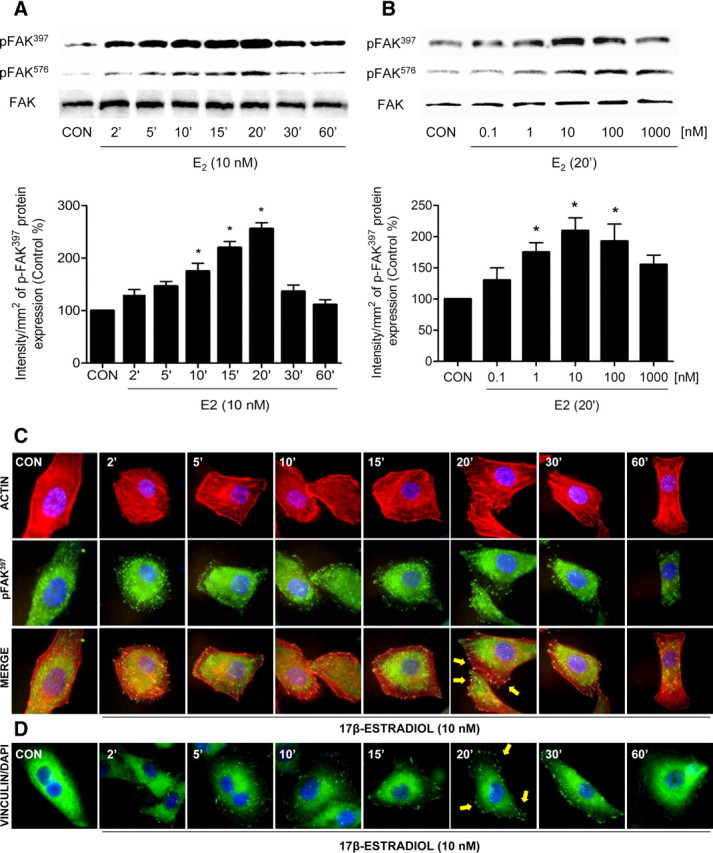
E2 activates FAK and induces rapid actin cytoskeleton rearrangement in T47-D cells. A and B, Time- and dose-dependent FAK activation in T47-D breast cancer cells after treatment with E2. Total cell amount of wild-type (FAK) or Tyr397-phosphorylated FAK (p-FAK397) and Tyr576-phosphorylated FAK (p-FAK576) are shown with Western blot. Revised and corrected. Phospho-FAK397 densitometry values were adjusted to FAK intensity and then normalized to the control sample. *, P < 0.05 vs. corresponding control. C, T47-D cells were treated with E2 (10 nm) for the indicated time. Cells were stained with antiphospho-Tyr397 FAK (p-FAK397) linked to fluorescein isothiocyanate, actin was stained with phalloidin linked to Texas Red, and nuclei were counterstained with DAPI. Yellow arrows indicate membrane-localized p-FAK397. D, Cells were treated with E2 (10 nm) for different times and were stained with antivinculin. Nuclei were counterstained with DAPI. CON, Control. Yellow arrows indicate membrane-localized vinculin. All experiments were repeated three times with consistent results, and representative images are shown.
Treatment with E2 (10 nm) led to a rapid change of the spatial organization of actin fibers and FAK subcellular localization. Actin translocated from the cytoplasm toward the edge of the cell membrane where it colocalized with phosphorylated FAK, forming typical focal adhesion complexes in association with specialized membrane structures, such as filopodia and membrane ruffles (Fig. 1C).
To ascertain the role of FAK in the formation of focal adhesion complexes, we studied the localization of vinculin, a major component of focal adhesion complexes (31). E2 rapidly induced vinculin membrane localization at sites where cortical actin complexes were formed (Fig. 1D).
To test whether FAK is required for the estrogen-dependent cytoskeletal rearrangement in breast cancer cells, we silenced FAK with small interfering RNAs (siRNAs) (Supplemental Fig. C). In FAK-silenced cells, E2 failed to induce actin reorganization (Supplemental Fig. D).
Estrogen activates FAK via ERα
FAK phosphorylation induced by E2 was prevented by the addition of the ER antagonist ICI 182,780 (ICI, 100 nm) (Fig. 2A). T47-D breast cancer cells express both ER isoforms, ERα and ERβ. To identify which isoform mediates the signaling of E2 to FAK we silenced ERα or ERβ with targeted siRNAs. Transfection of ERα siRNAs resulted in a clear reduction of ERα expression, along with a dramatic decrease in FAK phosphorylation on tyrosine 397 and 576 during exposure to estrogen (Fig. 2B), in the absence of modifications of the expression of FAK (Fig. 2B). On the contrary, silencing of ERβ did not influence FAK phosphorylation by E2 (Fig. 2C).
Fig. 2.

Estrogen signals to FAK via ERα. A, Breast cancer cells were exposed for 20 min to 10 nm E2 in the presence or absence of the ER antagonist ICI 182,780 (ICI; 100 nm). Phosphorylation of FAK was assayed with Western analysis. B and C, Breast cancer cells were transfected with siRNA vs. ERα (siRNA ERα) or ERβ (siRNA ERβ) or with vehicle, and protein analysis for ERα, ERβ, actin, total immunoreactive FAK (FAK) or phospho-FAK397, and phospho-FAK576 was performed on cell lysates after treatment for 20 min with 10 nm E2. CON, Control. All experiments were repeated three times with consistent results, and representative images are shown.
ERα signals to FAK through a Gαi/Gβ-dependent signaling pathway that involves c-Src and PI3K
FAK phosphorylation during E2 exposure was prevented by the G protein inhibitor, pertussis toxin (PTX) (Fig. 3A) and by interfering with the signaling of the G proteins Gαi and Gβ, but not of Gα13 (Fig. 3B). With coimmunoprecipitation experiments, we also found a ligand-induced interaction of ERα with Gαi1 and Gβ1 (Fig. 3, C–E), which was prevented by ICI 182,780. Consistent with the previous results indicating that ERβ is not implicated in the signal transduction of estrogen to FAK, we did not find any ligand-induced increase in the interaction of this receptor with Gαi1 (Fig. 3D).
Fig. 3.

ERα signaling to FAK requires Gαi/Gβ. A, Breast cancer cells were exposed for 20 min to 10 nm E2, in the presence or absence of the G protein inhibitor PTX (100 ng/ml), of the c-Src inhibitor, PP2 (0.2 μm), or of the PI3K inhibitor WM (30 nm), and Tyr397 FAK phosphorylation was assayed with Western analysis. B, Breast cancer cells were treated with E2 (10 nm) after transfection with dominant-negative Gα13 or Gαi constructs or siRNAs vs. Gβ1. Gα13, Gαi, Gβ1, actin, FAK, or phospho-FAK397 was assayed in cell extracts. C–E, T47-D cell protein extracts were immunoprecipitated with antibodies toward ERα, Gαi1, or Gβ1, and coimmunoprecipitation of Gαi1 (C), ERα and ERβ (D), and ERα and Gαi1 (E) was tested by Western analysis. F, Breast cancer cells were exposed for 20 min to 10 nm E2, in the presence or absence of the MAPK inhibitor PD98059 (PD; 5 mm), and Tyr397 FAK phosphorylation was assayed with Western analysis. CON, Control; IP, Immunoprecipitation. All experiments were performed three times, and representative blots are presented.
To clarify the signaling intermediates implicated in FAK activation by ERα in T47-D breast cancer cells, we used different pharmacological inhibitors in cells exposed to E2. The Src kinase inhibitor PP2 (10 μm), and wortmannin (WM, 30 nm), an inhibitor of phosphatidylinositol 3-OH kinase (PI3K), significantly inhibited FAK Tyr397 phosphorylation induced by E2 (Fig. 3A), indicating that in the presence of E2 ERα signals to FAK via c-Src and PI3K. PD98059 (5 mm) effectively inhibited ERK1/2 MAPK activation by E2 (Supplemental Fig. E), but was unable to reduce the activation of FAK by E2 (Fig. 3F), suggesting that the ERK 1/2 MAPK cascade not is involved in FAK phosphorylation.
With multiple immunoprecipitation assays we found that, in the presence of E2, ERα increases its interaction with c-Src, p85α and FAK and that this interaction is disrupted by the use of ICI 182,780 (Fig. 4A). The E2-dependent formation of a multiprotein complex between ERα, Src, p85α, and FAK was confirmed by separate immunoprecipitations of each component of the complex (Fig. 4, B–D).
Fig. 4.

ERα signals to FAK via interaction with Src and PI3K. A–D, T47-D cells were exposed to 10 nm E2 for 20 min, in the presence or absence of the ER antagonist ICI 182,780 (ICI; 100 nm). Cell protein extracts were immunoprecipitated with an antibody. vs. ERα (A), c-Src (B), p85α (C), and FAK (D). The immunoprecipitates (IPs) were assayed for coimmunoprecipitation of ERα, c-Src, p85α, and FAK. The membranes were reblotted for the immunoprecipitated protein to show equal input. E, T47-D cells were exposed to 10 nm E2 for 20 min, in the presence or absence of the Src kinase inhibitor, PP2 (10 μm). Cell protein extracts were immunoprecipitated with an antibody. vs. FAK. The IPs were assayed for Tyr397 or Tyr576 FAK phosphorylation, as well as for overall Tyr FAK phosphorylation with a general antiphosphotyrosine antibody (4G10). The membranes were reblotted for FAK to show equal input. F, Breast cancer cells were exposed for 20 min to E2 (10 nm) in the presence or absence of ICI 182,780 (100 nm), of the G protein inhibitor, PTX (100 ng/ml), or of the Src kinase inhibitor, PP2 (10 μm). Cell content of wild-type or phosphorylated c-Src Tyr416 is shown. G, Cells were treated with 10 nm E2 for 20 min with or without ICI 182,780, PTX, PP2, and the PI3K inhibitor WM (30 nm). Cell content of total immunoreactive or Thr308-phosphorylated Akt is shown. The experiments were performed in triplicates, and representative images are shown. CON, Control.
The estrogen-dependent signaling to c-Src mediates the phosphorylation of Tyr397 and Tyr576 on FAK (Fig. 4E). However, overall FAK phosphorylation, tested with a nonspecific antiphosphotyrosine antibody (4G10), did not change in the presence of E2, and when Src was inhibited, E2 triggered a global tyrosine dephosphorylation of FAK (Fig. 4E). This is consistent with the findings of a previous study (32) and suggests that estrogen may control both tyrosine phosphorylation as well as dephosphorylation via different pathways.
Increased c-Src Tyr416 phosphorylation was observed in the presence of E2, and this was abolished with the use of ICI 182,780 and PTX (Fig. 4F). Furthermore, in the presence of E2 the PI3K downstream effector, Akt, was functionally activated, as shown by enhanced phosphorylation on Thr308. This was inhibited by ICI 182,780, PTX, PP2, and WM (Fig. 4G), supporting the concept that E2 triggers, through a G protein-mediated cascade, the formation of an integrated signaling complex involving Src and PI3K.
Estrogen induces N-WASP phosphorylation by FAK via cdc42 in breast cancer cells
FAK activates N-WASP, a protein involved in actin nucleation, by inducing N-WASP phosphorylation on a conserved tyrosine residue by recruiting the small GTPase cdc42 (12, 33, 34, 35). During treatment with E2, FAK, cdc42, and N-WASP were silenced with specific siRNAs. Blockade of FAK resulted in impaired N- WASP Ser484/485 phosphorylation (Fig. 5A). Blockade of cdc42 impaired N-WASP phosphorylation by E2 but not FAK Tyr397 phosphorylation (Fig. 5A), and silencing of N-WASP did not alter FAK Tyr397 phosphorylation (Fig. 5A), suggesting that ERα signals to N-WASP via FAK and cdc42.
Fig. 5.

ER signals to FAK, cdc42, and N-WASP. Breast cancer cells were incubated in the presence of 10 nm E2 for 20 min with or without silencing of FAK, cdc42, or N-WASP with specific siRNAs. A, Actin, FAK, Tyr397 phospho-FAK, cdc42, N-WASP, and Ser484/485 phospho-N-WASP were assayed in cell extracts. B, Cells were stained with anti-phospho-Ser484/485 N-WASP (p-N-WASP) linked to fluorescein isothiocyante (green), and anti-phospho-Tyr397 FAK (p-FAK) linked to Texas Red (red). Nuclei were counterstained with DAPI. Double staining (yellow signal) highlights areas of colocalization. C and D, The bar graphs show the quantification of the membrane-localized Tyr397 phospho-FAK and Ser484/485 phospho-N-WASP in the different conditions. *, P < 0.05 vs. control. Membrane-localized Tyr397 p-FAK/Ser484/485 p-N-WASP complexes were counted in 40 different cells for condition. All the experiments were repeated three times with consistent results, and the representative images are shown. CON, Control.
To confirm this, we studied the subcellular localization of phosphorylated FAK Tyr397 and N-WASP Ser484/485. In control cells, phospho-FAK Tyr397 and phospho-N-WASP Ser484/485 were diffusely distributed throughout the cytoplasm (Fig. 5B). Short-term exposure to E2 (10 nm) led to an increase in FAK Tyr397 and N-WASP Ser484/485 phosphorylation and translocation to the plasmatic membrane and to colocalization of the two proteins at the edge of the membrane (Fig. 5B). This colocalization was prevented by the use of FAK, cdc42, and N-WASP siRNAs (Fig. 5B).
Estrogen signaling to FAK and N-WASP leads to regulation of the Arp2/3 complex
In response to cdc42, the Arp2/3 complex is regulated by N-WASP and initiates actin branching at the cell membrane and hence the formation of lamellipodia. N-WASP acts as a scaffolding complex relaying signals from small GTPases, such as cdc42, to the Arp2/3 complex. Because N-WASP has a conserved verprolin-homology, cofilin-homology, acidic domain that directly binds to and activates the Arp2/3 complex (36, 37, 38), we studied whether estrogen-phosphorylated N-WASP may directly regulate Arp-2 in breast cancer cells.
We therefore examined the subcellular localization of Arp-2 with immunofluorescence in the presence of E2. Breast cancer cells treated with E2 displayed membrane translocation of Arp-2 (Fig. 6A), which was consistent with the phosphorylation of FAK and N-WASP. Blockade of FAK or N-WASP with siRNAs abrogated the estrogen-induced membrane translocation of Arp-2, suggesting that estrogen signals to Arp-2 through a FAK/cdc42/N-WASP cascade.
Fig. 6.

Estrogen signaling to N-WASP turns into a membrane relocalization of the Arp2/3 complex. A, Breast cancer cells were treated for 20 min with 10 nm E2 in the presence or absence after transfection with siRNAs vs. FAK, N-WASP, and Arp-2. Cells were stained with an antibody against Arp-2 (fluorescein isothiocyante, green), actin fibers were stained with phalloidin linked to Texas Red (red), and nuclei were counterstained with DAPI. Yellow arrows indicate membrane-localized Arp-2. B, Quantification of the membrane-localized Arp-2 complexes in different conditions. Results are expressed as percent vs. control cells (mean ± sd). *, P < 0.05 vs. control. Membrane-localized Arp-2 complexes were counted in 40 different cells. The experiments were repeated three times with consistent results. CON, Control.
Estrogen-induced breast cancer cell migration and invasion require FAK and N-WASP phosphorylation
To address the question of the relevance of the ERα/FAK/cdc42/N-WASP signaling cascade on breast cancer cell migration and invasion, we pretreated T47-D cells with cytosine arabinoside [1-(β-d-arabinofuranosyl] cytosine hydrochloride-Ara-C, 100 μm), an inhibitor of DNA strand separation that prevents cell division (so to dissect the actions of estrogen on movement from those on cell proliferation), and we performed horizontal migration assays. Treatment with E2 (10 nm) significantly increased the number of T47-D breast cancer cells that migrated through the starting line, as well as the mean length of migration compared with control (Fig. 7, A and B). This was blocked by silencing ERα, c-Src, FAK, cdc42, N-WASP, and Arp-2 (Fig. 7A) and was also blocked by PTX and WM but not by PD98059 (Fig. 7, A and B).
Fig. 7.
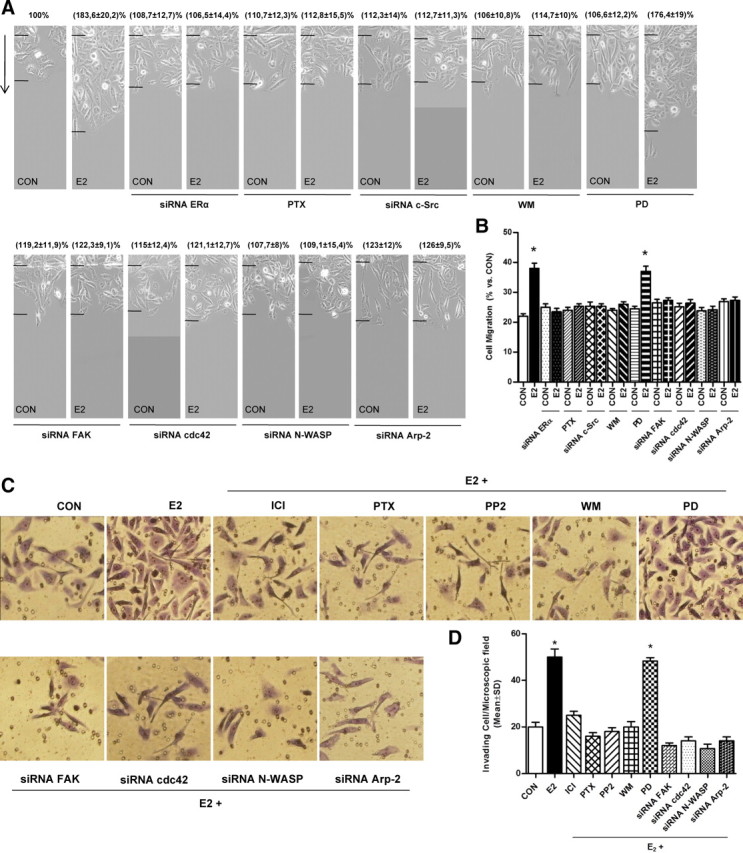
Intracellular signaling mechanisms involved in ER-enhanced T47-D cell migration and invasion. A, Cells were treated with estrogen (10 nm) for 48 h, in the presence or absence of PTX (100 ng/ml), of PD98059 (PD, 5 mm), or WM (30 nm). Other cells were transfected with siRNAs toward ERα, c-Src, FAK, cdc42, N-WASP, and Arp-2. Representative images are shown. The arrows indicate the direction of migration. The upper black lines indicate the starting line, and the lower black lines indicate the mean migration distance. B, Cell migration distances were measured, and values are presented as percent of control (CON). *, P < 0.01 vs. control. The experiments were performed in triplicate. C, T47-D cells were treated with 10 nm E2 in the presence or absence of different inhibitors or siRNAs as indicated in panel A. Breast cancer cell invasion through Matrigel was assayed with invasion chambers. Representative images in chambers with Matrigel are shown. D, Invading cells were counted in the central field of triplicate membranes. *, P < 0.05 vs. control.
Finally, we checked the role of FAK and N-WASP on breast cancer cell invasion of Matrigel. Ara-C-pretreated cells showed enhanced invasion in the presence of E2 (Fig. 7, C and D). This was blocked by silencing FAK, cdc42, N-WASP, and Arp-2 (Fig. 7, C and D). The enhancement of cancer cell invasion induced by E2 was also prevented by blocking ER with ICI 182,780, by blocking G proteins with PTX, c-Src with PP2, and PI3K with wortmannin, but not by the MAPK inhibitor PD98059 (Fig. 7, C and D).
Discussion
The key finding of this work is that estrogen induces rapid changes of breast cancer cell membrane morphology that are linked to enhanced motility and invasion. This is achieved through an ERα-dependent signaling to the actin regulators FAK and N-WASP, which activate a remodeling of the actin cytoskeleton toward the membrane and the formation of specialized structures linked to cell movement. In the presence of E2, ERα recruits a Gαi/Gβ-dependent signaling that triggers the formation of a multiprotein complex where ERα, c-Src, PI3K, and FAK interact. Within this complex FAK is hyperphosphorylated on Tyr397 and Tyr576, and this leads to the later recruitment of cdc42. Activated cdc42 turns into N-WASP Ser484/485 phosphorylation and translocation to sites where the actin cytoskeleton and the cell membrane are actively remodeled and into membrane localization of the Arp2/3 protein complex. Arp2/3 is responsible for branching of actin filaments and thus for cell membrane remodeling induced by estrogens.
Estrogens act as promoters of cell movement in different tissues, including the breast (39). This action is particularly relevant in estrogen receptor positive (ER+) breast cancers that are driven to invade and metastasize by endogenous or exogenous estrogens (6, 10). For this reason, understanding the basis through which estrogens drive cancer cells to interact with the extracellular environment to enact movement and invasion heralds profound biological and medical implications.
We show that estrogen rapidly activates FAK via phosphorylation on Tyr397/576, leading to the formation of focal adhesion complexes. FAK is a nonreceptor tyrosine kinase that recruits Src family kinases and phosphatidylinositol-3-OH kinase via autophosphorylation and is a pivotal modulator of adhesion turnover (40, 41). We identify the recruitment of PI3K along with the activation of c-Src by estrogen, and we show that this step is required for FAK phosphorylation on Tyr397/576. This is consistent with previous reports showing that the c-Src/PI3K pathway is implicated in Tyr397 FAK phosphorylation (40, 41). Based on our results and on those from previous reports (32), estrogen seems to control FAK tyrosine phosphorylation through multiple mechanisms, inducing Tyr397/576 phosphorylation via Src, while triggering a dephosphorylation of other tyrosine residues, through alternative, unidentified pathways. FAK contains at least six identified tyrosine phosphorylation sites that have been shown to play regulatory roles during the signaling of this kinase (Tyr397/407/576/577/861/925). The current understanding is that autophosphorylation on Tyr397 creates a docking site for Src and other SH2-containing proteins (42). Src would thus trigger the phosphorylation of the other Tyr residues, modulating FAK activity (42). The present and previous (32) evidence that estrogen induces tyrosine dephosphorylation of FAK when Src is inhibited stands for the existence of alternate mechanisms recruited by estrogens that may play a role in controlling FAK activity.
ERα-dependent FAK activation results in enhanced motility and invasion of breast cancer cells. FAK has been recently established as a central controller of cell migration, particularly in the setting of tumor metastasis. Overexpression of FAK is related to the metastatic behavior of various tumors, such as lung cancer (43), ovarian cancer (44), and melanoma (45). In human breast cancer, high FAK expression is associated with aggressive phenotype (46). In animal models, inhibition of FAK activity in a rat breast cancer metastasis model abrogates cancer diffusion to the lung (47), and targeted deletion of FAK in mouse mammary epithelium reduces the pool of cancer stem/progenitor cells in primary tumors and their self-renewal and migration (48). In contrast, silencing of FAK in human and mouse mammary tumor cells results in cell senescence and in loss of invasive ability (49).
In support of the mechanistic model of estrogen-dependent activation of FAK and of breast cancer cell motility and invasion that we delineate in this paper, a recent publication shows that in tamoxifen-resistant variants and in metastases of recurrent hormone-treated breast cancers, ER binds to FAK and estrogen modulates FAK autophosphorylation in a c-Src-dependent manner (50). In addition, inhibition of c-Src and FAK activity blocked the proliferation of all tamoxifen-resistant variants (50). Overall, these findings highlight the relevance of the activity of FAK for cancer progression. The identification of the mechanistic basis of FAK regulation by estrogen may thus offer important insights to better understand the role of this hormone on breast cancer metastasis and for potential future therapeutic strategies (26).
Our data shows that estrogen controls FAK and cell movement by regulating cdc42 and its effector N-WASP (12). Interestingly, in the presence of estrogen, FAK only associates with cdc42-activated N-WASP and does not activate N-WASP itself. Although FAK phosphorylation of N-WASP does not affect N-WASP activity toward Arp2/3, it seems important for maintaining the cytoplasmic distribution of N-WASP and for promoting cell motility (12). Because cdc42 regulates actin dynamics in cell membrane projections, interaction of FAK with cdc42-activated N-WASP might couple actin polymerization with membrane protrusion during cell movement (18).
In conclusion, the present results show that within the broader range of actions of ERs, rapid extranuclear signaling to the actin cytoskeleton through the FAK/cdc42/N-WASP/Arp-2/3 cascade is relevant for the generation of estrogen-dependent breast cancer cell movement and invasion. Through this cascade E2 leads to rapid changes of cell membrane morphology, with a rearrangement of the actin cytoskeleton and the formation of focal adhesion complexes at sites where structures related with cell movement are formed. The identification of these actions increases our understanding of the effects of estrogens on breast cancer progression and might be useful to develop new tools to interfere with the ability to diffuse locally or at distant sites of breast tumors.
Materials and Methods
Cell cultures and treatments
The human breast carcinoma cell line T47-D was obtained from the American Type Culture Collection. T47-D cells were grown in RPMI 1640 supplemented with l-glutamine (2 mm), 10% fetal bovine serum (FBS). Before treatments, breast cancer cells were kept 24 h in medium containing steroid-deprived FBS. Before experiments investigating nontranscriptional effects, cancer cells were kept in medium containing no FBS for 8 h. E2, PTX, PD98059, and wortmannin were from Sigma-Aldrich (Saint-Louis, MO); PP2 was from Calbiochem (La Jolla, CA); ICI 182,780 was from Tocris Cookson (Avonmouth, UK). Whenever an inhibitor was used, the compound was added 30 min before start of the active treatments.
Immunoblottings
Cell lysates were separated by SDS-PAGE. Antibodies used were: p-FAK (Y397) (611807), FAK (610088) (BD Transduction Laboratories, Lexington, KY); FAK (Tyr576) (07-157, UPSTATE, Lake Placid, NY); ERα (TE111, NeoMarkers, Union City, CA), actin (C-11), ERα (H-184), ERβ (N-19), Gα13 (A-20), Gαi1 (R4), Gβ1 (C-16), c-Src (sc-5266), p-FAK (Tyr397)-R (sc-11765-R), cdc42 (B-B) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), P85α (N-18), Vinculin (C-20), c-Src (H-12), Tyr204-P-ERK (sc-7969) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), Akt (9272), phospho-Src (Tyr416), N-WASP (30D10) phospho-Akt Thr308 (9275S) (Cell Signaling Technology, Beverly, MA), antivinculin mouse monoclonal antibody (V284), ERK1/ERK2 (444944), phospho-WASP Ser484/485 (AB1964) (Chemicon International, Temecula, CA). Primary and secondary Antibodies were incubated with the membranes using a standard technique. Immunodetection was accomplished using enhanced chemiluminescence.
Cell immunofluorescence
T47-D cells were grown on coverslips. Cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton for 5 min. Blocking was performed with PBS containing 3% BSA for 30 min. Cells were incubated with antibodies against FAK, phospho-FAK Tyr397 (Transduction Laboratories), Vinculin (sc-7648), phospho-WASP Ser484/485 (AB1964) (Chemicon International), and Arp2 (1:900; H-84, Santa Cruz Biotechnology) overnight at 4 C followed by incubation with a fluorescein-conjugated goat antirabbit/mouse secondary antibody (1:200; Vector Laboratories, Inc., Burlingame, CA). Cells were then incubated with Texas Red-phalloidin (Sigma) for 30 min. After washing, the nuclei were counterstained with or 4′-6-diamidino-2-phenylindole (DAPI) (Sigma) and mounted with Vectashield mounting medium (Vector Laboratories). Immunofluorescence was visualized using an Olympus BX41 microscope and recorded with a high-resolution DP70 Olympus digital camera (Olympus Corp., Lake Success, NY).
Immunoprecipitations
Breast cancer cells were washed with ice-cold PBS and lysed with: 20 mm Tris-HCl, pH 7.4, 10 mm EDTA, 100 mm NaCl, 1% Igepal, 1 mm Na3VO4, 50 mm NaF, 0.1 mg/liter phenylmethylsulfonylfluoride, 0.3 mg/liter aprotinin, and 0.01% protease inhibitor mixture (Sigma-Aldrich) before addition of the immunoprecipitating antibodies vs. ERα, Gαi1, Gβ1, and c-Src in 500 μl of lysis buffer for 1 h at 4 C with gentle rocking. Subsequently, 40 μl of 1:1 Protein-A-agarose was added and gently rocked for 2 additional hours at 4 C. The mixture was then centrifuged at 12,000 × g for 5 min at 4 C. The supernatant was removed, and the immunoprecipitates were washed with 500 ml of: 20 mm Tris-HCl, pH 7.4, 10 mm EDTA, 150 mm NaCl, 1% Igepal, 1 mm Na3VO4, 50 mm NaF, 0.1 mg/liter phenylmethylsulfonylfluoride, 0.3 mg/liter aprotinin, and 0.01% protease inhibitor mixture (Sigma-Aldrich). Immunoprecipitated proteins were separated under reducing and denaturing conditions by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA). Nonspecific binding was blocked with 5% skim milk in PBS-Tween. Membranes were incubated with anti-FAK, p85α, ERα, c-Src, Gαi1, and Gβ1antibodies.
Cell migration assays
Cell migration was assayed with razor-scrape assays as previously described (17). Briefly, a razor blade was pressed through the confluent T47-D breast cancer cell monolayer into the plastic plate to mark the starting line. T47-D cells were swept away on one side of that line. Cells were washed, and 2.0 ml of DMEM containing steroid-deprived FBS and gelatin (1 mg/ml) were added. Cytosine β-d-arabinofuranoside hydrochloride (Sigma) (10 μm), a selective inhibitor of DNA synthesis that does not inhibit RNA synthesis, was used 1 h before the test substance was added. Migration was monitored for 48 h. Every 12 h fresh medium and treatments were replaced. Migration distance was measured by phase-contrast microscopy.
Cell invasion assays
Cell invasion was assayed using BD BioCoat growth factor reduced (GFR) Matrigel Invasion Chambers (BD Biosciences, Palo Alto, CA). In brief, after rehydrating the GFR Matrigel inserts, the test substance was added to the wells. An equal number of control inserts (no GFR Matrigel coating) were prepared as control. T47-D cell suspension (0.5 ml; 2.5 × 104 cells/ml) was added to the inside of the inserts. The chambers were incubated for 24 h at 37 C, 5% CO2 atmosphere. After incubation, noninvading cells were removed from the upper surface of the membrane using cotton-tipped swabs. The cells on the lower surface of the membrane were stained with Diff-Quick. Cells were counted in the central field of triplicate membranes.
Gene silencing with RNA interference
Synthetic siRNAs targeting ERα (siRNA SMARTpool ESR1), ERβ (siRNA SMARTpool ESR2), c-SRC (siRNA SMARTpool SRC), FAK (siRNA SMARTpool FAK), and control siRNAs (D-001810–01-05) were purchased from Dharmacon (Lafayette, CO). Gβ1 siRNAs, N-WASP, and Arp-2 siRNAs were from Santa Cruz Biotechnology (Santa Cruz, CA). The siRNAs were used at the final concentration of 50 nm. Breast cancer cells were treated 48 h after siRNAs transfection. Efficacy of gene silencing was checked with Western analysis and found to be optimal at 48 h.
Transfection experiments
Dominant-negative constructs for Gαi1 (Gαi1 G202T) and Gα13 (Gα13 Q226L/D294N) were from the Guthrie cDNA Resource Center (www.cdna.org). The inserts were cloned in pcDNA3.1+. The plasmids (10 μg) were transfected into T47-D cells using Lipofectamine (Invitrogen, Carlsbad, CA). Parallel cells were transfected with empty pcDNA3.1+ plasmid. Cells (60–70% confluent) were treated 24 h after transfection.
Statistical analysis
All values are expressed as mean ± sd. Statistical analyses and graphics were done using InStat from GraphPad Prism Software. Statistical differences between mean values were determined by ANOVA, followed by the Fisher’s protected least significance difference.
NURSA Molecule Pages:
Ligands: 17β-estradiol | Fulvestrant;
Nuclear Receptors: ERα.
Footnotes
This work was supported by Progetti di Ricerca di Interesse Nazionele (PRIN) Grant 2004057090_007 by the Italian University and Scientific Research Ministry (to T.S.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online September 29, 2010
A.M.S. and M.I.F. contributed equally to this work.
Abbreviations: DAPI, 4′-6-Diamidino-2-phenylindole; E2, 17β-Estradiol; ER, estrogen receptor; FAK, focal adhesion kinase; FBS, fetal bovine serum; GFR, growth factor reduced; N-WASP, neural Wiskott-Aldrich syndrome protein; PI3K, phosphatidylinositol 3-OH kinase; PTX, pertussis toxin; siRNA, small interfering RNA; WASP, Wiskott-Aldrich syndrome protein; WM, wortmannin.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ2007. Cancer statistics, 2007. CA Cancer J Clin 57:43–66 [DOI] [PubMed] [Google Scholar]
- 2.Giretti MS, Fu XD, De Rosa G, Sarotto I, Baldacci C, Garibaldi S, Mannella P, Biglia N, Sismondi P, Genazzani AR, Simoncini T2008. Extra-nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodelling, migration and invasion. PLoS One 3:e2238 [DOI] [PMC free article] [PubMed]
- 3.Horwitz KB2008. The year in basic science: update of estrogen plus progestin therapy for menopausal hormone replacement implicating stem cells in the increased breast cancer risk. Mol Endocrinol 22:2743–2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma H, Bernstein L, Pike MC, Ursin G2006. Reproductive factors and breast cancer risk according to joint estrogen and progesterone receptor status: a meta-analysis of epidemiological studies. Breast Cancer Res 8:R43 [DOI] [PMC free article] [PubMed]
- 5.Verkooijen HM, Bouchardy C, Vinh-Hung V, Rapiti E, Hartman M2009. The incidence of breast cancer and changes in the use of hormone replacement therapy: a review of the evidence. Maturitas 64:80–85 [DOI] [PubMed] [Google Scholar]
- 6.Yager JD, Davidson NE2006. Estrogen carcinogenesis in breast cancer. N Engl J Med 354:270–282 [DOI] [PubMed] [Google Scholar]
- 7.Folkerd EJ, Dowsett M2010. Influence of sex hormones on cancer progression. J Clin Oncol 28:4038–4044 [DOI] [PubMed] [Google Scholar]
- 8.Fuqua SA2001. The role of estrogen receptors in breast cancer metastasis. J Mammary Gland Biol Neoplasia 6:407–417 [DOI] [PubMed] [Google Scholar]
- 9.Dunbier AK, Anderson H, Ghazoui Z, Folkerd EJ, A'hern R, Crowder RJ, Hoog J, Smith IE, Osin P, Nerurkar A, Parker JS, Perou CM, Ellis MJ, Dowsett M2010. Relationship between plasma estradiol levels and estrogen-responsive gene expression in estrogen receptor-positive breast cancer in postmenopausal women. J Clin Oncol 28:1161–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Platet N, Cathiard AM, Gleizes M, Garcia M2004. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol 51:55–67 [DOI] [PubMed] [Google Scholar]
- 11.Acconcia F, Barnes CJ, Kumar R2006. Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cancer cells. Endocrinology 147:1203–1212 [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Suetsugu S, Cooper LA, Takenawa T, Guan JL2004. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J Biol Chem 279:9565–9576 [DOI] [PubMed] [Google Scholar]
- 13.Pollard TD, Borisy GG2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112:453–465 [DOI] [PubMed] [Google Scholar]
- 14.Flamini MI, Sanchez AM, Goglia L, Tosi V, Genazzani AR, Simoncini T2009. Differential actions of estrogen and SERMs in regulation of the actin cytoskeleton of endometrial cells. Mol Hum Reprod 15:675–685 [DOI] [PubMed] [Google Scholar]
- 15.Fu XD, Giretti MS, Baldacci C, Garibaldi S, Flamini M, Sanchez AM, Gadducci A, Genazzani AR, Simoncini T2008. Extra-nuclear signaling of progesterone receptor to breast cancer cell movement and invasion through the actin cytoskeleton. PLoS One 3:e2790 [DOI] [PMC free article] [PubMed]
- 16.Sanchez AM, Flamini MI, Fu XD, Mannella P, Giretti MS, Goglia L, Genazzani AR, Simoncini T2009. Rapid signaling of estrogen to WAVE1 and moesin controls neuronal spine formation via the actin cytoskeleton. Mol Endocrinol 23:1193–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simoncini T, Scorticati C, Mannella P, Fadiel A, Giretti MS, Fu XD, Baldacci C, Garibaldi S, Caruso A, Fornari L, Naftolin F, Genazzani AR2006. Estrogen receptor α interacts with Gα13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol Endocrinol 20:1756–1771 [DOI] [PubMed] [Google Scholar]
- 18.Mitra SK, Hanson DA, Schlaepfer DD2005. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6:56–68 [DOI] [PubMed] [Google Scholar]
- 19.Robles E, Gomez TM2006. Focal adhesion kinase signaling at sites of integrin-mediated adhesion controls axon pathfinding. Nat Neurosci 9:1274–1283 [DOI] [PubMed] [Google Scholar]
- 20.van Nimwegen MJ, van de Water B2007. Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol 73:597–609 [DOI] [PubMed] [Google Scholar]
- 21.Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, Simkins S, Xu L2000. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res 6:2417–2423 [PubMed] [Google Scholar]
- 22.Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, Liu ET, Cance WG1995. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res 55:2752–2755 [PubMed] [Google Scholar]
- 23.Weiner TM, Liu ET, Craven RJ, Cance WG1993. Expression of focal adhesion kinase gene and invasive cancer. Lancet 342:1024–1025 [DOI] [PubMed] [Google Scholar]
- 24.Xu LH, Yang X, Craven RJ, Cance WG1998. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ 9:999–1005 [PubMed] [Google Scholar]
- 25.Lightfoot Jr HM, Lark A, Livasy CA, Moore DT, Cowan D, Dressler L, Craven RJ, Cance WG2004. Upregulation of focal adhesion kinase (FAK) expression in ductal carcinoma in situ (DCIS) is an early event in breast tumorigenesis. Breast Cancer Res Treat 88:109–116 [DOI] [PubMed] [Google Scholar]
- 26.Fu XD, Goglia L, Sanchez AM, Flamini M, Giretti MS, Tosi V, Genazzani AR, Simoncini T2010. Progesterone receptor enhances breast cancer cell motility and invasion via extra-nuclear activation of focal adhesion kinase. Endocr Relat Cancer 17:431–443 [DOI] [PubMed] [Google Scholar]
- 27.Sood AK, Coffin JE, Schneider GB, Fletcher MS, DeYoung BR, Gruman LM, Gershenson DM, Schaller MD, Hendrix MJ2004. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol 165:1087– 1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailly M, Macaluso F, Cammer M, Chan A, Segall JE, Condeelis JS1999. Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation. J Cell Biol 145: 331–345 [DOI] [PMC free article] [PubMed]
- 29.Mullins RD, Heuser JA, Pollard TD1998. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA 95:6181–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takenawa T, Suetsugu S2007. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 8:37–48 [DOI] [PubMed] [Google Scholar]
- 31.Ziegler WH, Liddington RC, Critchley DR2006. The structure and regulation of vinculin. Trends Cell Biol 16:453–460 [DOI] [PubMed] [Google Scholar]
- 32.Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, Goddard S, Gobert-Gosse S, Corbo L2008. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell 31:212–221 [DOI] [PubMed] [Google Scholar]
- 33.Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK2000. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature 404:151–158 [DOI] [PubMed] [Google Scholar]
- 34.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97:221–231 [DOI] [PubMed] [Google Scholar]
- 35.Schlaepfer DD, Mitra SK2004. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev 14:92–101 [DOI] [PubMed] [Google Scholar]
- 36.Bompard G, Caron E2004. Regulation of WASP/WAVE proteins: making a long story short. J Cell Biol 166:957–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prehoda KE, Scott JA, Mullins RD, Lim WA2000. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science 290:801–806 [DOI] [PubMed] [Google Scholar]
- 38.Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G2004. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol 14:303–311 [DOI] [PubMed] [Google Scholar]
- 39.Ricketts D, Turnbull L, Ryall G, Bakhshi R, Rawson NS, Gazet JC, Nolan C, Coombes RC1991. Estrogen and progesterone receptors in the normal female breast. Cancer Res 51:1817–1822 [PubMed] [Google Scholar]
- 40.Reiske HR, Kao SC, Cary LA, Guan JL, Lai JF, Chen HC1999. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem 274:12361– 12366 [DOI] [PubMed] [Google Scholar]
- 41.Thamilselvan V, Craig DH, Basson MD2007. FAK association with multiple signal proteins mediates pressure-induced colon cancer cell adhesion via a Src-dependent PI3K/Akt pathway. FASEB J 21:1730–1741 [DOI] [PubMed] [Google Scholar]
- 42.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC2005. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer 5:505–515 [DOI] [PubMed] [Google Scholar]
- 43.Fong YC, Liu SC, Huang CY, Li TM, Hsu SF, Kao ST, Tsai FJ, Chen WC, Chen CY, Tang CH2009. Osteopontin increases lung cancer cells migration via activation of the αvβ3 integrin/FAK/Akt and NF-κB-dependent pathway. Lung Cancer 64:263–270 [DOI] [PubMed] [Google Scholar]
- 44.Hu XW, Meng D, Fang J2008. Apigenin inhibited migration and invasion of human ovarian cancer A2780 cells through focal adhesion kinase. Carcinogenesis 29:2369–2376 [DOI] [PubMed] [Google Scholar]
- 45.Kaneda T, Sonoda Y, Ando K, Suzuki T, Sasaki Y, Oshio T, Tago M, Kasahara T2008. Mutation of Y925F in focal adhesion kinase (FAK) suppresses melanoma cell proliferation and metastasis. Cancer Lett 270:354–361 [DOI] [PubMed] [Google Scholar]
- 46.Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC, Geradts J, Iacocca M, Cowan D, Little D, Craven RJ, Cance W2005. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol 18:1289–1294 [DOI] [PubMed] [Google Scholar]
- 47.van Nimwegen MJ, Verkoeijen S, van Buren L, Burg D, van de Water B2005. Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Res 65:4698–4706 [DOI] [PubMed] [Google Scholar]
- 48.Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, Wicha MS, Guan JL2009. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res 69:466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG2009. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest 119:252–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Planas-Silva MD, Bruggeman RD, Grenko RT, Stanley Smith J2006. Role of c-Src and focal adhesion kinase in progression and metastasis of estrogen receptor-positive breast cancer. Biochem Biophys Res Commun 341:73–81 [DOI] [PubMed] [Google Scholar]