

Abstract
Cushing’s disease is caused by glucocorticoid-resistant pituitary corticotroph adenomas. We have previously identified the loss of nuclear Brg1 as one mechanism that may lead to partial glucocorticoid resistance: this loss is observed in about 33% of human corticotroph adenomas. We now show that Brg1 loss of function correlates with cyclin E expression in corticotroph adenomas and with loss of the cell cycle inhibitor p27Kip1 expression. Because Brg1 is thought to have tumor suppressor activity, the present study was undertaken to understand the putative contribution of cyclin E derepression produced by loss of Brg1 expression on adenoma development. Overexpression of cyclin E in pituitary proopiomelanocortin cells leads to abnormal reentry into cell cycle of differentiated proopiomelanocortin cells and to centrosome instability. These alterations are consistent with the intermediate lobe hyperplasia and anterior lobe adenomas that were observed in these pituitaries. When combined with the p27Kip1 knockout, overexpression of cyclin E increased the incidence of pituitary tumors, their size, and their proliferation index. These results suggest that cyclin E up-regulation and p27Kip1 loss-of-function act cooperatively on pituitary adenoma development.
Upregulation of cyclin E expression cooperates with p27Kip1 loss in corticotroph adenomas.
Cyclin-dependent kinases (CDKs) and their activating subunits, the cyclins, are essential for proper cell cycle regulation in eukaryotes. The active cyclin-CDK complexes regulate by phosphorylation a unique set of protein substrates that are essential for progression through different phases of the cell cycle. CDK inhibitors (CDKIs) tightly regulate the cell cycle to ensure appropriate progression of a cell through the different phases of the cycle and arrest (1). To evaluate their specific roles as cell cycle regulators, genetic ablations of cell cycle control proteins were made in mice. Unexpectedly, the loss of function of a subset of these cell cycle regulators preferentially resulted in pituitary tumor development, like ablation of the CDKIs p18INK4c (2, 3), p27Kip1 (4, 5, 6, 7), and also Rb+/− mice (8). These mutant mice all develop pituitary intermediate lobe (IL) tumors with nearly complete penetrance and also frequent anterior lobe (AL) tumors.
The human pituitary is also very sensitive to spontaneous tumor formation with a prevalence of pituitary adenomas evaluated to be up to 16% (9). Most human pituitary tumors are benign, never diagnosed, and without significant clinical consequence. Some tumors are nonsecreting but others, depending on the cell types affected, produce hormones. Thus, corticotroph adenomas cause Cushing’s disease because these tumors express the proopiomelanocortin (POMC) gene and produce excess amounts of ACTH. ACTH secretion from these tumors is increased in part because they are partially resistant to glucocorticoids (Gc) negative feedback regulation (10). The feedback exerted by Gc and their receptor [glucocorticoid receptor (GR)] is necessary to repress POMC gene transcription and inhibit ACTH release (11). Gc also inhibit the growth of corticotroph cells (12). We have previously identified two proteins that are essential for GR trans-repression of POMC transcription. Indeed, we showed that brahma related-gene 1 (Brg1) and histone deacetylase 2 (HDAC2) are forming a multiprotein complex together with GR and the orphan nuclear receptor nuclear growth factor IB (NGFI-B), and further that all these proteins are required for repression of POMC transcription (13). In view of the essential role of Brg1 and HDAC2 in Gc feedback, we assessed their expression in a panel of corticotroph adenomas and found that they are misexpressed in about 50% of Cushing’s disease patients, thus providing a molecular mechanism for the Gc resistance of these tumors (13). Brg1 was also known to have tumor suppressor activity (14, 15) and was implicated in the control of the cell cycle. Indeed, it was suggested that Brg1 might act as repressor of cyclin E expression (16). Thus, the loss of nuclear Brg1 activity may contribute to the pathogenesis of Cushing’s disease in at least two ways: by causing Gc resistance and by disruption of cell cycle control through derepression of cyclin E expression.
Cyclin E is cyclically expressed during the cell cycle (17). Its expression begins at the late G1 phase of the cycle and lasts till the end of the S phase. Regulation of cyclin E levels is insured through protein degradation by the proteosome pathway and through transcriptional regulatory mechanisms. Activation and repression of cyclin E gene transcription are regulated by members of the E2F family of transcription factors and involve recruitment of corepressors pRb and HDACs (16). Cyclin E binds to the serine/threonine protein kinase cdk2 and activates it to regulate the G1-S phase transition. The activity of this complex is preferentially inhibited by its interaction with the CDKI p27Kip1 (1). On the other hand, the cyclin E/cdk2 complex phosphorylates p27Kip1, which causes its dissociation from the complex and targets p27Kip1 for degradation by the ubiquitin-proteasome pathway (18). Cyclin E is also important to initiate DNA replication by direct activation of S-phase specific genes and also participates in the control of genome stability and the centrosome cycle (19). Deregulation of these processes may contribute to development of malignancies.
There is evidence supporting a primary role for cyclin E in cancer, suggesting that deregulation of this protein may be critical to alter regulation of the G1-S transition contributing to tumor development. Overexpression of cyclin E has been associated with progression of some cancers like breast carcinomas, leukemia, and lymphomas (20, 21). Also, transgenic mouse models of cyclin E overexpression have a tendency to develop malignancies in different tissues, supporting the notion of cyclin E as a dominant oncogene. With regard to Cushing’s disease, it was observed that cyclin E is preferentially increased in corticotroph adenomas compared with other pituitary tumors (22).
Here we show that cyclin E expression is repressed by Brg1 in the corticotroph model cells AtT-20. In agreement with this, we show that cyclin E is up-regulated in 100% of corticotroph adenomas that are deficient in nuclear Brg1. Further, these adenomas are very frequently deficient for p27Kip1 expression. We observed that forced pituitary expression of cyclin E leads to increased cell proliferation, centrosome instability, and sporadic hyperplasia and/or tumors. Significantly, we also found that cyclin E collaborates with p27Kip1 loss to increase the frequency, size, and proliferation index of pituitary tumors in mice.
Results
Cyclin E expression is correlated with p27Kip1 loss in Brg1-negative Cushing’s disease adenomas
Brg1 has been implicated in regulation of cellular proliferation and is a potential tumor suppressor. In particular, it was shown to induce growth arrest, in part, by down-regulation of select E2F target genes such as cyclin E (23). To investigate whether loss of Brg1 correlates with cyclin E expression in human corticotroph adenomas (Fig. 1A), we analyzed the expression of cyclin E by immunohistochemistry in a panel of 25 adenomas from Cushing’s disease patients (Fig. 1B and Supplemental Table 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). We used Tpit as a marker of corticotroph cells to delineate tumor and normal pituitary tissue in the pathology specimens (24). As described earlier (13), Brg1 is detected in the nucleus of all cells in normal pituitary tissue but absent from about 33% of corticotroph adenomas (e.g. Fig. 1B). In contrast, cyclin E is normally undetectable by immunohistochemistry in human pituitary but it appears in a fraction of adenomas, whereas expression of p27Kip1 is lost in a tumor subset (Fig. 1B). Strikingly, all Brg1-negative adenomas are cyclin E positive (100%) in contrast to 70% of Brg1-positive adenomas (Fig. 1A). In addition, a much higher frequency of p27Kip1-negative tumors is found in the Brg1-negative group (88% compared with 41% or 18%, for cyclin E-positive and -negative adenomas, respectively). Consistent with the fact that all adenomas are from Cushing’s disease patients, the bulk of cells in all tumors were positive for ACTH, Tpit, and GR, despite the patient’s relative resistance to the dexamethasone suppression test (Fig. 1B).
Fig. 1.

Cyclin E up-regulation and loss of p27Kip1 expression in corticotroph adenomas. A, A panel of 25 human corticotroph adenomas was studied by immunohistochemistry for Brg1, cyclin E, and p27Kip1 expression. All samples were also positive for ACTH, Tpit, and GR expression. Normal pituitary tissue is always positive for nuclear Brg1 and p27Kip1 and negative for cyclin E. The upper panels represent data for adenomas that are Wt for Brg1 (i.e. positive) whereas the lower panels represent data for the Brg1-negative subset. For each, the left panel indicates percentage of tumors that are either cyclin E negative (Wt) or cyclin E positive, and the right panels indicate the proportion of each tumor group that has lost expression of p27Kip1. No tumor was ever observed to be Brg1 negative and cyclin E negative. B, Immunohistochemical analyses for a representative human Brg1-negative, cyclin E-positive, p27Kip1-negative adenoma. Normal pituitary is shown for comparison. C, The putative repressor activity of Brg1 on cyclin E expression was verified in AtT-20 cells using RNA interference with a short hairpin loop RNA target against Brg1 (shpBrg1) in comparison with a scrambled-sequence control RNA (shpCTRL). Western blot analysis of Brg1, cyclin E, and GAPDH indicated successful knockdown of Brg1 resulting in up-regulation of cyclin E. D, Sections from two representative tumors of the Brg1-negative, cyclin E-positive, and p27Kip1-negative group showing colabeling of POMC-positive cells with pHH3. All tumor samples of this group showed similar double-positive cells, but no pHH3-positive, ACTH-negative cells. IB, Immunoblotting.
Brg1 represses cyclin E expression
To test whether the loss of Brg1 in corticotroph cells, as observed in some Cushing patients (13), could result in up-regulation of cyclin E, we performed RNA knockdown of Brg1 in AtT-20 cells. In contrast to a control short hairpin RNA (shpRNA), a shpRNA directed against Brg1 efficiently reduced Brg1 protein levels, and this was accompanied by an increase in cyclin E as revealed by Western blotting (Fig. 1C). Thus, Brg1 represses cyclin E expression in pituitary corticotroph cells.
Pituitary overexpression of cyclin E increases differentiated cell proliferation
Proliferation of terminally differentiated cells is rare. Transformation of such cells leading to reentry into cell cycle could lead to adenoma formation. In most corticotroph adenomas, proliferating cells revealed using the mitosis marker phosphohistone H3 (pHH3), are differentiated because they express ACTH (Fig. 1D). Thus, cycling differentiated corticotroph cells could directly contribute to adenoma development. To test the hypothesis that cyclin E may be sufficient to drive corticotroph cells into the cell cycle and/or induce tumors, we generated transgenic mice overexpressing cyclin E in pituitary corticotrophs and melanotrophs (the POMC-expressing lineages) under control of the POMC promoter (Fig. 2A). We established four founder lines that express cyclin E (Tg-PCE) at different levels as shown by Western blot analysis (Fig. 2B). Most data presented here were obtained using line 337, but line 649 behaved similarly. To verify that the POMC-cyclin E transgene is expressed as expected, we assessed transgene expression by immunofluorescence at embryonic d 18.5. At this developmental stage, POMC-positive cells do not usually express detectable levels of nuclear cyclin E (Fig. 2C), and they do not proliferate as assessed by the mitosis marker pHH3 (Fig. 2E). In Tg-PCE pituitaries, cyclin E protein was detectable in most POMC-positive cells (Fig. 2D), and double-positive cells for ACTH and pHH3 were present (Fig. 2F, inset); we also observed double-positive cells by colabeling for Ki67 (data not shown). We conclude that cyclin E overexpression is sufficient to drive differentiated POMC cells to reenter the cell cycle.
Fig. 2.
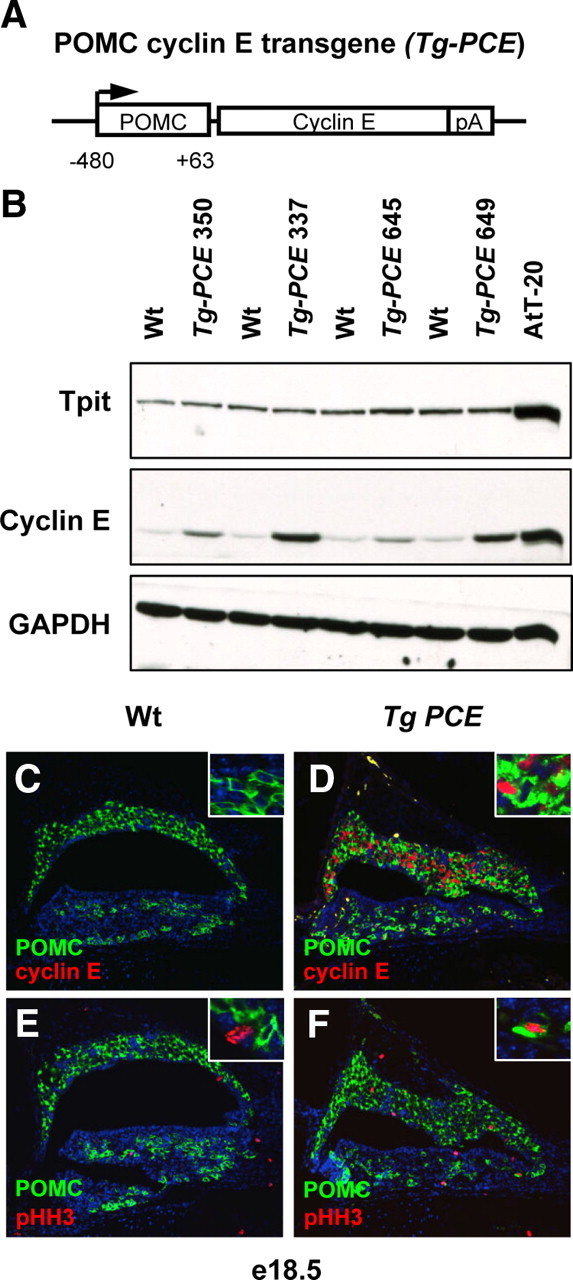
Forced expression of cyclin E in mouse pituitary. A, Structure of cyclin E-expressing transgene driven by the rat POMC promoter (Tg-PCE). B, Analysis of four different transgenic lines compared with their Wt sibs for expression of Tpit, cyclin E, and GAPDH analyzed by Western blot of adult pituitary extracts. The number on top refers to the identification number of each transgenic line. Line 337 was mostly used for further studies, but line 649 also produced similar results. C, Characterization of embryonic d 18.5 (e18.5) Tg-PCE transgenic mice. Expression of the transgene was assessed on pituitary sections by immunofluorescence. Cyclin E overexpression is detected in a large number of IL melanotroph cells as well as in some AL cells (D), whereas no cyclin E was detected in the ACTH-positive cells of sib control embryos (C). Analysis of pHH3-positive cells showed few positive cells (usually POMC-negative; E, inset) for this marker of mitosis in control pituitary (E) but much more frequent positive cells in the transgenic pituitary (F), including cells that are positive for both pHH3 and ACTH (F, inset).
Hyperplasia, adenomas, and centrosome instability in POMC-cyclin E transgenic mice
No significant morphological differences were observed between Wt and Tg-PCE pituitaries at 8 months or 1 yr of age (data not shown). However by 2 yr of age, some Tg-PCE pituitaries (three of 17) exhibited hyperplasia of the IL (Tpit, ACTH, Brg1, and p27Kip1-positive cells) and another three mice had AL adenomas (Fig. 3A). The frequency of hyperplasia or adenomas was low at 18% each (three of 17), but combined, they represented 36% of mice compared with none in wild-type (Wt) siblings (sibs). It is not clear why hyperplasias were only observed in the IL (Fig. 3C) and tumors in the AL. However, this may correlate with activity of the POMC promoter used (Langlais D. and Drouin J. in preparation) and with the persistent increase in Ki67-positive cells observed in both 8-month and 2-yr transgenic ILs (Fig. 3B). In contrast, POMC-cyclin E transgene expression decreased in AL of older mice (data not shown).
Fig. 3.

Cyclin E expression increases pituitary proliferation index. A, Cohorts of Tg-PCE mice and their control sibs (Wt) were investigated at 2 yr of age for a pituitary phenotype. Six transgenic pituitaries of a total of 17 were found to have abnormal pituitaries, three with IL hyperplasia (C) and three with AL adenomas (D–F). B, Quantitation of cyclin E and Ki67-positive cells in adult ILs of Tg-PCE transgenic pituitaries compared with their Wt control sibs. Cyclin E-positive and Ki67-positive (a marker of proliferation) cells were identified by immunohistochemistry on pituitary sections of 8-month-old and 2-yr-old mice as indicated. For each group, the number of positive cells was counted on duplicate sections from eight to 10 different mice. F, Summary of marker expression in the three AL adenomas. Only one tumor was positive for the differentiation markers Pit-1, PRL, and GH.
Two adenomas were small and well delineated (Fig. 3D), and the third was much larger (Fig. 3E). We investigated the nature of the AL adenomas using markers (Fig. 3F). Surprisingly, no tumor was positive for Tpit or ACTH. All adenomas expressed Pitx1, a marker of the ectodermal origin of the pituitary and were positive for Brg1, cyclin E, and Ki67. Whereas two adenomas were nonsecreting and negative for differentiation markers Tpit, Pit1, and SF1, the third adenoma was positive for prolactin (PRL), GH, and Pit1 (Fig. 3F).
It was previously reported (25, 26, 27, 28) that high cyclin E expression leads to centrosome instability and genomic instability (27). To assess whether this may occur in pituitary cells, we investigated centrosome integrity in IL of 2 yr-old Tg-PCE mice (Fig. 4). The increased cell proliferation observed in Tg-PCE pituitaries (Fig. 3B) is reflected in higher occurrence of cells with duplicated γ-tubulin-positive (19) centrosomes (Fig. 4A). In addition, transgenic pituitaries exhibit significant increases in number of cells with three or more centrosomes (Fig. 4, A and C) and with structurally abnormal centrosomes (Fig. 4, B and C). The increased occurrence of abnormal centrosomes likely contributes to genome instability (28, 29) and may predispose to tumor development.
Fig. 4.
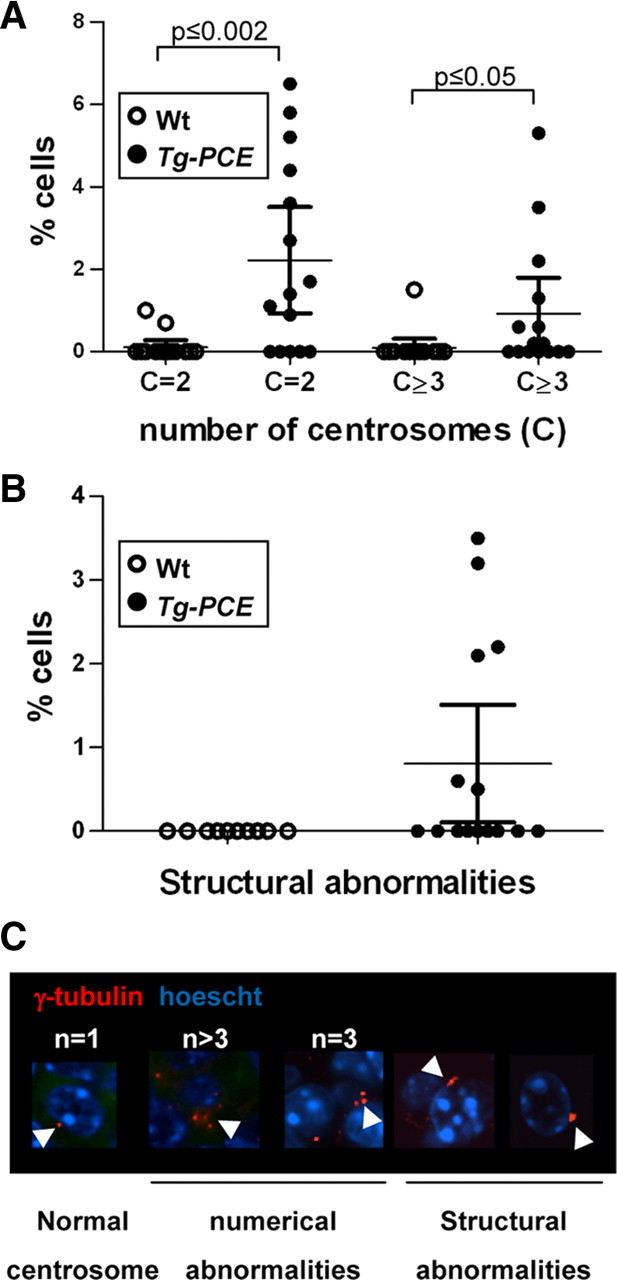
Cyclin E overexpression causes centrosome abnormalities. Centrosomes (C) revealed using γ-tubulin immunofluorescence (19 ) were analyzed on two sections from each mouse reported in Fig. 3A; analyses were only performed on IL tissues with normal appearance, i.e. not in hyperplastic or tumor areas. Panel A, Dividing cells have two visible centrosomes (C = 2), and abnormal numbers of centrosomes are reported as C ≥ 3. Each dot represents data for a different mouse. Bar represents the means ± sem. Panel B, Quantitation of structural centrosome abnormalities. These were never observed in control tissues. Panel C, Examples of centrosomes observed in this study.
Cooperation between cyclin E and p27Kip1 loss of function
Tumor initiation is a multistep process. The limited effect of overexpressing cyclin E might be due to high levels of cell cycle inhibitors in differentiated cells, and particularly p27Kip1 in adult pituitary (30). Because p27Kip1 is a key regulator of the cyclin E-cdk2 complex and because activation of this complex leads to inactivation of p27Kip1, we assessed the impact of cyclin E expression on the expression and status of p27Kip1. Using the Brg1 knockdown paradigm in AtT-20 cells (Fig. 1C), we observed the resulting cyclin E up-regulation at both mRNA (Fig. 5A) and protein levels (Figs. 1C and 5B). Oddly, opposing effects were observed on p27Kip1 expression, with increased mRNA levels (Fig. 5A) associated with stable or slightly decreased total p27Kip1 (Fig. 5B). However, a convincing decrease of Thr187-phosphorylated p27Kip1 is observed, possibly reflecting increased turnover of p27Kip1. Because p27Kip1 loss of function leads to IL pituitary tumors (4, 5, 6, 7), we investigated whether cyclin E up-regulation would enhance the effect of loss of p27Kip1 on pituitary tumor formation. Representative pituitaries at 8 months of age from each genotype suggest that the combined loss of p27Kip1 with overexpression of cyclin E results in larger pituitary tumors (Fig. 6A). The average tumor weight (Fig. 6B) of Tg-PCE;p27Kip1−/− mice is about 3 times that of p27Kip1−/− mice (P ≤ 0.04) and more than 11-fold the size of Wt pituitaries (P ≤ 0.008). The size increase between Tg-PCE;p27Kip1−/− and p27Kip1−/− tumors may be explained, in part, by higher proliferation rates stimulated by cyclin E overexpression (Fig. 3B). Indeed, the abundance of Ki67-positive cells appeared greater in Tg-PCE;p27Kip1−/− mice (Fig. 6C); it is noteworthy that the loss of p27Kip1 has the largest effect on the number of Ki67-positive cells. Ki67-positive cells were counted in both IL and AL of each pituitary in the panel (Fig. 6D). This analysis showed that the increase is mostly evident in the AL (P ≤ 0.03) where most of the Tg-PCE;p27Kip1−/− samples have more than 5% Ki67-positive cells compared with the p27Kip1−/− tumors that predominantly have less than 5%.
Fig. 5.
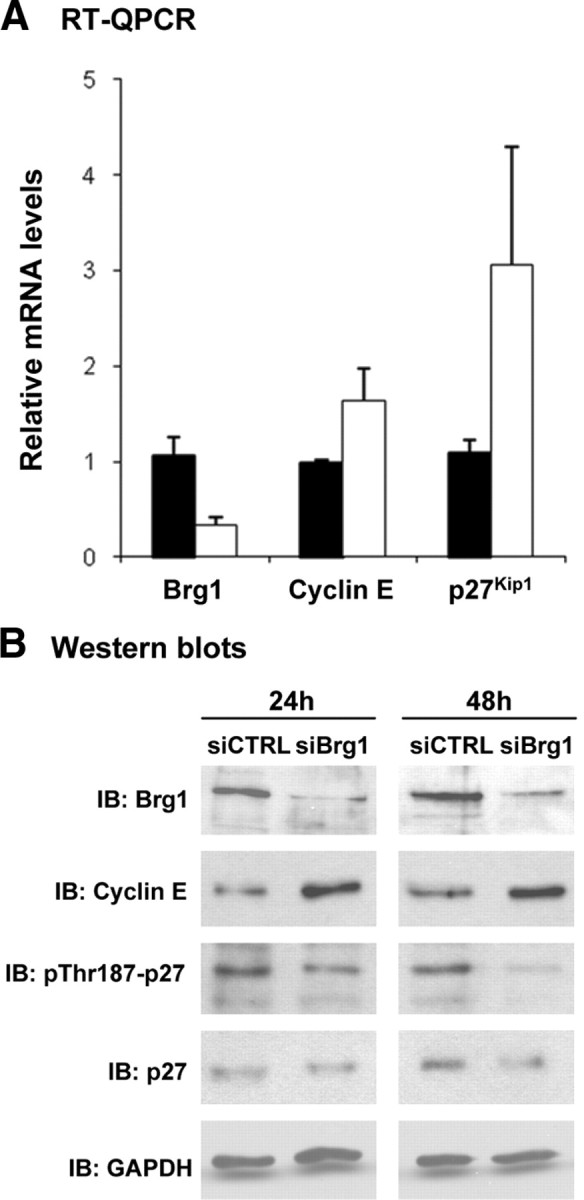
Effect of Brg1 knockdown [small interfering RNA (siRNA)] in AtT-20 cells on cyclin E and p27Kip1 mRNA and protein levels. A, Quantitative real-time PCR (RT-QPCR) analysis of Brg1, cyclin E, and p27Kip1 transcripts in AtT-20 after Brg1 knockdown. Brg1 transcripts are decreased 3.1-fold (P ≤ 0.0001) whereas cyclin E and p27Kip1 transcripts are increased 1.7-fold (P ≤ 0.005) and 2.8-fold (P ≤ 0.05), respectively (n = 3). B, Levels of Brg1, cyclin E, p27Kip1, phospho-p27Kip1 (Thr187), and GAPDH in extracts from AtT-20 cells treated with siCTRL and siBrg1 for 24 and 48 h revealed by Western blot. IB, Immunoblotting.
Fig. 6.
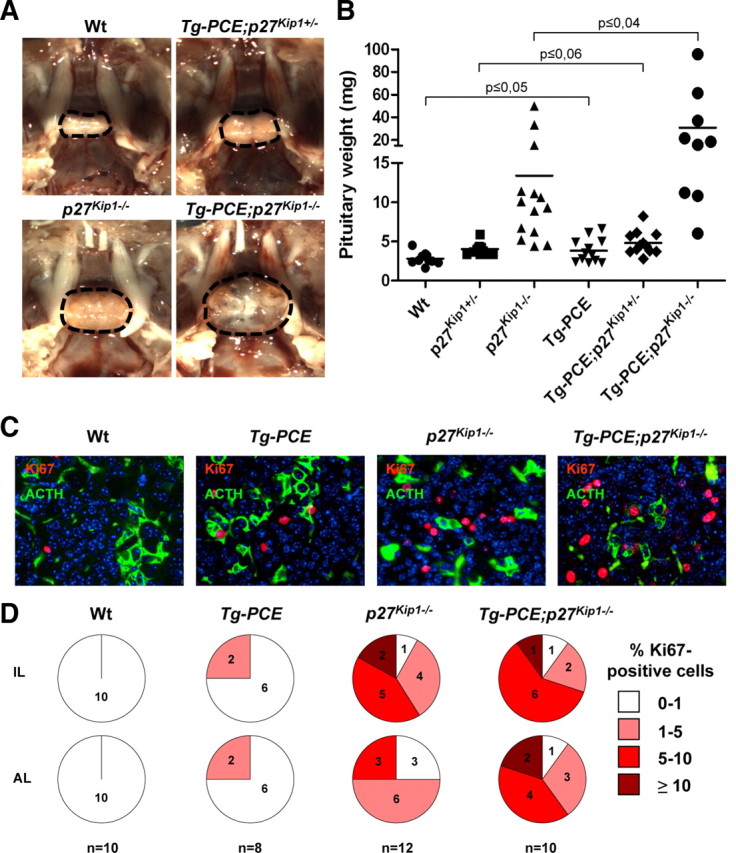
Cyclin E cooperates with p27Kip1 loss of function for pituitary tumor development. A, Photographs of representative in sellae pituitaries from 8-month-old mice: Wt, Tg-PCE and p27Kip1 heterozygote (Tg-PCE;p27Kip1+/−), p27Kip1 knockout (p27Kip1−/−), and Tg-PCE transgenic p27Kip1 knockout (Tg-PCE;p27Kip1−/−). B, Distribution of pituitary weights for mice of the different genotypes. The number of pituitaries in each group is indicated in Table 1. Bars represent the medians. C, Colabeling of representative anterior pituitary sections for mice of the indicated genotypes using Ki67 as marker of proliferating cells and ACTH to identify corticotrophs. D, Quantitation of Ki67-positive cells in the IL and AL of pituitaries from each genotype. The number of mice studied in each group is indicated at the bottom and for each mouse, two sections were quantitated. In each case, the percentage of Ki67-positive cells was subdivided into four categories as indicated. The increased Ki67 index observed in AL of Tg-PCE;p27Kip1−/− compared with p27Kip1−/− pituitaries is statistically significant (P ≤ 0.03).
In addition, the frequency of both IL and AL tumor formation was increased in Tg-PCE;p27Kip1−/− compared with p27Kip1−/−, with frequencies of 100% compared with 72% in the IL and 40% compared with 7% in the AL (Table 1). It was reported that the penetrance of p27Kip1-dependent pituitary tumors varies with genetic background (31); our observation of 70% IL tumors in mice of mixed background is consistent with previous observations. It is noteworthy that IL and AL adenomas are quite different in these models: indeed, the IL adenoma cells of both p27Kip1−/− and Tg-PCE;p27Kip1−/− are always positive for the POMC lineage marker Tpit, indicating that they are differentiated (Table 2). In contrast, Tpit-positive cells were not observed in AL adenomas of p27Kip1−/− mice, and only one adenoma from Tg-PCE;p27Kip1−/− pituitaries contained a cluster of Tpit-positive cells. One pituitary adenoma of each genotype contained dispersed cells positive for the gonadotroph marker steroidogenic factor 1 (SF-1), and none were found to have Pit1-positive cells (Table 2). These observations are consistent with prior work that described p27Kip1−/− pituitary AL tumors as poorly differentiated (32). Because the bulk of adenoma cells are negative for differentiation markers except Pitx1, we assessed expression of two transcription factors that have been associated with pituitary stem or progenitor cells, Sox2 and Sox9 (33). Neither is expressed in p27Kip1−/− or Tg-PCE;p27Kip1−/− AL or IL adenomas (Table 2). In summary, the overexpression of cyclin E cooperates with loss of p27Kip1 to increase the frequency, size, and proliferation index of pituitary tumors, notwithstanding the very different differentiation status of IL and AL adenoma cells.
Table 1.
Pituitary phenotype of 8-month-old mice of the indicated genotypes
| Phenotype | Wt | Tg-PCE | p27 Kip1−/− | Tg-PCE;p27 Kip1−/− |
|---|---|---|---|---|
| Normal | 100% (12/12) | 88% (7/8) | 21% (3/14) | — |
| IL hyperplasia | — | — | 7% (1/14) | — |
| IL adenoma | — | — | 72% (10/14) | 100% (10/10) |
| AL hyperplasia | — | 12% (1/8) | — | — |
| AL adenoma | — | — | 7% (1/14) | 40% (4/10) |
| IL and AL adenoma | — | — | 7% (1/14) | 40% (4/10) |
The percentage and numbers of observed phenotypes are indicated over the total number of mice in each group. —, Not observed.
Table 2.
Expression of pituitary markers in adenomas from p27Kip1−/− and Tg-PCE;p27Kip1−/− pituitaries
| Wt pituitary | p27Kip1 −/− | Adenomas | Tg-PCE;p27Kip1−/− | Adenomas | ||
|---|---|---|---|---|---|---|
| Staining | IL | AL | IL | AL | IL | AL |
| Pitx1 | + | + | + | + | + | + |
| Brg1 | + | + | + | + | + | + |
| Tpit | + | + (5–10%) | + | − | + | One cell cluster (1/4) |
| SF-1 | − | + (5–10%) | − | Dispersed cells ∼ 5% | − | Dispersed cells ∼ 5% (1/4) |
| Pit-1 | − | + (>60%) | − | − | − | − |
| CyclinE | − | − | − | − | + | + (90%) |
| Sox2 | Around lumen | − | − | − | − | |
| Sox9 | Around lumen | − | − | − | − | |
| n = 12 | n = 12 | n = 9 | n = 1 | n = 10 | n = 4 |
The number of pituitary adenomas analyzed for marker expression by immunohistochemistry are indicated at bottom and correspond to adenomas described in Table 1. IL adenomas were positive for Tpit in both genotypes and negative for all other lineage markers. The single AL adenoma from p27Kip1−/− pituitary stained positively for SF-1 (∼5% cells). One Tg-PCE;p27Kip1−/− pituitary AL adenoma exhibited a similar SF-1-positive adenoma, and another AL adenoma contained a cluster of Tpit-positive cells and a mass of undifferentiated cells. All other AL adenomas cells appeared undifferentiated. +, >95% positive cells; –, no positive cells.
Discussion
Glucocorticoid resistance and corticotroph adenomas
Although up-regulation of cyclin E was observed in a variety of tumors (20, 21) including pituitary tumors, the present work suggests a causal relationship between up-regulation of cyclin E and a presumably earlier event, the loss of nuclear Brg1 that likely causes glucocorticoid resistance (13). Indeed all Brg1-negative corticotroph adenomas were found to express cyclin E although Brg1-positive tumors also frequently exhibit cyclin E up-regulation, indicating that other mechanisms may account for cyclin E misexpression. These observations on human adenomas are consistent with Brg1-dependent derepression of cyclin E expression in AtT-20 cells (Fig. 1C) and with the unaltered Brg1 expression in cyclin E-expressing mouse pituitaries (Table 2).
No gene or protein has so far been implicated in both Gc resistance and tumorigenesis of pituitary corticotroph adenomas. By documenting a Brg1-dependent up-regulation of cyclin E, the present work clearly supports the idea that Brg1 may be involved in both processes of Gc resistance and corticotroph tumorigenesis. The Brg1-dependent loss of Gc-negative feedback may also directly contribute to corticotroph adenoma development by derepression of other tumor suppressors and/or by activation of tumor-promoting genes.
Cyclin E and p27Kip1 in control of pituitary cell growth
The control of G1-S transition is of tremendous importance for cell cycle regulation. In particular, cyclin E expression is essential for cell cycle reentry of quiescent cells (34). We showed previously that most adult POMC cells have exited the cell cycle, do not express detectable cyclin E, and express p27Kip1 (30). Cyclin E expression, particularly associated with loss of p27Kip1, could allow POMC cells to leave their quiescent state and enter S phase. The cyclin E/p27Kip1 balance may thus regulate corticotroph and melanotroph proliferation acting as gatekeeper to protect them from cell cycle reentry. Cyclin E up-regulation, on its own, is unlikely to be sufficient for tumor formation, but we show that it leads to cell cycle reentry of differentiated pituitary POMC cells (Figs. 2F and 3B). Increased cyclin E/cdk2 would lead to enhanced progression through cell cycle but also to inactivation of p27Kip1 by phosphorylation of Thr187 (18, 35, 36). This phosphorylation targets p27Kip1 for degradation through the ubiquitin proteasome pathway (18). Consistent with enhanced p27Kip1 turnover and degradation, we observed lower steady-state levels of phosphorylated p27Kip1 and total p27Kip1 (Fig. 5B) in AtT-20 cells that exhibit high cyclin E expression. This decrease was observed despite elevated p27Kip1 mRNA (Fig. 5A) in the Brg1 knockdown paradigm used in these experiments. Increased p27Kip1 mRNA may have been caused directly by the Brg1 knockdown or mediated through another regulator. Because p27Kip1−/− pituitaries did not show increased cyclin E expression (Table 2), the loss of p27Kip1 is unlikely responsible for cyclin E up-regulation. Rather, cyclin E up-regulation is most likely due to the loss of Brg1, possibly mediated through E2F (23).
We observed a strong inverse correlation between cyclin E up-regulation and p27Kip1 expression in the subgroup of Brg1-negative corticotroph adenomas (Fig. 1A, lower panels). In a multistep model of tumor development, the enhanced proliferation caused by cyclin E up-regulation may be an early event, predisposing cells to the effect of a second hit, such as the loss of p27Kip1 expression. The p27Kip1 gene is rarely mutated in pituitary adenomas (37, 38). It was shown in some instances to be silenced through DNA methylation but in most cases, p27Kip1 expression appears to be down-regulated at the protein level (32, 39). Whereas cyclin E up-regulation may lead to decreased steady-state p27Kip1 as discussed above, other mechanism(s) may also contribute to establishment of tumors by down-regulation of p27Kip1.
Decreased p27Kip1 was preferentially associated with corticotroph and metastatic pituitary adenomas (40) and further, corticotroph adenomas were found to have higher proliferative index compared with other pituitary adenomas (41, 42). Accordingly in mouse pituitaries, ectopic cyclin E enhanced tumor frequency (Table 1), size (Fig. 6, A and B) and proliferation index (Fig. 6, C and D) of p27Kip1-deficient mice, revealing a synergism between these two events. In addition to its effect on proliferation, cyclin E overexpression may also promote tumorigenesis through other mechanisms.
Up-regulation of cyclin E and centrosome instability
In addition to its role in cell cycle control, cyclin E has also been involved in DNA replication, apoptosis, and DNA repair (43). Its expression must be tightly regulated and begins at the G1 phase until the end of the S phase. During these phases, centrosome duplication and DNA replication are initiated. The presence of two centrosomes at mitosis is critical for the formation of bipolar mitotic spindles that ensure proper DNA partition between two daughter cells. This critical coordination between centrosome and DNA duplication is achieved, in part, by the activation of the cyclin E-cdk2 complex. Perturbation of the control exerted by this complex might result in abnormal multiplication of centrosomes and lead to abnormal mitoses and chromosome segregation errors. Centrosome amplification is a feature frequently seen in various cancer cells, and a mechanism was recently proposed to link centrosome amplification to chromosomal instability (29). In human cancers, centrosome and genomic instability have been associated with cyclin E-overexpressing tumors (19, 44). Interestingly, human pituitary tumors have also been shown to exhibit signs of genome instability (45, 46, 47). In our Tg-PCE mice, centrosome instability was observed and may contribute to tumor development. It is therefore reasonable to propose that up-regulation of cyclin E expression may predispose to pituitary adenoma formation by producing genome instability.
Differentiation status of pituitary adenoma cells
The analyses of differentiation markers in p27Kip1−/− pituitaries indicated that the frequent IL hyperplasia and adenomas contain differentiated POMC- and Tpit-positive cells (Table 2 and Ref. 32). In contrast, the rare AL adenomas are poorly differentiated with a minority of cells expressing only SF-1, a marker of the gonadotroph lineage (Table 2). This pattern was not significantly altered by introduction of the POMC-cyclin E transgene: indeed despite their increased frequency, size, and proliferation index, AL adenomas only occasionally contain Tpit-positive cells.
The appearance of POMC-negative tumors in Tg-PCE transgenics may reflect cyclin E activities other than its well known cell cycle-regulatory functions. Indeed, kinase-independent functions of cyclin E have been reported recently (48) and, in particular, cyclin E has been implicated in asymmetric cell division in Caenorrhabitis elegans (49) and in cell fate determination in Drosophila (50). Thus, cyclin E overexpression may promote low-frequency corticotroph dedifferentiation. Notwithstanding the possibility that transgene expression may be leaky in non-POMC cells (at levels below immunofluorescence or green fluorescent protein detection) (51), the appearance of non-POMC-expressing tumors in the transgenics may indicate that upon cyclin E-dependent transformation, the pituitary tumor cells dedifferentiated. Intriguingly, there is no indication of similar dedifferentiation in the IL where transgene expression is maintained throughout adult life (Fig. 3B). To assess the possibility that undifferentiated cells of AL adenomas may have features of pituitary stem or progenitor cells, we tested these cells for expression of Sox2 and Sox9 expression (Table 2). Neither marker is expressed in these adenomas: the differentiation status of these cells remains unclear. These undifferentiated cells may represent a parallel with human tumors that progress to a nonsecreting status.
Recent advances in stem cells and tumor biology led to the hypothesis of cancer stem cells. According to this model, a small number of undifferentiated progenitor cells proliferate and give rise to most of the tumor mass. This does not appear to be the case for many corticotroph adenomas, because we found that proliferating cells are POMC-positive in Brg1-negative/cyclin E-positive tumors (Fig. 1D). Cycling corticotroph adenoma cells are thus differentiated, but they grow slowly compared with more aggressive tumors. These relatively benign tumors may represent a unique example of early tumor development; indeed, they might have gone undetected if it was not for their endocrine consequences. In this context, cyclin E expression with loss of p27Kip1 may constitute sufficient imbalance to allow cell cycle reentry of differentiated cells and adenoma development. Progression to a more aggressive tumor state may be accompanied by appearance of proliferating undifferentiated cells of the cancer stem cell type.
Materials and Methods
Generation of transgenic mice
For generation of the Tg-PCE transgene, the mouse cyclin E1 cDNA was inserted downstream of the −480-bp POMC promoter, and transgenic mice were generated as described (52); they were back-crossed into the C57Bl/6 background. p27Kip1-null mice (CD1 genetic background) were obtained from The Jackson Laboratory (Bar Harbor, ME); the series of mice carrying the p27Kip1 mutant allele and/or the Tg-PCE transgene (Fig. 6 and Table 1) were analyzed in a mixed genetic background (C57Bl/6;CD1). The genotyping was performed on tail DNA with the following primers; 5′-GAACTAGGCCTGCCTCACAC-3′ (POMC promoter) and 5′-AGGGCTGACTGCTATCCTCGCTTT-3′ (cyclin E) for Tg-PCE; and 5-CTTGGGTGGAGAGGCTATTC-3′ (neomycin), 5′-AGGTGAGATGACAGGAGATC-3′ (neomycin), 5′-CTCCTGCCATTCGTATCTGC-3′ (p27Kip1), and 5′-GATGGACGCCAGACAAGC-3′ (p27Kip1) for p27Kip1-null mice. All animal experimentation was approved by The Animal Ethics Review Committee of the Institut de recherches cliniques de Montréal (IRCM).
Western blot and RNA knockdown
Brg1 shpRNA and siBrg1 (Dharmacon, Lafayette, CO) were used in AtT-20 culture cells as previously described (13). The following antibodies were used for Western blotting; mouse-anti-Brg1 1:500 (Chemicon, Temecula, CA), rabbit-anticyclin E 1:1000 (M-20, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), mouse-anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 1:50 000 (Abcam, Inc., Cambridge, MA), mouse-anti-p27 1:2000 (BD Biosciences, Palo Alto, CA), goat-anti-p-p27 (Thr187) 1:500 (Santa Cruz Biotechnology), and antirabbit-Tpit (53).
Quantitative Real-time PCR
Quantitative real-time PCR analysis of Brg1, cyclin E, and p27Kip1 transcripts was performed as previously described (54) on AtT-20 RNA after Brg1 knockdown using small interfering RNA against Brg1 (siBrg1) in comparison with random sequence control RNA (siCRTL). The primers used were as follows. Brg1: sense, AGATGGTGAGCCTCTGGATGA; antisense, TCATACCCTGGGTTCATTTCAAGC.; Cyclin E: sense, AAATCAGACCACCCAGAGCCT; antisense, TGGAGCTTATAGACTTCGCACACC; p27Kip1: sense, TTGGTGGACCAAATGCCTGA; antisense, TCTTCTGTTCTGTTGGCCCTT.
Histology analysis
Pituitaries were dissected from the skull, weighed, and fixed in 4% paraformaldehyde for 4 h before paraffin embedding. Embryos were fixed in 4% paraformaldehyde and embedded in paraffin (55). Pathology specimens from human corticotroph adenomas were previously described (13). Only six adenomas of the series were macroadenomas (≥10 mm), and their marker distribution did not differ significantly from other adenomas (Supplemental Table 1). Studies of human adenomas were approved by the IRCM Human Ethics Review Committee.
Immunochemistry and immunofluorescence
Primary antibodies diluted in 10% normal goat serum-0.2% Tween 20 in PBS were incubated overnight and used at the following dilutions: rabbit-anti-Brg1 1:10 (H-88, Santa Cruz Biotechnology), rabbit-anticyclin E 1:100 (M-20, Santa Cruz Biotechnology), rabbit-anti-Ki67 1:100 (Labvision, Fremont, CA), mouse-anti-p27Kip1. 1: 400 (BD Biosciences), rabbit-anti-Pitx1 (55), rabbit-anti-Tpit 1:200, rabbit-anti-phospho-Histone H3 1:200 (Ser10, Upstate Biotechnology, Inc., Lake Placid, NY), rabbit-anti-SF-1 1:100 (gift from Dr. K. Morohashi), rabbit-anti-Sox2 1:200 (Chemicon, Temecula, CA), rabbiti-anti-Sox9 1:200 (gift from Dr. F. Poulat), rabbit-anti-Ki67 1:50 (Labvision), rabbit-anti-Pit1 1:250 (provided by Simon Rhodes), guinea pig-anti-PRL 1:200 (National Hormone and Peptide Program) (53). All secondary antibodies were used 1:150 (Vector Laboratories, Inc., Burlingame, CA). For colocalization, primary antibodies were incubated overnight, after which mouse-anti-POMC 1:200 (Cortex Biochemicals, Concord, MA) and biotinylated antirabbit-IgG 1:150 (Vector Laboratories) were then incubated for 1 h followed by antimouse-fluorescein 1:150 (ImmunoPure Antibody) and streptavidin-fluorescein 1:150 (Molecular Probes, Inc., Eugene, OR). For γ-tubulin/Hoescht staining, a rabbit anti-γ-tubulin antibody (gift of Dr. K. Kukasawa) was used at 1:100 dilution.
Acknowledgments
We thank all colleagues from the laboratory for their critical comments. We also thank Lise Laroche (Institut de recherches cliniques de Montréal, Montréal, Canada)for expert secretarial assistance; Simon Rhodes (Indiana University School of Medicine, Indianapolis, IN), for his generous gift of Pit1 antibody; Dr. K. Morohashi (National Institute for Basic Biology, Okazaki, Japan), for the SF-1 antibody; Dr. Francis Poulat (Centre National de la Recherche Scientifique, Montpellier, France), for the Sox9 antibody. We also thank Dr. K. Fukasawa (H. Lee Moffitt Cancer Center, Tampa, FL), for the γ-tubulin antibody, and Dr. A. F. Parlow (Research Professor of Obstetrics and Gynecology, University of California Los Angeles School of Medicine, National Hormone and Peptide Program) for the GH and PRL antibody.
Footnotes
This work was supported by research grants (to J.D.) from the Canadian Institutes of Health Research and National Cancer Institute of Canada.
Disclosure Summary. The authors have nothing to disclose.
First Published Online July 21, 2010
Abbreviations: AL, Anterior lobe; CDK, cyclin-dependent kinase; CDKI, CDK inhibitor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Gc, glucocorticoids; GR, glucocorticoid receptor; HDAC, histone deacetylase; IL, intermediate lobe; pHH3, phosphohistone H3; POMC, proopiomelanocortin; PRL, prolactin; sibs, siblings; SF-1, steroidogenic factor 1; shpRNA, short hairpin RNA; Wt, wild type.
References
- 1.Sherr CJ, Roberts JM1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501–1512 [DOI] [PubMed] [Google Scholar]
- 2.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y1998. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 12:2899–2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Latres E, Malumbres M, Sotillo R, Martín J, Ortega S, Martín-Caballero J, Flores JM, Cordón-Cardo C, Barbacid M2000. Limited overlapping roles of p15(INK4b) and p18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J 19:3496–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM1996. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 85:733–744 [DOI] [PubMed] [Google Scholar]
- 5.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A1996. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85:721–732 [DOI] [PubMed] [Google Scholar]
- 6.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K1996. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85:707–720 [DOI] [PubMed] [Google Scholar]
- 7.Chien WM, Rabin S, Macias E, Miliani de Marval PL, Garrison K, Orthel J, Rodriguez-Puebla M, Fero ML2006. Genetic mosaics reveal both cell-autonomous and cell-nonautonomous function of murine p27Kip1. Proc Natl Acad Sci USA 103:4122–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA1992. Effects of an Rb mutation in the mouse. Nature 359:295–300 [DOI] [PubMed] [Google Scholar]
- 9.Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, McCutcheon IE2004. The prevalence of pituitary adenomas: a systematic review. Cancer 101:613–619 [DOI] [PubMed] [Google Scholar]
- 10.Arnaldi G, Angeli A, Atkinson AB, Bertagna X, Cavagnini F, Chrousos GP, Fava GA, Findling JW, Gaillard RC, Grossman AB, Kola B, Lacroix A, Mancini T, Mantero F, Newell-Price J, Nieman LK, Sonino N, Vance ML, Giustina A, Boscaro M2003. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 88:5593–5602 [DOI] [PubMed] [Google Scholar]
- 11.Philips A, Maira M, Mullick A, Chamberland M, Lesage S, Hugo P, Drouin J1997. Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol Cell Biol 17:5952–5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svec F1984. Glucocorticoids inhibit the growth of AtT-20 mouse pituitary tumor cells. Mol Cell Endocrinol 35:33–40 [DOI] [PubMed] [Google Scholar]
- 13.Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, Lacroix A, Batista D, Stratakis C, Hanson J, Meij B, Drouin J2006. Role of Brg1 and HDAC2 in GR trans-repression of pituitary POMC gene and misexpression in Cushing disease. Genes Dev 20:2871–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE2003. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res 63:560–566 [PubMed] [Google Scholar]
- 15.Gunduz E, Gunduz M, Ouchida M, Nagatsuka H, Beder L, Tsujigiwa H, Fukushima K, Nishizaki K, Shimizu K, Nagai N2005. Genetic and epigenetic alterations of BRG1 promote oral cancer development. Int J Oncol 26:201–210 [PubMed] [Google Scholar]
- 16.Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC2000. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101:79–89 [DOI] [PubMed] [Google Scholar]
- 17.Sherr CJ, Roberts JM2004. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18:2699–2711 [DOI] [PubMed] [Google Scholar]
- 18.Vlach J, Hennecke S, Amati B1997. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 16:5334–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanashiro K, Kanai M, Geng Y, Sicinski P, Fukasawa K2008. Roles of cyclins A and E in induction of centrosome amplification in p53-compromised cells. Oncogene 27:5288–5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iida H, Towatari M, Tanimoto M, Morishita Y, Kodera Y, Saito H1997. Overexpression of cyclin E in acute myelogenous leukemia. Blood 90:3707–3713 [PubMed] [Google Scholar]
- 21.Erlanson M, Landberg G2001. Prognostic implications of p27 and cyclin E protein contents in malignant lymphomas. Leuk Lymphoma 40:461–470 [DOI] [PubMed] [Google Scholar]
- 22.Jordan S, Lidhar K, Korbonits M, Lowe DG, Grossman AB2000. Cyclin D and cyclin E expression in normal and adenomatous pituitary. Eur J Endocrinol 143:R1–R6 [DOI] [PubMed]
- 23.Hendricks KB, Shanahan F, Lees E2004. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol 24:362–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallette-Kasic S, Figarella-Branger D, Grino M, Pulichino AM, Dufour H, Grisoli F, Enjalbert A, Drouin J, Brue T2003. Differential regulation of proopiomelanocortin and pituitary-restricted transcription factor (TPIT), a new marker of normal and adenomatous human corticotrophs. J Clin Endocrinol Metab 88:3050–3056 [DOI] [PubMed] [Google Scholar]
- 25.Kronenwett U, Castro J, Roblick UJ, Fujioka K, Ostring C, Faridmoghaddam F, Laytragoon-Lewin N, Tribukait B, Auer G2003. Expression of cyclins A, E and topoisomerase II alpha correlates with centrosome amplification and genomic instability and influences the reliability of cytometric S-phase determination. BMC Cell Biol 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koutsami MK, Tsantoulis PK, Kouloukoussa M, Apostolopoulou K, Pateras IS, Spartinou Z, Drougou A, Evangelou K, Kittas C, Bartkova J, Bartek J, Gorgoulis VG2006. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J Pathol 209:512–521 [DOI] [PubMed] [Google Scholar]
- 27.Spruck CH, Won KA, Reed SI1999. Deregulated cyclin E induces chromosome instability. Nature 401:297–300 [DOI] [PubMed] [Google Scholar]
- 28.Fukasawa K2005. Centrosome amplification, chromosome instability and cancer development. Cancer Lett 230:6–19 [DOI] [PubMed] [Google Scholar]
- 29.Ganem NJ, Godinho SA, Pellman D2009. A mechanism linking extra centrosomes to chromosomal instability. Nature 460:278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bilodeau S, Roussel-Gervais A, Drouin J2009. Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle re-entry of differentiated cells. Mol Cell Biol 29:1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chien WM, Garrison K, Caufield E, Orthel J, Dill J, Fero ML2007. Differential gene expression of p27Kip1 and Rb knockout pituitary tumors associated with altered growth and angiogenesis. Cell Cycle 6:750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lloyd RV, Ruebel KH, Zhang S, Jin L2002. Pituitary hyperplasia in glycoprotein hormone α subunit-, p18(INK4C)-, and p27(kip-1)-null mice: analysis of proteins influencing p27(kip-1) ubiquitin degradation. Am J Pathol 160:1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC2008. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci USA 105:2907–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P2003. Cyclin E ablation in the mouse. Cell 114:431–443 [DOI] [PubMed] [Google Scholar]
- 35.Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE1997. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 11:1464–1478 [DOI] [PubMed] [Google Scholar]
- 36.Müller D, Bouchard C, Rudolph B, Steiner P, Stuckmann I, Saffrich R, Ansorge W, Huttner W, Eilers M1997. Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene 15:2561–2576 [DOI] [PubMed] [Google Scholar]
- 37.Ikeda H, Yoshimoto T, Shida N1997. Molecular analysis of p21 and p27 genes in human pituitary adenomas. Br J Cancer 76:1119–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka C, Yoshimoto K, Yang P, Kimura T, Yamada S, Moritani M, Sano T, Itakura M1997. Infrequent mutations of p27Kip1 gene and trisomy 12 in a subset of human pituitary adenomas. J Clin Endocrinol Metab 82:3141–3147 [DOI] [PubMed] [Google Scholar]
- 39.Yoshino A, Katayama Y, Ogino A, Watanabe T, Yachi K, Ohta T, Komine C, Yokoyama T, Fukushima T2007. Promoter hypermethylation profile of cell cycle regulator genes in pituitary adenomas. J Neurooncol 83:153–162 [DOI] [PubMed] [Google Scholar]
- 40.Lidhar K, Korbonits M, Jordan S, Khalimova Z, Kaltsas G, Lu X, Clayton RN, Jenkins PJ, Monson JP, Besser GM, Lowe DG, Grossman AB1999. Low expression of the cell cycle inhibitor p27Kip1 in normal corticotroph cells, corticotroph tumors, and malignant pituitary tumors. J Clin Endocrinol Metab 84:3823–3830 [DOI] [PubMed] [Google Scholar]
- 41.Mastronardi L, Guiducci A, Spera C, Puzzilli F, Liberati F, Maira G1999. Ki-67 labelling index and invasiveness among anterior pituitary adenomas: analysis of 103 cases using the MIB-1 monoclonal antibody. J Clin Pathol 52:107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korbonits M, Chahal HS, Kaltsas G, Jordan S, Urmanova Y, Khalimova Z, Harris PE, Farrell WE, Claret FX, Grossman AB2002. Expression of phosphorylated p27(Kip1) protein and Jun activation domain-binding protein 1 in human pituitary tumors. J Clin Endocrinol Metab 87:2635–2643 [DOI] [PubMed] [Google Scholar]
- 43.Mazumder S, DuPree EL, Almasan A2004. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr Cancer Drug Targets 4:65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishimura T, Takahashi M, Kim HS, Mukai H, Ono Y2005. Centrosome-targeting region of CG-NAP causes centrosome amplification by recruiting cyclin E-cdk2 complex. Genes Cells 10:75–86 [DOI] [PubMed] [Google Scholar]
- 45.Szymas J, Schluens K, Liebert W, Petersen I2002. Genomic instability in pituitary adenomas. Pituitary 5:211–219 [DOI] [PubMed] [Google Scholar]
- 46.Chesnokova V, Melmed S2009. Pituitary tumour-transforming gene (PTTG) and pituitary senescence. Horm Res 71(Suppl 2):82–87 [DOI] [PubMed] [Google Scholar]
- 47.Dworakowska D, Grossman AB2009. The pathophysiology of pituitary adenomas. Best Pract Res Clin Endocrinol Metab 23:525–541 [DOI] [PubMed] [Google Scholar]
- 48.Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, Roberts JM, Kaldis P, Clurman BE, Sicinski P2007. Kinase-independent function of cyclin E. Mol Cell 25:127–139 [DOI] [PubMed] [Google Scholar]
- 49.Fujita M, Takeshita H, Sawa H2007. Cyclin E and CDK2 repress the terminal differentiation of quiescent cells after asymmetric division in C. elegans. PLoS ONE 2:e407 [DOI] [PMC free article] [PubMed]
- 50.Berger C, Pallavi SK, Prasad M, Shashidhara LS, Technau GM2005. A critical role for cyclin E in cell fate determination in the central nervous system of Drosophila melanogaster Nat Cell Biol 7:56–62 [DOI] [PubMed] [Google Scholar]
- 51.Lavoie PL, Budry L, Balsalobre A, Drouin J2008. Developmental dependence on NurRE and EboxNeuro for expression of pituitary POMC. Mol Endocrinol 22:1647–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tremblay Y, Tretjakoff I, Peterson A, Antakly T, Zhang CX, Drouin J1988. Pituitary-specific expression and glucocorticoid regulation of proopiomelanocortin fusion gene in transgenic mice. Proc Natl Acad Sci USA 85:8890–8894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamolet B, Pulichino AM, Lamonerie T, Gauthier Y, Brue T, Enjalbert A, Drouin J2001. A pituitary cell-restricted T-box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell 104:849–859 [DOI] [PubMed] [Google Scholar]
- 54.Rambaud J, Desroches J, Balsalobre A, Drouin J2009. TIF1β/KAP-1 is a coactivator of the orphan nuclear receptor NGFI-B/Nur77. J Biol Chem 284:14147–14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lanctôt C, Lamolet B, Drouin J1997. The bicoid-related homeoprotein Ptx1 defines the most anterior domain of the embryo and differentiates posterior from anterior lateral mesoderm. Development 124:2807–2817 [DOI] [PubMed] [Google Scholar]