

Abstract
Glucokinase (GK) plays a crucial role as glucose sensor in glucose-induced insulin secretion in pancreatic β-cells. The bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) acts as an endogenous GK activator. Therefore, the goal of this study was the analysis of GK-PFK-2/FBPase-2 complex formation and its effect on metabolic stimulus-secretion coupling in β-cells in dependence upon glucose. The interaction between GK and PFK-2/FBPase-2 was analyzed in insulin-secreting MIN6 cells with a new fluorescence-based mammalian two-hybrid system. In contrast to the commonly used mammalian two-hybrid systems that require sampling before detection, the system used allows monitoring of the effects of environmental changes on protein-protein interactions on the single-cell level. Increasing the glucose concentration in the cell culture medium from 3 to 10 and 25 mmol/liter amplified the interaction between the enzymes stepwise. Importantly, in line with these results, overexpression of PFK-2/FBPase-2 in MIN6 cells evoked only at 10 and 25 mmol/liter, an increase in insulin secretion. Furthermore, a PFK-2/FBPase-2 mutant with an abolished GK-binding motif neither showed a glucose-dependent GK binding nor was able to increase insulin secretion. The results obtained with the mammalian two-hybrid system could be confirmed by fluorescence resonance energy transfer experiments in COS cells. Furthermore, the established interaction between GK and the liver GRP served in all experiments as a control. Thus, this study clearly showed that binding and activation of GK by PFK-2/FBPase-2 in β-cells is promoted by glucose, resulting in an enhancement of insulin secretion at stimulatory glucose concentrations, without affecting basal insulin secretion.
Live-cell imaging studies show that binding and activation of glucokinase by PFK-2/FBPase-2 in beta-cells is promoted by glucose resulting in an enhancement of insulin secretion.
The glucose sensor enzyme glucokinase (GK) plays a pivotal role in coupling millimolar changes of the extracellular glucose concentration to metabolism both in pancreatic β-cells and liver (1, 2, 3, 4). In β-cells glucose phosphorylation by GK is the rate-limiting step of glucose metabolism and thus of glucose-induced insulin secretion (5, 6, 7). Glucose is the most potent activator of GK, inducing a comprehensive conformational change. Furthermore, for short-term regulation of GK activity, posttranslational mechanisms including protein-protein interactions proved to be of particular importance. The liver-specific GK-regulatory protein (GRP), which competitively inhibits GK activity, was the first GK-binding protein to be identified (8). The interaction between GK and GRP takes place at low glucose concentrations and is stimulated in the presence of fructose-6-phosphate (8, 9). Furthermore the GK-GRP complex is translocated to the nucleus (10, 11, 12, 13). In previous experiments binding between GK and the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) has been documented (14, 15, 16, 17). The GK-binding motif is localized in the FBPase-2 domain and consists of the amino acids LKVWT for the β-cell isoform (15). Yeast two-hybrid studies have shown that both the liver and β-cell isoform of PFK-2/FBPase-2 are able to bind to the GK protein (15). PFK-2/FBPase-2 determines intracellular levels of fructose-2,6-bisphosphate by synthesizing and degrading this important regulator of carbohydrate metabolism (18, 19, 20). Overexpression of PFK-2/FBPase-2 results in a significant activation of GK enzyme activity in insulin-producing cells (16). In pancreatic β-cells the PFK-2/FBPase-2 acts not only as an endogenous activator of GK enzyme activity but also significantly improves the coupling between glycolysis and oxidative flux (14). Importantly, the activation of GK by PFK-2/FBPase-2 affects only the Vmax value of the enzyme, but not the S0.5 for glucose (16). Recently discovered small chemical GK activators allosterically activate the enzyme by lowering the glucose S0.5 (21, 22, 23, 24, 25, 26, 27, 28, 29). Some of the compounds were able to additionally increase the Vmax (21, 22, 25). GK activators enhance glucose-stimulated insulin release from pancreatic islets as well as glucose disposal by the liver (26, 28, 30). Further elucidation of the physiological GK activation by PFK-2/FBPase-2 will help to advance the concept of GK activation based on physiological needs for treatment of type 2 diabetes.
Because detection of protein-protein interactions is a major challenge in proteome research, two-hybrid assays have been established and modified in various ways in the past decade (31, 32, 33, 34, 35, 36, 37). Thus, a two-hybrid system was further adapted in this study to analyze interactions in insulin-producing cells. Both in mammalian cells and in yeast these assays make use of the fact that many eukaryotic transcriptional activators consist of two separable domains, which only initiate reporter gene transcription when they are closely approximated (38, 39, 40). The required proximity is established through direct binding between the proteins the interaction of which is studied. Solubility, folding, and posttranslational modifications required for specific protein interactions of mammalian proteins are partially insufficient in yeast. Thus, only the mammalian system provides a natural environment. In the systems used in this study the transcriptional activator domains are a herpes simplex virus VP16 transcriptional activation domain (AD) and the yeast GAL4 DNA-binding domain (BD), each of which is fused to one of the proteins of interest. Association of GAL4-DNA-BD and VP16-AD, driven by the interaction of the fused proteins of interest, results in binding to the promoter region and transcriptional activation of the reporter gene. In most mammalian two-hybrid experiments analyzing strong interactions of viral proteins, luciferase or chloramphenicol acetyltransferase activity was determined as a reporter signal (34, 35). However, in some studies fluorescent proteins, namely green fluorescent protein and enhanced yellow fluorescent protein (EYFP), have already been applied successfully as a reporter providing a higher resolution of signal detection (36, 41). The use of EYFP as a reporter gene, together with enhanced cyan fluorescent protein (ECFP) expression under control of a constitutive promoter, allows normalization for differences in transfection efficiency and enables quantitative analysis of protein-protein interactions on the single-cell level (41).
The present study applied such a fluorescence-based mammalian two-hybrid system in combination with a semiautomated microscopy approach to elucidate the regulatory principle of the interaction between GK and its activator PFK-2/FBPase-2 in insulin-secreting MIN6 cells. Moreover, this system was compared with the analysis of protein-protein interactions by the use of fluorescent fusion proteins and intracellular fluorescence resonance energy transfer (FRET). Finally, the effect of the GK-PFK-2/FBPase-2 interaction on the threshold of glucose-induced insulin secretion was determined.
Results
Glucose-dependent interaction between GK and the GRP in the mammalian two-hybrid system
The well-characterized interaction between GK and the GRP (42) could be assayed with the fluorescence-based mammalian two-hybrid system. The system was initially performed in COS cells, a previously established cellular model for the GK-GRP interaction (10). Transient transfection of COS cells with the GK and GRP two-hybrid plasmids whereas culturing the cells in medium containing 25 mmol/liter glucose resulted in a significant interaction signal, given as the ratio of EYFP over ECFP that was 70% higher than in negative controls. Because a two-hybrid signal is only produced by direct binding of the fusion proteins to each other, this indicates complex formation of GK and GRP. Incubation of the transfected cells with medium containing 5 mmol/liter glucose resulted in a 50% higher EYFP/ECFP ratio than with 25 mmol/liter glucose (Fig. 1). This is consistent with the role of GRP to bind GK in hepatocytes at low glucose concentrations. Thus, this mammalian two-hybrid system, which allows the calculation of the interaction strength on the single-cell level, is a suitable tool for analyzing protein-protein interactions in a quantitative manner while different extracellular parameters can be set.
Fig. 1.

Glucose-dependent modulation of GK binding to the GRP. The interaction strength of the fusion proteins DNA-BD-GRP and AD-GK was measured in a fluorescence-based mammalian two-hybrid assay. DNA-BD and AD-GK (white bars) or DNA-BD-GRP and AD (dashed bars) or DNA-BD-GRP and AD-GK (black bars) were coexpressed in COS cells. The cells were preincubated in the presence of 5 mmol/liter or 25 mmol/liter glucose. Thereafter, the fluorescence intensities of ECFP and EYFP were determined. Data are expressed as means ± sem of three to five individual experiments with a total of 142–266 nuclei analyzed. **, P < 0.01; ***, P < 0.001 compared with negative controls; ##, P < 0.01 compared with cells incubated with 5 mmol/liter glucose (ANOVA/Bonferroni’s test).
Glucose-dependent binding of islet PFK-2/FBPase-2 to GK
The two-hybrid system was established in MIN6 cells to study the interaction between GK and PFK-2/FBPase-2 in insulin-secreting cells. Furthermore, a triple mutation was inserted in the GK-binding motif of PFK-2/FBPase-2, and the GK-binding behavior of this mutant (PFK-2/FBPase-2-Mut) was compared with the wild type. The islet PFK-2/FBPase-2 isoform fused to the GAL4-DNA-binding domain showed a significant interaction with GK fused to the VP16 AD when the cells were incubated with standard medium containing 25 mmol/liter glucose. The EYFP/ECFP ratio was 2.3 times higher than the ratio of the negative controls. Thus, the interaction of GK with PFK-2/FBPase-2 can be regarded as a weak interaction. At a low glucose concentration of 3 mmol/liter the EYFP reporter signal expression in relation to constitutively expressed ECFP was diminished to a value only slightly above the negative control (Fig. 2A). This suggests that an even smaller fraction of GK is bound in the complex with PFK-2/FBPase-2 when the glucose concentration is decreased. The PFK-2/FBPase-2-Mut showed at 3 mmol/liter glucose with 108% a minor GK interaction comparable to the wild-type PFK-2/FBPase-2 (Fig. 2B). The significant increase in GK binding of the PFK-2/FBPase-2 wild type to 146% at 25 mmol/liter glucose was not detectable for the mutant. Interaction between GK and PFK-2/FBPase-2-Mut at 25 mmol/liter glucose was with 114% significantly lower compared with wild-type PFK-2/ FBPase-2 (Fig. 2B).
Fig. 2.
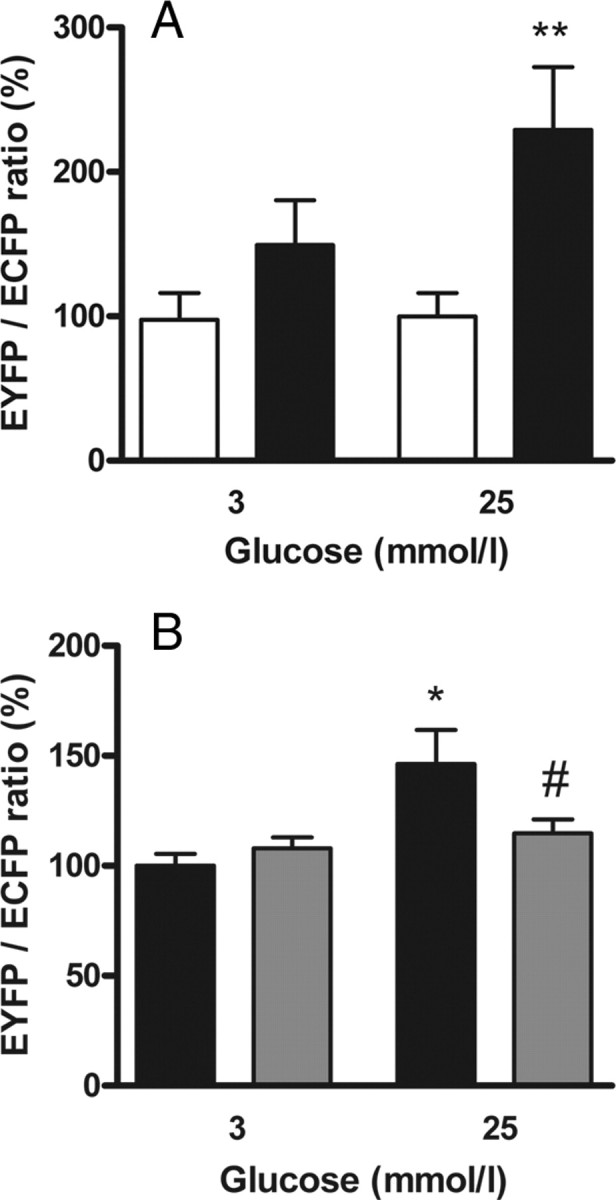
Glucose promoted binding of islet PFK-2/FBPase-2 to GK. The interaction strength of the fusion proteins DNA-BD-PFK-2/FBPase-2 or DNA-BD-PFK-2/FBPase-2-Mut and AD-GK was measured in a fluorescence-based mammalian two-hybrid assay in insulin-secreting MIN6 cells. A, DNA-BD-PFK-2/FBPase-2 and AD (white bars) or DNA-BD-PFK-2/FBPase-2 and AD-GK (black bars) were coexpressed in MIN6 cells and preincubated with 3 mmol/liter or 25 mmol/liter glucose. Thereafter, the fluorescence intensities for ECFP and EYFP were determined. Data are expressed as means ± sem of three individual experiments with a total of 60–70 nuclei analyzed. **, P < 0.01 compared with negative control (ANOVA/Bonferroni’s test). B, DNA-BD-PFK-2/FBPase-2 and AD-GK (black bars) or DNA-BD-PFK-2/FBPase-2-Mut and AD-GK (gray bars) were coexpressed in MIN6 cells and preincubated with 3 mmol/liter or 25 mmol/liter glucose. Thereafter, the fluorescence intensities for ECFP and EYFP were determined. Data are expressed as means ± sem of three individual experiments with a total of 44–152 nuclei analyzed. *, P < 0.05 compared with cells incubated with 3 mmol/liter glucose; #, P < 0.05 compared with DNA-BD-PFK-2/FBPase-2 wild type (ANOVA/Bonferroni’s test).
A semiautomated approach to analyze protein-protein interactions with the mammalian two-hybrid system
The use of a semiautomated microscopy approach made it possible to simultaneously monitor the interaction strength of GK binding to PFK-2/FBPase-2 at three different glucose concentrations over a time period of 40 h. The signals were highly variable for 10 h after adjusting the different glucose concentrations. The number of cells expressing fluorescent protein was low during this 10-h time period. Then the values ranged around a plateau before the negative controls started to increase slightly from 50 h after transfection. Generally, the negative controls showed less variability than the samples in which GK and islet PFK-2/FBPase-2 were coexpressed. It is noteworthy that the EYFP/ECFP ratio exhibited an oscillating profile particularly in the coexpressing cells and slightly in the negative controls, possibly due to batch expression of the proteins (Fig. 3A). Therefore, the values were averaged over the period from 36–48 h post transfection for final analysis. GK and islet PFK-2/FBPase-2 showed a significant interaction signal when the cells were preincubated at 25 mmol/liter glucose. Also at 10 mmol/liter glucose, the proteins showed a distinct interaction. At 3 mmol/liter glucose only a slightly higher EYFP/ECFP ratio than in the negative controls was detected. The interaction strength at 3 mmol/liter glucose was significantly lower than at 25 mmol/liter (Fig. 3B).
Fig. 3.

The interaction strength between GK and PFK-2/FBPase-2 rises with increasing glucose concentrations. The interaction of the fusion proteins DNA-BD-PFK-2/FBPase-2 and AD-GK was measured in a semiautomated fluorescence-based mammalian two-hybrid assay. DNA-BD-PFK-2/FBPase-2 and AD (dashed line in A and white bars in B) or DNA-BD-PFK-2/FBPase-2 and AD-GK (solid line in A and black bars in B) were coexpressed in insulin-secreting MIN6 cells. In cells cultured at 3, 10, or 25 mmol/liter glucose, respectively, the degree of interaction between DNA-BD and AD proteins was analyzed every hour as the mean value for the nuclear EYFP/ECFP ratio. A, Images were taken hourly over a period of 40 h. The time lapse of the calculated EYFP/ECFP ratio is exemplary, shown for a representative single experiment for the interaction of GK with islet PFK-2/FBPase-2 at 25 mmol/liter glucose (solid line) together with the respective negative control at 25 mmol/liter glucose (dashed line). Data are expressed as means ± sem of 2–120 nuclei analyzed per time point. B, The mean nuclear EYFP/ECFP ratios obtained between 36 and 48 h after transfection were determined together with the respective sem for 341–1369 cells per sample. The means ± sem of the EYFP/ECFP ratio are shown for three to five independent experiments with a total of 1331–5905 nuclei analyzed. *, P < 0.05 compared with negative control, #, P < 0.05 compared with cells incubated with 3 mmol/liter glucose (ANOVA/Bonferroni’s test).
Effect of low vs. high glucose on colocalization between GK and PFK-2/FBPase-2
The morphology and size of COS cells allow a standardized cytoplasmic region analysis necessary for colocalization as well as FRET studies. Thus, COS cells were cotransfected with expression vectors encoding the fluorescent fusion proteins ECFP-GK and wild-type or mutant EYFP-PFK-2/FBPase-2 and preincubated in medium containing 3 mmol/liter or 25 mmol/liter glucose. After both preincubation conditions, cells showed comparable cell viability and cytoplasmic localization of ECFP-GK and wild-type or mutant EYFP-PFK-2/FBPase-2. GK and wild-type PFK-2/FBPase-2 as well as GK and PFK-2/FBPase-2-Mut exhibited distinct colocalization at both glucose concentrations indicating cytoplasmic cocompartmentalization (Fig. 4). The degree of colocalization between ECFP-GK and EYFP-PFK-2/FBPase-2 wild type was 7.8% at 3 mmol/liter glucose and 11.0% at 25 mmol/liter glucose (Fig. 4C). Mutation of the GK-binding motif of PFK-2/FBPase-2 resulted in degrees of colocalization of 6.9% at 3 mmol/liter glucose and 8.1% at 25 mmol/liter glucose (Fig. 4C).
Fig. 4.
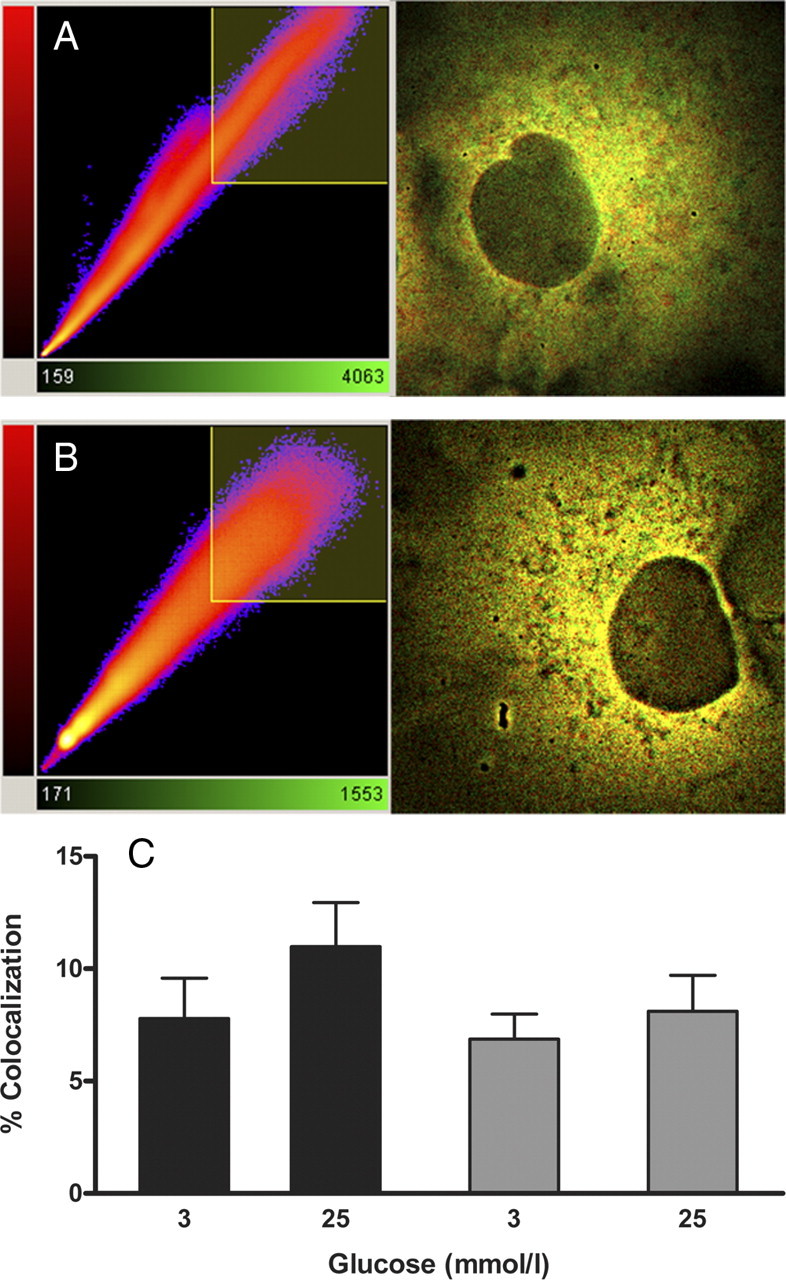
Effect of low vs. high glucose on colocalization between GK and islet PFK-2/FBPase-2. ECFP-GK and EYFP-PFK-2/FBPase-2 or EYFP-PFK-2/FBPase-2-Mut fusion proteins were co-overexpressed in COS cells and precultured in medium containing 3 mmol/liter or 25 mmol/liter glucose, respectively. Fluorescence images were taken, and the degree of colocalization was calculated with the Imaris software (Bitplane). A typical image overlay of one cell co-overexpressing ECFP-GK and EYFP-PFK-2/FBPase-2 incubated at 3 mmol/liter glucose (A) and one at 25 mmol/liter glucose (B) is shown with the associated correlation diagram. ECFP-GK is depicted in green and EYFP-PFK-2/FBPase-2 is shown in red. C, Colocalization of ECFP-GK and EYFP-PFK-2/FBPase-2 (black bars), and of ECFP-GK and EYFP-PFK-2/FBPase-2-Mut (gray bars) is expressed as means ± sem of three individual experiments with a total of five to eight cells analyzed.
Effect of low vs. high glucose on FRET between GK and GRP or PFK-2/FBPase-2
Sensitized emission-based FRET efficiency (FRETN) was measured between ECFP and EYFP fused to each other to determine the FRETN detection range of the microscopy setup. FRETN was 9.1 and therefore by the factor of 31 higher than the negative control in which ECFP and EYFP were expressed separately. A low FRET rate was also detectable in the negative control because of a residual dimerization tendency of ECFP and EYFP as well as bleed-through that happens when the donor ECFP is excited by the acceptor EYFP excitation wavelength or vice versa (Fig. 5A). ECFP-GK and EYFP-GRP showed a significant FRET signal both at 5 mmol/liter and 25 mmol/liter glucose. At low glucose concentration, FRETN was 2.3 times higher than the negative control and 50% higher in comparison to cells incubated at 25 mmol/liter glucose.
Fig. 5.

Effect of low vs. high glucose on FRET between GK and its binding partners GRP and islet PFK-2/FBPase-2. Fluorescent proteins were coexpressed in COS cells and precultured with the indicated glucose concentrations. Thereafter, fluorescence images were taken in living cells, and the sensitized emission-based FRET efficiency (FRETN) was calculated from the ECFP and EYFP emission intensities. A, FRETN between separately expressed ECFP and EYFP (white bars) or the fluorescent fusion protein ECFP-EYFP (black bars). B, FRETN between EYFP-GRP and ECFP (white bars) or ECFP-GK (black bars). C, FRETN between EYFP-PFK-2/FBPase-2 and ECFP (white bars) or ECFP-GK (black bars), and between EYFP-PFK-2/FBPase-2-Mut and ECFP (dashed bars) or ECFP-GK (gray bars). Data are expressed as means ± sem of three individual experiments with a total of 8–22 cells analyzed. *, P < 0.05; ***, P < 0.001 compared with negative control; ###, P < 0.001 compared with cells incubated with 3 mmol/liter or 5 mmol/liter glucose; §§§, P < 0.001 compared with EYFP-PFK-2/FBPase-2 wild type, respectively (panel A, Student’s t test; panels B and C, ANOVA/Bonferroni’s test).
Although the fusion proteins ECFP-GK and EYFP-PFK-2/FBPase-2 wild type showed only a slightly higher colocalization at 25 mmol/liter glucose than at 3 mmol/liter glucose when they were co-overexpressed in COS cells, the enzymes exhibited FRET exclusively at 25 mmol/liter glucose. At 25 mmol/liter glucose FRETN was, with 0.82, 2.5 times higher than the negative control. In contrast, no significant FRETN efficiency was detectable between ECFP-GK and EYFP-PFK-2/FBPase-2-Mut at 25 mmol/liter glucose. When the cells were preincubated with medium containing 3 mmol/liter glucose, no FRET was observed between ECFP-GK and EYFP-PFK-2/FBPase-2, neither for the wild type nor for the mutant PFK-2/FBPase-2 (Fig. 5C).
Islet PFK-2/FBPase-2 expression evoked a glucose-dependent increase in insulin secretion
MIN6 cells were transfected with EYFP as a control or EYFP-PFK-2/FBPase-2 or EYFP-PFK-2/FBPase-2-Mut and insulin secretion were determined after 1-h starvation and subsequent stimulation with 3, 10, or 25 mmol/liter glucose. Comparable transfection efficiency and protein expression level of EYFP, EYFP-PFK-2/FBPase-2, and EYFP-PFK-2/FBPase-2-Mut were verified by fluorescence microscopy. Control cells showed a glucose-induced insulin secretion with a threshold above 3 mmol/liter glucose (Fig. 6). Whereas at 3 mmol/liter glucose insulin secretion in EYFP-PFK-2/FBPase-2 transfected cells was comparable to control cells, at 10 and 25 mmol/liter EYFP-PFK-2/FBPase-2 expression induced a higher insulin secretion. At 10 mmol/liter glucose insulin secretion of EYFP-PFK-2/FBPase-2-transfected cells was significantly higher in comparison to control cells (Fig. 6). In contrast, transfection with EYFP-PFK-2/FBPase-2-Mut did not evoke an increase in insulin secretion in MIN6 cells. Thus, insulin secretion of wild-type PFK-2/FBPase-2-expressing cells was at 10 and 25 mmol/liter glucose significantly higher compared with mutant PFK-2/FBPase-2-expressing cells (Fig. 6).
Fig. 6.

Effect of islet PFK-2/FBPase-2 on glucose-induced insulin secretion. Insulin-secreting MIN6 cells were transfected with EYFP (white bars), EYFP-PFK-2/FBPase-2 (black bars), or EYFP-PFK-2/FBPase-2-Mut (gray bars) and cultured for 48 h. Cells were starved for 1 h and thereafter stimulated for 1 h with 3, 10, or 25 mmol/liter glucose, respectively. Insulin secretion is shown as insulin secreted per cellular insulin content and protein. Data are expressed as means ± sem of four individual experiments. *, P < 0.05 compared with negative control; #, P < 0.05; ##, P < 0.01 compared with EYFP-PFK-2/FBPase-2-Mut stimulated with the same glucose concentration (ANOVA/Bonferroni’s test).
Discussion
The glucose sensor enzyme GK plays a crucial role during stimulus-secretion coupling in pancreatic β-cells. In contrast to the transcriptional regulation of liver GK by insulin and glucagons, the β-cell isoenzyme protein levels are only marginally affected by these hormones (43, 44). Posttranslational regulation mechanisms of GK, including protein-protein interaction and compartmentalization processes, therefore make an important contribution to the complex network maintaining glucose homeostasis (44, 45, 46). In β-cells the GK protein is exclusively localized in the cytoplasm. The cytoplasmic bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2), a key regulator of glucose metabolism, proved to be a binding partner of GK (15, 17). Unlike the GRP, which competitively inhibits GK activity only in liver (10, 11, 12, 13), PFK-2/FBPase-2 proved to be an activating GK-binding partner in both liver and pancreatic β-cells (14, 16, 17). Furthermore, PFK-2/FBPase-2 amplified the increase of intrinsic GK activity by glucose, whereas both the affinity of GK for glucose and the positive cooperativity were unaffected (16). Thus, it becomes apparent, that the endogenous GK activator PFK-2/FBPase-2 plays an important role in glucose-induced insulin secretion in pancreatic β-cells.
The objective of this study was to analyze the interaction of GK with the β-cell isoform of PFK-2/FBPase-2 with special emphasis on glucose dependency. For the purpose of detecting differences in the interaction of the enzymes in a physiological environment, fusion proteins of the wild type and a mutant PFK-2/FBPase-2 were generated to perform mammalian two-hybrid and FRET experiments. The GK-binding motif is located within the FBPase-2 domain of PFK-2/FBPase-2 (15). Three of the five amino acids forming this motif were mutated to abolish the GK-binding characteristics in this mutant PFK-2/FBPase-2.
Mammalian two-hybrid and FRET experiments were similarly applied to the interaction of GK with its regulatory protein (GRP) the modulation of which by glucose has been described earlier (10, 11, 12, 13). Thus, the action of both GK interaction partners can be compared with respect to the liver metabolism. FRET efficiency strongly depends on the distance of the donor-acceptor pair of fluorophores but also on the relative orientation of the dipole moments of donor and acceptor. Although the performed sensitized emission-based FRET measurements must be considered as semiquantitative, the obtained values for the interaction of GK both with PFK-2/FBPase-2 and the GRP in the cytoplasm of COS cells in relation to the internal positive control support the classification of both interactions as weak. However, it must be taken into account that in the positive control ECFP and EYFP are directly fused to each other, providing a 1:1 ratio of donor and acceptor; an exact 1:1 stoichiometry, however, cannot be achieved by expression of proteins from different vectors, in this case ECFP-GK and EYFP-GRP or EYFP-PFK-2/FBPase-2. Although there is a significant interaction between GK and wild-type PFK-2/FBPase-2 at high glucose, the binding to the GRP was minor. At low glucose the GK-PFK-2/FBPase-2 interaction was diminished, but a significant binding between GK and GRP was detectable. Comparable results were obtained with the mammalian two-hybrid system.
Crystal structure analysis of GK protein revealed a closed conformation of catalytically active GK and a superopened conformation in which GK is inactive (27). From kinetic modeling there is strong evidence that up to three additional conformational intermediates exist between the two already established ones and that the percentage of each conformation depends upon the glucose concentration (27, 47, 48). Transferring this multiple conformational state model to the situation in the liver, this study provides evidence for an increase of the closed conformation at high glucose through interaction of GK with PFK-2/FBPase-2. In contrast, at low glucose PFK-2/FBPase-2 did not interact with GK, and the enzyme switches to the superopened conformation. In this conformation GRP is able to bind GK and to translocate the enzyme to the nucleus (10, 13). Thus, both GK binding partners seem to be important to adjust liver metabolism to the needs of fasting and refeeding. As posttranslational modifications of PFK-2/FBPase-2 (17, 18, 19, 20) are not taken into account here, the hypothesis must be further tested in future experiments with special emphasis on the liver metabolism.
Colocalization in COS cells was highest at 25 mmol/liter between GK and wild-type PFK-2/FBPase-2 but not significantly lower at 3 mmol/liter glucose or between GK and the mutant PFK-2/FBPase-2. Assuming a compartmentalization of the cytoplasm for macromolecular channelling of glycolytic intermediates, this indicates that both proteins are located in close proximity, which is a prerequisite for a direct interaction. The direct interaction, determined by FRET, was detectable only between GK and wild-type PFK-2/FBPase-2 at 25 mmol/liter. Thus, GK-PFK-2/FBPase-2 complex formation seems to be directly dependent upon the glucose concentration and feasible only with an intact binding motif in the FBPase-2 domain. Interestingly, mammalian two-hybrid experiments in insulin-secreting MIN6 cells provided further evidence for an only very weak, and thus not significant, GK interaction at 3 mmol/liter glucose with both the wild-type and the mutant PFK-2/FBPase-2. At 25 mmol/liter glucose, a significant interaction was detectable exclusively between GK and wild-type PFK-2/FBPase-2.
Two-hybrid systems offer a great advantage for measurement of direct binary interactions. Furthermore, implementation of the fluorescent protein EYFP as a reporter in the mammalian two-hybrid system allows detection of weak protein-protein interactions in living mammalian cells in a quantitative manner, not achievable through analysis of the reporter gene alkaline phosphatase in the supernatant medium of cultured cells. Outlier cells that would strongly influence the mean value of a data set can be easily identified by single-cell analysis. However, fluorescence microscopy-based data acquisition, as well as analysis, is time consuming. Thus, the number of analyzed cells within a manually performed two-hybrid experiment is limited. Furthermore protein-protein interactions could be monitored only during a narrow time period. To increase both the number of analyzed cells and the recorded time points, an open fluorescence microscope scanning system was used in a semiautomated approach. In addition, high content data analysis was performed including background correction, automated cell detection, and cell tracking. Finally the EYFP/ECFP ratio was directly available on the single-cell level. Cells with a low or high constitutive ECFP expression were excluded from the final analysis using gates in the scan^R analysis software. This procedure has significantly reduced the se of the measurement. Thus, this approach made it possible to determine the interaction between GK and PFK-2/FBPase-2 at three different glucose concentrations in parallel for a time period of 40 h in insulin-secreting MIN6 cells. The data obtained with this approach, in fact, suggest a stepwise amplification of interaction strength between the proteins with increasing glucose concentrations. Thus, it could be shown for the first time that the GK-PFK-2/FBPase-2 binding process itself is glucose dependent.
Together with the previous observation that overexpression of β-cell PFK-2/FBPase-2 resulted in a greater stimulatory effect of glucose on GK enzyme activity when the glucose concentration was increased (14), the present study provides direct evidence for a shift of the GK conformational equilibrium to its enzymatically active closed conformation by PFK-2/FBPase-2 binding. Thus, complex formation prevents the slow conformational change of GK into the superopened conformation. Finally, the interaction leads to an increase in GK enzyme activity, thereby facilitating the role of GK as a sensor for glucose-induced insulin secretion. Actually, in line with the increase in GK-PFK-2/FBPase-2 binding, an increase in insulin secretion was obtained at 10 and 25 mmol/liter glucose through overexpression of PFK-2/FBPase-2 in MIN6 cells. Furthermore, an increase in insulin secretion was not detectable after overexpression of the mutant PFK-2/FBPase-2 not able to interact with GK. The results fit with previous observations in β-cells with respect to the threshold of glucose-induced insulin secretion (2, 4, 5, 6), because at 3 mmol/liter glucose, overexpression of PFK-2/FBPase-2 did not evoke an increase in insulin secretion. In contrast, treatment of β-cells with small synthetic GK activators (21, 22, 24, 25) caused an increase in insulin secretion already at such glucose concentrations, thus changing the threshold of glucose-induced insulin secretion.
The present study underlines the physiological importance of posttranslational GK regulation by protein-protein interactions. Because GK activation has been described as a promising therapeutic strategy in the treatment of type 2 diabetes, further elucidation of the endogenous GK activation mechanisms will contribute to a better understanding of the metabolic coupling between GK activation and glucose-stimulated insulin secretion in β-cells. Furthermore, a viable method for the intracellular detection of a weak protein-protein interaction and its posttranslational modification was established.
Materials and Methods
Materials
Restriction enzymes and modifying enzymes for the cloning procedures were from New England Biolabs (Beverly, MA) or Fermentas (St. Leo-Rot, Germany). Custom oligonucleotides were synthesized by MWG Biotech (Ebersberg, Germany). The reporter vector pHASH-3 was a kind gift of Dr. S. Herlitze (Department of Neurosciences, Case Western Reserve University, Cleveland, OH). All reagents of analytical grade were from Merck (Darmstadt, Germany). All tissue culture equipment was from Invitrogen (Karlsruhe, Germany) and Greiner-Bio One (Frickenhausen, Germany).
Cell culture and transient transfection
COS cells and MIN6 cells were grown in DMEM supplemented with 25 mmol/liter glucose, 10% (vol/vol) fetal calf serum, 10 U/ml penicillin, 10 μg/ml streptomycin, and 2 mmol/liter glutamine in a humidified atmosphere at 37 C and 5% CO2. For the mammalian two-hybrid experiments, COS cells (passages 13–35) were seeded in six-well microplates at a density of 3 × 104 cells and grown for 1 d. MIN6 cells (passages 37–44) were seeded in six-well microplates at a density of 8 × 104 cells and grown for 5 d. Thereafter, cells were transfected with the mammalian two-hybrid vectors with jetPEI (Qbiogene, Montreal, Quebec, Canada) according to the manufacturer’s instructions. For each transfection 0.4 pmol of the reporter vector pHASH-3 was used in combination with 0.1 pmol of the expression vector pM (negative control), pM-GRP, pM-PFK-2/FBPase-2 or pM-PFK-2/FBPase-2-Mut, and 0.1 pmol of the expression vector pVP16 (negative control) or pVP16-GK. COS cells were incubated for 21 h in the presence of 5 mmol/liter or 25 mmol/liter glucose, respectively, before image acquisition. MIN6 cells were incubated for 20 h after transfection with standard DMEM containing 25 mmol/liter glucose. Thereafter, cells were incubated in the presence of 3 mmol/liter or 25 mmol/liter glucose, respectively, for another 24 h before image acquisition. For the semiautomated scan^R approach MIN6 cells (passages 39–40) were seeded at a density of 1.5 × 105 in 12-well microplates and grown for 3 d. Then the cells were transfected with equimolar amounts of the vectors pGL4.EYFP, pBIND.ECFP-PFK-2/FBPase-2, and pACT or pACT-GK with a total amount of 1 μg DNA and grown for 24 h in standard DMEM. Thereafter, the medium was replaced, and cells were incubated in DMEM containing 3, 10, or 25 mmol/liter glucose. Image acquisition was started 1 h after medium replacement. For colocalization and FRET studies, COS cells (passages 5–20) were seeded at a density of 2 × 104 cells on 35-mm glass-bottom dishes (MatTek, Ashland, MA) and grown for 1 d. Thereafter, cells were transfected as described above with either 0.5 μg of pECFP or pECFP-GK plasmid and 0.5 μg pEYFP-GRP or 0.5 μg of pEYFP-PFK-2/FBPase-2 or 0.5 μg of pEYFP-PFK-2/FBPase-2-Mut, or with 0.5 μg pECFP and 0.5 μg pEYFP, or with 1 μg pECFP-EYFP alone. The culture medium was replaced 1 d after transfection, and the cells were incubated for another 24 h with medium containing 3, 5, or 25 mmol/liter glucose, respectively, before image acquisition.
Mammalian two-hybrid analysis
Mammalian two-hybrid analysis was performed using the cloning vectors pM and pVP16 from the BD Matchmaker Mammalian Assay Kit 2 (CLONTECH Laboratories, Inc., Palo Alto, CA) to create the GAL4 DNA-BD and VP16 AD fusion constructs, respectively. The cDNA sequences of rat β-cell PFK-2/FBPase-2 and rat liver GRP were amplified by PCR and subcloned in frame (SalI and HindIII restriction sites) to the GAL4-DNA-BD into pM. Human β-cell GK coding cDNA was amplified by PCR and subcloned in frame (SalI and HindIII restriction sites) to the VP16-AD into pVP16 (CLONTECH). The fluorescence-based reporter plasmid pHASH-3 (41) was used for transfection in combination with the pM and pVP16 constructs. pHASH-3 contains the gene for the yellow fluorescent protein (EYFP) under control of an inducible promoter containing GAL4-binding sites. Furthermore, pHASH-3 carries the cyan fluorescent protein (ECFP) gene, which is constitutively expressed. Both EYFP and ECFP carry a nuclear localization signal so that fluorescence is detectable in the nucleus of transfected cells. COS cells or MIN6 cells were transfected with the two-hybrid vectors for expression of the fusion proteins and the reporter vector.
For a semiautomated microscope approach of mammalian two-hybrid analysis, vectors of the CheckMate/Flexi Vector Mammalian Two-Hybrid System (Promega Corp., Madison, WI) were modified as follows. Human β-cell GK coding cDNA was amplified by PCR and subcloned in frame (SgfI and PmeI restriction sites) to the VP16-AD into pFN10A (ACT) Flexi Vector to generate pACT-GK. The cDNA sequence of rat β-cell PFK-2/FBPase-2 was amplified by PCR and subcloned in frame (SgfI and PmeI restriction sites) to the GAL4-DNA-BD into pFN11A (BIND) Flexi Vector to generate pBIND-PFK-2/FBPase-2. To generate pECFP-Nuc, the vectors pECFP-N1 and pEYFP-Nuc (CLONTECH) were digested with the restriction enzymes AgeI and BsrGI, and the EYFP was replaced by ECFP. The cDNA of ECFP-Nuc, including the cytomeglovirus promoter and simian virus 40 early polyadenylation signal, was amplified by PCR using composite primers to introduce PvuII (5′) and ClaI (3′) restriction sites. Renilla Luciferase coding cDNA was removed from pBIND-PFK-2/FBPase-2 by digestion with PvuII and ClaI, and ECFP-Nuc was subcloned into the vector frame. The resulting vector was called pBIND.ECFP-PFK-2/FBPase-2. The cDNA sequence of EYFP-Nuc was amplified by PCR using composite primers to introduce HindIII (5′) and FseI (3′) restriction sites. The coding luc2P firefly luciferase cDNA was removed from the reporter vector pGL4.31 (Promega) by digestion with HindIII and FseI, and EYFP-Nuc was subcloned into the vector frame. The resulting reporter vector was called pGL4.EYFP. MIN6 cells were transfected with pGL4.EYFP, pBIND.ECFP-PFK-2/FBPase-2 and pACT (Promega) as negative control or with pACT-GK. For analysis of an interaction of the fusion proteins fluorescence intensities of EYFP and ECFP were determined in the nuclei of transfected cells. To quantify the interaction strength the ratio EYFP/ECFP was calculated for single cells from the background corrected average gray values of the EYFP and ECFP fluorescence intensities.
Colocalization and FRET analyses
The expression vectors pECFP-GK and pEYFP-GRP, as described previously (10), were used for overexpression of fluorescent fusion proteins. Rat β-cell PFK-2/FBPase-2 coding cDNA was amplified by PCR and subcloned in frame (KpnI and BamHI restriction sites) to the EYFP into pEYFP-C1 (CLONTECH). A plasmid encoding an ECFP-EYFP fusion protein with a two-amino acid spacer between ECFP and EYFP was generated as described elsewhere (49) and used as a positive control for FRET. COS cells were transfected with combinations of ECFP and EYFP plasmids. ECFP and EYFP fluorescence intensities were recorded in the cytoplasm of living cells by fluorescence microscopy. Colocalization of ECFP and EYFP was determined with the Imaris software (Bitplane, Zurich, Switzerland). The sensitized emission-based FRET efficiency (FRETN) was calculated from the ECFP emission with excitation at 436 nm, EYFP emission with excitation at 436 nm, and EYFP emission with excitation at 500 nm, based on the calculation of Vanderklish et al. (50).
Mutagenesis
The LKVWT GK-binding motif in the FBPase-2 domain of rat islet PFK-2/FBPase-2 (15) was changed to HKEWR (PFK-2/FBPase-2-Mut). Site-directed mutagenesis was performed in pM-PFK-2/FBPase-2 and pEYFP-PFK-2/FBPase-2 using the QuikChange II Site-Directed Mutagenesis Kit according to the manufacturer’s instructions (Stratagene, La Jolla, CA). The sequences of the specific mismatch oligonucleotides for generation of the triple mutant were g gag atc cag gac cac aaa gag tgg aga agc cag ttg aag ag (forward) and ct ctt caa ctg gct tct cca ctc ttt gtg gtc ctg gat ctc c (reverse). Mutagenesis was verified by sequence analysis.
Fluorescence microscopy
A cell^R/Olympus IX81 inverted microscope system equipped with a Cellcubator, as described previously (45), was used. D436/10-455DCLP-D480/40 and HQ500/20-530DCLP-D560/40 single-band filter sets were used for ECFP and EYFP, respectively (AHF Analysentechnik, Tübingen, Germany). For the mammalian two-hybrid analyses the cell culture plates with transfected, living cells were fixed on the microscope stage, and images were obtained with an UPLSAPO 20 × 0.75 numerical aperture objective (Olympus, Hamburg, Germany). For colocalization and FRET studies glass-bottom dishes were fixed on the microscope stage, and ECFP and EYFP images were obtained with an UPLSAPO 60 × 1.35 numerical aperture oil-immersion objective (Olympus). For FRET measurements, ECFP and EYFP emission were detected simultaneously using a DV-CC Dual View Simultaneous Imaging System (Optical Insights, LLC, Tucson, AZ) equipped with a 505 dcxr beam splitter and D465/30 and HQ535/30 emission filters (AHF Analysentechnik). For multiple image analysis of 12-well microplates, all motorized devices of the microscope system were synchronized by the scan^R acquisition software (Olympus Soft Imaging Solutions, Munich, Germany). The image focus was automatically determined with a gradient method from transmitted light images. In a 40-h time-lapse experiment 20 fluorescence images of each well were taken hourly with an UPLSAPO 20 × 0.75 numerical aperture objective (Olympus, Hamburg, Germany). Analysis parameters were specified and image processing was carried out with the scan^R analysis software. After background correction, individual cells were detected using an intensity threshold method and tracked over time. By setting gates, only cells with a mean ECFP fluorescence were finally considered to exclude artifacts. Thereafter the ECFP/EYFP ratio of each single cell was available over time.
Measurement of insulin secretion
MIN6 cells (passages 37–40) were seeded in six-well microplates at a density of 3.5 x 105 cells and grown for 3 d. Thereafter, cells were transfected with jetPEI (Qbiogene, Montreal, Quebec, Canada) according to the manufacturer’s instructions and 2 μg EYFP, EYFP-PFK-2/FBPase-2 or EYFP-PFK-2/FBPase-2-Mut and grown for 24 h. Thereafter, the medium was replaced by fresh standard DMEM. Transfection efficiency and protein expression were quantified using automated scan^R fluorescence microscopy. Cells were incubated for an additional 24 h in DMEM. Finally, cells were incubated for 1 h in bicarbonate-buffered Krebs-Ringer solution without glucose supplemented with 0.1% albumin and thereafter stimulated for 1 h with 3, 10, or 25 mmol/liter glucose, respectively. Thereafter, 1 ml of the incubation buffer from each well was carefully harvested and gently centrifuged to remove detached cells. In the final supernatants the secreted insulin was measured. Cells were homogenized by sonication in PBS (pH 7.4), and insulin content was measured in soluble fractions. Insulin was measured by ELISA, and the protein concentration was quantified by a Bradford protein assay.
Statistical analyses
Data are expressed as means ± sem. Statistical analyses were performed by ANOVA followed by Bonferroni’s test for multiple comparison or Student’s t test using the Prism analysis program (Graphpad, San Diego, CA).
Acknowledgments
We thank Dr. S. Herlitze (Department of Neurosciences, Case Western Reserve University, Cleveland, OH) for providing the reporter vector pHASH-3 and Dr. D. Krüger (Olympus Soft Imaging Solutions, Munich, Germany) for excellent technical support regarding the scan^R system. We also thank J. Kresse and R. Waterstradt for skillful technical assistance.
Footnotes
This work was supported by the European Union (Integrated Project EuroDia LSHM-CT-2006-518153 in the Framework Program 6 of the European-Community) and grants from the Dr. Buding Foundation and the German Diabetes Association (to S.B.).
Disclosure summary: The authors have nothing to disclose.
First Published Online August 11, 2010
Abbreviations: AD, Activation domain; BD, binding domain; ECFP, enhanced cyan fluorescent protein; EYFP, enhanced yellow fluorescent protein; FRET, fluorescence resonance energy transfer; FRETN, sensitized emission-based FRET efficiency; GK, glucokinase; GRP, glucokinase regulatory protein; PFK-2/FBPase-2, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase.
References
- 1.Baltrusch S, Tiedge M2006. Glucokinase regulatory network in pancreatic β-cells and liver. Diabetes 55(Suppl 2):S55–S64
- 2.Lenzen S, Panten U1988. Signal recognition by pancreatic B-cells. Biochem Pharmacol 37:371–378 [DOI] [PubMed] [Google Scholar]
- 3.Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL, Niswender K, Magnuson MA1993. Glucokinase as pancreatic β cell glucose sensor and diabetes gene. J Clin Invest 92:2092–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meglasson MD, Matschinsky FM1984. New perspectives on pancreatic islet glucokinase. Am J Physiol 246:E1–E13 [DOI] [PubMed]
- 5.Matschinsky FM1996. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45:223–241 [DOI] [PubMed] [Google Scholar]
- 6.Matschinsky FM2002. Regulation of pancreatic β-cell glucokinase: from basics to therapeutics. Diabetes 51(Suppl 3):S394–S404 [DOI] [PubMed]
- 7.Matschinsky FM, Glaser B, Magnuson MA1998. Pancreatic β-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes 47:307–315 [DOI] [PubMed] [Google Scholar]
- 8.Van Schaftingen E1989. A protein from rat liver confers to glucokinase the property of being antagonistically regulated by fructose 6-phosphate and fructose 1-phosphate. Eur J Biochem 179:179–184 [DOI] [PubMed] [Google Scholar]
- 9.Vandercammen A, Detheux M, Van Schaftingen E1992. Binding of sorbitol 6-phosphate and of fructose 1-phosphate to the regulatory protein of liver glucokinase. Biochem J 286:253–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baltrusch S, Francini F, Lenzen S, Tiedge M2005. Interaction of glucokinase with the liver regulatory protein is conferred by leucine-asparagine motifs of the enzyme. Diabetes 54:2829–2837 [DOI] [PubMed] [Google Scholar]
- 11.Bosco D, Meda P, Iynedjian PB2000. Glucokinase and glucokinase regulatory protein: mutual dependence for nuclear localization. Biochem J 348:215–222 [PMC free article] [PubMed] [Google Scholar]
- 12.de la Iglesia N, Veiga-da-Cunha M, Van Schaftingen E, Guinovart JJ, Ferrer JC1999. Glucokinase regulatory protein is essential for the proper subcellular localisation of liver glucokinase. FEBS Lett 456:332–338 [DOI] [PubMed] [Google Scholar]
- 13.Shiota C, Coffey J, Grimsby J, Grippo JF, Magnuson MA1999. Nuclear import of hepatic glucokinase depends upon glucokinase regulatory protein, whereas export is due to a nuclear export signal sequence in glucokinase. J Biol Chem 274:37125–37130 [DOI] [PubMed] [Google Scholar]
- 14.Baltrusch S, Langer S, Massa L, Tiedge M, Lenzen S2006. Improved metabolic stimulus for glucose-induced insulin secretion through GK and PFK-2/FBPase-2 coexpression in insulin-producing RINm5F cells. Endocrinology 147:5768–5776 [DOI] [PubMed] [Google Scholar]
- 15.Baltrusch S, Lenzen S, Okar DA, Lange AJ, Tiedge M2001. Characterization of glucokinase-binding protein epitopes by a phage-displayed peptide library. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel interaction partner. J Biol Chem 276:43915–43923 [DOI] [PubMed] [Google Scholar]
- 16.Massa L, Baltrusch S, Okar DA, Lange AJ, Lenzen S, Tiedge M2004. Interaction of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) with glucokinase activates glucose phosphorylation and glucose metabolism in insulin-producing cells. Diabetes 53:1020–1029 [DOI] [PubMed] [Google Scholar]
- 17.Smith WE, Langer S, Wu C, Baltrusch S, Okar DA2007. Molecular coordination of hepatic glucose metabolism by the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase:glucokinase complex. Mol Endocrinol 21:1478–1487 [DOI] [PubMed] [Google Scholar]
- 18.Claus TH, El-Maghrabi MR, Regen DM, Stewart HB, McGrane M, Kountz PD, Nyfeler F, Pilkis J, Pilkis SJ1984. The role of fructose 2,6-bisphosphate in the regulation of carbohydrate metabolism. Curr Top Cell Regul 23:57–86 [DOI] [PubMed] [Google Scholar]
- 19.Okar DA, Manzano A, Navarro-Sabatè A, Riera L, Bartrons R, Lange AJ2001. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci 26:30–35 [DOI] [PubMed] [Google Scholar]
- 20.Pilkis SJ, Claus TH, Kurland IJ, Lange AJ1995. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: a metabolic signaling enzyme. Annu Rev Biochem 64:799–835 [DOI] [PubMed] [Google Scholar]
- 21.Brocklehurst KJ, Payne VA, Davies RA, Carroll D, Vertigan HL, Wightman HJ, Aiston S, Waddell ID, Leighton B, Coghlan MP, Agius L2004. Stimulation of hepatocyte glucose metabolism by novel small molecule glucokinase activators. Diabetes 53:535–541 [DOI] [PubMed] [Google Scholar]
- 22.Efanov AM, Barrett DG, Brenner MB, Briggs SL, Delaunois A, Durbin JD, Giese U, Guo H, Radloff M, Gil GS, Sewing S, Wang Y, Weichert A, Zaliani A, Gromada J2005. A novel glucokinase activator modulates pancreatic islet and hepatocyte function. Endocrinology 146:3696–3701 [DOI] [PubMed] [Google Scholar]
- 23.Futamura M, Hosaka H, Kadotani A, Shimazaki H, Sasaki K, Ohyama S, Nishimura T, Eiki J, Nagata Y2006. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem 281:37668–37674 [DOI] [PubMed] [Google Scholar]
- 24.Fyfe MC, White JR, Taylor A, Chatfield R, Wargent E, Printz RL, Sulpice T, McCormack JG, Procter MJ, Reynet C, Widdowson PS, Wong-Kai-In P2007. Glucokinase activator PSN-GK1 displays enhanced antihyperglycaemic and insulinotropic actions. Diabetologia 50:1277–1287 [DOI] [PubMed] [Google Scholar]
- 25.Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, Marcus L, Qi L, Spence CL, Tengi J, Magnuson MA, Chu CA, Dvorozniak MT, Matschinsky FM, Grippo JF2003. Allosteric activators of glucokinase: potential role in diabetes therapy. Science 301:370–373 [DOI] [PubMed] [Google Scholar]
- 26.Guertin KR, Grimsby J2006. Small molecule glucokinase activators as glucose lowering agents: a new paradigm for diabetes therapy. Curr Med Chem 13:1839–1843 [DOI] [PubMed] [Google Scholar]
- 27.Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y2004. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 12:429–438 [DOI] [PubMed] [Google Scholar]
- 28.Leighton B, Atkinson A, Coghlan MP2005. Small molecule glucokinase activators as novel anti-diabetic agents. Biochem Soc Trans 33:371–374 [DOI] [PubMed] [Google Scholar]
- 29.Matschinsky FM2009. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov 8:399–416 [DOI] [PubMed] [Google Scholar]
- 30.Printz RL, Granner DK2005. Tweaking the glucose sensor: adjusting glucokinase activity with activator compounds. Endocrinology 146:3693–3695 [DOI] [PubMed] [Google Scholar]
- 31.Hoat TX, Bertin N, Ninomiya N, Fukuda S, Usui K, Kawai J, Hayashizaki Y, Suzuki H2009. Development of a high-throughput method for the systematic identification of human proteins nuclear translocation potential. BMC Cell Biol 10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikeda M, Inoue S, Muramatsu M, Minatogawa Y2007. Characterization and identification of a steroid receptor-binding protein, SRB-RGS. Biol Pharm Bull 30:1056–1064 [DOI] [PubMed] [Google Scholar]
- 33.Massoud TF, Paulmurugan R, De A, Ray P, Gambhir SS2007. Reporter gene imaging of protein-protein interactions in living subjects. Curr Opin Biotechnol 18:31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murakami Y, Fukazawa H, Kobatake T, Yamagoe S, Takebe Y, Tobiume M, Matsuda M, Uehara Y2002. A mammalian two-hybrid screening system for inhibitors of interaction between HIV Nef and the cellular tyrosine kinase Hck. Antiviral Res 55:161–168 [DOI] [PubMed] [Google Scholar]
- 35.Pan J, Peng X, Gao Y, Li Z, Lu X, Chen Y, Ishaq M, Liu D, Dediego ML, Enjuanes L, Guo D2008. Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PLoS One 3:e3299 [DOI] [PMC free article] [PubMed]
- 36.Shioda T, Andriole S, Yahata T, Isselbacher KJ2000. A green fluorescent protein-reporter mammalian two-hybrid system with extrachromosomal maintenance of a prey expression plasmid: application to interaction screening. Proc Natl Acad Sci USA 97:5220–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao HF, Kiyota T, Chowdhury S, Purisima E, Banville D, Konishi Y, Shen SH2004. A mammalian genetic system to screen for small molecules capable of disrupting protein-protein interactions. Anal Chem 76:2922–2927 [DOI] [PubMed] [Google Scholar]
- 38.Bartel P, Chien CT, Sternglanz R, Fields S1993. Elimination of false positives that arise in using the two-hybrid system. Biotechniques 14:920–924 [PubMed] [Google Scholar]
- 39.Chien CT, Bartel PL, Sternglanz R, Fields S1991. The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc Natl Acad Sci USA 88:9578–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fields S, Song O1989. A novel genetic system to detect protein-protein interactions. Nature 340:245–246 [DOI] [PubMed] [Google Scholar]
- 41.Hümmer A, Delzeith O, Gomez SR, Moreno RL, Mark MD, Herlitze S2003. Competitive and synergistic interactions of G protein β(2) and Ca(2+) channel β(1b) subunits with Ca (v) 2.1 channels, revealed by mammalian two-hybrid and fluorescence resonance energy transfer measurements. J Biol Chem 278:49386–49400. [DOI] [PubMed] [Google Scholar]
- 42.van Schaftingen E, Veiga-da-Cunha M, Niculescu L1997. The regulatory protein of glucokinase. Biochem Soc Trans 25:136–140 [DOI] [PubMed] [Google Scholar]
- 43.Iynedjian PB, Pilot PR, Nouspikel T, Milburn JL, Quaade C, Hughes S, Ucla C, Newgard CB1989. Differential expression and regulation of the glucokinase gene in liver and islets of Langerhans. Proc Natl Acad Sci USA 86:7838–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiedge M, Steffeck H, Elsner M, Lenzen S1999. Metabolic regulation, activity state, and intracellular binding of glucokinase in insulin-secreting cells. Diabetes 48:514–523 [DOI] [PubMed] [Google Scholar]
- 45.Baltrusch S, Lenzen S2007. Novel insights into the regulation of the bound and diffusible glucokinase in MIN6 β-cells. Diabetes 56:1305–1315 [DOI] [PubMed] [Google Scholar]
- 46.Rizzo MA, Magnuson MA, Drain PF, Piston DW2002. A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J Biol Chem 277:34168–34175 [DOI] [PubMed] [Google Scholar]
- 47.Antoine M, Boutin JA, Ferry G2009. Binding kinetics of glucose and allosteric activators to human glucokinase reveal multiple conformational states. Biochemistry 48:5466–5482 [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Li C, Chen K, Zhu W, Shen X, Jiang H2006. Conformational transition pathway in the allosteric process of human glucokinase. Proc Natl Acad Sci USA 103:13368–13373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He L, Bradrick TD, Karpova TS, Wu X, Fox MH, Fischer R, McNally JG, Knutson JR, Grammer AC, Lipsky PE2003. Flow cytometric measurement of fluorescence (Forster) resonance energy transfer from cyan fluorescent protein to yellow fluorescent protein using single-laser excitation at 458 nm. Cytometry A 53:39–54 [DOI] [PubMed] [Google Scholar]
- 50.Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM2000. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc Natl Acad Sci USA 97:2253–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]