

Abstract
The second messenger cAMP plays an important role in the regulation of renin gene expression. Nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) is known to stimulate renin gene transcription acting through PPARγ-binding sequences in renin promoter. We show now that activation of PPARγ by unsaturated fatty acids or thiazolidinediones drastically augments the cAMP-dependent increase of renin mRNA in the human renin-producing cell line Calu-6. The underlying mechanism involves potentiation of agonist-induced cAMP increase and up-regulation of adenylate cyclase 6 (AC6) gene expression. We identified a palindromic element with a 3-bp spacer (Pal3) in AC6 intron 1 (AC6Pal3). AC6Pal3 bound PPARγ and mediated trans-activation by PPARγ agonist. AC6 knockdown decreased basal renin mRNA level and attenuated the maximal PPARγ-dependent stimulation of the cAMP-induced renin gene expression. AC6Pal3 decoy oligonucleotide abrogated the PPARγ-dependent potentiation of cAMP-induced renin gene expression. Treatment of mice with PPARγ agonist increased AC6 mRNA kidney levels. Our data suggest that in addition to its direct effect on renin gene transcription, PPARγ “sensitizes” renin gene to cAMP via trans-activation of AC6 gene. AC6 has been identified as PPARγ target gene with a functional Pal3 sequence.
We describe the identification of adenylate cyclase 6 (AC6) as PPARγ-regulated gene. Induction of AC6 expression by PPARγ potentiates the cAMP-dependent stimulation of renin gene.
The protease renin is produced and secreted into circulation by the juxtaglomerular (JG) cells of the kidney (1). Renin is the rate-limiting factor of renin angiotensin system and plays a central role in the regulation of arterial blood pressure and sodium balance. Therefore, the transcription of renin gene is tightly regulated. Numerous transcription factors involved in the molecular control of renin gene have been identified so far (2). The interplay of these transcriptional regulators results in developmentally regulated and cell-specific expression of renin, which is finely tuned by various systemic and local cues.
Our group identified nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) as a transcription factor involved in the control of renin gene (3, 4, 5). PPARγ targets a proximal promoter Pal3 site (palindrome with a 3-bp spacer) and a distal enhancer PPAR-response element (PPRE)-like site. Because mouse renin (mRen)-Pal3 is functionally inactive, PPARγ regulates mRen expression through the PPRE-like sequence (4). In contrast, human renin (hRen) Pal3 plays a dominant role in the PPARγ-dependent trans-activation. The regulation of renin gene expression by PPARγ could be relevant for the development of hypertension associated with overweight, because PPARγ is activated and plasma renin activity is elevated during obesity (6, 7, 8, 9, 10, 11, 12). However, a possible role of PPARγ in the physiological control of renin transcription has still not been addressed.
cAMP is a second messenger that is believed to play a central role in the regulation of renin gene expression by mediating the stimulatory effect of sympathetic nerves and catecholamines (13). cAMP up-regulates both renin mRNA stability and the transcription of renin gene in vitro (14, 15, 16, 17, 18). In agreement with these data, JG-specific knockout of the stimulatory adenylate cyclase (AC) Gsα unit resulted in almost complete loss of renin (13).
Here, we demonstrate that PPARγ potentiates the stimulatory effect of cAMP on renin gene expression. The molecular mechanism involves a multiplication of cAMP increase upon agonist treatment and PPARγ-dependent trans-activation of AC6 gene through a Pal3 sequence located in the first intron.
Results
PPARγ agonists multiply the stimulatory effect of AC activators on renin gene expression
We first examined the effect of PPARγ agonists on the stimulation of renin gene expression by the AC activator forskolin in the hRen-producing cell line Calu-6. We used the monounsaturated oleic acid, which is an endogenous PPARγ ligand, and rosiglitazone, which is a pharmacological PPARγ ligand from the group of the thiazolidinedione (TZD) compounds (Fig. 1A). All three substances increased renin mRNA levels (forskolin, 17-fold; oleic acid, 2.4-fold; and rosiglitazone, >5-fold). However, the combination of a PPARγ ligand with forskolin resulted not in additive increase but rather multiplied the effect of forskolin. Thus, in the presence of oleic acid or rosiglitazone, forskolin up-regulated renin gene expression about 60-fold and 150-fold, respectively. Similar results were obtained when a membrane receptor-binding AC activator was applied. We used the neuropeptide pituitary AC-activating polypeptide (PACAP), which has been shown to increase renin expression in vivo and in vitro (19). PACAP increased renin mRNA levels and the addition of rosiglitazone multiplied this effect dose dependently (Fig. 1, B and C). Importantly, the potentiating effect was observed at nanomolar concentrations of PACAP and rosiglitazone, thus demonstrating that it is highly specific. Time-course experiments revealed that the potentiation becomes significant after 12 h and peaks after 16 h of incubation with PACAP and rosiglitazone (Fig. 1D). Next, we used the specific PPARγ antagonist GW9662 to test whether the TZDs rosiglitazone and pioglitazone act through PPARγ to multiply the PACAP-induced stimulation of renin gene expression (Fig. 1E). As expected, GW9662 abolished the effect of rosiglitazone and pioglitazone. In addition, GW9662 did not change the effect of PACAP and completely abrogated the potentiating effect of rosiglitazone and pioglitazone on the PACAP-dependent increase of renin mRNA levels. Altogether, the data presented in Fig. 1 demonstrate that the PPARγ activation multiplies the stimulatory action of AC agonists on renin gene expression in Calu-6 cells. For the following experiments, we used PACAP as AC activator and rosiglitazone as PPARγ ligand.
Fig. 1.

PPARγ agonists potentiate the cAMP-induced stimulation of renin gene expression. A, Effect of the PPARγ agonists oleic acid (250 μm) and rosiglitazone (500 nm) on the stimulation of renin gene expression by the direct AC activator forskolin (5 μm). B, Effect of the PPARγ agonist rosiglitazone (200 or 500 nm) on the stimulation of renin gene expression by the membrane receptor-binding AC activator PACAP (300 nm). C, Representative bands of amplified renin and β-actin cDNA fragments. Total RNA was isolated form Calu-6 cells incubated for 16 h as indicated (rosiglitazone was applied at 500 nm, PACAP at 300 nm). After reverse transcription, cDNA was amplified with the corresponding primers in real-time PCR. PCR was terminated in the linear phase of amplification, and the samples were loaded onto 2% agarose gel. St, Length standard. D, Time course of the effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on renin gene expression. E, The PPARγ antagonist GW9662 (1 μm) abrogates the potentiation of PACAP (300 nm)-induced renin gene expression by rosiglitazone (200 nm) or pioglitazone (1 μm). NS, Not significant. Renin and β-actin (internal control) mRNA levels were quantified by real-time PCR. Unless otherwise specified, Calu-6 cells were incubated with the indicated substances for 16 h. Control cells remained untreated throughout all experiments. Rosi, Rosiglitazone; Pio, pioglitazone. The data shown are means ± sem; *, P < 0.05.
Additive effects of rosiglitazone and PACAP on renin promoter activity
cAMP up-regulates renin gene expression by both stabilizing renin mRNA and inducing renin gene transcription (14, 15, 16, 17, 18). Therefore, we tested whether rosiglitazone potentiates the effect of PACAP on the transcriptional activity of renin gene promoter. To this end, we used a hRen promoter construct consisting of renin cAMP response element (CRE) (17) fused to the minimal hRen promoter (hRenMin) (CRE-hRenMin). The minimal promoter is featured by Pal3 PPARγ target motif in position −148 to −134 (3). PACAP or rosiglitazone alone trans-activated CRE-hRenMin up to about 2.5-fold in time-course experiments (Fig. 2A). The combined application of the two agonists resulted in almost 5-fold increase of transcriptional activity. Rosiglitazone did not significantly trans-activate a CRE-driven heterologous promoter construct (pCRE-Luc), whereas the combination of rosiglitazone and PACAP produced a comparable effect with PACAP alone (Fig. 2B). Conversely, PACAP did not induce the hRenMin, which lacks a functional CRE sequence, whereas rosiglitazone plus PACAP produced a similar effect as rosiglitazone alone (Fig. 2C). These data suggested that the combined effect of PACAP and rosiglitazone on renin gene trans-activation in Calu-6 cells was rather additive than potentiating.
Fig. 2.
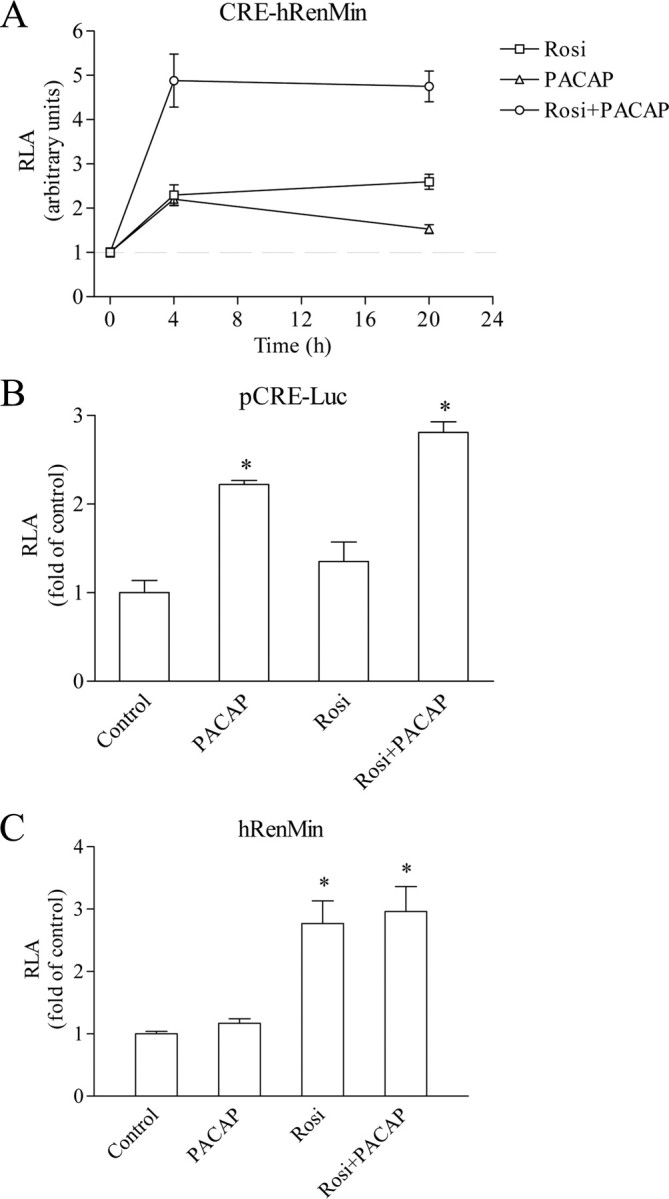
Summation of the trans-activating effects of PPARγ and cAMP on renin promoter. A, Time course of the effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on the activity of CRE-hRenMin promoter construct. B, Effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on the activity of a CRE-driven reporter construct (pCRE-Luc). C, Effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on the activity of hRenMin promoter construct that lacks functional CRE. Calu-6 cells were incubated for 16 h with the indicated substances. Control cells remained untreated. RLA, Relative luciferase activity; Rosi, rosiglitazone. The data shown are means ± sem; *, P < 0.05.
Rosiglitazone potentiates the PACAP-induced cAMP increase
In the next experiments, we examined the effect of rosiglitazone on the cAMP accumulation induced by PACAP in Calu-6 cells. Rosiglitazone increased the cAMP levels about 2-fold, PACAP 3-fold, and the combination of both 30-fold after 16 h of incubation. The cAMP levels were further elevated in the presence of the phosphodiesterase (PDE) inhibitor 3-isobutyl-1-methylxanthine (IBMX) (Fig. 3A). Thus, IBMX alone increased the cAMP levels 2.5-fold, rosiglitazone plus IBMX 8-fold, PACAP plus IBMX 23-fold, and the combination of rosiglitazone, PACAP, and IBMX more than 85-fold. Similarly to renin mRNA levels, the cAMP levels were not elevated by rosiglitazone in the presence of the PPARγ antagonist GW9662 (Fig. 3B). These results demonstrated that the cAMP accumulation correlates with the induction of renin gene expression upon treatment with rosiglitazone and PACAP. Furthermore, it appeared that the elevation of cAMP levels is caused by increased AC activity and not by decreased PDE activity, because it was present during PDE blockade with IBMX. The last data raised the intriguing possibility that rosiglitazone may act as an AC activator. However, short-term application (5 min) of rosiglitazone in the presence of IBMX neither influenced cAMP abundance nor induced further the effect of PACAP on the cAMP levels (Fig. 3C). Because under these experimental conditions the cAMP levels are direct marker of AC activity, we concluded that rosiglitazone is not a bona fide AC agonist. Notably, after 16 h of incubation, rosiglitazone also did not change basal cAMP levels or the PACAP-induced cAMP increase when the Calu-6 cells were concomitantly treated with the transcriptional inhibitor actinomycin D (Fig. 3D). The last two experiments suggested that the mechanism through which rosiglitazone potentiates the cAMP production is transcriptional.
Fig. 3.

Rosiglitazone potentiates the PACAP-induced cAMP increase. A and B, Effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on the cAMP accumulation. Calu-6 cells were incubated for 16 h with the above mentioned substances and without (A) or with the PPARγ antagonist GW9662 (1 μm) (B). Cells were incubated for additional 15 min with the PDE inhibitor IBMX (100 μm) where indicated. C, Rosiglitazone is not an AC activator. Effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on AC activity. Calu-6 cells were incubated for 5 min with the above mentioned substances after a 15-min preincubation with 100 μm IBMX. Under these conditions, the cAMP accumulation is directly proportional to AC activity. D, The potentiation of the PACAP-induced cAMP increase by rosiglitazone requires intact transcription. Effect of rosiglitazone (500 nm), PACAP (300 nm), or the combination of both on the cAMP levels in the presence of the transcriptional inhibitor actinomycin D (10 μm). Calu-6 cells were incubated for 16 h with the indicated substances. Rosi, Rosiglitazone. The data shown are means ± sem; *, P < 0.05 relative to control; #, P < 0.05 relative to the corresponding cAMP level in the absence of IBMX; **, P < 0.05 as indicated. NS, Not significant.
Rosiglitazone stimulates AC6 gene expression
Based on the results so far, we studied the effect of rosiglitazone on the mRNA abundance of ACs. Six AC isoforms were expressed in Calu-6 cells (AC1, AC3, and AC6 to AC9). Among these, only AC6 gene expression was significantly up-regulated by rosiglitazone (Fig. 4A). Rosiglitazone increased AC6 mRNA abundance dose dependently at nanomolar concentrations (Fig. 4B). The stimulation of AC6 gene expression became statistically significant after 12 h and reached its maximum after 16 h of incubation with rosiglitazone (Fig. 4C). This time course matched the time course of potentiation of PACAP-induced renin expression by rosiglitazone (Fig. 1D).
Fig. 4.
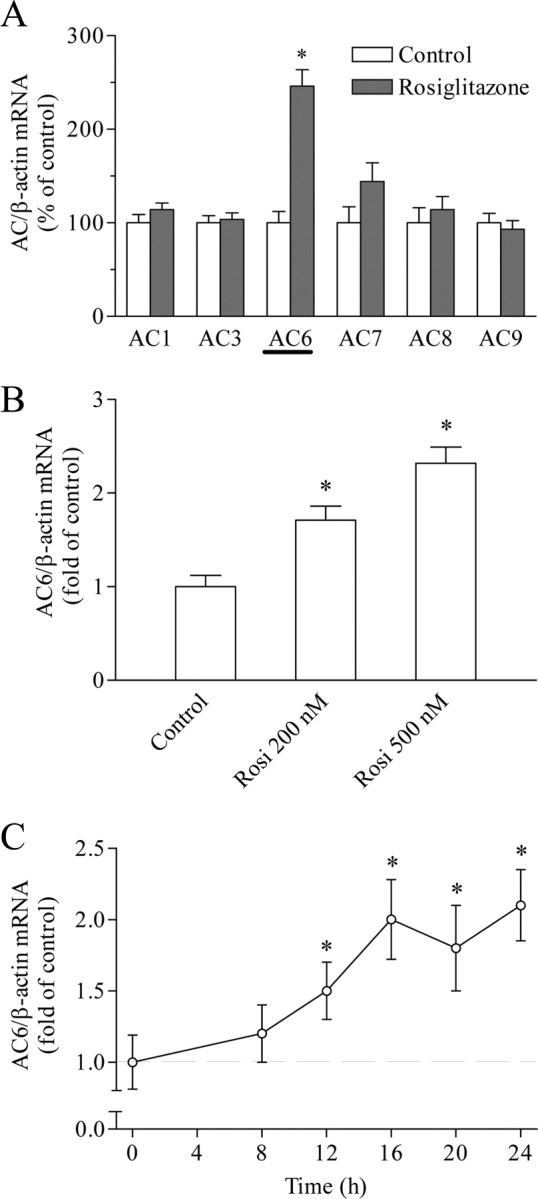
Expression of ACs in Calu-6 cells. A, Rosiglitazone was applied for 16 h at 500 nm. Control cells remained untreated. B, Regulation of AC6 gene expression by rosiglitazone (200 or 500 nm for 16 h). Control cells remained untreated. C, Time course of the effect of rosiglitazone (500 nm), on AC6 gene expression. ACs and β-actin (internal control) mRNA levels were quantified by real-time PCR. Rosi, Rosiglitazone. The data shown are means ± sem; *, P < 0.05.
AC6 is a PPARγ target gene: Identification of functional Pal3 site in AC6 intron 1
Because the multiplication effect on the cAMP level was dependent on intact transcription (Fig. 3D), we looked for possible PPARγ-binding sites (PPRE or Pal3) in the 5′-regulatory sequences (promoters) of human and mouse AC6 genes (20). Because there was no conserved PPARγ target sequence in the AC6 promoters, we next examined intron 1. We identified a completely conserved Pal3 element 5′-TGGTGAcctTTTCCA-3′ (AC6Pal3) located at +2149 to +2163 relative to the start of human AC6 intron 1 and at +225 to +239 relative to the start of mouse AC6 intron 1. To study the trans-activating properties of AC6Pal3, we cloned it in the hRenMin (Table 1). Construct hRenMin contains a functional Pal3 site (Fig. 5A). When this Pal3 site was inactivated by mutation, rosiglitazone did not trans-activate the reporter (construct Pal3mut-hRenMin) (Fig. 5B). We replaced hRen-Pal3 with AC6Pal3 (called below 5′-flanking) and cloned a second AC6Pal3 copy 3′-adjacent to the first base of the hRen promoter (construct AC6Pal3-hRenMin-AC6Pal3). In such a way, the latter copy of AC6Pal3 is located at the 3′-flank of the TATA-box of the reporter construct, thus resembling its natural position in intron 1 of AC6 gene. Rosiglitazone stimulated the activity of AC6Pal3-hRenMin-AC6Pal3 although to a lesser extent than the activity of hRenMin (Fig. 5C). When the 5′-flanking Pal3 was silenced by loss-of-function mutation (construct Pal3mut-hRenMin-AC6Pal3) (Fig. 5D), the 3′-flanking AC6Pal3 was sufficient to mediate rosiglitazone-induced trans-activation similarly the construct with two copies of AC6Pal3 (AC6Pal3-hRenMin-AC6Pal3). The PPARγ antagonist GW9662 abolished the induction of Pal3mut-hRenMin-AC6Pal3 by rosiglitazone (Fig. 5E). Mutation of AC6Pal3 in Pal3mut-hRenMin-AC6Pal3 to form construct Pal3mut-hRenMin-AC6Pal3mut also abrogated the stimulation by rosiglitazone (Fig. 5F).
Table 1.
Reporter gene plasmids
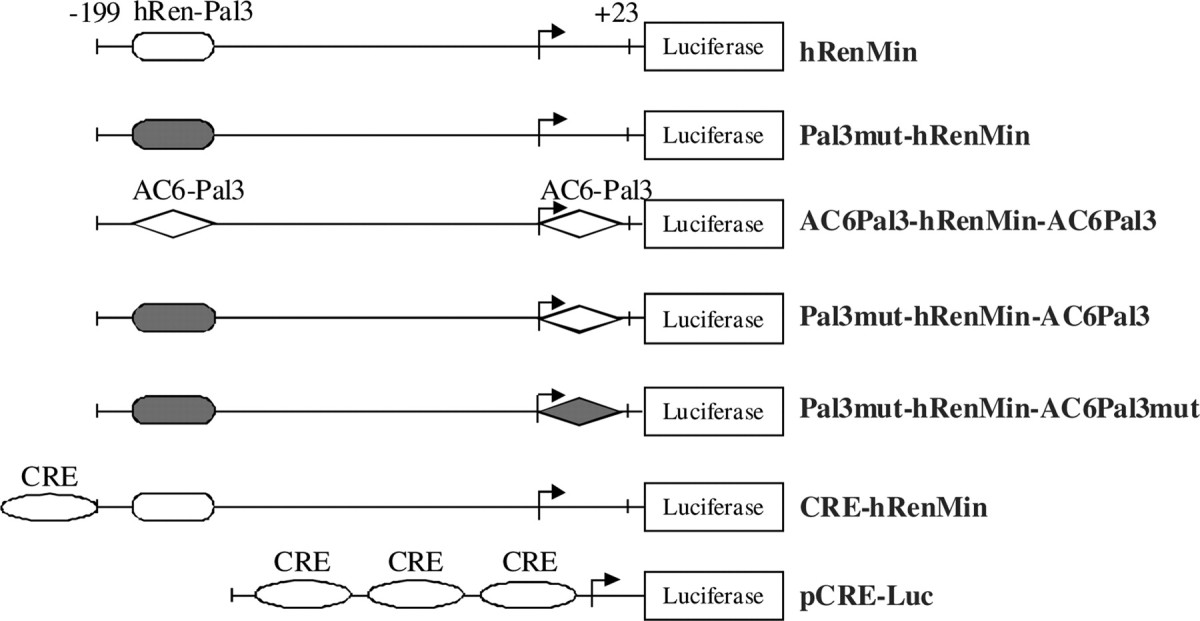
The cis-regulatory elements are illustrated with different geometric figures. A filled figure indicates a mutation.
Fig. 5.

Identification of functional Pal3 site in AC6 intron 1 (AC6Pal3). Transcriptional activity of hRenMin (A), Pal3mut-hRenMin (B), AC6Pal3-hRenMin-AC6Pal3 (C), Pal3mut-hRenMin-AC6Pal3 (D and E), and Pal3mut-hRenMin-AC6Pal3mut (F). Construct hRenMin represents the hRenMin that contains endogenous renin Pal3 PPARγ-binding site at −148 to −134. Construct Pal3mut-hRenMin does not contain functional PPARγ-binding site (because of silent mutation of Pal3). Construct AC6Pal3-hRenMin-AC6Pal3 contains AC6Pal3 site in place of the endogenous hRen-Pal3 and a second AC6Pal3 fused to the 3′-end of the hRenMin. Construct Pal3mut-hRenMin-AC6Pal3 contains mutated endogenous Pal3 and wild-type AC6Pal3 fused to the 3′-end of the hRenMin. Construct Pal3mut-hRenMin-AC6Pal3mut contains mutated endogenous Pal3 and mutated AC6Pal3 fused to the 3′-end of the hRenMin (see also Table 1). Calu-6 cells were treated with rosiglitazone (500 nm), GW9662 (1 μm), or the combination of both for 16 h as indicated. Control cells remained untreated. RLA, Relative luciferase activity; Rosi, rosiglitazone. The data shown are means ± sem; *, P < 0.05.
Chromatin immunoprecipitation (ChIP) experiments demonstrated binding of PPARγ to an approximately 200-bp native DNA region containing AC6Pal3 (Fig. 6). Two pairs of primers amplifying this region detected DNA precipitated with a PPARγ antibody but not with a nonspecific control antibody (Fig. 6, upper panel). Scarce signal was obtained from antibody-precipitates with a primer pair amplifying a nonrelated DNA fragment in hRen exon 10 (Fig. 6, upper panel), whereas clear signals with all three primer pairs were obtained when nonprecipitated input DNA was amplified (Fig. 6, lower panel). The summarized data from Figs. 5 and 6 showed that there is a conserved functional Pal3 cis-acting element in intron 1 of AC6 gene that binds PPARγ.
Fig. 6.

PPARγ binds to AC6 intron 1 in ChIP experiments with nuclear extract from Calu-6 cells. Two different primer pairs were used to amplify the Pal3 containing region of AC6 intron 1 (lanes 1 and 2). Primers amplifying a fragment from hRen exon 10 (Ren) were used as negative control (lane 3). Cross-linked Calu-6 nuclear extracts were precipitated with anti-PPARγ antibody (Santa Cruz Biotechnology, Inc.) or preimmune antigoat antibody where indicated (upper panel). After reversal of cross-linking, DNA was isolated and PCR amplified. The input samples (diluted 1:10) were not antibody-precipitated and served as a positive control (lower panel). St, Length standard (lane 4).
AC6 deficiency attenuates the maximal stimulation of renin gene expression induced by the combination of PACAP and rosiglitazone
Next, we studied the effect of AC6 deficiency on the rosiglitazone-induced potentiation of the PACAP-stimulated renin gene expression. AC6 was effectively knocked-down by RNA interference in Calu-6 cells (Fig. 7, A and B). The expression of AC1, AC3, and AC7, as well the inducibility of the classical PPARγ-regulated gene fatty acid binding protein 4 (FABP-4), remained unaffected thus confirming the specificity of the AC6 small interfering RNA (siRNA) (Fig. 7, C and D). Interestingly, the residual AC6 expression was still up-regulated by rosiglitazone during the knockdown (Fig. 7B). The stimulation of renin gene expression by PACAP, rosiglitazone, or the combination of both was generally milder in cells transfected with control nontargeting siRNA as compared with nontransfected cells (Fig. 7E, compare with Fig. 1B). These findings imply that the siRNA transfection exerts some nonspecific effects in Calu-6 cells. The AC6 deficiency decreased the basal expression of renin gene and attenuated significantly the increase of renin mRNA abundance by PACAP as well as the potentiating effect of rosiglitazone (Fig. 7E). Thus, AC6 is necessary for the basal expression of renin and for the maximal potentiation of the cAMP-induced renin gene expression by PPARγ agonists.
Fig. 7.

Knockdown of AC6 through RNA interference attenuates the maximal stimulation of renin gene expression induced by the combination of PACAP and rosiglitazone. Calu-6 cells were transfected with nontargeting siRNA as control (siControl) or with siAC6. A, Efficacy of the knockdown: AC6 and β-actin (internal control) immunoblot. B, Efficacy of the knockdown: AC6 mRNA expression. AC6 and β-actin (internal control) mRNA levels were quantified by real-time PCR. C, Specificity of the knockdown: AC1, AC3, and AC7 mRNA expression; ACs and β-actin (internal control) mRNA levels were quantified by real-time PCR. D, Specificity of the knockdown: preserved inducibility the classical PPARγ-regulated gene FABP-4. FABP-4 and β-actin (internal control) mRNA levels were quantified by real-time PCR. E, Effect of AC6 deficiency on renin mRNA levels. Renin and β-actin (internal control) mRNA levels were quantified by real-time PCR. Rosiglitazone was applied at 500 nm and PACAP at 300 nm. Calu-6 cells were incubated for 16 h as indicated. Rosi, Rosiglitazone. The data shown are means ± sem; *, P < 0.05 relative to siControl-Control; **, P < 0.05 relative to siAC6-Control; #, P < 0.05 relative to siControl-PACAP; §, P < 0.05 relative to siControl-PACAP + Rosi.
AC6Pal3 decoy oligonucleotide abrogates the potentiation of the cAMP-induced renin gene expression by PPARγ agonist
The PPARγ deficiency diminished the maximal stimulation of renin gene expression by the combination of PACAP and rosiglitazone. However, the potentiating effect was still observed because of the persisting stimulation of the residual AC6 mRNA expression by the PPARγ agonist. Therefore, we used a decoy oligonucleotide to target specifically the AC6Pal3-driven gene expression in Calu-6 cells. A control oligunucleotide containing scrambled AC6Pal3 sequence did not influence the increase of renin mRNA levels by PACAP and the further up-regulation by rosiglitazone (Fig. 8A). AC6Pal3 decoy DNA similarly did not affect the stimulation of renin gene expression by PACAP but abolished the amplifying effect of the PPARγ agonist rosiglitazone (Fig. 8A). These results were supported by the finding that AC6 mRNA levels were not elevated by the combination of PACAP and rosiglitazone in the presence of AC6Pal3 decoy oligonucleotide (Fig. 8B). The increase of FABP-4 mRNA expression was attenuated (but not abolished as seen for AC6) in the presence of AC6Pal3 decoy (Fig. 8C). Therefore, the decoy oligonucleotide appears to act predominantly on AC6 gene. We concluded that AC6Pal3 sequence is necessary for the potentiation of cAMP-induced renin gene expression by PPARγ. The last data provided also indirect evidence that AC6Pal3 is functional in its native genomic context.
Fig. 8.
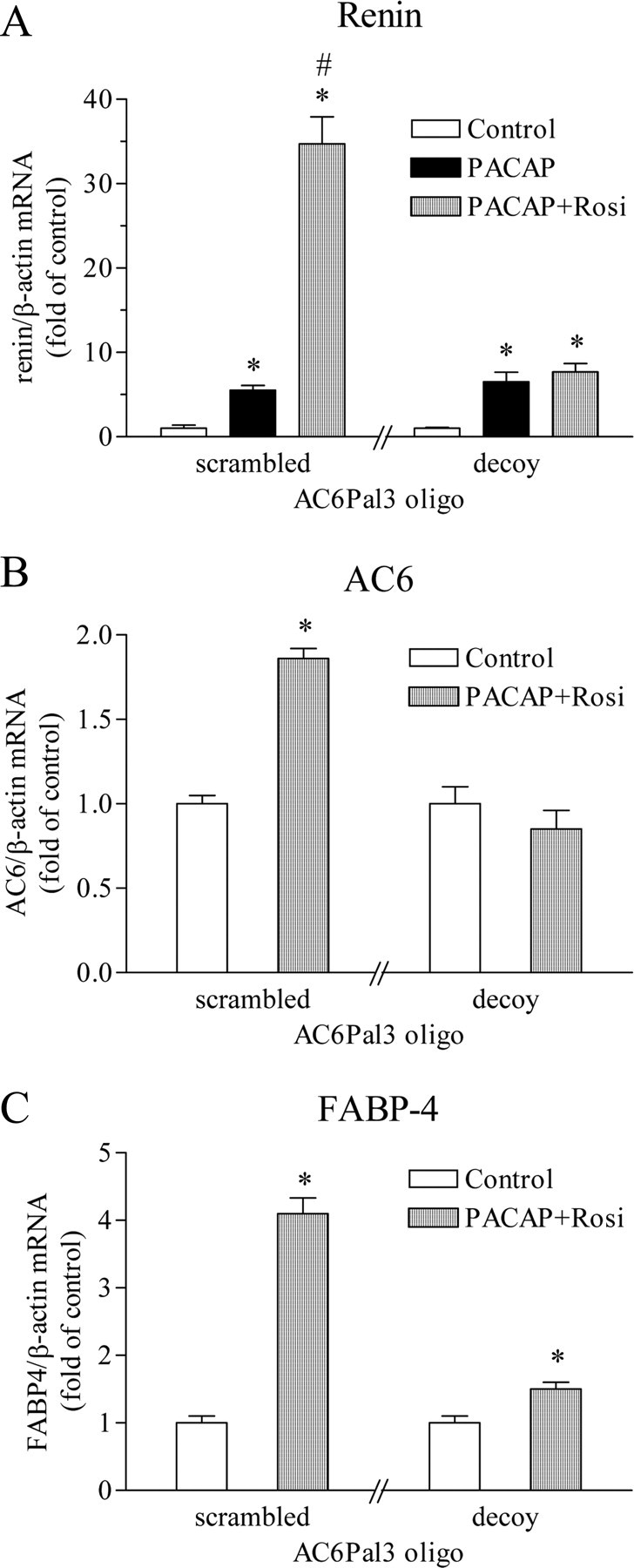
Targeting of AC6Pal3 by decoy oligonucleotide abrogates the potentiation of PACAP-induced renin mRNA expression by rosiglitazone. Calu-6 cells were incubated for 16 h with 25 μm double-stranded decoy oligonucleotide containing AC6Pal3 or double-stranded scrambled AC6Pal3 oligonucleotide, which served as control. Renin (A), AC6 (B), or FABP-4 (C), together with β-actin (internal control) mRNA levels, were quantified by real-time PCR. Rosiglitazone was applied at 500 nm and PACAP at 300 nm. Rosi, Rosiglitazone. The data shown are means ± sem; *, P < 0.05 relative to the corresponding control; #, P < 0.05 relative to scrambled-PACAP.
PPARγ targets AC6 gene in vivo
To evaluate the physiological relevance of the in vitro identification of AC6 as a PPARγ-regulated gene, we examined the effect of the PPARγ agonist pioglitazone on AC6 mRNA levels in mouse kidney. Treatment of mice with pioglitazone increased the renal mRNA expression of AC6 about 2-fold (Fig. 9A). There was no change in AC5, AC7, or Gsα mRNA levels, suggesting that the effect of pioglitazone is specific for AC6 (Fig. 9A).
Fig. 9.

Mice were fed with control (n = 6) or pioglitazone-containing diet (0.2 g/kg, n = 9). A, AC 5, 6, 7, Gsα, and β-actin (internal control) mRNA levels in kidneys were quantified by real-time PCR. The data shown are means ± sem; *, P < 0.05. B, PPARγ binds to AC6 intron 1 in ChIP experiments with mouse kidney nuclear extracts. Primers amplifying the Pal3 containing region of mouse AC6 intron 1 (AC6) or a fragment from mouse PPARγ exon 2 (exon) as negative control were used. Cross-linked mouse kidney nuclear extracts were precipitated with anti-PPARγ antibody (Santa Cruz Biotechnology, Inc.) (right panel). After reversal of cross-linking, DNA was isolated and PCR amplified. The input samples (diluted 1:10) were not antibody precipitated and served as a positive control (left panel). St, Length standard; AB, antibody.
Using mouse kidney nuclear extracts, we also found interaction of PPARγ with AC6Pal3 in ChIP (Fig. 9B). A primer pair amplifying a 200-bp native DNA region containing AC6Pal3 detected DNA precipitated with a PPARγ antibody (Fig. 9B, right panel). There was no signal from PPARγ-antibody-precipitates with a primer pair amplifying a nonrelated DNA fragment in mouse PPARγ exon 2 (Fig. 9B, right panel). Clear signals with both primer pairs were obtained when nonprecipitated input DNA was amplified (Fig. 9B, left panel).
Discussion
PPARγ has been previously found to target regulatory sequences in renin promoter to up-regulate the transcription of renin gene (3, 4). Now, we describe a second indirect mechanism through which PPARγ stimulates renin gene expression in Calu-6 cells. It involves a multiplication of the cAMP increase upon simultaneous AC agonist treatment by inducing AC6 gene. Because cAMP stimulates renin gene expression, the potentiation of cAMP increase by PPARγ agonists results in dramatic potentiation of renin gene expression. PPARγ agonists increased AC6 mRNA level up to 2.5-fold, whereas renin mRNA level raised more than 15-fold over the stimulation level induced by the receptor-binding AC agonist PACAP. Therefore, we could not formally rule out that further genes in the cAMP cascade upstream of ACs are targeted by PPARγ. cAMP regulates not only renin transcription, but also the stability of renin mRNA (14, 15, 16, 17, 18). Previous reports demonstrated that the effect cAMP on mRNA stability is important for the strong increase of renin mRNA levels by cAMP (16). Accordingly, our present data showed that the transcriptional effects of cAMP and PPARγ are additive, thus inferring that the potentiating effect of PPARγ on the cAMP-induced renin gene expression is rather due to enhanced stabilization of renin mRNA by the stronger increase of cAMP.
The signaling mechanism that we describe here could be physiologically important in vivo. Circulating catecholamines, sympathetic nerves, and prostaglandins (PGs) stimulate renin production by acting on G protein-coupled receptors, which upon ligand binding activate ACs and increase intracellular cAMP (21, 22). We used PACAP as a membrane receptor-binding cAMP agonist because it is relevant for the cAMP-dependent control of renin gene in vivo and because it stimulated renin gene expression in Calu-6 cells (19). In our hands, a β-adrenoreceptor agonist (isoproterenol) did not reproducibly stimulate renin gene expression in Calu-6 cells. Isoproterenol at different concentrations did also not significantly increase renin mRNA levels in primary cultures of JG cells, which are known to express β-adrenoreceptors (data not shown). One plausible explanation for these negative findings is that the β-adrenoreceptors are inactivated/internalized upon prolonged treatment with the agonist. Therefore, we could currently only speculate that the molecular mechanism we report of here is relevant for the physiological regulation of renin gene in vivo. It is unlikely that the levels of free fatty acids, which are endogenous PPARγ ligands, are subjected to fluctuations related to the physiological control of renin synthesis. On the other hand, a PGD2 derivative, 15-deoxy-Δ12,14-PG J2, is also an endogenous PPARγ ligand (23, 24). 15-Deoxy-Δ12,14-PG J2 production increases upon up-regulation of cyclooxygenase-2 (25), and cyclooxygenase-2 is well known to be involved in the physiological control of renin synthesis and release in vivo (26). PGD synthase is expressed along the nephron in the vicinity of the renin-producing JG cells (27), and PGD2-derived prostanoids are detectable in human urine (28). Because the PGD2-derived prostanoids are only several seconds stable in plasma, it is conceivable to imply that if excreted with the urine, they would be of renal origin. Thus, there is a potential source of physiologically regulated PPARγ activator within the kidney. Furthermore PPARγ mRNA was detected in native JG cells and PPARγ agonists stimulated renin gene expression in primary cultures of JG cells (3, 5). Whether AC6 is expressed in JG cells is a matter of controversy. Primary cultures of native mouse JG cells were enriched of AC6 mRNA (29), whereas AC6 protein was not detected by fluorescence-stained antibody in single isolated JG cells (30). However, we could not detect specific signal in the kidney, including collecting ducts where AC6 is known to be expressed, with the same AC6 antibody as well as with one further AC6 antibody (data not shown). We also did not observe consistent potentiation of renin gene expression in primary JG cell cultures, although forskolin or rosiglitazone alone increased renin mRNA level (M. Desch and V. T. Todorov, unpublished data) (3). Therefore, it remains to be verified whether the potentiation of cAMP-induced renin gene expression by PPARγ through up-regulation of AC6 transcription functions in native JG cells and is relevant in vivo.
In this study, we provide data that AC6 is a PPARγ-regulated gene. This mode of regulation of AC6 gene expression could be relevant in vivo at least in kidney. Although we do not currently provide clear evidence that AC6 is involved in the control of renin gene, it is feasible to hypothesize that the PPARγ-dependent stimulation of AC6 expression is important for other kidney functions. For instance, both AC6 and PPARγ are expressed in collecting duct cells where they act to promote water and sodium reabsorption. AC6 induces aquaporin-2 expression and membrane translocation (31). Importantly, pharmacological PPARγ agonists increase aquaporin-2 protein expression in collecting ducts (32). In accordance, fluid retention is a common side effect of the pharmacological PPARγ agonists (TZDs) (33). Thus, the PPARγ-induced increase of AC6 expression could be one of the underlying mechanisms.
We found that PPARγ targets a Pal3 element located in the first intron of AC6 gene (AC6Pal3), which is completely conserved between mouse and man. Binding of PPARγ to AC6Pal3 in its native genomic context has been demonstrated in the human Calu-6 cells and in mouse kidney, thus providing indirect evidence for the relevance of the PPARγ effect in vivo. AC6Pal3 delivered PPARγ inducibility to a heterologous promoter in transient transfection experiments. In addition, targeting of AC6Pal3 by decoy oligonucleotide also suggested that this cis-acting element is functional in its native position. Decoy oligonucleotides have been showed to efficiently silent gene expression (34).
AC6 is the second gene next to hRen (3), identified to be regulated by PPARγ through a Pal3 site and not through canonical PPRE. Localization of PPARγ-binding sites in introns was reported in 30–50% of the identified PPARγ target genes (35, 36). The same genome-wide studies showed that about 75% of the PPARγ target sequences are PPRE-like sites. Therefore, it could be predicted that further genes with functional Pal3 sites would be identified. PPARγ has been found to bind to a Pal3 sequence during screening for PPARγ-binding motifs with in vitro translated proteins in a cell-free system (37). In contrast to PPRE, which is a direct repeat with a 1-bp spacer, Pal3 is a palindrome of the consensus hormone response element 5′-A/GGGTCA-3′ with a 3-bp spacer. Although PPRE binds PPARγ as heterodimer with retinoid X receptor, Pal3 binds PPARγ also as homodimer (3, 4, 37). Our earlier studies demonstrated that hRen-Pal3 but not the originally described consensus Pal3 binds PPARγ as homodimer in nuclear extracts of Calu-6 cells (4). mRen-Pal3 is silent despite its homology to hRen-Pal3 (4). AC6Pal3 shares the same homology (60%) to consensus Pal3 and to hRen-Pal3. In contrast to hRen-Pal3 and similarly to consensus Pal3 (4), it binds a single PPARγ-containing protein complex in nuclear extracts of Calu-6 cells (data not shown). Single Pal3 binding activity of Calu-6 cells nuclear proteins was earlier demonstrated to be composed of PPARγ/retinoid X receptor α heterodimers (4). Sequence comparison among the known Pal3 elements suggests that the last two bases of the 5′-repeat are important for the protein-binding characteristics and hence for the mode of function of Pal3 (Table 2). These bases are variable, and their mutation in hRen-Pal3 abrogates the trans-activation by PPARγ agonist (4). On the other hand, although the first half of the 3′-repeat is also highly variable, its mutation in hRen-Pal3 did not influence trans-activation (4).
Table 2.
Comparison of consensus, AC6, hRen, and mRen-Pal3 sequences
| Pal3 sequences | |
| Consensus | GGGTCAcngTGACCC |
| AC6 | TGGTGAcctTTTCCA |
| hRen | GGGTACcctTCACCC |
| mRen | GCTTATcctATACCT |
The data presented in this paper raise the possibility that PPARγ could generally potentiate cAMP signaling by inducing AC6 transcription. This could be of particular relevance not only in kidney but also in other organs, such as heart or brain, where AC6 is highly expressed and has important functions.
Materials and Methods
Cell culture
Calu-6 cells (ATCC-HTB-56, hRen-producing cell line) were cultured in Eagle’s minimal essential medium supplemented with 10% fetal bovine serum, sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% nonessential amino acids at 37 C in a humidified atmosphere containing 5% CO2.
RNA isolation, reverse transcription, and quantitative LightCycler PCR
Total RNA was isolated from Calu-6 cells with the QIAGEN RNeasy Spin Columns. A standard protocol for reverse transcription was used.
Isolation of total RNA from mouse kidney tissue was performed after automated disruption and homogenization using the MagNa Lyser Instrument (Roche Diagnostics, Mannheim, Germany). Cleared lysate was used for further RNA purification (RNAeasy Mini kit; QIAGEN, Hilden, Germany). Subsequently, 1 μg of total RNA was reverse transcribed to cDNA using a reverse transcription system (Bioscience, Rockaway, NJ) with oligo(dT) primers according to the manufacturer’s protocol.
Real-time PCR (Roche, Indianapolis, IN) was performed as already described (3). The primers used are shown in Table 3.
Table 3.
Primers used in real-time PCR
| Gene | Sense primer | Antisense primer | Amplicon (bp) |
|---|---|---|---|
| hRen | atgaagggggtgtctgtggggtc | ggatatccatggcgtggatggcc | 304 |
| Human β-actin | cttctacaatgagctgcgtgtgg | gaggatcttcatgaggtagtcag | 313 |
| Human AC1 | ggcgagatgttgacatac | ctaagcctccttcccagctg | 223 |
| Human AC3 | gaagagagatgaggagctg | gccattggtgttgacatc | 262 |
| Human AC6 | cacatcagcagcatcgg | cagtccagcccatcaaaggtctc | 164 |
| Human/mouse AC7 | gggcctgtgattgctggagtg | cggtaacctggattttccc | 121 |
| Human AC8 | cagaatactctggctgccc | cttgtgctccaggctggagtc | 204 |
| Human AC9 | ggaacatcatcccctacc | catctccttggcgaactcg | 338 |
| Mouse AC5 | atgaagctgatggac | ctggccacattcactgtattgc | 155 |
| Mouse AC6 | cacatcagcagcatcgg | cagtccagcccatcaaaggtctc | 164 |
| Mouse Gsα | aaccgcctgcaggaggctc | caggagtggtgtagcgagc | 175 |
| Mouse β-actin | ccgccctaggcaccagggtg | ggctggggtgttgaaggtctcaaa | 285 |
The sequences are in 5′ to 3′ orientation.
Plasmids
Plasmid constructs used in this study are shown in Table 1. hRenMin represents modified pGL3 vector (Promega, Madison, WI) encoding firefly luciferase under the control of the hRenMin (bases −199 to +23 relative to the transcription starting site) (38). A 44-bp-long hRen enhancer fragment containing CRE was cloned into the 5′-end of hRenMin to form CRE-hRenMin. The endogenous Pal3 site of hRenMin at −148 to −134 was inactivated by mutation in construct Pal3mut-hRenMin (3). Construct AC6Pal3-hRenMin-AC6Pal3 contains two AC6 intron 1 Pal3 sequences (AC6Pal3, 5′-TGGTGAcctTTTCCA-3′): one replaces the endogenous hRen-Pal3 and the other is inserted adjacent to the 3′-end of the hRen promoter to resemble its natural position in AC6 gene. In construct Pal3mut-hRenMin-AC6Pal3, AC6Pal3 is cloned adjacent to the 3′-end of the hRen promoter within construct Pal3mut-hRenMin. Mutated AC6Pal3 was cloned adjacent to Pal3mut-hRenMin in construct Pal3mut-hRenMin-AC6Pal3mut. Vector pCRE-Luc (Clontech, Mountain View, CA) contains firefly luciferase reporter under the control of multiple CRE sequences.
Transient transfection and luciferase assay
Firefly luciferase reporter vector (0.2 μg) and 0.01 μg of renilla luciferase (pRL-0 vector; Promega) were transfected with FuGENE 6 transfection reagent (Roche) (39). Twenty-four hours after transfection, the medium was replaced with a fresh medium without (control) or with the active substances. Relative luciferase activity was calculated as firefly luciferase normalized to renilla luciferase activity.
cAMP measurement
cAMP levels in confluent Calu-6 cells plated onto 24-well plates were determined with Direct cAMP Immunoassay kit (Assay Designs, Ann Arbor, MI) following the instructions of the manufacturer.
Chromatin immunoprecipitation
DNA:protein complexes isolated from rosiglitazone-treated Calu-6 cells or from homogenized mouse kidneys were analyzed in ChIP assays using the ChIP-IT kit from Active Motif as already described (Active Motif, Carlsbad, CA) (3, 4). In short, DNA:protein cross-linked complexes were precleared with protein G agarose beads (input samples), or were further precipitated with preimmune antigoat antiserum, or anti-PPARγ antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). After reversal of the cross-linking, digestion of RNA and proteins, DNA was eluted and PCR-amplified for 28 cycles. The primers used for the amplification of human AC6 intron 1 fragments containing the Pal3 site were: sense, 5′-cttgcccaaggcagtgtggctag-3′; antisense, 5′-caccatctggaaggacagatcagg-3′ (amplicon length 177 bp); and sense, 5′-gaaggtactgacacagagagg-3′; antisense, 5′-caccatctggaaggacagatcagg-3′ (amplicon length 206 bp). hRen exon 10 fragment (116 bp) was amplified as negative control with primers: sense, 5′-gctgtgcacactggccat-3′; antisense, 5′-ggttgttacgccgatcaaactc-3′. Mouse AC6 intron 1 and PPARγ exon 2 (negative control) fragments were amplified with primers: sense, 5′-tcccctctcgtacttcagg-3′; antisense, 5′-caccatcttgaaggacagatcag-3′ (amplicon size 200 bp); and sense, 5′-gtggatctctccgtaatg-3′; antisense, 5′-ggtactcttgaagtttcaggtc-3′ (amplicon size 166 bp), respectively.
RNA interference
Ready-to-use double-stranded siRNAs were synthesized by Dharmacon to target human AC6 (ON-TARGETplus siRNA SMARTpool L-006636–00) (Dharmacon, Lafayette, CO). Fifty nanomol double-stranded nontargeting siControl siRNA (Dharmacon, used as control) or AC6-specific siRNA (siAC6) were transfected into Calu-6 cells using Dharmafect 2 (Dharmacon) according to the manufacturer’s protocol. The cells were harvested 72 h after transfection.
Western blot analysis
Calu-6 total cellular protein was isolated in lysis buffer [10 mm Tris, 1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40, and 5 mm Pefablock]. Protein concentration was determined (MicroProtein Determination kit; Sigma, St. Louis, MO). The samples were boiled in Laemmli buffer and loaded on 10% SDS-polyacrylamide gels. After electrophoresis, proteins were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA) in transfer buffer (48 mm Tris, 39 mm glycine, 0.037% SDS, and 20% methanol). The membranes were blocked and then incubated with 1:500-diluted rabbit polyclonal anti-AC6 (FabGennix, Inc., Frisco, TX) or mouse anti-β-actin antibody (Santa Cruz Biotechnology, Inc.). The bound antibodies were visualized with horseradish peroxidase-conjugated antirabbit or antimouse IgGs (DiaNova, Hamburg, Germany), respectively, followed by enhanced chemiluminescence detection (Santa Cruz Biotechnology, Inc.) and exposure to Kodak Biomax MS film (Kodak, Rochester, NY).
Animal experiments
All animal experiments were conducted in accord with accepted standards of humane animal care, as outlined in the Ethical Guidelines and the German law for the welfare of animals and were approved by the local authorities; 129 wild-type mice were fed with a control diet (C1000; Altromin, Lage, Germany) and had free access to tap water. After an adaptation period, the food pellets were replaced by custom-made pellets containing 0.2 g/kg pioglitazone with otherwise identical composition (C1000 + Pioglitazone; Altromin) as already described (40). The drug intake was estimated to be 20–30 mg/kg−1 · d−1. Control animals remained on control diet.
Statistics
Experiments were performed in duplicate with at least three samples per condition. Levels of significance were estimated by ANOVA followed by the Student’s unpaired t test. P < 0.05 was considered significant.
Acknowledgments
We thank Dr. Anelia Todorova for the excellent technical assistance.
NURSA Molecule Pages:
Ligands: GW 9662 | Pioglitazone | Rosiglitazone;
Nuclear Receptors: PPAR-γ.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft Grant SFB 699/B1 (to V.T.T.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online September 22, 2010
Abbreviations: AC, Adenylate cyclase; ChIP, chromatin immunoprecipitation; CRE, cAMP response element; FABP-4, fatty acid binding protein 4; hRen, human renin; hRenMin, minimal hRen promoter; IBMX, 3-isobutyl-1-methylxanthine; JG, juxtaglomerular; mRen, mouse renin; PACAP, pituitary AC-activating polypeptide; PDE, phosphodiesterase; PG, prostaglandin; PPARγ, peroxisome proliferator-activated receptor-γ; PPRE, PPAR-response element; SDS, sodium dodecyl sulfate; siAC6, AC6-specific siRNA; siRNA, small interfering RNA; TZD, thiazolidinedione.
References
- 1.Castrop H, Höcherl K, Kurtz A, Schweda F, Todorov V, Wagner C2010. Physiology of kidney renin. Physiol Rev 90:607–673 [DOI] [PubMed] [Google Scholar]
- 2.Pan L, Gross KW2005. Transcriptional regulation of renin: an update. Hypertension 45:3–8 [DOI] [PubMed] [Google Scholar]
- 3.Todorov VT, Desch M, Schmitt-Nilson N, Todorova A, Kurtz A2007. Peroxisome proliferator-activated receptor-γ is involved in the control of renin gene expression. Hypertension 50:939–944 [DOI] [PubMed] [Google Scholar]
- 4.Todorov VT, Desch M, Schubert T, Kurtz A2008. The Pal3 promoter sequence is critical for the regulation of human renin gene transcription by peroxisome proliferator-activated receptor-γ. Endocrinology 149:4647–4657 [DOI] [PubMed] [Google Scholar]
- 5.Desch M, Schreiber A, Schweda F, Madsen K, Friis UG, Weatherford ET, Sigmund CD, Sequeira Lopez ML, Gomez RA, Todorov VT2010. Increased renin production in mice with deletion of peroxisome proliferator-activated receptor-γ in juxtaglomerular cells. Hypertension 55:660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonen A, Tandon NN, Glatz JF, Luiken JJ, Heigenhauser GJ2006. The fatty acid transporter FAT/CD36 is upregulated in subcutaneous and visceral adipose tissues in human obesity and type 2 diabetes. Int J Obes 30:877–883 [DOI] [PubMed] [Google Scholar]
- 7.Hall JE1994. Louis K. Dahl Memorial Lecture. Renal and cardiovascular mechanisms of hypertension in obesity. Hypertension 23:381–394 [DOI] [PubMed] [Google Scholar]
- 8.Hall JE2003. The kidney, hypertension, and obesity. Hypertension 41:625–633 [DOI] [PubMed] [Google Scholar]
- 9.Minehira K, Bettschart V, Vidal H, Vega N, Di Vetta V, Rey V, Schneiter P, Tappy L2003. Effect of carbohydrate overfeeding on whole body and adipose tissue metabolism in humans. Obes Res 11:1096–1103 [DOI] [PubMed] [Google Scholar]
- 10.Shea J, French CR, Bishop J, Martin G, Roebothan B, Pace D, Fitzpatrick D, Sun G2009. Changes in the transcriptome of abdominal subcutaneous adipose tissue in response to short-term overfeeding in lean and obese men. Am J Clin Nutr 89:407–415 [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Eckel RH2009. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab 297:E271–E288 [DOI] [PubMed]
- 12.Westerbacka J, Kolak M, Kiviluoto T, Arkkila P, Sirén J, Hamsten A, Fisher RM, Yki-Järvinen H2007. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 56:2759–2765 [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, Chen M, Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J2007. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsα in juxtaglomerular cells. Am J Physiol Renal Physiol 292:F27–F37 [DOI] [PubMed]
- 14.Chen M, Schnermann J, Smart AM, Brosius FC, Killen PD, Briggs JP1993. Cyclic AMP selectively increases renin mRNA stability in cultured juxtaglomerular granular cells. J Biol Chem 268:24138–24144 [PubMed] [Google Scholar]
- 15.Klar J, Sandner P, Müller MW, Kurtz A2002. Cyclic AMP stimulates renin gene transcription in juxtaglomerular cells. Pflugers Arch 444:335–344 [DOI] [PubMed] [Google Scholar]
- 16.Lang JA, Ying LH, Morris BJ, Sigmund CD1996. Transcriptional and posttranscriptional mechanisms regulate human renin gene expression in Calu-6 cells. Am J Physiol 271:F94–F100 [DOI] [PubMed]
- 17.Pan L, Black TA, Shi Q, Jones CA, Petrovic N, Loudon J, Kane C, Sigmund CD, Gross KW2001. Critical roles of a cyclic AMP responsive element and an E-box in regulation of mouse renin gene expression. J Biol Chem 276:45530–45538 [DOI] [PubMed] [Google Scholar]
- 18.Ying L, Morris BJ, Sigmund CD1997. Transactivation of the human renin promoter by the cyclic AMP/protein kinase A pathway is mediated by both cAMP-responsive element binding protein-1 (CREB)-dependent and CREB-independent mechanisms in Calu-6 cells. J Biol Chem 272:2412–2420 [DOI] [PubMed] [Google Scholar]
- 19.Hautmann M, Friis UG, Desch M, Todorov V, Castrop H, Segerer F, Otto C, Schütz G, Schweda F2007. Pituitary adenylate cyclase-activating polypeptide stimulates renin secretion via activation of PAC1 receptors. J Am Soc Nephrol 18:1150–1156 [DOI] [PubMed] [Google Scholar]
- 20.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942 [DOI] [PubMed] [Google Scholar]
- 21.Bader M, Ganten D2000. Regulation of renin: new evidence from cultured cells and genetically modified mice. J Mol Med 78:130–139 [DOI] [PubMed] [Google Scholar]
- 22.Friis UG, Stubbe J, Uhrenholt TR, Svenningsen P, Nüsing RM, Skøtt O, Jensen BL2005. Prostaglandin E2 EP2 and EP4 receptor activation mediates cAMP-dependent hyperpolarization and exocytosis of renin in juxtaglomerular cells. Am J Physiol Renal Physiol 289:F989–997 [DOI] [PubMed]
- 23.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM1995. 15-Deoxy-δ 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 83:803–812 [DOI] [PubMed] [Google Scholar]
- 24.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM1995. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell 83:813–819 [DOI] [PubMed] [Google Scholar]
- 25.García-Bueno B, Madrigal JL, Pérez-Nievas BG, Leza JC2008. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-γ activation after stress in rats. Endocrinology 149:1969–1978 [DOI] [PubMed] [Google Scholar]
- 26.Harris RC2003. Interactions between COX-2 and the renin-angiotensin system in the kidney. Acta Physiol Scand 177:423–427 [DOI] [PubMed] [Google Scholar]
- 27.Vitzthum H, Abt I, Einhellig S, Kurtz A2002. Gene expression of prostanoid forming enzymes along the rat nephron. Kidney Int 62:1570–1581 [DOI] [PubMed] [Google Scholar]
- 28.Hirata Y, Hayashi H, Ito S, Kikawa Y, Ishibashi M, Sudo M, Miyazaki H, Fukushima M, Narumiya S, Hayaishi O1988. Occurrence of 9-deoxy-δ 9,δ 12–13,14-dihydroprostaglandin D2 in human urine. J Biol Chem 263:16619–16625 [PubMed] [Google Scholar]
- 29.Grünberger C, Obermayer B, Klar J, Kurtz A, Schweda F2006. The calcium paradoxon of renin release: calcium suppresses renin exocytosis by inhibition of calcium-dependent adenylate cyclases AC5 and AC6. Circ Res 99:1197–1206 [DOI] [PubMed] [Google Scholar]
- 30.Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH2007. Adenylyl cyclase isoform v mediates renin release from juxtaglomerular cells. Hypertension 49:618–624 [DOI] [PubMed] [Google Scholar]
- 31.Nielsen S, Frøkiaer J, Marples D, Kwon TH, Agre P, Knepper MA2002. Aquaporins in the kidney: from molecules to medicine. Physiol Rev 82:205–244 [DOI] [PubMed] [Google Scholar]
- 32.Tiwari S, Blasi ER, Heyen JR, McHarg AD, Ecelbarger CM2008. Time course of AQP-2 and ENaC regulation in the kidney in response to PPAR agonists associated with marked edema in rats. Pharmacol Res 57:383–392 [DOI] [PubMed] [Google Scholar]
- 33.Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, Le Winter M, Porte D, Semenkovich CF, Smith S, Young LH, Kahn R2003. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation 108:2941–2948 [DOI] [PubMed] [Google Scholar]
- 34.Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, Robbins PD, Gadiparthi S, Burke NA, Watkins SF, Grandis JR2003. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci USA 100:4138–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert Jr CJ, Liu XS, Lazar MA2008. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev 22:2941–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Børgesen M, Francoijs KJ, Mandrup S, Stunnenberg HG2008. Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev 22:2953–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okuno M, Arimoto E, Ikenobu Y, Nishihara T, Imagawa M2001. Dual DNA-binding specificity of peroxisome-proliferator-activated receptor γ controlled by heterodimer formation with retinoid X receptor α. Biochem J 353:193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mrowka R, Steege A, Kaps C, Herzel H, Thiele BJ, Persson PB, Blüthgen N2007. Dissecting the action of an evolutionary conserved non-coding region on renin promoter activity. Nucleic Acids Res 35:5120–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Todorov V, Müller M, Schweda F, Kurtz A2002. Tumor necrosis factor-α inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol 283:R1046–R1051 [DOI] [PubMed]
- 40.Artunc F, Sandulache D, Nasir O, Boini KM, Friedrich B, Beier N, Dicks E, Pötzsch S, Klingel K, Amann K, Blazer-Yost BL, Scholz W, Risler T, Kuhl D, Lang F2008. Lack of the serum and glucocorticoid-inducible kinase SGK1 attenuates the volume retention after treatment with the PPARγ agonist pioglitazone. Pflugers Arch 456:425–436 [DOI] [PubMed] [Google Scholar]