

Abstract
Impairment of insulin and IGF-I signaling in the brain is one of the causes of dementia associated with diabetes mellitus and Alzheimer’s disease. However, the precise pathological processes are largely unknown. In the present study, we found that SH2-containing inositol 5′-phosphatase 2 (SHIP2), a negative regulator of phosphatidylinositol 3,4,5-trisphosphate-mediated signals, is widely expressed in adult mouse brain. When a dominant-negative mutant of SHIP2 was expressed in cultured neurons, insulin signaling was augmented, indicating physiological significance of endogenous SHIP2 in neurons. Interestingly, SHIP2 mRNA and protein expression levels were significantly increased in the brain of type 2 diabetic db/db mice. To investigate the impact of increased expression of SHIP2 in the brain, we further employed transgenic mice overexpressing SHIP2 and found that increased amounts of SHIP2 induced the disruption of insulin/IGF-I signaling through Akt. Neuroprotective effects of insulin and IGF-I were significantly attenuated in cultured cerebellar granule neurons from SHIP2 transgenic mice. Consistently, terminal deoxynucleotide transferase-mediated dUTP nick end labeling assay demonstrated that the number of apoptosis-positive cells was increased in cerebral cortex of the transgenic mice at an elderly age. Furthermore, SHIP2 transgenic mice exhibited impaired memory performance in the Morris water maze, step-through passive avoidance, and novel-object-recognition tests. Importantly, inhibition of SHIP2 ameliorated the impairment of hippocampal synaptic plasticity and memory formation in db/db mice. These results suggest that SHIP2 is a potent negative regulator of insulin/IGF-I actions in the brain, and excess amounts of SHIP2 may be related, at least in part, to brain dysfunction in insulin resistance with type 2 diabetes.
Lipid phosphatase SHIP2 is increased in brain in diabetic mice and causes the impairment of insulin/IGF1 action responsible for maintaining brain functions.
Insulin and IGF-I contribute to the modulation of various brain functions, including neuronal survival, learning and memory, and cognitive function (1, 2). These actions are mediated by the insulin and IGF-I receptors widely distributed in the central nervous system (1, 2). In mammals, the brain is considered to utilize insulin and IGF-I from both locally produced and peripheral sources (2, 3). Growing evidence indicates that the risk of age-related dementia and Alzheimer’s disease is increased in type 2 diabetes mellitus (4, 5, 6), and the disturbance in insulin/IGF-I signaling may be a biological basis underlying the link between these diseases (7, 8). In fact, the contents of insulin, IGF-I, and their receptors are decreased in the brains of patients with Alzheimer’s disease (9), and intranasal insulin, delivered with direct access to the brain, has beneficial effects on the cognitive performance in healthy subjects (10) and Alzheimer’s disease patients (11). However, the risk factors and mechanisms that link diabetes and cognitive decline to dementia still remain unclear (4), because brain function is slowly and modestly changed in diabetic patients and animal models.
Insulin and IGF-I receptor signalings are mediated by the phosphatidylinositol (PI) 3-kinase and MAPK pathways (12, 13). PI3-kinase activates Akt through the production of PI(3,4,5)P3 from PI(4,5)P2, resulting in the phosphorylation and inhibition of glycogen synthase kinase 3 (GSK3). Dysregulation of GSK3 activity is associated with the neuropathology of Alzheimer’s disease, because GSK3 induces neuronal cell death, hyperphosphorylation of τ protein relevant to neurofibrillary tangle formation, and production of amyloid β-peptide (14, 15, 16). The cell survival properties of IGF-I are mediated by the activation of the PI3-kinase/Akt, but not MAPK pathway, in the hippocampal neurons (17). Thus, the impairment of insulin/IGF-I signaling through Akt and GSK3 appears to be one of the critical factors for the development of neurodegeneration in brain.
SH2-containing inositol 5′-phosphatase 2 (SHIP2), a lipid phosphatase that hydrolyzes PI(3,4,5)P3 to PI(3,4)P2, has been shown to inhibit peripheral insulin signaling (18, 19, 20). Along this line, SHIP2 knockout mice showed an increased sensitivity to insulin in the liver and skeletal muscle and are highly resistant to weight gain on high-fat diet (21). Interestingly, endogenous SHIP2 expression is elevated in the skeletal muscle and adipose tissue of type 2 diabetic db/db mice (22). We previously generated transgenic mice overexpressing SHIP2 and found that the mice exhibited impaired insulin signaling through Akt in peripheral tissues and showed glucose intolerance and reduced insulin sensitivity (23). In adult mouse brain, the expression of SHIP2 mRNA and protein has been detected (23, 24). An in vitro study demonstrated that depletion of SHIP2 enhanced nerve growth factor-induced PI(3,4,5)P3 production, leading to the neurite outgrowth in human neuroblastoma PC12 cells deficient in the inositol 3′-phosphatase, phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (25). However, the functional role of SHIP2 in the central nervous system has not been intensively studied.
In the present study, we found that SHIP2 expression was elevated in the brain of diabetic db/db mice, in addition to the periphery as mentioned above (22). We therefore employed SHIP2 transgenic mice to determine the impact of overexpression of SHIP2 on insulin signaling in the hippocampus, cerebral cortex, and cerebellar granule (CG) neurons. We further investigated whether the increased SHIP2 expression affects insulin/IGF-I-mediated neuroprotection and memory formation. Finally, to reveal the pathophysiological significance of elevated brain levels of SHIP2 in the diabetic state, we examined whether SHIP2 inhibition ameliorates the impaired synaptic plasticity and memory in db/db mice.
Results
Enhancement of insulin/IGF-I signaling by expression of dominant-negative mutant of SHIP2 in cultured neurons
A previous semiquantitative RT-PCR study demonstrated that SHIP2 mRNA is highly expressed in the adult mouse brain (24), although an in situ hybridization study failed to show the significant expression of SHIP2 in the adult rat brain except for cerebellum (26). Therefore, we precisely analyzed the expression levels of SHIP2 mRNA in the hippocampus, cerebral cortex, cerebellum, hypothalamus, and liver of male C57BL/6J mice (14 wk of age), using a quantitative real-time PCR method. The PCR products revealed a single band of the expected size (186 bp) (Fig. 1A). The SHIP2 mRNA expression was highest in the cerebellum among four brain regions tested, which was comparable to that in the liver, and moderate expression of SHIP2 mRNA was detected in the hippocampus, cerebral cortex, and hypothalamus.
Fig. 1.
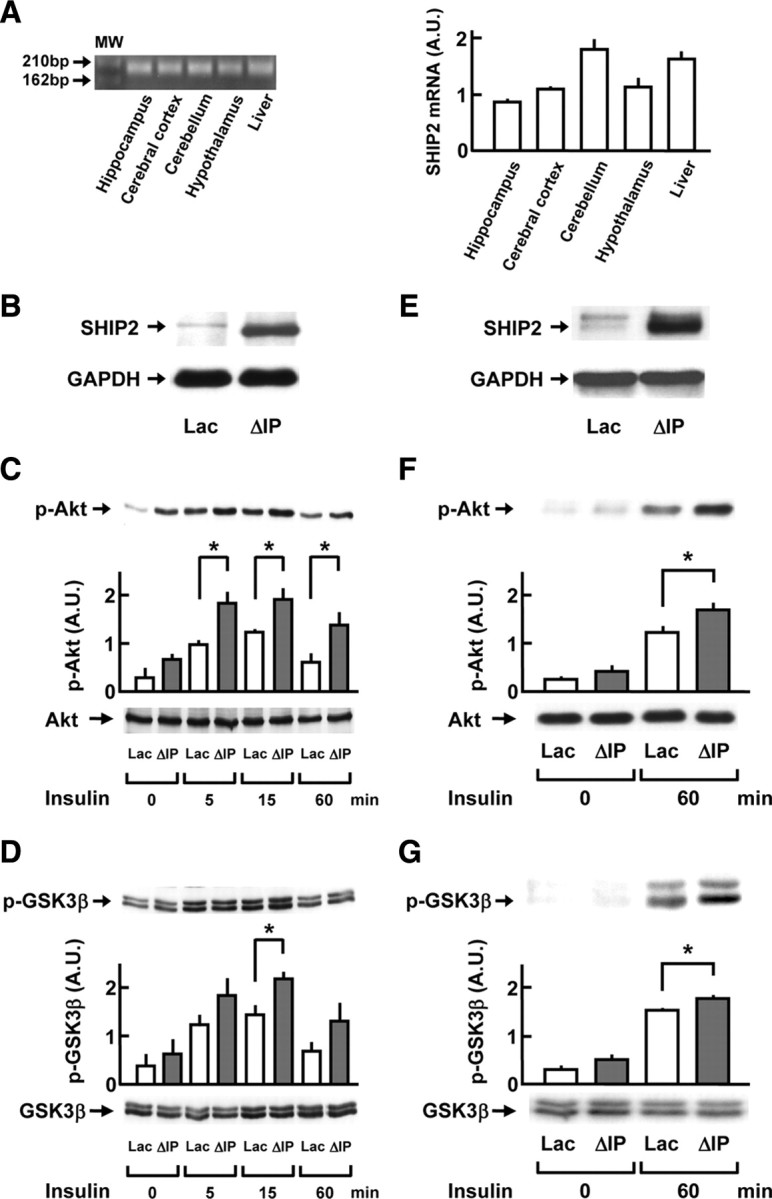
Enhancing effect of dominant-negative SHIP2 mutant on insulin signaling in primary cultured neurons. A, Quantitative real-time RT-PCR was performed to analyze the expression levels of SHIP2 mRNA in the hippocampus, cerebral cortex, cerebellum, hypothalamus, and liver of mice. Left, The RT-PCR product of SHIP2 showing a single band of the expected size of 186 bp on agarose gel electrophoresis. Right, The expression levels of endogenous SHIP2 mRNA in each brain region and liver in C57BL/6J mice (14 wk of age). B–G, ΔIP-SHIP2, a dominant-negative SHIP2 mutant, was expressed in mouse CG neurons and rat cortical neurons by adenovirus-mediated gene transfer. The cells infected with adenovirus containing LacZ gene were used as controls. The serum-starved CG neurons and cortical neurons were stimulated with insulin at 1.7 and 0.5 nm, respectively. The cell lysates were subjected to immunoblot analysis. B, Expression of ΔIP-SHIP2 in CG neurons detected with anti-SHIP2 antibody, which recognizes C-terminal domain of SHIP2. C and D, Increase in the insulin-induced phosphorylations of Akt at Thr308 (C) and GSK3β at Ser9 (D) in the CG neurons expressing ΔIP-SHIP2 (ΔIP), in comparison with those of control cells (Lac). E, Expression of ΔIP-SHIP2 in rat cortical neurons. F and G, Increase in the insulin-induced phosphorylations of Akt at Thr308 (F) and GSK3β at Ser9 (G) in the cortical neurons expressing ΔIP-SHIP2. The amount of total Akt or GSK3β remained unchanged. The phosphorylation level in each sample was analyzed by densitometry, and averaged. Values represent means ± se of four or six separate experiments. *, P < 0.05. p-, Phosphorylated; A.U., arbitrary unit; M.W., molecular weight.
We examined the effect of a dominant-negative SHIP2 mutant (5′-phosphatase-dead mutant of SHIP2, ΔIP-SHIP2) expression on the insulin and IGF-I signaling in CG neurons from C57BL/6J mice and cortical neurons from Sprague Dawley rats to reveal the physiological significance of endogenous SHIP2 expression in the brain neuronal cells. Adenovirus-mediated expression of ΔIP-SHIP2 in CG neurons was confirmed by Western blotting, as shown in Fig. 1B. When the CG neurons expressing ΔIP-SHIP2 were treated with 1.7 nm insulin for 5, 15, and 60 min, the levels of phospho-Thr308-Akt were 1.5- to 2.2-fold increased throughout the observation periods, compared with cells expressing LacZ (Fig. 1C). In addition, the phosphorylation levels of GSK3β at Ser9 were 1.5-fold higher at 15 min in CG neurons expressing ΔIP-SHIP2 than those in cells expressing LacZ (Fig. 1D). Similarly, after a 15-min stimulation with IGF-I (6.5 nm), the levels of phospho-Thr308-Akt in CG neurons expressing ΔIP-SHIP2 were greatly higher than those of LacZ-expressing cells (data not shown). Furthermore, when rat cortical neurons expressing ΔIP-SHIP2 (Fig. 1E) were treated with 0.5 nm insulin for 60 min, the levels of phospho-Thr308-Akt (Fig. 1F) and Ser9-GSK3β (Fig. 1G) were 1.4- and 1.2-fold increased, respectively, compared with cells expressing LacZ. The expression levels of total Akt, GSK3β, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were unaltered under these conditions (Fig. 1).
Increase in SHIP2 mRNA and protein levels in diabetic db/db mice
Previous studies demonstrated that the insulin receptor system is impaired in the brains of diabetic or aged rodents (5, 27). Therefore, we investigated the SHIP2 mRNA expression in these pathophysiological states. Surprisingly, in the cerebral cortex, the levels of SHIP2 mRNA were slightly increased in db/m+ mice, and significantly (2.4-fold) increased in db/db mice when compared with control m+/m+ mice at 8 wk of age (Fig. 2A), although we could not detect significant changes in the hippocampus (data not shown). The expression levels of insulin receptor mRNA in the hippocampus and cerebral cortex did not differ among db/db, db/m+, and m+/m+ mice (Fig. 2B). The levels of SHIP2 protein were significantly (1.4-fold) increased in cerebral cortex of db/db mice, compared with m+/m+ mice (Fig. 2C), whereas there were no differences in the levels of insulin receptor protein in cerebral cortex among db/db, db/m+, and m+/m+ mice (Fig. 2D). Furthermore, in aged C57BL/6J mice (24–27 months of age), the hippocampal and cerebrocortical expression levels of SHIP2 mRNA were 2.7-fold and 1.9-fold increased, compared with those in young mice (14 wk of age), respectively, whereas the levels of insulin receptor mRNA in these two brain regions were significantly lower than those in the young mice (data not shown).
Fig. 2.

Increased expression of SHIP2 in the brain of diabetic db/db mice. A and B, The mRNA levels of SHIP2 (A) and insulin receptor (IR; panel B) in the cerebral cortex of m+/m+, db/m+, and db/db mice at 8 wk of age. C and D, The protein levels of SHIP2 (C) and IR (D) in the cerebral cortex of m+/m+, db/m+, and db/db mice at 8 wk of age. Relative expression level was calculated as a ratio to the amount of GAPDH in each sample. Values represent means ± se of 10 mice (A and B) and six mice (C and D). *, P < 0.05. A.U., Arbitrary unit.
Impairment of insulin/IGF-I signaling in the brain of SHIP2 transgenic mice
We have previously reported that the expression levels of SHIP2 protein were greatly elevated in SHIP2 transgenic mice used in the present study (23). To further characterize the expression profiles, we analyzed the levels of SHIP2 protein in the hippocampus, cerebral cortex, cerebellum, and hypothalamus by Western blotting. As shown in Fig. 3A, the protein expression was apparently detected in the four brain regions in wild-type mice, and the expression levels were remarkably elevated in SHIP2 transgenic mice in a region-independent manner. The brain weight was not significantly different between SHIP2 transgenic mice (442.4 ± 6.5 mg, seven males) and wild-type littermates (444.8 ± 3.8 mg, seven males) at 6 months of age.
Fig. 3.
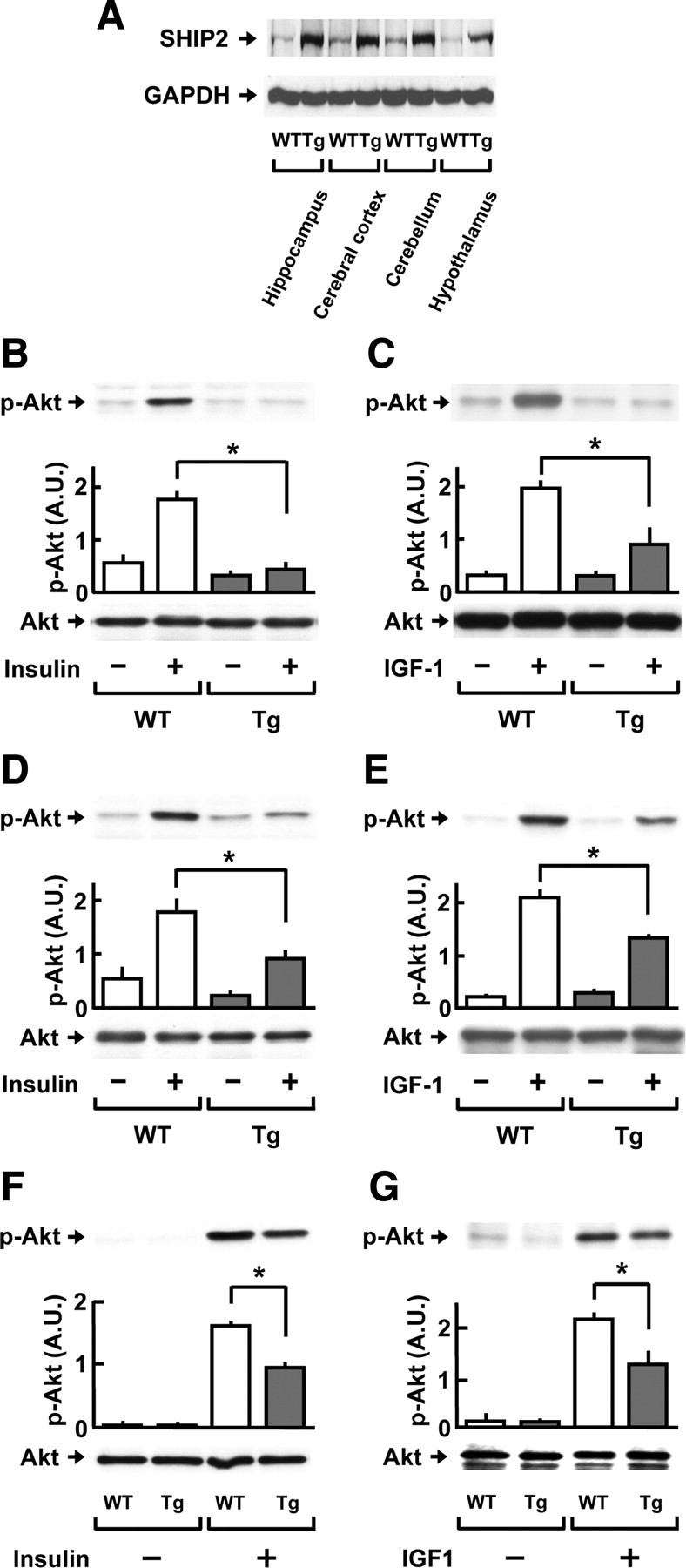
Reduction of insulin and IGF-I signaling in the hippocampus, cerebral cortex, and CG neurons of SHIP2 transgenic mice. A, The protein expression levels of SHIP2 in the hippocampus, cerebral cortex, cerebellum, and hypothalamus of wild-type (WT) and SHIP2 transgenic (Tg) mice at 3–6 months of age were analyzed by immunoblot analysis. GAPDH protein is shown as a loading control. B–E, Insulin/IGF-I signaling in the hippocampus and cerebral cortex of Tg mice and WT mice at 3–6 months of age. Tissues were isolated after i.c.v. injection of insulin (25 mU, 30 min) or IGF-I (10 μg, 60 min) and subjected to immunoblot analysis. Graphs show the insulin- (B) and IGF-I- (C) induced phosphorylation of Akt at Thr308 in the hippocampus, and the insulin- (D) and IGF-I- (E) induced phosphorylation of Akt at Thr308 in the cerebral cortex. F and G, Insulin/IGF-I signaling in the CG neurons isolated from Tg mice and WT mice. Cells were starved in serum-free BME containing 5 mm KCl and then treated with 1.7 nm insulin or 6.5 nm IGF-I for 5 min. The insulin- (F) and IGF-I- (G) induced phosphorylations of Akt at Thr308 were compared between the cells from Tg and WT mice. The amount of total Akt protein remained unchanged. The phosphorylation level in each sample was analyzed by densitometry and averaged. Values represent means ± se of three or five mice. *, P < 0.05. p-, Phosphorylated; A.U., arbitrary unit.
To investigate the influence of SHIP2 overexpression on insulin signaling in the hippocampus and cerebral cortex, SHIP2 transgenic mice and wild-type mice (3–6 months of age) were injected intracerebroventricularly (i.c.v.) with insulin (25 mU per mouse) or IGF-I (10 μg per mouse), and the Akt phosphorylation levels at Thr308 were measured 30 and 60 min after the injection. In the hippocampus, insulin efficiently induced phosphorylation of Akt in wild-type mice, but the increase in Akt phosphorylation was greatly diminished in SHIP2 transgenic mice (Fig. 3B). Similar results were obtained after the treatment with IGF-I (Fig. 3C). In the cerebral cortex of SHIP2 transgenic mice, the phosphorylation levels of Akt after the injection of either insulin (Fig. 3D) or IGF-I (Fig. 3E) were nearly halved, as compared with those of wild-type mice.
We analyzed the changes in insulin/IGF-I signaling by neuronal overexpression of SHIP2 more directly, using cultured mouse CG neurons. Higher levels of SHIP2 protein were observed in CG neurons from SHIP2 transgenic mice than those from wild-type mice (data not shown). After the stimulation with insulin (1.7 nm), the levels of phospho-Thr308-Akt (Fig. 3F) and phospho-Ser9-GSK3β (data not shown) were greatly increased in the cells from wild-type mice; however, the phosphorylation levels of Akt and GSK3β after the insulin stimulation were decreased by 41% and 21% in the cells from SHIP2 transgenic mice, respectively, compared with wild-type controls. In addition, IGF-I (6.5 nm)-induced phosphorylations of Akt (Fig. 3G) and GSK3β (data not shown) were significantly reduced in CG neurons from SHIP2 transgenic mice, compared with those from wild-type mice. Under the present conditions, the total protein levels of Akt (Fig. 3) and GSK3β (data not shown) were not altered between these mice.
We further investigated the PI(3,4,5)P3 levels in the hippocampal cells by using immunocytochemical staining. Although the basal immunoreactivity of PI(3,4,5)P3 was undetectable (data not shown), the hippocampal CA1 region and the dentate gyrus of wild-type mice exhibited apparent immunoreactivity after i.c.v. injection of insulin. In contrast, only weak immunoreactivity was seen in the hippocampal region of SHIP2 transgenic mice and db/db mice (Supplemental Fig. 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org).
Impairment of insulin- and IGF-I-induced neuroprotection in CG neurons from SHIP2 transgenic mice
To investigate whether overexpression of SHIP2 affects neuronal cell survival, CG neurons were treated with insulin and IGF-I, after which cell viability was measured by the 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Cell death was induced by switching from medium supplemented with 25 mm KCl and 10% fetal bovine serum (FBS) to serum-free medium containing 5 mm KCl (28). Low K+-induced cell death in CG neurons from SHIP2 transgenic mice was greater than that from wild-type mice (Fig. 4A). This neurotoxicity was improved by treatment with insulin in a concentration-dependent manner (1.7–170 nm). However, the neuroprotective effects of insulin were significantly reduced in CG neurons from SHIP2 transgenic mice, compared with those from wild-type mice. Similarly, the neuroprotective effects of IGF-I (13 and 130 nm) were reduced by approximately 25% in the cells from SHIP2 transgenic mice, compared with those from wild-type mice (Fig. 4B).
Fig. 4.
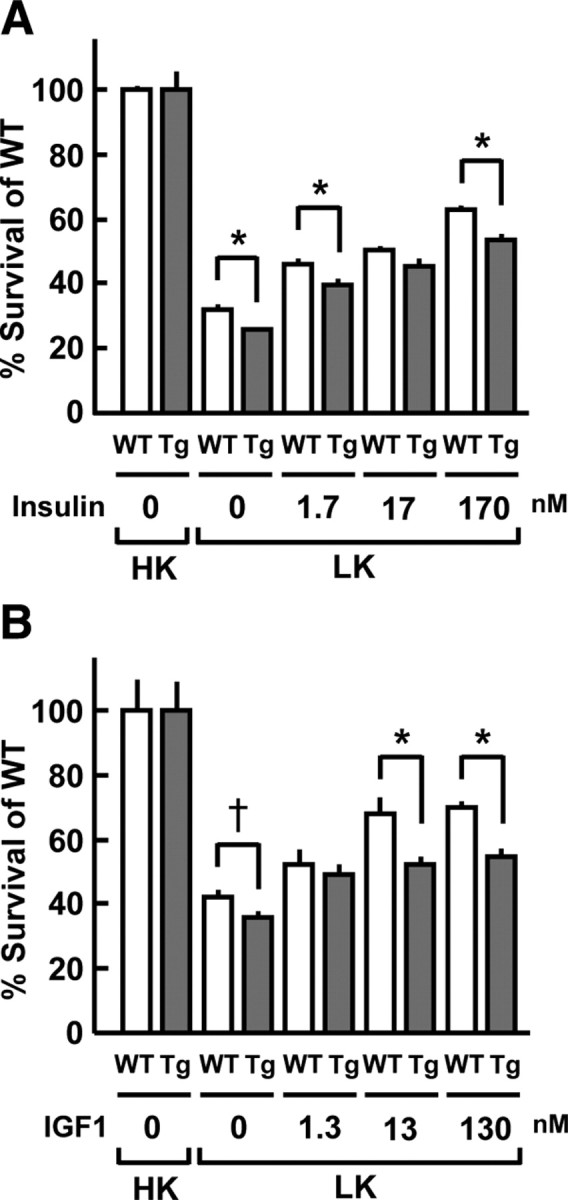
Impairment of insulin- and IGF-I-elicited neuroprotection in CG neurons from SHIP2 transgenic (Tg) mice. CG neurons isolated from SHIP2 Tg and wild-type (WT) mice were preincubated in BME containing 10% FBS and 25 mm KCl (high K+: HK) with insulin (0, 1.7, 17, and 170 nm) or IGF-I (0, 1.3, 13, and 130 nm) for 24 h and then incubated in serum-free BME containing 5 mm KCl (low K+: LK) with insulin (A) or IGF-I (B) at respective concentrations for 36 h. As a negative control, cells were incubated in BME containing 10% FBS and 25 mm KCl for 60 h. Cell viability was measured by MTT assay. The viable cell numbers obtained in four or six separate experiments were averaged and presented as means ± se. *, P < 0.05; †, P = 0.0598.
Increase in apoptosis-positive cells in the cerebral cortex of aged SHIP2 transgenic mice
To elucidate the influence of SHIP2 overexpression on apoptosis in brain neuronal cells, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assays were performed in sagittal sections including the cerebral cortex and hippocampus of young adult (3- to 6-month-old) and aged (12- to 15-month-old) SHIP2 transgenic mice. At young ages, no difference in the numbers of TUNEL-positive cells was observed either in the hippocampus or cerebral cortex of SHIP2 transgenic mice when compared with those of wild-type mice (data not shown). In contrast, the elderly transgenic mice had greater numbers of apoptosis-positive cells in the cerebral cortex than wild-type mice (Fig. 5, A and B), although no such abnormality was detected in the hippocampus (data not shown).
Fig. 5.
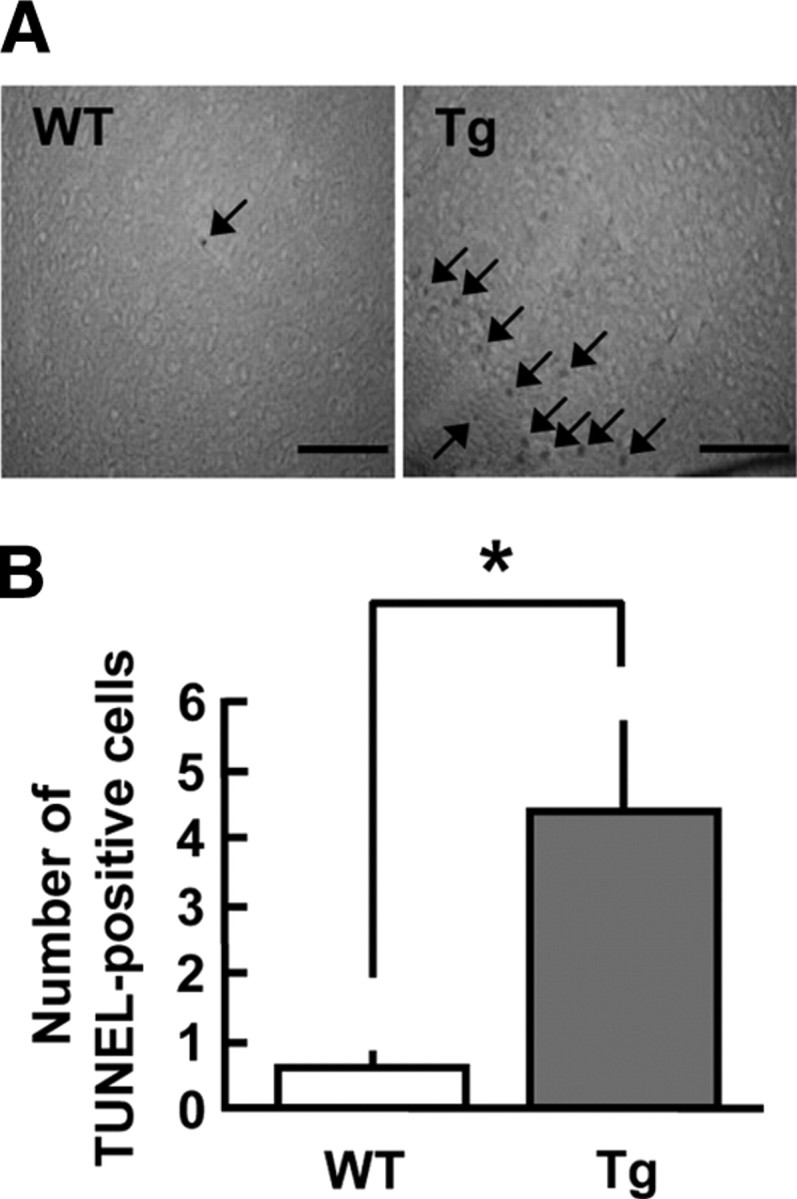
Increase in apoptosis-positive cells in the cerebral cortex of SHIP2 transgenic (Tg) mice. Sagittal sections (40 μm) including cerebral cortex were obtained from the brain of SHIP2 Tg and wild-type (WT) mice at 12–15 months of age, and apoptotic cells were stained by in situ TUNEL method. A, Typical microscopic images demonstrating TUNEL-positive cells (arrow) in the cerebral cortex of WT and Tg mice. Scale bars, 200 μm. Similar results were obtained in seven or eight separate experiments. B, Graph shows the number of TUNEL-positive cells/mm2 in the cerebral cortex of Tg and WT mice. Values represent means ± se of seven or eight mice. *, P < 0.05.
Impairment of memory function in SHIP2 transgenic mice
Insulin and IGF-I have been shown to exert a memory-enhancing action in rodents (27, 29). We therefore investigated whether overexpression of SHIP2 affects spatial learning and memory behavior in mice, using the Morris water maze. In the hidden-platform trials, escape latency to find the submerged platform became progressively shorter over 5 d in both wild-type and SHIP2 transgenic mice. However, on d 5, the escape latency was 1.6-fold longer in SHIP2 transgenic mice than that of wild-type mice (Fig. 6A). We further performed probe trials 4 h after the end of hidden-platform trials, in which the platform was removed and each mouse was allowed to swim freely for 120 sec inside the pool. When compared with wild-type mice, the numbers of crossing at original platform location were reduced by 30% in SHIP2 transgenic mice (Fig. 6B). In the visible-platform trials, there was no difference in escape latencies between these two genotypes over 5 d (Fig. 6C). These results demonstrate that the ability of spatial learning and memory was impaired in SHIP2 transgenic mice.
Fig. 6.

Impairment of memory function in SHIP2 transgenic (Tg) mice. A–C, The Morris water maze tests were conducted to assess the spatial learning and memory in SHIP2 Tg and wild-type (WT) mice at 5–6 months of age. A, Hidden-platform trials. The escape latencies to reach the hidden platform in the water maze were measured in four trials per day and averaged. B, Probe tests conducted 4 h after the hidden-platform trials on d 5. Number of crossings at the original platform location for 120 sec was measured. C, Escape latencies in the visible-platform trials. Values represent means ± se of 27 or 28 (A and B) and eight mice (C). D, The step-through passive-avoidance tests were conducted in SHIP2-Tg and WT mice at 5 months old. Graphs show latency to enter the dark compartment on d 1 (open column, initial session) and d 2 (gray column, retention session). On d 1, the mice received a foot shock on the initial trial and then given an i.c.v. injection of 30 mU insulin. Values represent means ± se of nine or 10 mice. E, The novel-object recognition tests were conducted in SHIP2-Tg and WT mice at 2–3 months of age. Graphs show percent exploratory time of object 1 (open column) and object 2 (gray column) during the training session on d 1, and that of object 1 (open column) and the novel object 3 (black column) during the retention session on d 2. Values represent means ± se of 10 or 11 mice. *, P < 0.05; **, P < 0.01.
Next we analyzed the learning and memory performance of mice on step-through passive avoidance test. In wild-type mice, the entrance latencies were constantly increased in the retention session 24 h after the initial session with a foot-shock (Fig. 6D). Moreover, the entrance latencies in the retention session were significantly (2.1-fold) longer in wild-type mice injected i.c.v. with insulin than those in the vehicle-treated wild-type mice. In contrast, the i.c.v. injection of insulin failed to increase the entrance latencies in SHIP2 transgenic mice. These results indicate that the central effect of insulin on passive avoidance learning and memory was reduced in SHIP2 transgenic mice.
We further examined the alteration of cognitive performance of SHIP2 transgenic mice in the novel object recognition test. During the training session, both wild-type and SHIP2 transgenic mice exhibited equal exploratory time for each object (objects 1 and 2). Interestingly, in the retention session, wild-type mice spent more time with the novel (object 3) than the familiar object (object 1); however, no preference for the novel object was observed in SHIP2 transgenic mice (Fig. 6E). Thus, SHIP2 transgenic mice exhibited significantly impaired novel object recognition performance.
SHIP2 inhibitor improves the impairment of synaptic plasticity and memory formation in diabetic db/db mice
Long-term potentiation (LTP) of synaptic transmission in the hippocampal slices is regarded as an in vitro model of memory. To analyze LTP, we performed electrophysiological recordings in mouse hippocampal slices. Figure 7 illustrates the field excitatory postsynaptic potentials (fEPSPs) before and after high-frequency tetanic stimulation. In control (m+/m+) mice, the tetanic stimulation led to a long-lasting increase in the slope of fEPSPs, indicating a robust induction of LTP in the hippocampal CA1 neurons (Fig. 7A). In diabetic db/db mice, however, the increase in the slope of fEPSPs returned to baseline approximately 40 min after the tetanic stimulation, and no maintained LTP was observed, as reported previously (30, 31). We then investigated the impact of SHIP2 inhibition on the impaired LTP in db/db mice, using a specific inhibitor of SHIP2, AS1949490 (32). Interestingly, a sustained increase in the slope of fEPSPs appeared in the hippocampal slices of db/db mice after the treatment with 10 μm AS1949490 (Fig. 7A). The slopes of fEPSPs 60 min after tetanic stimulation were 16-fold larger in the presence of AS1949490, compared with those of vehicle controls in db/db mice and were comparable to those of vehicle controls in m+/m+ mice (Fig. 7B). AS1949490 did not affect the slope of fEPSPs in m+/m+ mice. These results indicate that SHIP2 inhibition can improve the impairment of LTP induction in the hippocampal neurons of diabetic db/db mice.
Fig. 7.
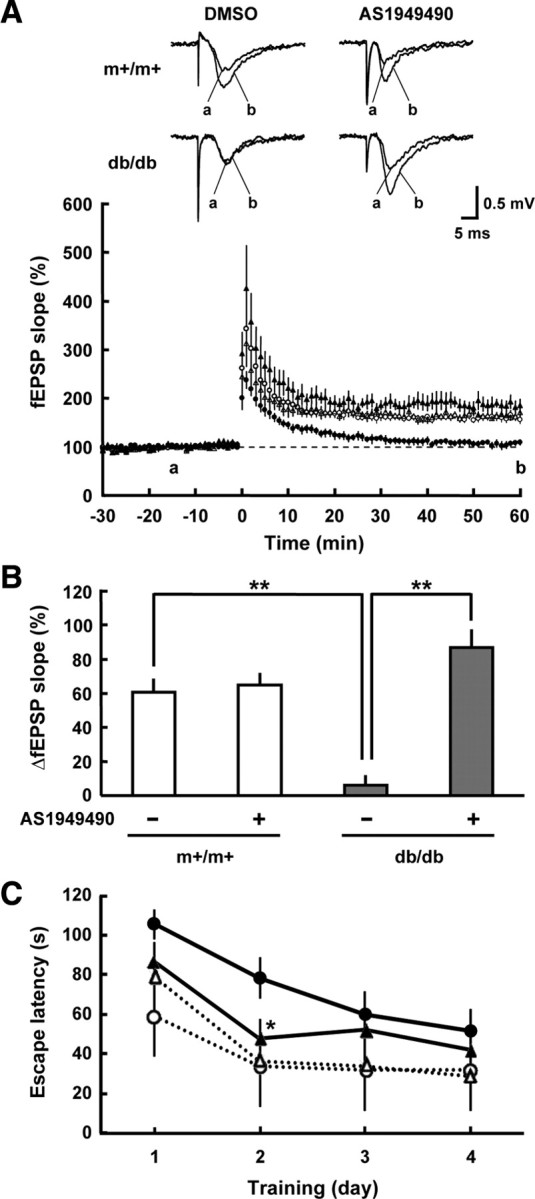
Inhibition of SHIP2 improves the impairment of hippocampal LTP and memory function in diabetic db/db mice. A, Recordings of fEPSPs before and after tetanic stimulation in the CA1 in slices from m+/m+ mice treated with vehicle (open circles) or 10 μm AS1949490 (open triangles), and db/db mice treated with vehicle (filled circles) or 10 μm AS1949490 (filled triangles). Tetanic stimulation (100 Hz, for 1 sec) was delivered at time 0. Inset traces show typical fEPSP recorded during the baseline (a) and 60 min after tetanic stimulation (b). B, The increase in averaged fEPSP slope (55–60 min after tetanic stimulation) over the baseline in slices from m+/m+ mice and db/db mice in the absence or presence of AS1949490. Values represent means ± se of three to five slices. **, P < 0.01. C, The Morris water maze tests were conducted in db/db mice treated with vehicle (filled circles) or AS1949490 (filled triangles) and age-matched control C57BL/6J mice treated with vehicle (open circles) or AS1949490 (open triangles) at 8–12 wk of age. The escape latencies to find the hidden platform were measured in four trials per day and averaged. AS1949490 (10 μg) or vehicle (DMSO) was i.c.v. injected after the end of the last training trial each day. Values represent means ± se of five to 10 mice. *, P < 0.05, significantly different between db/db mice treated with or without AS1949490.
We further performed the Morris water maze trials to examine the effects of AS1949490 on the spatial learning and memory in mice. In the hidden platform trials, the escape latency was markedly longer in db/db mice than age-matched C57BL/6J control mice (Fig. 7C), as reported previously (31). When C57BL/6J mice were treated with an i.c.v. injection of AS1949490 (10 μg per mouse), no change in the memory performance was observed. However, the escape latency on day 2 was significantly reduced in db/db mice treated with AS1949490 than those treated with vehicle (Fig. 7C). Swimming speed was not altered by the treatment with AS1949490 in both mice (data not shown). These indicate that SHIP2 inhibition can ameliorate the impaired memory function in diabetic db/db mice.
Discussion
The insulin and IGF-I actions control numerous brain functions, including the regulation of neuronal survival, neurogenesis, learning and memory, cognitive function, and lifespan (2, 6, 29). Recent evidence establishes a strong connection between type 2 diabetes and the risk of Alzheimer’s disease, and insulin resistance is considered to be a potential mechanism underlying the connection (4, 5, 6, 7, 8). In addition, age-related decrease in serum IGF-I in the elderly populations (29) and defects in IGF-I receptor system in Alzheimer’s disease (33) indicate that IGF-I resistance also contributes to the development of age- related brain dysfunctions. However, the pathological processes inducing the aberrant insulin/IGF-I signaling in brain have not been clarified. In metabolic insulin signaling pathway in peripheral tissues, several critical nodes exist, and the lipid phosphatase SHIP2 has been identified as an important negative regulator of the PI3-kinase node at the level of PI(3,4,5)P3 (13, 19, 34). In the present study, we demonstrated that endogenous SHIP2 contributes to the fine-tune regulation of insulin/IGF-I signaling in brain. Furthermore, we found that the expression of SHIP2 in mouse brain was significantly increased in the diabetic state, and abnormally increased expression of SHIP2 caused the impairment of brain insulin/IGF-I signaling, the attenuation of neuroprotective effects of insulin/IGF-I, and the decline in learning and memory in mice.
Several studies have shown that hyperinsulinemia, a consequence of obesity and insulin resistance, is related to a higher risk of Alzheimer’s disease (35). In addition, memory function is impaired in obese and type 2 diabetic db/db mice (30, 31). In this study, we found that db/db mice exhibited selectively increased expression of SHIP2 in the cerebral cortex, whereas no change in the expression of insulin receptor was observed. Although the mechanism behind the abnormal SHIP2 expression remains unclear, hyperinsulinemia or dyslipidemia developed in db/db mice might be relevant to the alteration, because cerebral expression of SHIP2 was not altered after 4 wk of hyperglycemia under insulin-deficient conditions in streptozotocin-induced diabetic mice (data not shown). These raised a possibility that increased expression of SHIP2 may be implicated in brain dysfunctions in insulin resistance with type 2 diabetes.
Diabetic patients are at increased risk for brain aging associated with cognitive disorders and dementia (4, 36). In addition, both diabetic and aged rats show persistent inhibition of long-term potentiation and facilitation of long-term depression in the hippocampus and neocortex, indicating that diabetes and aging act on synaptic plasticity through common mechanisms (37). The present RT-PCR experiments demonstrated that nondiabetic C57BL/6J mice displayed the decreased expression of insulin receptor mRNA in the hippocampus and cerebral cortex at an elderly age, in agreement with a previous in situ hybridization study in rat brain (27). Furthermore, we found that the expression levels of SHIP2 mRNA in these brain regions were higher in aged than in young mice. It has been reported that i.c.v. injection of insulin induces phosphorylation of Akt and GSK3β in rat brain at 3–8 months of age but not at 24 months (38). Therefore, we suppose that abnormal expression of SHIP2 and insulin receptor synergistically impairs insulin/IGF-I signaling in brain with aging, and the impact of SHIP2 is particularly pronounced in type 2 diabetes.
Excessive SHIP2 expression is known to reduce the content of PI(3,4,5)P3, resulting in decline of Akt and GSK3β phosphorylation in peripheral insulin’s target cells (19, 20, 39). In this study, we examined the influence of SHIP2 on insulin/IGF-I signaling in neuronal cells using SHIP2 transgenic mice and demonstrated that overexpression of SHIP2 decreased hippocampal and cerebrocortical Akt phosphorylation in response to i.c.v. injection of insulin and IGF-I. Similar results were observed in cultured CG neurons. These changes were not due to the decrease in the upstream signaling molecules of PI3-kinase, because the expression levels of insulin receptor, insulin receptor substrate 1, and insulin receptor substrate 2 were not altered in SHIP2 transgenic mice (data not shown). Accordingly, it is likely that overexpression of SHIP2 induces impairment of insulin/IGF-I signaling via lowering of PI(3,4,5)P3 level even in the central nervous system.
Insulin and IGF-I are known to exert antiapoptotic neuroprotective action via PI3-kinase/Akt pathway, and their cell survival effects were inhibited by wortmannin and LY294002, PI3-kinase inhibitors, in CG neurons (40, 41). This neuroprotective mechanism involves the phosphorylation of the Bcl-2 family member Bad by Akt, thereby suppressing caspase-3-dependent apoptosis (42). In addition, neuroprotection correlates with inhibition of GSK3β activity, which can be achieved by Akt-induced phosphorylation. Indeed, GSK3β-overexpressing mice showed increased neuronal apoptosis in the hippocampus (43), and constitutive active GSK3β (Ser9Ala) phosphorylated and translocated a proapoptotic Bcl-2 family member Bax to mitochondria in CG neurons, resulting in induction of apoptosis (15). In this study, we observed that neuroprotective effects of insulin and IGF-I were impaired by overexpression of SHIP2 in CG neurons. Moreover, SHIP2 transgenic mice showed an increase in the number of apoptosis-positive cells in cerebral cortex at an elderly age. Therefore, the observed cell death associated with overexpression of SHIP2 appears to be due to the impairment of insulin/IGF-I signaling via Akt and its downstream cascades.
In this study, we identified the impairment of memory function of SHIP2 transgenic mice by three different methods: the Morris water maze test, passive-avoidance test, and novel-object-recognition test. The Morris water maze test can evaluate hippocampus-dependent spatial learning and memory in rodents (31). SHIP2 transgenic mice showed impaired memory performance both in the hidden platform and probe trials. Because the swimming speed during the task (data not shown) and latency under visible platform conditions did not differ from those of wild-type mice, SHIP2 transgenic mice appear to have no deficits in swimming and visual abilities. The step-through passive avoidance response is also known to require hippocampus-dependent learning (44). We observed that central action of insulin augmented passive avoidance learning and memory in wild-type mice, in agreement with a previous study in rats (45). However, the augmenting effect of insulin was remarkably reduced in SHIP2 transgenic mice. These suggest that overexpression of SHIP2 has the great impact on insulin-induced memory facilitation in the hippocampus. In this study, we further found that SHIP2 transgenic mice were significantly impaired in their novel object recognition ability. Recognition memory relies on two functionally distinct memory processes, recollection and familiarity, which are supported by medial temporal lobe structures (46). Evidence has indicated that recollection depends on the hippocampus, whereas familiarity depends on the adjacent perirhinal cortex, although consensus has not been reached (46, 47). We therefore suggest that an excess amount of SHIP2 disturbs physiological functions of insulin/IGF-I system in multiple brain regions including the hippocampus and medial temporal lobe and impairs the memory performance in mice.
The PI3-kinase signaling is well known to be involved in the induction and maintenance of hippocampal LTP, the prominent cellular model for memory formation (48). Inhibition of the inositol 3′-phosphatase, PTEN, has been shown to augment AMPA-type glutamate receptor synaptic expression and excitatory synaptic strength through the increase in intracellular PI(3,4,5)P3 levels (49). It has been reported that diabetic db/db mice show impairments in LTP in hippocampal CA1 neurons as well as deficits in spatial memory (30, 31). Importantly, the present experiments demonstrate that treatment with SHIP2 inhibitor ameliorated the impairment of both hippocampal LTP and spatial memory in db/db mice, whereas the memory functions in control mice were not affected by this treatment. Also in the hippocampal slices of SHIP2 transgenic mice, there was a trend toward increasing LTP by the SHIP2 inhibitor treatment (data not shown). These suggest that inhibition of SHIP2 restores the reduced PI(3,4,5)P3 levels and ameliorates the deficits in hippocampal synaptic plasticity in diabetic db/db mice. Although apparent changes in SHIP2 expression could not be detected in the hippocampus, unlike in the cerebral cortex, of db/db mice under the present conditions, a subtle changes at some specific subcellular compartment might be sufficient to affect the hippocampal function. Interestingly, the more severe impairment of LTP and spatial memory performance, as well as hyperglycemia and insulin resistance, was observed in db/db mice, compared with SHIP2 transgenic mice. It is possible that the phosphatase activity might be increased under the long-term hyperinsulinemic condition, because it has been suggested that insulin-induced tyrosine phosphorylation of SHIP2 modulates its phosphatase activity (18).
Insulin and IGF-I receptors are present in particularly high concentrations in neurons, and in lower levels in glia (3, 33), and little is known about the functional significance of insulin/IGF-I signaling in glial cells. In this study, we detected the expression of SHIP2 protein in glial cells in addition to neuronal cells (data not shown). Therefore, we cannot completely rule out the possibility that the observed changes in SHIP2 transgenic mice are attributed to overexpression of SHIP2 in glial cells in brain. However, excessive SHIP2 expression directly causes disruption of insulin/IGF-I signaling and neuroprotective mechanism in cultured neuronal cells. Moreover, despite the fact that SHIP2 overexpression in glioblastoma cells induces cell cycle arrest and inhibits DNA synthesis (50), expression of glial fibrillary acidic protein, a marker of astrocyte, in cerebral cortex and hippocampus of SHIP2 transgenic mice did not decrease compared with that of wild-type mice (data not shown). These support our view that overexpression of SHIP2 in neuron has a crucial impact on brain functions. Further investigations are needed to elucidate the precise role of SHIP2 in neurons, using transgenic mice with neuron-specific overexpression of SHIP2, in our future study.
In conclusion, we demonstrated that the inositol phosphatase SHIP2 has a great impact on the regulation of insulin/IGF-I signaling responsible for neuroprotection and higher-order functions of the brain. In particular, the present finding that expression of SHIP2 in brain was markedly enhanced in the diabetic state provides a new insight into the pathophysiological mechanisms of brain dysfunction in diabetes. Because SHIP2 inhibitor exhibited the ameliorating effect on the impaired memory function in diabetic mice, a better understanding of SHIP2 function may lead to the development of new therapeutic approaches to brain dysfunctions in insulin resistance with type 2 diabetes.
Materials and Methods
Materials
Human regular insulin (Humalin R) was provided by Eli Lilly (Indianapolis, IN). Human recombinant IGF-I (Somazon) was provided by Astellas Pharma, Inc. (Tokyo, Japan). The polyclonal anti-Thr308 phospho-specific Akt antibody, anti-Ser21/9 phospho-specific GSK3α and β-antibody and antiinsulin receptor antibody were purchased from Cell Signaling Technology (Beverly, MA). The monoclonal anti-Akt antibody was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The monoclonal anti-GSK3β antibody was from BD Transduction Laboratory (Lexington, KY). The polyclonal anti-SHIP2 antibody was prepared as previously described (18). AS1949490 was synthesized according to a previous report (32). All other reagents were of analytical grade and purchased from Sigma (St. Louis, MO) or Wako Pure Chemical Industries (Osaka, Japan).
Animals
SHIP2 transgenic mice were generated as described previously (23) and mated with C57BL/6J mice (CLEA Japan, Inc., Tokyo, Japan) to prepare F2-F6 mice. BKS.Cg-m+/m+/J (m+/m+), BKS.Cg-m+/+Leprdb/J (db/m+), and BKS.Cg-+Leprdb/+Leprdb/J (db/db) mice were purchased from Charles River Japan, Inc. (Yokohama, Japan). All experimental procedures used in this study were approved by the Committee of Animal Experiments at University of Toyama.
Cell culture
CG neurons were cultured as described previously (51). In brief, CG neurons were obtained from dissociated cerebellum of mice at 7–8 d of age. Cells were plated in Basal Medium Eagle (BME; Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 25 mm KCl (high K+), 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin on culture dishes coated with poly-d-lysine. Cytosine arabinofuranoside (10 μm) was added to the culture medium 16–20 h after plating to prevent replication of nonneuronal cells. Cells were then cultured for 4–5 d. A primary culture of neuronal cells from the cerebral cortices of 17-d-old Sprague Dawley rat (Japan SLC, Hamamatsu, Japan) embryos was prepared by the same method as described previously (52). In experiments to analyze Akt and GSK3β phosphorylation, CG neurons were incubated in BME supplemented with 5 mm KCl (low K+), 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin for 2 h and then stimulated with 1.7 nm insulin or 6.5 nm IGF-I. Rat cortical neurons were serum starved for 3 d and then stimulated with 0.5 nm insulin.
Infection with recombinant adenoviruses
Adenoviruses having a gene encoding ΔIP-SHIP2 were prepared as described previously (20). Culture neurons were infected with adenoviruses for 2 h and then washed in PBS. Subsequently, CG neurons were incubated in BME containing 25 mm KCl and 10% FBS for 4–5 d, and cortical neurons were incubated in serum-free medium for 3 d. These cells were then stimulated with insulin or IGF-I.
Quantitative real-time RT-PCR
Hippocampus, cerebral cortex, cerebellum, hypothalamus, and liver were isolated from 14-wk-old or 24- to 27-month-old C57BL/6J mice and 8-wk-old m+/m+, db/m+, and db/db mice. RNA extraction, reverse transcription, and quantitative real-time PCR using a Mx3000p Real-Time PCR System (Stratagene, La Jolla, CA) were performed as previously described (23). The primer pairs used were as follows. SHIP2: 5′-TTGAACCCAAGGCTGAAGTC-3′ and 5′-GCCACTTTGTTCTTGGTGGC-3′; insulin receptor: 5′-AATGGCAACATCACACACTACC-3′ and 5′-CAGCCCTTTGAGACAATAATCC-3′; GAPDH: 5′-AATGTGTCCGTCGTGGATCTGA-3′ and 5′-GATGCCTGCTTCACCACCTTCT-3′. The relative mRNA expression levels of SHIP2 and insulin receptor were calculated as a ratio to those of GAPDH.
i.c.v. injection
i.c.v. injection was performed according to a previously described method (53). In brief, a guide cannula was implanted into the lateral ventricle of anesthetized mice at 3–6 months of age. After a 7- to 8-d recovery period, the mice were fasted overnight and i.c.v. injected with 2.0 μl of vehicle [PBS or dimethylsulfoxide (DMSO)], human regular insulin (25 mU dissolved in 2 μl PBS), IGF-I (10 μg dissolved in 2 μl PBS), or AS1949490 (10 μg dissolved in 2 μl DMSO). Each injection was performed manually over a period of 20 sec (at a rate of ∼0.1 μl/sec) using a 10-μl Hamilton syringe, and the syringe needle was held in position for an additional 30 sec to ensure the injection. Then, hippocampus and cerebral cortex were isolated from anesthetized mice 30 min after the insulin injection or 60 min after the IGF-I injection.
Western blotting
Hippocampus, cerebral cortex, cerebellum, and hypothalamus isolated from mice and cultured mouse CG and rat cortical neurons were lysed and subjected to Western blot analysis according to the protocol described previously (23).
Cell viability
CG neurons were preincubated in BME containing 10% FBS and 25 mm KCl with insulin (0, 1.7, 17, and 170 nm) or IGF-I (0, 1.3, 13, and 130 nm) for 24 h and then transferred to serum-free BME containing 5 mm KCl with insulin or IGF-I at respective concentrations for 36 h. The cell viability was assessed by MTT assay (28).
TUNEL assay
Mice at 12–15 months of age were deeply anesthetized with pentobarbital and transcardially perfused with 4% paraformaldehyde for 5 min. The brains were postfixed in 4% paraformaldehyde for 4 h and placed in 30% sucrose in PBS for 24 h at 4 C. Sagittal sections (40 μm) were cut in a freezing microtome. The DNA fragmentation characteristic of apoptosis was detected by TUNEL method according to the manufacturer’s instructions (DeadEnd Colorimetric TUNEL System; Promega Corp., Madison, WI).
The Morris water maze test
The Morris water maze test was performed, as described previously (30). In brief, the water-maze apparatus (Muromachi Kikai, Tokyo, Japan) consists of a circular pool (diameter, 120 cm) filled with water (25 C), and invisible platform (diameter, 10 cm) 1 cm under the surface of the water. Extramaze visual cues were located around the pool. In hidden-platform trials, mice were trained to escape from water by swimming (up to 120 sec) to the platform, for four trials per day (interval of 15–30 min) over 5 d. The escape latency and the swimming velocity were recorded by a computer system (Muromachi Kikai). If the animal had not found the platform within 120 sec, it was hand guided to the platform. In either case, mice were allowed to stay on the platform for 15 sec before being placed back in their home cage. In probe trials, the platform was removed and conducted 4 h after the last training session on d 5, mice were allowed to swim freely for 120 sec. During this period, the number of crossings at original platform location was recorded. In visible-platform trails, extra-maze visual cues were removed, and a blue cylinder (diameter, 4 cm; height, 20 cm) was put on the platform. Mice were trained to escape to the platform by swimming (up to 120 sec) for four trials per day over 5 d. For the drug treatment, the animals were given an i.c.v. injection of vehicle (DMSO) or AS1949490 (10 μg) in a total volume of 2 μl after the end of the last training trial each day.
Step-through passive avoidance test
We performed a step-through passive avoidance test to evaluate learning and memory in mice, according to the method as described previously (45). In brief, in the initial session on d 1, mice were placed in the light compartment of a passive avoidance apparatus (MPB-M001; Melquest, Toyama, Japan), and latency to enter the dark compartment was measured. The animals received a mild foot shock (0.05 mA, 2 sec) by a shock generator (SG-100, Melquest). The animals were then given an i.c.v. injection of vehicle (PBS) or insulin (30 mU) in a total volume of 2 μl. Blood glucose levels were not affected by the i.c.v. injection (data not shown). After 24 h, memory retention session was carried out to measure the latency to enter the dark compartment in the same apparatus. If the animal had not entered the dark compartment within 3 min, it was given a score of 180 sec.
Novel-object recognition test
The behavioral paradigm was designed based on that described in Wang et al. (54). In brief, each mouse was individually habituated to an open-field chamber (35 × 30 × 16 (H) cm) for 10 min in the absence of objects on d 0 (habituation session). During the training session on d 1, two objects (objects 1 and 2) were placed in the chamber, and each mouse was allowed to explore them for 5 min. During the retention session on d 2, each mouse was placed back into the same chamber, in which one of the familiar objects (object 2) used during training was replaced by a novel object (object 3) at the same position and allowed to explore freely for 5 min. Animal behavior was recorded, and the duration of exploration of each object was analyzed. The mouse was regarded as exploring an object when its head was oriented to the object within a distance of 2 cm. The percentage of time spent with either two new objects (objects 1 and 2) or one familiar and one novel object (objects 1 and 3) over the total exploration time was calculated to evaluate novel-object recognition memory.
Electrophysiology
Electrophysiolosical recordings were performed using m+/m+ and db/db mice (male, 8 wk of age). Animals were decapitated, and their brains were dissected in cold artificial cerebrospinal fluid (ACSF) containing 125 mm NaCl, 3.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 2.0 mm MgSO4, 2.5 mm CaCl2 and 25 mm glucose, bubbled with a mixture of 95% O2 and 5% CO2. Hippocampal slices (300 μm) were obtained with a slicer (LinearSlicer Pro7; Dosaka, Kyoto, Japan) and kept submerged at 30 C for at least 1 h in ACSF. For recording, slices were transferred to a submerged recording chamber and continuously perfused with ACSF at a rate of 1–2 ml/min at 30 C. Shaffer collaterals/commissural fibers in CA3 were stimulated using a bipolar stainless steel wire (diameter, 50 μm). fEPSPs were recorded in the stratum radiatum of CA1 using sharp glass pipettes (2 mΩ, filled with 2 m NaCl). Stimulus intensity was adjusted to produce responses at 30–50% of the maximal fEPSP amplitude and test stimuli were delivered at 20-sec intervals throughout the experiment. LTP was induced by tetanic stimulation delivered at 100 Hz for 1 sec. AS1949490 was dissolved in DMSO and diluted with ACSF. AS1949490 or the vehicle (0.1% DMSO) was bath applied at least 90 min before the tetanic stimulation. Data were analyzed using Scope software (AD Instruments, Colorado Springs, CO). The slope of the initial rising phase of fEPSP was measured and expressed as a percentage of the averaged baseline. LTP was defined as >20% increase in the fEPSP slope 60 min after the tetanic stimulation, compared with the baseline.
Statistical analysis
Data are expressed as means ± se. The significance of differences between two groups was assessed by Student’s t test, and differences between multiple groups were assessed by one-way ANOVA and Bonferroni test, using Statview software (SAS Institute, Cary, NC). P < 0.05 was considered statistically significant.
Acknowledgments
We thank Mr. Ryo Oshita, Ms. Emi Tokai, Mr. Kentaro Okamoto, Dr. Yukihisa Murakami (University of Toyama, Toyama, Japan), and Dr. Hajime Ishihara (Sainou South Hospital, Toyama, Japan) for their technical assistance.
Footnotes
This study was supported in part by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, grants from the Naito Foundation, Foundation for Growth Science, and Novo Nordisk Pharma for Insulin Research (to T.S.); grants from the Takeda Science Foundation, the Smoking Research Foundation, and the Tamura Foundation for Promotion of Science and Technology (to H.T.); a grant from the Sasakawa Scientific Research Grant (to Y.S.); Grant-in-Aid for Scientific Research (A, 22240051) from the Ministry of Education, Science, Sports and Culture; grants from Japan Society for the Promotion of Science Asian Core Program and the Core Research for Evolutionary Science and Technology program of Japan Science and Technology Agency (to H.N.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online September 9, 2010
Y.S and H.T. contributed equally to this work.
Abbreviations: ACSF, Artificial cerebrospinal fluid; BME, Basal Medium Eagle; CG, cerebellar granule; ΔIP-SHIP2, 5′-phosphatase-dead mutant of SHIP2; DMSO, dimethylsulfoxide; FBS, fetal bovine serum; fEPSP, field excitatory postsynaptic potential; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSK3, glycogen synthase kinase 3; i.c.v., intracerebroventricular; LTP, long-term potentiation; MTT, 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; PI, phosphatidylinositol; PTEN, phosphatase and tensin homolog deleted on chromosome 10; SHIP2, SH2-containing inositol 5′-phosphatase 2; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
References
- 1.van der Heide LP, Ramakers GM, Smidt MP2006. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol 79:205–221 [DOI] [PubMed] [Google Scholar]
- 2.Broughton S, Partridge L2009. Insulin/IGF-like signalling, the central nervous system and aging. Biochem J 418:1–12 [DOI] [PubMed] [Google Scholar]
- 3.Gerozissis K2008. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur J Pharmacol 585:38–49 [DOI] [PubMed] [Google Scholar]
- 4.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P2006. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5:64–74 [DOI] [PubMed] [Google Scholar]
- 5.Li ZG, Zhang W, Sima AA2007. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 56:1817–1824 [DOI] [PubMed] [Google Scholar]
- 6.Craft S2007. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res 4:147–152 [DOI] [PubMed] [Google Scholar]
- 7.Jones A, Kulozik P, Ostertag A, Herzig S2009. Common pathological processes and transcriptional pathways in Alzheimer’s disease and type 2 diabetes. J Alzheimers Dis 16:787–808 [DOI] [PubMed] [Google Scholar]
- 8.Han W, Li C2010. Linking type 2 diabetes and Alzheimer’s disease. Proc Natl Acad Sci USA 107:6557–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM2005. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease. Is this type 3 diabetes? J Alzheimers Dis 7:63–80 [DOI] [PubMed] [Google Scholar]
- 10.Benedict C, Hallschmid M, Schmitz K, Schultes B, Ratter F, Fehm HL, Born J, Kern W2007. Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32:239–243 [DOI] [PubMed] [Google Scholar]
- 11.Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey WH 2nd, Craft S2008. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J Alzheimers Dis 13:323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondy CA, Cheng CM2004. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol 490:25–31 [DOI] [PubMed] [Google Scholar]
- 13.Taniguchi CM, Emanuelli B, Kahn CR2006. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96 [DOI] [PubMed] [Google Scholar]
- 14.Jope RS, Johnson GV2004. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29:95–102 [DOI] [PubMed] [Google Scholar]
- 15.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA2004. Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24:9993–10002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J2001. Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J 20:27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng WH, Quirion R2004. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem 89:844–852 [DOI] [PubMed] [Google Scholar]
- 18.Ishihara H, Sasaoka T, Hori H, Wada T, Hirai H, Haruta T, Langlois WJ, Kobayashi M1999. Molecular cloning of rat SH2-containing inositol phosphatase 2 (SHIP2) and its role in the regulation of insulin signaling. Biochem Biophys Res Commun 260:265–272 [DOI] [PubMed] [Google Scholar]
- 19.Sasaoka T, Wada T, Tsuneki H2006. Lipid phosphatases as a possible therapeutic target in cases of type 2 diabetes and obesity. Pharmacol Ther 112:799–809 [DOI] [PubMed] [Google Scholar]
- 20.Wada T, Sasaoka T, Funaki M, Hori H, Murakami S, Ishiki M, Haruta T, Asano T, Ogawa W, Ishihara H, Kobayashi M2001. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5′-phosphatase catalytic activity. Mol Cell Biol 21:1633–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sleeman MW, Wortley KE, Lai KM, Gowen LC, Kintner J, Kline WO, Garcia K, Stitt TN, Yancopoulos GD, Wiegand SJ, Glass DJ2005. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat Med 11:199–205 [DOI] [PubMed] [Google Scholar]
- 22.Hori H, Sasaoka T, Ishihara H, Wada T, Murakami S, Ishiki M, Kobayashi M2002. Association of SH2-containing inositol phosphatase 2 with the insulin resistance of diabetic db/db mice. Diabetes 51:2387–2394 [DOI] [PubMed] [Google Scholar]
- 23.Kagawa S, Soeda Y, Ishihara H, Oya T, Sasahara M, Yaguchi S, Oshita R, Wada T, Tsuneki H, Sasaoka T2008. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology 149:642–650 [DOI] [PubMed] [Google Scholar]
- 24.Schurmans S, Carrió R, Behrends J, Pouillon V, Merino J, Clément S1999. The mouse SHIP2 (Inppl1) gene: complementary DNA, genomic structure, promoter analysis, and gene expression in the embryo and adult mouse. Genomics 62:260–271 [DOI] [PubMed] [Google Scholar]
- 25.Aoki K, Nakamura T, Inoue T, Meyer T, Matsuda M2007. An essential role for the SHIP2-dependent negative feedback loop in neuritogenesis of nerve growth factor-stimulated PC12 cells. J Cell Biol 177:817–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muraille E, Dassesse D, Vanderwinden JM, Cremer H, Rogister B, Erneux C, Schiffmann SN2001. The SH2 domain-containing 5-phosphatase SHIP2 is expressed in the germinal layers of embryo and adult mouse brain: increased expression in N-CAM-deficient mice. Neuroscience 105:1019–1030 [DOI] [PubMed] [Google Scholar]
- 27.Zhao WQ, Chen H, Quon MJ, Alkon DL2004. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol 490:71–81 [DOI] [PubMed] [Google Scholar]
- 28.Canu N, Tufi R, Serafino AL, Amadoro G, Ciotti MT, Calissano P2005. Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J Neurochem 92:1228–1242 [DOI] [PubMed] [Google Scholar]
- 29.Sonntag WE, Ramsey M, Carter CS2005. Growth hormone and insulin-like growth factor-1 (IGF-1) and their influence on cognitive aging. Ageing Res Rev 4:195–212 [DOI] [PubMed] [Google Scholar]
- 30.Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T2002. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 113:607–615 [DOI] [PubMed] [Google Scholar]
- 31.Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP2008. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11:309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suwa A, Yamamoto T, Sawada A, Minoura K, Hosogai N, Tahara A, Kurama T, Shimokawa T, Aramori I2009. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. Br J Pharmacol 158:879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C2010. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 31:224–243 [DOI] [PubMed] [Google Scholar]
- 34.Lazar DF, Saltiel AR2006. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat Rev Drug Discov 5:333–342 [DOI] [PubMed] [Google Scholar]
- 35.Luchsinger JA, Gustafson DR2009. Adiposity, type 2 diabetes, and Alzheimer’s disease. J Alzheimers Dis 16:693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Launer LJ2005. Diabetes and brain aging: epidemiologic evidence. Curr Diab Rep 5:59–63 [DOI] [PubMed] [Google Scholar]
- 37.Artola A2008. Diabetes-, stress- and ageing-related changes in synaptic plasticity in hippocampus and neocortex–the same metaplastic process? Eur J Pharmacol 585:153–162 [DOI] [PubMed] [Google Scholar]
- 38.García-San Frutos M, Fernández-Agulló T, De Solís AJ, Andrés A, Arribas C, Carrascosa JM, Ros M2007. Impaired central insulin response in aged Wistar rats: role of adiposity. Endocrinology 148:5238–5247 [DOI] [PubMed] [Google Scholar]
- 39.Blero D, De Smedt F, Pesesse X, Paternotte N, Moreau C, Payrastre B, Erneux C2001. The SH2 domain containing inositol 5-phosphatase SHIP2 controls phosphatidylinositol 3,4,5-trisphosphate levels in CHO-IR cells stimulated by insulin. Biochem Biophys Res Commun 282:839–843 [DOI] [PubMed] [Google Scholar]
- 40.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME1997. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275:661–665 [DOI] [PubMed] [Google Scholar]
- 41.D'Mello SR, Borodezt K, Soltoff SP1997. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci 17:1548–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241 [DOI] [PubMed] [Google Scholar]
- 43.Engel T, Hernández F, Avila J, Lucas JJ2006. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26:5083–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stubley-Weatherly L, Harding JW, Wright JW1996. Effects of discrete kainic acid-induced hippocampal lesions on spatial and contextual learning and memory in rats. Brain Res 716:29–38 [DOI] [PubMed] [Google Scholar]
- 45.Park CR, Seeley RJ, Craft S, Woods SC2000. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav 68:509–514 [DOI] [PubMed] [Google Scholar]
- 46.Kafkas A, Migo EM2009. Familiarity and recollection in the medial temporal lobe. J Neurosci 29:2309–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Squire LR, Wixted JT, Clark RE2007. Recognition memory and the medial temporal lobe: a new perspective. Nat Rev Neurosci 8:872–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelly A, Lynch MA2000. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology 39:643–651 [DOI] [PubMed] [Google Scholar]
- 49.Moult PR, Cross A, Santos SD, Carvalho AL, Lindsay Y, Connolly CN, Irving AJ, Leslie NR, Harvey J2010. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J Neurosci 30:4088– 4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor V, Wong M, Brandts C, Reilly L, Dean NM, Cowsert LM, Moodie S, Stokoe D2000. 5′ phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol Cell Biol 20:6860–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA2000. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3β. Mol Cell Biol 20:9356–9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yasuda M, Fukuchi M, Tabuchi A, Kawahara M, Tsuneki H, Azuma Y, Chiba Y, Tsuda M2007. Robust stimulation of TrkB induces delayed increases in BDNF and Arc mRNA expressions in cultured rat cortical neurons via distinct mechanisms. J Neurochem 103:626–636 [DOI] [PubMed] [Google Scholar]
- 53.Tsuneki H, Murata S, Anzawa Y, Soeda Y, Tokai E, Wada T, Kimura I, Yanagisawa M, Sakurai T, Sasaoka T2008. Age-related insulin resistance in hypothalamus and peripheral tissues of orexin knockout mice. Diabetologia 51:657–667 [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Noda Y, Zhou Y, Mouri A, Mizoguchi H, Nitta A, Chen W, Nabeshima T2007. The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in β amyloid25–35 i.c.v.-injected mice: involvement of dopaminergic systems. Neuropsychopharmacology 32:1261–1271 [DOI] [PubMed] [Google Scholar]