

Abstract
The Gram-negative bacterial endotoxin lipopolysaccharide (LPS) elicits a variety of biological responses. Na+/I− symporter (NIS)-mediated iodide uptake is the main rate-limiting step in thyroid hormonogenesis. We have recently reported that LPS stimulates TSH-induced iodide uptake. Here, we further analyzed the molecular mechanism involved in the LPS-induced NIS expression in Fisher rat thyroid cell line 5 (FRTL-5) thyroid cells. We observed an increase in TSH-induced NIS mRNA expression in a dose-dependent manner upon LPS treatment. LPS enhanced the TSH-stimulated NIS promoter activity denoting the NIS-upstream enhancer region (NUE) as responsible for the stimulatory effects. We characterized a novel putative conserved κB site for the transcription factor nuclear factor-κB (NF-κB) within the NUE region. NUE contains two binding sites for the transcription factor paired box 8 (Pax8), main regulator of NIS transcription. A physical interaction was observed between the NF-κB p65 subunit and paired box 8 (Pax8), which appears to be responsible for the synergic effect displayed by these transcription factors on NIS gene transcription. Moreover, functional blockage of NF-κB signaling and site-directed mutagenesis of the κB cis-acting element abrogated LPS stimulation. Silencing expression of p65 confirmed its participation as an effector of LPS-induced NIS stimulation. Furthermore, chromatin immunoprecipitation corroborated that NIS is a novel target gene for p65 transactivation in response to LPS. Moreover, we were able to corroborate the LPS-stimulatory effect on thyroid cells in vivo in LPS-treated rats, supporting that thyrocytes are capable of responding to systemic infections. In conclusion, our results reveal a new mechanism involving p65 in the LPS-induced NIS expression, denoting a novel aspect in thyroid cell differentiation.
Na+/I− Symporter constitutes a novel target of the transcription factor p65 in response to the bac-terial endotoxin lipopolysaccharide in the thyroid cell.
Iodide plays a key role in thyroid physiology as an essential constituent of the thyroid hormones and a major regulator of thyroid function (1). The first step in thyroid hormonogenesis comprises the active transport of iodide across the basolateral plasma membrane, a process mediated by the Na+/I− symporter (NIS) (2). NIS plays an important role in iodide homeostasis (3, 4, 5). TSH is the main regulator of thyroid growth and differentiation. All steps in thyroid hormonogenesis are under TSH regulation, including iodide uptake and NIS gene expression (2).
The rat NIS gene has a minimal promoter region located within −199 and −110 bp (2) and an NIS upstream enhancer (NUE) between −2495 and −2264 bp (2, 6, 7). The NUE region stimulates transcription in a thyroid-specific, cAMP-dependent manner and is involved in the most relevant aspects of NIS regulation. It has been reported that the NUE region contains two thyroid transcription factor-1 (TTF-1)-binding sites that have no apparent effect on NIS transcription, two paired box 8 (Pax8)-binding sites, and a degenerate cAMP-response element (CRE-L) (6). Full TSH/cAMP-dependent transcription requires at least one Pax8-binding site and the integrity of the CRE-L sequence (6).
Nuclear factor-κB (NF-κB) refers to a family of ubiquitous and inducible cellular transcription factors that play a critical role in many cellular processes by regulating the expression of genes involved in inflammation, cell adhesion, cell cycle regulation, angiogenesis, and apoptosis (8, 9). There are five known members of the mammalian NF-κB/Rel family: p65 (also named RelA), c-Rel, RelB, p50, and p52. These proteins share a conserved N-terminal region named “Rel homology domain” (RHD), which is responsible for DNA binding, dimerization, and nuclear translocation (10). In addition, p65, c-Rel, and RelB contain a C-terminal transactivation domain (TAD), thus constituting the transcriptionally active members of the NF-κB family, whereas p50 and p52 primarily repress transcription unless associated with a TAD-containing NF-κB family member, or other proteins capable of coactivator recruitment (8, 10, 11). In most cells, Rel family members form homodimers and heterodimers with different specificities in several combinations. The most well studied and predominant form of NF-κB consists of a heterodimer composed of p65/p50 subunits, even though p65 constitutes the main effector and the most studied subunit of the classic NF-κB pathway (12). The importance of p65 is underscored by the observation that macrophages derived from p65 knockout mice display a severe defect in the expression of inflammatory cytokines upon proinflammatory stimulation (13).
Pathogen-sensing Toll-like receptors (TLRs) mediate downstream activation of NF-κB through an evolutionary conserved signaling pathway (14, 15), thus controlling the expression of several proinflammatory genes in immune cells (16). Among the TLR members, TLR-4 has been well characterized for its capability to recognize the typical pathogen-associated molecular pattern from Gram-negative bacteria, lipopolysaccharide (LPS) (17). A common feature regarding the regulation of NF-κB signaling is its sequestration in the cytoplasm as an inactive complex by physical association with a class of inhibitory molecules known as IκBs (10). Exposure of cells to a variety of inducers such as LPS, phorbol esters, IL-1, or TNF-α results in phosphorylation, ubiquitination, and degradation of the IκB proteins (11, 18). The absence of IκB proteins exposes the nuclear localization sequence in the remaining NF-κB subunit, which in turn leads to a rapid translocation to the nucleus, where it activates a plethora of specific target genes by binding to cognate DNA-regulatory elements known as κB sites in the promoter/enhancer regions of NF-κB-dependent genes (8, 19).
The bacterial endotoxin LPS is an essential outer membrane glycolipid in virtually all Gram-negative bacteria (20). LPS exhibits a wide array of biological effects, mainly on innate immune cells inducing an inflammatory response (21). However, evidence of LPS effects on endocrine cells has been reported in recent years (22, 23, 24). Studies from our group have demonstrated that the endotoxin exerts a direct action on the thyroid cells by up-regulating TSH-stimulated thyroglobulin and NIS gene expression, which reveals the capacity of LPS to alter thyroid-specific gene expression (25, 26). In addition, we recently described the ability of thyroid cells to specifically recognize LPS through TLR-4 (25).
Based on our previous results, we sought to examine the molecular mechanism involved in LPS-induced NIS expression in thyroid cells. Here, we described that LPS treatment increased TSH-induced NIS mRNA expression. By testing internal deletions of the NIS promoter, we determined that the NUE region is responsible for the LPS-stimulatory effect. Bioinformatic analysis over the NUE region looking for conserved sites of LPS canonical effectors showed a novel conserved cis-acting element for the transcription factor NF-κB. In addition, site-directed mutagenesis of the κB site abrogated LPS-induced NIS stimulation. Silencing of endogenous p65 by small interfering RNA (siRNA) confirmed its participation as mediator of LPS action. Remarkably, a physical interaction was observed between the NF-κB p65 subunit and Pax8, main regulator of NIS expression, which appears to be responsible for the synergic effect displayed by these transcription factors on NIS gene transcription. Furthermore, using chromatin immunoprecipitation (ChIP) we corroborated that NIS is a novel target gene for p65 transactivation in response to LPS. Finally, we were able to reproduce the LPS-stimulatory effect on thyroid cells of LPS-treated rats, which revealed that thyrocytes has the potential capability to respond to systemic infections. Overall, our results uncovered the role of the transcription factor p65 in LPS-induced NIS expression, and they established a novel mechanistic model for LPS-induced gene expression in thyrocytes.
Results
LPS stimulates TSH-induced NIS mRNA levels
To investigate the molecular mechanism involved in the LPS-stimulatory effect on NIS gene expression, we analyzed LPS-mediated effects at the transcriptional level. Starved Fisher rat thyroid cell line 5 (FRTL-5) cells were treated with LPS (10 and 100 ng/ml) in the presence or absence of TSH (0.5 mIU/ml) for 6 h, and NIS mRNA levels were analyzed by quantitative RT-PCR assay. As expected (27, 28), TSH stimulated NIS mRNA expression (Fig. 1). In agreement with previous observations indicating that LPS increases TSH-induced iodide uptake and NIS protein expression (25), LPS treatment augmented TSH-stimulated NIS mRNA levels in a dose-dependent manner (Fig. 1). Incubation with LPS in the absence of TSH did not modify NIS mRNA levels in comparison with those of nonstimulated cells. These data suggest that LPS modulates TSH-induced NIS transcription.
Fig. 1.

LPS increases TSH-stimulated NIS mRNA level in thyroid cells. Quiescent FRTL-5 cells (5 d of TSH and serum deprivation) were treated with LPS (10–100 ng/ml) in the presence or absence of TSH (0.5 mIU/ml) for 6 h. Total RNA was extracted and reverse transcribed. Real-time PCR analysis was performed to relatively quantify NIS mRNA levels to those of β-actin under the different conditions. The expression level of untreated cells was set to 1. Values are indicated as fold of change to the mRNA levels of nontreated cells. #, P < 0.001 vs. basal; *, P < 0.05; **, P < 0.001 vs. TSH (ANOVA; Student-Newman-Keuls).
LPS up-regulates the TSH-dependent transcriptional activation of the NIS gene
To study whether LPS exerts a transcriptional regulation of the NIS gene, we assessed the effect of LPS on the activity of the rat NIS promoter. The luciferase reporter construct pNIS-2.8 was transiently transfected into FRTL-5 cells, and its transcriptional activity in response to LPS (100ng/ml) was assayed in the presence or absence of TSH (0.5 mIU/ml) for 24 h. TSH stimulated pNIS-2.8 activity, as has been extensively reported (28, 29), whereas LPS induced a significant increase of the TSH-mediated transcriptional activation (Fig. 2A). Consistent with the data presented in Fig. 1, the NIS promoter activity was not affected by LPS in the absence of TSH (data not shown).
Fig. 2.

LPS stimulation of NIS promoter activity depends on the NUE region and involves a NUE-C-binding site. A, FRTL-5 cells were transiently transfected with different NIS promoter constructs linked to the reporter gene luciferase (Luc). The left panel shows transfected constructs, and the right panel shows transcriptional activity in response to the indicated stimulus. Starved cells were treated with LPS (100 ng/ml) in the presence of TSH (0.5 mIU/ml) for 24 h. Results are expressed as Luc activity normalized to β-galactosidase and relative to basal activity for each construct. #, P < 0.001 vs. basal; *, P < 0.005 vs. TSH (ANOVA; Student-Newman-Keuls). B, FRTL-5 cells were transiently transfected with different NUE constructs linked to Luc. The left panel shows transfected constructions in which the shapes refer to transcription factor-binding sites. The right panel shows transcriptional activity in response to the indicated stimulus. Cells were treated after starvation as indicated in the figure for 24 h. Results are expressed as Luc activity normalized to β-galactosidase and relative to basal activity for each construct. #, P < 0.001; ##, P < 0.01 vs. basal; *, P < 0.005 vs TSH (ANOVA; Student-Newman-Keuls). TK, Thymidine kinase.
To identify the NIS promoter region responsible for the LPS-induced regulation, we analyzed the transcriptional activity of the 5′-deleted constructs lacking the NUE region i.e. pNIS-2.0, pNIS-1.2, and pNIS-0.5. As described, NUE is required for full TSH stimulation of NIS gene (7, 29), and LPS had no effect on the transcriptional activity of any NUE-deleted construct (Fig. 2A). Transfection of the internal deleted NUE-carrying constructs, pNIS-1.2 NUE and pNIS-0.5 NUE, showed significant activity in response to TSH, as reported (29). Moreover, an increase of the TSH-induced transcriptional activity was observed when cells were treated with LPS (Fig. 2A). In summary, the results indicate that LPS augments the TSH-dependent transcriptional activation of the NIS gene and that cis-elements present within the NUE region seem to be required.
The NUE region contains at least three crucial sites whose sequence substitutions lead to severe loss of enhancer activity in thyroid cells (6). One of them is the overlapped binding sites of Pax8/TTF-1 (NUE-A), and the other two are the CRE-L site (NUE-B) and the second binding site for Pax8 (NUE-C). To determine the sequence directly responsible for the LPS-mediated enhancer activity, we tested site-directed mutants of the wild-type rat NUE region linked to the thymidine kinase promoter (pNUE) (6, 7). To this end, transiently transfected FRTL-5 cells were treated with LPS (100 ng/ml) in the presence of TSH (0.5 mIU/ml) for 24 h. As reported, TSH induced the enhancer activity of the reported vector pNUE (6, 7). In agreement with the results stated above, under LPS stimulation pNUE displayed a significant increase in the reporter gene activity induced by TSH. By contrast, the site-directed mutants, pNUE-B MT and pNUE-A+C MT, did not respond to treatment with TSH in the presence or absence of the endotoxin (Fig. 2B). Mutagenesis of Pax8 sites (NUE-A or NUE-C) showed different results. Despite the fact that both site-directed mutants respond to TSH treatment, NUE-A did not impair LPS-mediated stimulation of NUE activity, whereas disruption of NUE-C site interfered with the LPS effect (Fig. 2B). Hence, although NUE-C plays a role in mediating the LPS-induced stimulation of the NUE region, it also requires cooperation of a fully TSH-responsive promoter.
A novel NF-κB-binding site mediates NIS up-regulation by LPS
A bioinformatical study of the rat NUE region sequence was undertaken with MatInspector version 8.0 software (Genomatix, Munich, Germany) to identify possible transcription factor-binding sites involved in the effect of LPS. The analysis revealed a putative κB-binding site (5′-GGACAGTCCC-3′) between nucleotides −2284 and −2275 minus strand (core similarity = 1; matrix similarity = 0.93) strikingly similar to the current NF-κB consensus motif (5′-GGGRNNTYCC-3′ R =A/G, Y=T/C) (19), and in close proximity with NUE-C site (−2296 to −2288) (Fig. 3A).
Fig. 3.

A novel κB consensus site at the NUE region of NIS promoter is required for the LPS effect. A, Comparison of the NIS upstream enhancer (NUE) sequences of human (hNIS), rat (rNIS), and mouse (mNIS). Alignment was performed by using ClustalW2 Multiple Sequence Alignment (http://www.ebi.ac.uk/tools/clustalw2) and manually fitted as described elsewhere (2 77 ). Shaded boxes denote conserved cis-acting regions experimentally tested along the sequences. Note that one Pax8-binding site (NUE-C) is missing on the human NUE, and a putative κB sequence is surprisingly close to the NUE-A element (33 ). Numbers on the left side represent their location in the NIS promoter considering as +1 the first nucleotide where transcription starts. B, Representative EMSA showing that LPS treatment increases a recruitment of the NF-κB complex to the region −2291 to −2272 within NUE region (NUE-κB probe) (lines 1–3). Specificity as achieved by performing competition reactions in the presence of a 100-fold excess of cold oligonucleotide NUE-κB (lines 4–6), and by the absence of shifted complex using the cognate sequence-mutated oligonucleotide NUE-κB MT (lines 10–12). Incubations performed in the presence of an anti-p65 antibody supershifted the specific NF-κB complex (lines 7–9), thus denoting the ability of the p65 subunit to recognize the putative κB cis-acting element observed within NUE region. C, FRTL-5 cells were transiently transfected with the pNUE constructs linked to Luc shown in the left panel. The right panel shows transcriptional activity in response to the indicated stimulus. Cells were treated after starvation as indicated for 24 h. Results are expressed as Luc activity normalized to β-galactosidase and relative to basal activity for each construct. #, P < 0.001; ##, P < 0.01 vs. basal; *, P < 0.01 vs. TSH; †, P < 0.01 vs. TSH in pNUE; ††, P < 0.001 vs. TSH + LPS in pNUE (ANOVA; Student-Newman-Keuls). TK, Thymidine kinase.
The alignment and comparison of the NUE region in different species (human, rat, and mouse) showed a remarkable conservation of the sequence (70% homology) and position of putative cis-acting elements, including the κB site discovered here, uncovering the potential importance of this binding site (30, 31). In the rodent NUE region, two Pax8 elements are present (7, 32), whereas in the human NUE, the Pax8 element downstream to CRE-L is missing (33) (Fig. 3A). The NIS gene promoter/enhancer is regulated by these trans-acting factors, although with some variation among species (7, 33, 34, 35). Surprisingly, we found that the κB region is strongly conserved along the tested species and strictly positioned close to a Pax8 element, raising the question of whether NF-κB is a functionally important trans-acting factor related to NIS transcriptional regulation.
Using EMSA, we examined the binding of FRTL-5 nuclear extracts to the radiolabeled NUE-κB oligonucleotide. As shown in Fig. 3B (lines 1–3) a specific single NF-κB complex binds to the probe. The observed complex was highly responsive to the indicated treatment. Binding specificity was confirmed by complete displacement of the shifted band using an excess of the unlabeled self oligonucleotide (Fig. 3B, lines 4–6). Conversely, the observed DNA-NF-κB complex was not competed by the mutated κB probe (NUE- κB MT, which contains the underlined mutations GGACAGTCCC to AAGCAGCTTC in the κB-binding core motif) (data not shown). Moreover, 32P-labeled NUE-κB MT did not form a complex when incubated with FRTL-5 nuclear extracts (Fig. 3B, lines 10–12). When we assessed the participation of the NF-κB p65 subunit in the observed complex by supershift assay using a specific monoclonal anti-p65 antibody, a visible although partial displacement was obtained (Fig. 3B, lines 7–9).
To confirm the role of NF-κB in the described effect, we disrupted by site-directed mutagenesis the κB-binding site core found in the rat NUE. FRTL-5 cells were transiently transfected with pNUE and the κB mutant reporter construct (pNUE-κB MT). After transfection, cells were treated with LPS (100 ng/ml) in the presence of TSH (0.5 mIU/ml), and luciferase activity was assayed after 24 h. Results demonstrated that disruption of the κB-binding sequence completely abrogates the ability of the NIS enhancer to respond to the endotoxin (Fig. 3C). Similar results were obtained in the context of the pNIS-2.8 promoter mutated at the κB site (data not shown). This indicates that the NF-κB site is indeed functional and essential for LPS-induced NIS promoter activity. Moreover, mutating of the κB-binding site also reduces, but does not abrogate, the induction exerted by TSH on the NIS promoter (Fig. 3C), suggesting that this region may be involved in the physiological TSH responsiveness.
p65 and Pax8 synergistically stimulate NIS transcription
Our evidence suggests that NF-κB is a novel player involved in NIS transcriptional regulation. We explored which NF-κB members are able to induce NIS transcriptional activity. Expression vectors encoding p65, c-Rel, RelB, p52, or p50 were cotransfected into Cos-7 cells along with the reporter construct pNUE or a NF-κB-responsive vector containing five multimerized κB-binding sites linked to luciferase (5×κB-luc) as control. Pax8 expression plasmid, transfected as positive control, activated pNUE transcriptional activity as described elsewhere (6). Interestingly, we observed that the transactivation domain carrying subunits p65 and cRel, but not RelB, enhanced pNUE transcriptional activity, albeit to a different extent (Fig. 4A, black bars). In addition to p65, cRel and RelB activated the NF-κB-driven reporter gene (Fig. 4A, gray bars). As expected, such an effect was not evident for p50 or p52 (11).
Fig. 4.

p65 activates NIS gene expression and interacts with Pax8. A, Cos-7 cells were transiently cotransfected with an expression vector encoding the five different NF-κB members along with the indicated reporter vectors. Pax8 expression plasmid was used to monitor pNUE activity as positive control. 5×κB-luc reporter vector was used as a positive control to ensure functional expression of the transfected NF-κB proteins. pNUE construct was used as a reporter gene of NIS transcription. The same amounts of DNA were transfected under all the assayed conditions by adding an empty pcDNA3.1 vector. Luciferase activity was assayed 48 h after transfection. Results are expressed as Luc activity normalized to β-galactosidase and relative to the activity of the empty vector. #, P < 0.005; ##, P < 0.01 vs. vector; *, P < 0.001 vs. vector (ANOVA; Student-Newman-Keuls). B, p65N and p65C are expression vectors encoding p65 protein segments N-terminal (aa 1-313) and C-terminal (aa 314-551), respectively. pNUE plasmid was used as a reporter gene. Results are expressed as Luc activity normalized to β-galactosidase and relative to basal activity for each construct. *, P < 0.01; **, P < 0.005; ***, P < 0.001 vs. vector; †, P < 0.01 vs. p65 + Pax8 (ANOVA; Student-Newman-Keuls). C, Upper panel, Schematic representation of full-length p65 and p65-deleted constructs linked to GST used as bait proteins. Locations of the N-terminal RHD, and the C-terminal TAD are indicated. Lower panel, GST pull-down assay demonstrates p65 and p65N interaction with the paired domain transcription factor Pax-8. FRTL-5 protein extracts were incubated with Sepharose-bound GST (lane 1) or GST-p65 constructs (lanes 2–4), and the interacting proteins were visualized by Western blot (WB) using a monoclonal anti-Pax8 antibody. A fraction (10%) of input whole-cell lysates (WCL) used as a prey protein was analyzed for loading comparison (lanes 1–4, lower panel). D, Cos-7 cells were transiently cotransfected with the expression vectors encoding the transcription factors p65 and Pax8 along with the indicated pNUE reporter vectors bearing site-directed mutations. Results are expressed as Luc activity normalized to β-galactosidase and relative to the activity displayed by the empty expression vector arbitrarily set as 1.0. *, P < 0.01 vs. vector; #, P < 0.05 vs. Pax8 or p65 alone (ANOVA; Student-Newman-Keuls).
The assembly of NF-κB with different coactivator and corepressor proteins is crucial in orchestrating its ultimate biological effects. NF-κB can associate with different coactivators, such as the CREB-binding protein (CBP) or p300, thus sharply increasing the transcriptional activity of the NF-κB complex (17, 36, 37). Several lines of evidence indicate that Pax8 is a transcription factor strictly required for NIS gene transcription in thyroid and nonthyroid cells (38, 39). Physical interactions of Pax8 with TTF-1 and p300 have been demonstrated to regulate thyroid gene expression (40, 41). Although site-directed mutation of the Pax8-binding site NUE-C did not alter the nucleotide sequence of the adjacent κB-binding site, this disruption abrogated LPS stimulation; as a consequence, we further hypothesized that Pax8 and p65 may act together to induce NIS expression. To test this hypothesis, Cos-7 cells were cotransfected with p65 and Pax8 expression vectors together with the reporter construct pNUE. Each transcription factor, when transfected separately, enhanced NIS transcription (Fig. 4, A and B). Interestingly, coexpression of p65 and Pax8 led to a marked synergism in the activation of the NIS promoter (Fig. 4B).
We sought next to determine the region of p65 involved in NIS transactivation and Pax8 cooperation. We generated vectors encoding p65-truncated proteins containing its N-terminal (p65N) or C-terminal (p65C) regions. Mutants were cotransfected along with the Pax8 expression vector and pNUE reporter into Cos-7 cells. As expected, deletion of the TAD impaired the capacity of p65 to activate NIS transcription. Thus, p65 lacking its N-terminal portion activated NIS transcription, although it did not synergize with Pax8 (Fig. 4B).
Considering the close proximity of the κB- and NUE-C-binding sites within the rat NIS enhancer and the synergic stimulus exerted by p65 and Pax8 on NIS transcriptional activity, we studied the potential physical interaction between these transcription factors using glutathione-S-transferase (GST) pull-down assays with total FRTL-5 protein extracts as a prey (Fig. 4C). A physical interaction between p65 and Pax8 was demonstrated by the complexes formed with GST-p65 beads and endogenous Pax8 (Fig. 4D, lane 2). These findings were corroborated by GST pulldown using GST-Pax8 beads and endogenous p65 (data not shown). As shown in Fig. 4C (line 4), Pax8 seems to interact with p65 RHD because it was retained by the construct GST-p65N. In contrast, the TAD-carrying construct GST-p65C did not precipitate Pax8 above background levels (line 3). The lower panel shows the input extracts used for the pull-down assays, as probed for Pax8. These data demonstrate that p65 could interact through its N-terminal domain [amino acids (aa) 1-313] with Pax8 in thyroid cells to regulate NIS gene expression. To further confirm this finding, FRTL-5 nuclear cell lysates were immunoprecipitated with anti-p65 monoclonal antibody, and then bound Pax8 was detected by immunoblot (Supplemental Fig. 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org).
Next, we analyzed whether the described binding sites were involved in the observed synergic effect in transfected COS-7 cells. As displayed in Fig. 4D, site-directed mutagenesis of the κB as well as NUE-C binding site abrogated the trans-activating individual effect of the transcription factors. Interestingly, binding site mutation disrupted the cooperation of p65 and Pax8 in activating NIS enhancer function. Supplemental Fig. 2 shows p65 and Pax8 interaction with its cognate binding site present within NUE region. Lastly, the obtained evidence seems to indicate a cooperative role displayed by p65 and Pax8 on NUE function. This cooperative association might involve not only a direct protein-protein interaction but also a direct interaction with its cis-elements.
NF-κB proteins are involved in LPS-induced NIS gene expression
NF-κB subunits normally reside in the cytoplasm of nonstimulated cells and are imported to the nucleus in response to LPS signaling to regulate target gene transcription (12, 42). Because the nuclear entry of NF-κB subunits is a key step in signal transduction, we analyzed the accumulation of p65 in the nucleus of FRTL-5 cells after LPS treatment in the presence of TSH. We observed a strong induction of p65 nuclear translocation after LPS treatment compared with that in TSH-treated cells (Fig. 5A), confirming activation of the NF-κB signaling pathway and supporting the participation of p65 as an intracellular effector of LPS action in thyroid follicular cells.
Fig. 5.

NF-κB-mediated pathway is involved in the LPS-stimulating effect. A, Representative Western blot showing nuclear recruitment of p65 after treatment. Basal FRTL-5 cells were treated with TSH (0.5 mIU/ml) in the presence or absence of LPS (100 ng/ml) for different periods of time. Nuclear protein fraction was recovered as described in Materials and Methods. The nuclear protein histone H1 was used to correct loading differences. The lack of staining for the cytoplasmic protein α-tubulin shows no cytoplasmic contamination in the nuclear preparation. Densitometric analysis was performed to determine the relative nuclear increase (fold increase) of p65 normalized to histone H1. The value of nonstimulated cells was set up arbitrarily as 1.0. B, Relative activity of the NF-κB response element (5×κB-luc) in response to the indicated treatments. The NF-κB inhibitor SZ (20 μm) was used as specificity control. #, P < 0.01; ##, P < 0.001 vs. basal; †, P < 0.01 vs. same condition in the absence of SZ (ANOVA; Student-Newman-Keuls). C, Relative quantification of NIS mRNA levels (RT/qPCR) normalized to the levels of β-actin. Sulfasalazine was added to the media 1 h before treatment. Values are indicated as fold of change to the mRNA levels of nontreated cells. #, P < 0.005; ##, P < 0.01 vs. basal; *, P < 0.01 vs. TSH; †, P < 0.01 vs. same condition in the absence of inhibitor (ANOVA; Student-Newman-Keuls). D, FRTL-5 cells were transiently transfected with pNUE construction linked to Luc. Cells were treated after starvation as indicated in the figure for 24 h. Results are expressed as Luc activity normalized to β-galactosidase and relative to basal activity for each construct. #, P < 0.001; ##, P < 0.005 vs. basal; *, P < 0.005 vs. TSH; †, P < 0.01 vs. same condition in the absence of inhibitor (ANOVA; Student-Newman-Keuls).
We subsequently analyzed the ability of NF-κB to act as a transcription factor to regulate the expression of its target genes in response to LPS in thyrocytes. FRTL-5 cells were transiently transfected with the NF-κB reporter 5×κB-luc. Cells were treated with LPS (100 ng/ml) for 24 h in the absence or presence of TSH (0.5 mIU/ml). As shown in Fig. 5B, these results suggest that LPS potently induced activation of NF-κB signaling in TSH-treated cells. However, a slight increase was observed in the absence of TSH. Assay specificity was verified by inhibiting the activation of 5×κB-luc by performing the assay in the presence of the NF-κB-specific inhibitor sulfasalazine (SZ) (43) (Fig. 5B). It is noteworthy that TSH treatment slightly recruits p65 to the nuclear compartment (Fig. 5A) and induces NF-κB-mediated transcriptional activation (Fig. 5B), thus underscoring the potential ability of TSH to induce NF-κB signal pathway, as previously postulated (44, 45).
To gain insights into the mechanisms involved in LPS-induced NIS expression, we investigated NF-κB pathway activation using the chemical inhibitor SZ. Induction of NIS mRNA expression (Fig. 5C) and NIS promoter activity (Fig. 5D) by LPS in the presence of TSH was blunted by the presence of SZ (20 μm). The inhibitor abolished the effect of LPS but did not significantly affect TSH stimulation on the analyzed parameters at the assayed concentration. Thus, our results confirm the involvement of the NF-κB signaling in the stimulatory effect triggered by LPS on NIS transcriptional expression in thyroid cells.
Silencing of p65 by siRNA abrogates LPS effect on NIS gene expression
Present data seem to indicate that p65 is potentially involved in LPS-induced NIS transcriptional expression. To corroborate the role of p65 in NIS transcriptional stimulation by LPS, we silenced endogenous p65 protein expression by a specific siRNA pool. FRTL-5 cells were transfected with nonrelated siRNA (scramble) or specific rat p65 siRNA. An immunoblotting assay showed that p65 was efficiently knocked down after 72 h of siRNA transfection. In addition, p65 siRNA, but not scramble siRNA, specifically reduced the expression of p65 but not β-actin (Fig. 6A). Functional analysis demonstrated that the knockdown of p65 blocked the stimulatory effect of LPS on NIS mRNA expression (Fig. 6B). The specificity of this approach was confirmed by transfecting p65 siRNA in combination with an expression vector encoding a human p65 cDNA that is not targeted by the rat-specific siRNA pool. As shown in Supplemental Fig. 3, under these conditions, p65 expression levels were recovered. Reconstitution of p65 expression in thyroid cells reversed the NIS mRNA repression, displaying an expression level comparable to that observed in wild-type cells (Fig. 6B). When we analyzed the transcriptional effect displayed by LPS in the absence of p65 on NIS promoter activity, a similar abrogation was observed (Fig. 6C). These findings indicate that the NF-κB p65 subunit plays an important role in LPS-induced NIS up-regulation.
Fig. 6.
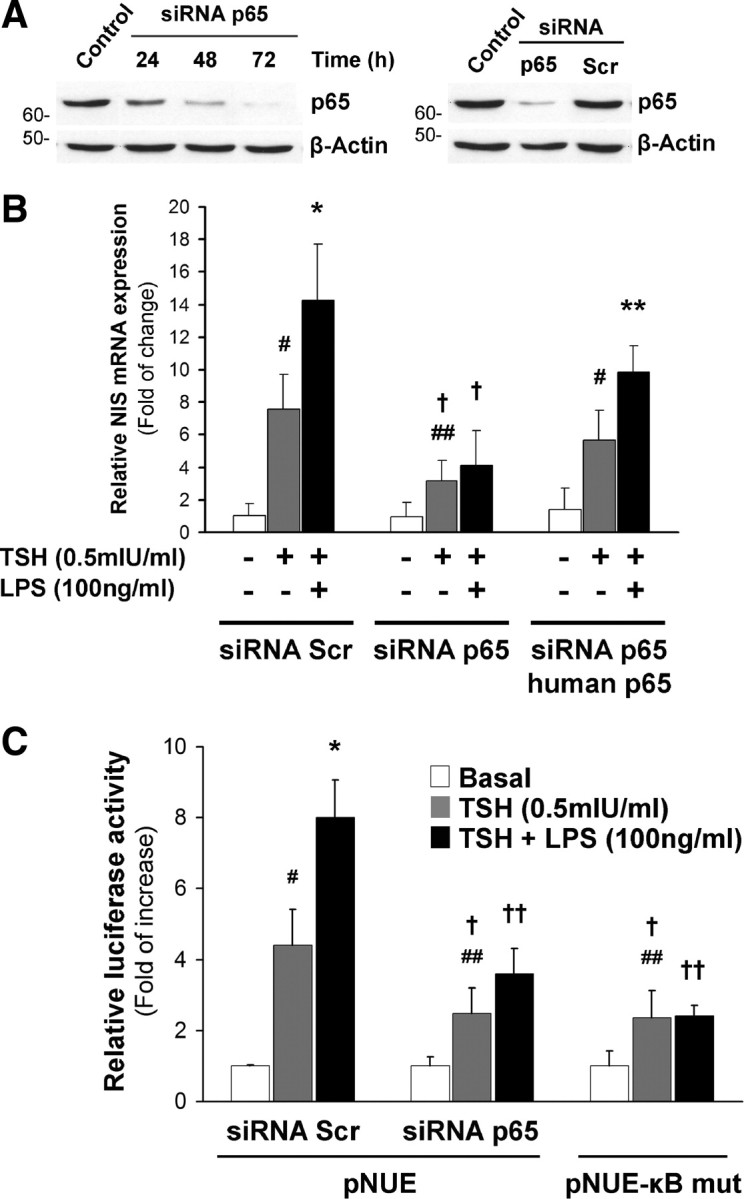
Silencing of endogenous p65 by siRNA blocks the LPS effect on NIS expression. A, Representative Western blot evaluating p65 protein levels after siRNA [scrambled (Scr) or p65-directed] transfection in FRTL-5 cells. B, Quiescent FRTL-5 cells transfected with scrambled siRNA, p65-targeted siRNA (p65 siRNA), and p65 siRNA together with human p65 expression vector (p65 siRNA/human p65) were treated after 72 h from siRNA transfection with TSH (0.5 mIU/ml) in the presence or absence of LPS (100 ng/ml) for 6 h. Relative quantification of NIS mRNA levels was performed by RT/qPCR and normalized to the levels of β-actin. Values are indicated as fold of change to the mRNA levels of nontreated cells. #, P < 0.005; ##, P < 0.05 vs. basal; *, P < 0.01; **, P < 0.05 vs. TSH; †, P < 0.001 vs. same treatment in scr-transfected cells (ANOVA; Student-Newman-Keuls). C, FRTL-5 cells were transiently cotransfected with scrambled or p65-specific siRNA and the pNUE reporter construction. pNUE-κB MT was used as LPS-nonresponsive control. Cells were treated for 12 h after starvation as indicated in the figure. Results are expressed as Luc activity normalized to β-galactosidase and relative to basal activity for each construct. #, P < 0.005; ##, P < 0.05 vs. basal; *, P < 0.01 vs. TSH; †, P < 0.05; ††, P < 0.005 vs. same condition in Scr-transfected cells (ANOVA; Student-Newman-Keuls).
p65 binds to the NIS enhancer region
To further prove the mechanism by which p65 promotes NIS expression, we examined the association in vivo between endogenous p65 and the NUE region using ChIP assays. FRTL-5 cells were stimulated with LPS (100 ng/ml) in the presence or absence of TSH (0.5 mIU/ml) for 1 h. Soluble cross-linked and sonicated chromatin was immunoprecipitated using a monoclonal ChIP-suitable antibody against the p65 subunit. The DNA content of immunoprecipitates was then analyzed by real-time PCR. Quantitative ChIP analysis revealed enrichment in sequences spanning the κB site (P1) predicted in the NUE region (Fig. 7B, black bars) but not sequences close to the transcriptional start site (P2) of the NIS gene (Fig. 7B, gray bars). To rule out nonspecific background of the ChIP assay, we performed the immunoprecipitation using nonrelated mouse IgG (mock). Therefore, the measured association between p65 and the NUE DNA region is specific. Interestingly, ChIP analysis showed that enrichment in p65 immunoprecipitates was consistently reduced in the presence of the NF-κB inhibitor SZ (Fig. 7B). Hence, these experiments establish that, in response to LPS stimulation, p65 binds to the NIS promoter in an inducible fashion and confirm that p65 directly binds to the NUE region carrying a putative κB consensus site.
Fig. 7.

p65 binds to the NUE region in response to LPS treatment. A, Schematic drawing of the rat NIS promoter (PP, proximal promoter). Positions are relative to the start of the first exon denoted as +1. The location of primer sets (P1 and P2) used in ChIP experiments are shown. Transcription factor-binding sites are indicated. B, Basal FRTL-5 cells were treated under the indicated conditions for 1 h before cross-linking and further ChIP assay. Sulfasalazine was added to the media 1 h before treatment. DNA amount was evaluated by qPCR. Results are expressed as relative fold of increase (p65 IP/Total input), taking arbitrarily the basal relation as 1.0. Relative fold of increase were calculated according to the equation Fold Change = 2[(Ct.input − Ct.target) − (Ct.input − Ct.mock)]; #, P < 0.01 vs. basal; *, P < 0.01 vs. TSH; †, P < 0.05; ††, P < 0.005 vs. the same condition in the absence of inhibitor (ANOVA; Student-Newman-Keuls).
Subseptical dose of LPS induces NIS mRNA expression in rat thyroid cells
To study the role of LPS in thyroid physiology in vivo, we measured the response displayed by the gland after a single injection of LPS in a subseptical concentration (10 μg/kg). After the treatment we observed a rapid stimulation of NIS mRNA expression in the thyroid gland. By 3 h after LPS application, specific NIS mRNA expression level was 2.2-fold higher than in thyroids of untreated controls; surprisingly, as soon as 1 h after injection, NIS mRNA levels were also increased, although levels were not of statistical significance. Six hours after LPS treatment, NIS transcript levels reach basal levels, and 24 h after treatment NIS mRNA level remained unchanged (Fig. 8, upper panel). Plasmatic levels of thyroid hormones were evaluated after the endotoxin challenge. Serum T3 and T4 were significantly decreased 6 and 24 h after injection, compared with control animals, thus assuming the characteristics of the nonthyroidal illness (NTI) syndrome elicited by LPS (46). Consistently, data concerning the serum levels of the antiinflammatory agent cortisol show a significant increase 6 h after the inflammatory stimulus (Supplemental Fig. 4).
Fig. 8.

LPS injection induces NIS mRNA expression in vivo. Male Wistar rats were divided in five groups (n = 8 per group). Animals were treated as described in Materials and Methods for the indicated periods of time with a subseptical dose of LPS (10 μg/kg). Total RNA was extracted from thyroid and liver tissue and reverse transcribed. Real-time PCR analysis was performed to relatively quantify mRNA levels. Upper panel shows NIS, IκB-α, and IL-6 mRNA expression in the thyroid gland, whereas lower panel demonstrate D1, IκB-α, and IL-6 mRNA expression in the liver. The expression level of control animals was arbitrarily set to 1. Data are indicated as fold of change to the mRNA levels of control animals and presented as box plots. *, P < 0.05 vs. control (Kruskal-Wallis; Dunn).
A time-delaying strategy that control unrestricted NF-κB activity relies on the principle that negative regulators are themselves highly regulated NF-κB target genes. Particularly, NF-κB inhibitor-α (IκB-α) is one of the earliest induced NF-κB targets and plays an important role in removing NF-κB from its cognate DNA sequence ensuring termination of the early NF-κB response (47). We observed an up-regulation of the mRNA expression levels of the NF-κB inhibitor IκB-α in thyroid gland, the expression of which reliably, although indirectly, indicates NF-κB activation after LPS treatment. Moreover, in the thyroid lysate we observed a strong increase in the expression of the proinflammatory cytokine IL-6 gene, a known target of NF-κB activation recently described to respond specifically to p65 regulation (48) (Fig. 8, upper panel).
Additionally, we analyze the expression of well-described markers of systemic inflammation in the liver as control of the response to the endotoxin treatment (49, 50). As expected, we observed a rapid and transient reduction in the mRNA levels of type 1 deiodinase (D1) as well as a strong increase of the cytokine IL-6 and the NF-κB-regulator IκB-α subunit (Fig. 8, lower panel).
Discussion
Our results provide direct evidence that LPS increases the TSH-induced NIS gene expression at the transcriptional level in thyroid cells. This is consistent with our previous reports indicating that LPS stimulates TSH-dependent iodide uptake and NIS protein expression (25) as well as thyroglobulin gene expression (26), all of which are well-defined markers of thyroid differentiation. Taken together, these data favor the ability of the endotoxin to stimulate different aspects of thyroid hormone biosynthesis modifying thyroid homeostasis. This notion is strongly supported by our recent observations indicating that a functional TLR-4 conferring LPS responsiveness is expressed in thyroid cells (25). The special ability of thyroid follicular cells to specifically recognize and respond to LPS by increasing the expression of proteins involved in thyroid function argues for a possible role of this endotoxin as a potential modifier of thyroid homeostasis with pathophysiological implications.
NIS is a membrane protein responsible for active transport of iodide in the thyroid, gastric mucosa, intestinal epithelium, and lactating mammary glands (1, 3). Iodide uptake in thyroid follicular cells is an essential step in the biosynthesis of thyroid hormones, a process up-regulated by TSH through either transcriptional or posttranslational modification of NIS (1, 51). Despite the molecular cloning of the NIS enhancer and its 5′-flanking region, the detailed mechanism by which NIS gene expression is regulated by TSH and other cell-specific cues is largely unknown (6, 7, 34). The proximal promoter region of the NIS gene contains binding sites for thyroid-specific transcription factors (34), but the complete activation of the gene by TSH requires the further upstream enhancer (6, 7, 28, 52). In rat thyroid cells, the regulatory region that confers the strongest TSH/cAMP response was mapped to the NUE, a 232-bp region located between −2495 and −2264 nucleotides that contains clusters of several binding sites for TTF-1 and Pax8, as well as a degenerate cAMP-responsive element (CRE-L) (6). To analyze the role of LPS in the regulation of NIS gene transcription, we used a 2.8-kb DNA fragment of the rat NIS promoter that includes NUE region (29). Our results of transient transfection assays agree with the requirement of the NUE region for a strong functional response to TSH, as previously described (2, 7). To pinpoint the region involved in TSH-dependent LPS-induced stimulation, we studied several deletions of the NIS promoter. The region between −2854 and −1941 bp, which includes the NUE region, was responsible for the LPS-stimulatory effect.
Surprisingly, using a bioinformatical search to find potential cis-elements related to LPS transcriptional effect, we detected a novel κB site in the NUE region of the NIS 5′-flanking region (−2284 to −2275). Moreover, the site appears to act effectively as a functional mediator of the LPS stimulation of NIS expression, as demonstrated here by site-directed mutagenesis. Interestingly, we observed that the κB region is strongly conserved among different species i.e. rats, mice, and humans, which led to the question of whether NF-κB is a functional and important trans-acting factor for NIS gene regulation. In concordance, recent reports have suggested that genome comparison of closely related species could lead to correct predictions of regulatory sequences (30, 31, 53).
These findings seem to indicate that the LPS signal interacts at the level of the TSH-stimulated NUE-dependent activity. In addition to CRE-L, TSH-dependent transcriptional activation of NUE requires participation of at least one of the two existing Pax8-binding sites (6, 33). Coincidentally, by site-directed mutagenesis, we observed the involvement of the Pax8-binding site NUE-C in the LPS action. Consistent with this observation, an earlier report from our group proposed the mediation of Pax8 in the LPS action on thyroid gene regulation by demonstrating the functional relevance of TTF-1 and Pax8-binding activity to the C-site present in the thyroglobulin promoter responsible for LPS-stimulated thyroglobulin gene expression (26). It is interesting that we demonstrated a novel ability of the NF-κB p65 subunit to synergize with Pax8 (Fig. 4). Because the Pax8 protein is expressed in a tissue-specific manner and, particularly in the thyroid, it is induced and activated by TSH (54, 55), such a synergistic mechanism involving p65 and Pax8 helps us to understand how LPS can induce NUE-mediated transcriptional activity in the presence but not the absence of TSH. However, the observation of cAMP-induced transcription mediated by protein kinase A-induced phosphorylation of p65 in some cell types could also account for the necessity of TSH to elicit the LPS effect (56, 57, 58). In agreement, serine 276 of p65 is specifically phosphorylated by protein kinase A during IκB degradation and is strikingly necessary for the recruitment of coactivators to p65 for active transcription (59, 60). Although the binding of p65 and Pax8 to their adjacent sites in NUE could create a strong synergy, such cooperation seems to involve direct protein-protein interactions between p65 RHD, but not the C-terminal region containing the TAD domain, and Pax8, as suggested by the GST pull-down assay. Moreover, functional cooperation of p65 with Pax8 was not observed in the absence of the RHD, suggesting the importance of the observed DNA-protein interaction. These findings, together with the observation that the proximity of p65 and Pax8 sites in NUE is conserved in rats, mice, and humans, suggest a functional interaction between both proteins and a potential role of this interaction in thyroid-specific gene expression and differentiation. Similarly, in different systems, the RHD of the NF-κB p65 subunit interacts with several diverse coactivator proteins, such as CREB-binding protein (CBP). As mentioned before p300, BRCA1, and members of the basic region-leucine zipper (bZIP) family, to activate transcription (17, 36, 37, 61).
Here we used different approaches to characterize the functional role of NF-κB as the mediator of LPS action in thyroid cells. The induction of p65 nuclear translocation as well as the binding to its cognate κB sequence observed after LPS treatment favors the participation of p65 as an intracellular effector of LPS in FRTL-5 cells. Concordantly, chemical exposure to the NF-κB-specific inhibitor sulfasalazine or specific knockdown of the NF-κB p65 subunit by siRNA prevented LPS-induced up-regulation of NIS expression. Finally, LPS induced binding of p65 to the enhancer region of the NIS promoter, as we successfully demonstrated by ChIP analysis (Fig. 7B). Consistent with this finding, transcriptional activity stimulated by direct binding of p65 to a target gene promoter in response to LPS has been widely described in other genes (62, 63, 64). All these findings underline a possible role of p65 as an important mediator of the endotoxin action in thyroid cells.
According to the results obtained here, the molecular basis for the stimulation of iodide transport by the proinflammatory stimuli LPS appears to rely on the activation of NF-κB pathway in thyroid cells. This probably occurs downstream from the TLR-4 activation, as we recently proposed (25). Although much has been learned about the function and signaling of TLRs in macrophages and dendritic cells, the mechanisms of recognition of microbes by epithelial cells are still poorly understood. Scarce information regarding the role of NF-κB pathway activation in response to TLR inducers in thyroid cells has been reported. Nevertheless, it was suggested that treatment with LPS as well as with the TLR-3 agonist polyinosinic:polycytidylc acid (Poly-I:C) activates NF-κB pathway and p65 nuclear recruitment in thyroid cells (65). The present study provides evidence that, in thyrocytes, LPS stimulates TSH-induced NIS gene expression through a specific transcriptional mechanism that involves a direct interaction of the NF-κB p65 subunit within the NIS promoter-enhancer region NUE.
Previous outstanding results indicated that the NUE region can integrate diverse classes of cell signals, which have been described particularly for the CRE-L site (6, 28, 52, 66). This apparent redundancy makes it possible for TSH as well as cAMP and other cell signals to reach NUE to modify NIS expression through multiple routes during development and under certain physiological or pathological conditions (2, 51). More interestingly, we have demonstrated that p65 and, to a lesser degree, c-Rel stimulate NUE transcriptional activity. Hence, future studies on the regulation of a possible synergism of p65 and other factors at the NUE region are likely to provide more insights into the transcriptional regulation of the NIS gene. As observed, when the κB site was mutated or p65 was silenced, TSH failed to fully activate NIS expression. In this context, the activation of the NF-κB pathway by TSH or by stimulating anti-TSH receptor antibodies in thyroid cells has been recently reported (44). However, little is still known about the role of NF-κB in thyroid physiology. Our results raise the question of whether some members of the NF-κB family could be physiological regulators of NIS gene expression. Experiments are currently in progress to address this hypothesis.
Radioactive iodide uptake has been largely exploited in the diagnosis and treatment of differentiated thyroid cancers and their metastases (1). Unfortunately, a number of thyroid carcinomas have lost this capacity, mainly due to a defective localization or expression of NIS (1, 2), which has been the major therapeutic limitation. Although the molecular mechanism that contributes to the reduced or absent NIS expression is still poorly understood, recovery of NIS function in neoplastic transformed thyrocytes is crucial to make them susceptible to effective radioiodine treatment. Advances in understanding the mechanisms responsible for NIS gene regulation, such as those described here, could contribute to creating new approaches to achieve NIS induction in cancer cells, thus allowing for effective radioiodide therapy.
Here, we demonstrate the stimulatory effect of LPS on NIS mRNA expression in vivo in thyroid cells of LPS-treated rats, which confirms that thyrocytes have a potential capability to respond to systemic infections. The physiological parallel increase of the inhibitory IκB-α subunit levels favors the activation of NF-κB-mediated transcriptional response stimulated by LPS. It is known that the induction of the IκB-α gene expression is a characteristic sign of the nuclear response to NF-κB (11). Genes activated in response to an inflammatory stimulus can be divided into primary and secondary response genes. Primary response genes are usually activated most rapidly and are formally defined as those genes induced without de novo protein synthesis (67). The induction of NIS by LPS seems to be a primary response to inflammatory stimulus favoring the mediation of NF-κB in the LPS action. Even though the role of several host proteins upon infection has been described, we do not have yet an integrated model to understand the actual physiological implication of our findings during the inflammatory response. A priori we hypothesized that the LPS-induced NIS expression appears not to be part of the defense cell response to the inflammatory stimulus but rather a concurrent action of LPS with unknown pathophysiological consequences.
Our observations of diminished thyroid hormone levels after LPS treatment are in accordance with the initial induction of the NTI syndrome. It is known that bacterial infections are able to elicit the NTI syndrome, a clinical condition of thyroid dysfunction-related disturbance in the hypothalamic-pituitary-thyroid axis not primarily originating in the thyroid (46). Coincidentally, the increase of NIS expression induced by LPS is apparently in contrast to a reduction of thyroid cell function due to the inflammatory stimulus. However, in more advanced stages a down-regulation of the hypothalamic function or locally generated proinflammatory factors may further contribute to the decreased secretion of thyroid hormone. The reduction of D1 and the increase of IL-6 in liver confirm the ability of a single low dose of LPS to induce the stage of NTI, suggesting that subseptical concentrations of circulating LPS could have unexplored pathophysiological implication.
Interestingly, the current observed increment in thyroidal IL-6 mRNA expression, which is a representative marker of the inflammatory response, reveals the active participation of thyroid tissue in the inflammatory response. In accordance, the mRNA expression of the proinflammatory cytokine IL-1β peaked very rapidly in thyroid and liver after LPS administration (49). In agreement, it has been demonstrated that bacterial and viral thyroid infections are associated with subacute thyroiditis and autoimmune diseases. Interestingly, a crucial role of TLRs in these processes has been proposed (65, 68). Taken together, these findings suggest a role of the LPS/TLR-4/NF-κB pathway activation in the thyrocyte with the development of thyroid gland dysfunction.
In summary, we have demonstrated the existence of a novel and functional conserved binding site for the transcription factor NF-κB within the enhancer region present in NIS promoter that seems to be responsible for the LPS-enhanced NIS gene expression in thyroid cells. Evidence that LPS induces activation of the NF-κB pathway, in turn increasing the expression of NIS, has been obtained. This effect is due, at least in part, to a direct interaction of p65 with a specific κB site at the NIS enhancer. These findings shed light on a possible new regulatory mechanism in NIS transcriptional expression involving a NF-κB site present in the NUE region of NIS promoter.
Materials and Methods
Reagents and antibodies
Phenol-extracted LPS from Escherichia coli 055:B5 containing less than 1% protein was from Sigma-Aldrich (Saint Louis, MO). Bovine TSH was a generous gift from the National Institute of Diabetes, Digestive and Kidney Diseases National Pituitary Hormone Program and Dr. A. F. Parlow, National Institutes of Health (Torrance, CA). Goat polyclonal antihuman Pax8, monoclonal antihuman Pax8, monoclonal antihuman p65, and monoclonal antihuman Histone H1 antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal antibodies antimouse α-tubulin, antimouse β-actin, and nonspecific mouse IgG were from Sigma-Aldrich.
Plasmids
The −2854 to +13 bp DNA fragment (pNIS-2.8) of the rat NIS promoter and the 5′-deletion constructs with NUE region (pNIS-NUE-1.2 and pNIS-NUE-0.5) or without the enhancer (pNIS-2.0, pNIS-1.2, and pNIS-0.5) were described previously (28). The −2386 to −2153 bp DNA fragment of the rat NIS promoter cloned 5′ upstream to the thymidine kinase promoter (pNUE), and the site-directed mutants pNUE-A, pNUE-B, and pNUE-A+C have been reported (6).
To generate bacterial expression vectors for GST-p65 full length (GST-p65FL, aa 1-551), GST-p65 N-terminal (GST-p65N, aa 1-313), and GST-p65 C-terminal (GST-p65C, aa 314-551), the corresponding rat p65 cDNA fragments were obtained by PCR and cloned in frame into pGEX-4T vector. Full-length human Pax8 and GST-Pax8 expression vectors have been reported (41). p65 expression vectors (p65FL, p65N, and p65C) were generated by PCR amplification of the corresponding rat p65 cDNA and subsequently cloned into the expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA). Expression plasmids encoding c-Rel, RelB, p52, and p50 were as reported elsewhere (69). The expression vector encoding human p65 cDNA has been previously described (70). pNUE-C MT and pNUE-κB MT were generated by site-directed mutagenesis using the QuikChange kit (Stratagene, La Jolla, CA). The design of the mutagenic primers and mutant oligonucleotides was based on the transcription factor consensus-binding sequences found in the TRANSFACT version 7.0 database (BIOBASE, Wolfenbüttel, Germany). The nucleotide sequence of all constructs obtained by PCR was confirmed by DNA sequencing (Macrogen, Inc., Seoul, South Korea). 5×κB-luc reporter vector containing five κB consensus sites linked to the luciferase-coding sequence was obtained from CLONTECH Laboratories, Inc. (Mountain View, CA). pCMV-β-galactosidase normalizator plasmid was purchased from Promega Corp. (Madison, WI).
Cell culture and transient transfections
The rat thyroid cell line FRTL-5 was grown in DMEM/Ham F-12 medium, supplemented with 5% (vol/vol) heat-inactivated calf bovine serum (Gibco, Langley, OK), 1 mIU/ml bovine TSH, 10 μg/ml bovine insulin, 5 μg/ml bovine transferrin, 2 μmol/ml glutamine, and antibiotics (Sigma-Aldrich) (71). When cells reached 50–60% of confluence, they were shifted to same medium without TSH and containing 0.2% (vol/vol) calf serum (starvation medium-basal condition-). Cells were maintained for 5–7 d in this medium, allowing cells to be quiescent before the experiments (72). TSH-starved cells were treated with the indicated concentrations of LPS in the absence or presence of 0.5 mIU/ml TSH for different periods of time. After the treatments, cell viability was higher than 95%, as determined by the Trypan blue dye exclusion assay.
For transient transfection, FRTL-5 cells were cultured in six-well plates and transfected using LipofectAMINE Plus reagent (Invitrogen) as specified by the manufacturer. The day after transfection, cells were split into 24-well plates at 80% confluence. The next day, growth media were replaced by basal media, and transfected cells were starved and treated as stated above.
Cos-7 cells were grown in DMEM supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Natocor Laboratories, Córdoba, Argentina). Cells were transiently cotransfected in suspension at a density of 3 × 104 cells/96-well with 10 ng of luciferase reporter-promoter constructs, 10 ng of p65 or p65-deleted expression vectors, 20 ng of Pax8 expression vector, and 1 ng of β-galactosidase reporter vector using Lipofectamine 2000 (Invitrogen) as described in the manufacturer’s protocol. Each transfection was done in triplicate with an equal amount of total DNA by adding appropriate amounts of the empty vector pcDNA3.1. After transfection, cells were cultured for 48 h and assayed for luciferase activity using a luciferase reporter assay system (Promega), according to the manufacturer’s recommended protocol and normalized relative to the levels of β-galactosidase activity (26).
Animals and treatment
Male Wistar WKAH/Hok rats (250–300 g) were obtained from Laboratory Animal Facility, Facultad de Ciencias Veterinarias, Universidad Nacional de La Plata (Buenos Aires, Argentina). Rats were housed on a 12-h light, 12-h dark cycle in a temperature-controlled room. Animals received commercial rat chow and water ad libitum. Rats were allocated randomly into five groups (eight animals per group), four of which received a subseptical dose of LPS (E. coli 055:B5, 10 μg/kg) dissolved in sterile 0.9% saline solution in a single ip injection. The fifth group of rats or control group was injected ip with 0.5 ml saline solution. Animals were killed by CO2 asphyxiation at the indicated time points (1, 3, 6, or 24 h). Blood was taken by cardiac puncture, and tissues were removed and further processed for total RNA extraction. Animal protocols and procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and local institutional animal care committee guidelines.
RNA extraction and RT/quantitative PCR (qPCR)
Total RNA purification and cDNA synthesis were performed as described (71). qPCR analysis was performed using an ABI Prism 7500 detection system (Applied Biosystems, Foster City, CA) and SYBR green chemistry. Reactions were carried out in triplicate using, for each 15-μl reaction, 5 μl of cDNA template (equivalent to 250 ng of total RNA), 0.3 μm forward and reverse primers, 2× Brilliant SYBR Green qPCR Master Mix (Stratagene), and 30 nm Rox as passive reference dye. Gene-specific primer sets are included in Supplemental Table 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org. To quantify changes in gene expression, the 2−ΔΔCt method was used to calculate the relative changes normalized against the housekeeping gene β-actin used as internal control (73). For each pair of primers a dissociation plot resulted in a single peak, indicating that single cDNA product was amplified. Specific target amplification was confirmed by automatic sequencing. All primers were from Sigma-Genosys (Houston, TX).
Protein extraction and Western blot
Total and nuclear protein extracts were prepared as described elsewhere (26). Proteins (50 μg) were resolved by SDS-PAGE on a 10% Tris-glycine gel and transferred to nitrocellulose membranes (Whatman, Inc., Clifton, NJ). Membranes were blocked in 5% skimmed milk, 0.1% Tween 20, Tris-buffered saline (TBS-Tween). Blots were incubated with 1 μg/ml anti-p65 or 2 μg/ml anti-Pax8 antibody in TBS-Tween overnight at 4 C. Equal loading was assessed by stripping and reprobing the same blot with 0.5 μg/ml anti-β-actin, 0.2 μg/ml anti-α-tubulin, or 1 μg/ml anti-histone H1 antibodies. Band intensities were measured densitometrically using ImageJ Image Software (National Institutes of Health, Bethesda, MD).
EMSA
Assays were performed as previously described (26). Synthesized double-stranded oligonucleotides (sequences available in Supplemental Table 1) were labeled with α-32P-dATP using the Klenow fragment of DNA polymerase. Nuclear extracts (5 μg) were incubated in a 20-μl reaction volume on ice in binding mix buffer containing 40 mm HEPES (pH 7.9), 100 mm KCl, 0.5 mm dithiothreitol, 0.2 mm EDTA, 5% glycerol, 200 μg/ml BSA (Fraction V), and 50 ng/μl sonicated salmon sperm DNA. Labeled oligonucleotide probe (1 ng DNA) was added, and incubation continued at room temperature for 30 min. The resulting DNA-protein complexes were separated from free labeled-DNA on a 5% native polyacrylamide gel in 0.25× Tris-borate-EDTA buffer [22 mm Tris (pH 8.5), 22 mm boric acid, 0.5 mm EDTA], then vacuum dried and exposed to x-ray film. Autoradiograms the signal intensity of which was not in an oversaturated range were analyzed. For competition assays, a 100-fold excess of cold oligonucleotides was added to the reaction mixture 30 min before addition of the labeled oligonucleotide. In supershift experiments, nuclear extracts were incubated with monoclonal anti-p65 or polyclonal anti-Pax8 antibodies (0.5 μg) 1 h on ice before the labeled probe was added and then processed as indicated above.
GST pull-down
For GST pull-downs, GST fusion constructs encoding p65 and its deletions or Pax8 were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside in E. coli BL21. Recombinant GST-proteins were purified using the GST pull-down assay kit from Pierce Biotechnology (Rockford, IL). Equal amounts of GST or GST-tagged proteins bound to glutathione-Sepharose beads (GSBs) were incubated with GSB-precleared FRTL-5 extracts (200 μg) overnight at 4 C under constant rotation. GSBs were then washed four times with 1:1 TBS:ProFound Lysis Buffer and protease inhibitors. Specifically bound proteins were analyzed by immunoblot as described above.
Chromatin immunoprecipitation
ChIP assays were performed as described (64). Briefly, after stimulation, the cross-linker formaldehyde was added directly to the cell culture media to a final concentration of 1%. The fixation proceeded for 10 min and was stopped by the addition of glycine to a final concentration of 125 mm. FRTL-5 cells were rinsed twice with cold PBS plus 1 mm phenylmethylsulfonylfluoride, and then scraped. Nuclei were purified and resuspended in nuclear lysis buffer [50 mm Tris-HCl (pH 8), 10 mm EDTA, 1% sodium dodecyl sulfate (SDS), and protease inhibitors cocktail] and incubated on ice for 10 min. Samples were broken by sonication into chromatin fragments of an average length of 500/1000 bp and then centrifuged at 10,000 × g. The supernatant was diluted 5-fold in Immunoprecipitation (IP) dilution buffer [1% Triton X-100, 5 mm EDTA, 50 mm Tris-HCl (pH 7.5), 0.5% Nonidet P-40, 150 mm NaCl] and precleared by adding salmon sperm DNA-saturated Protein A/G Plus Agarose (Santa Cruz Biotechnology) for 30 min at 4 C. Precleared chromatin from 2 × 106 cells was incubated with 2 μg of affinity-purified monoclonal anti-p65 antibody [sc-8008, Santa Cruz Biotechnology; suitable for ChIP analysis (74)] or control mouse IgG overnight at 4 C under rotation. Immune complexes were allowed to bind to salmon sperm DNA-saturated Protein A/G PLUS-Agarose during 4 h at 4 C under rotation. Immunoprecipitates were washed four times with IP wash buffer [0.1% SDS, 1% Triton X-100, 5 mm EDTA, 50 mm Tris-HCl (pH 7.5), 0.5% Nonidet P-40, 150 mm NaCl]; twice with high-salt IP wash buffer [0.1% SDS, 1% Triton X-100, 5 mm EDTA, 500 mm NaCl, 50 mm Tris-HCl (pH 7.5)], and once with TE [10 mm Tris-HCl (pH 8), 1 mm EDTA]. DNA was purified using Chelex-100 (Bio-Rad Laboratories, Hercules, CA) as described elsewhere (75). DNA was quantitated by qPCR as mentioned above. Relative fold increases were calculated according to the equation: 2 −[(Ct.input − Ct.target) − (Ct.input − Ct.mock)] (64, 75).
RNA interference
siRNA pool (sc-61876) targeting specifically rat NF-κB p65 subunit was acquired from Santa Cruz Biotechnology (76). siRNA (200 pmol) was transfected into FRTL-5 cells seeded onto six-well plates using LipofectAMINE 2000 reagent according to the manufacturer’s protocol. After transfection, cells were starved in basal media supplemented with 5% calf serum for 72 h and then treated with the indicated agents. A nonspecific siRNA duplex (scrambled siRNA) was used as a control. The expression of p65 was monitored by Western blotting of total protein extracts isolated from control and siRNA-transfected cells 72 h after transfection.
Statistical analysis
Results are presented as the mean ± se from at least three independent experiments unless indicated. Multiple group analysis was conducted by one-way ANOVA. As posttest, the Student-Newman-Keuls multiple-comparisons test was used. Comparisons between two groups were made using unpaired Student’s t test. Data derived from RT-qPCR assays performed in animal tissues are given as median and range. Results were analyzed by the nonparametric Kruskal-Wallis and Dunn’s multiple comparison post hoc tests. Statistical tests were performed using Prism 3.0 software (GraphPad Software, San Diego, CA). Differences were considered significant at P < 0.05.
Acknowledgments
We thank Nancy Carrasco (Albert Einstein College of Medicine, Bronx, NY) for critical review of the manuscript and for helpful discussions. We thank Dr. Pilar Santisteban (Instituto de Investigaciones Biomédicas Alberto Sols, Madrid, Spain) for kindly providing the NIS-promoter constructs pNIS-2.8 and its deletions; and Dr. Roberto Di Lauro (CEINGE Biotecnologie Avanzate, Naples, Italy) for pNUE reporter and its site-directed mutants. We thank Dr. Mariastella Zannini (Istituto di Endocrinologia e Oncologia Sperimentale G. Salvatore-CNR, Naples, Italy) for providing the Pax8 expression vector and the GST-Pax8 bacterial expression vector. We thank Drs. Thomas Wirth, Bernd Baumann (Institute of Physiological Chemistry, Ulm University, Ulm, Germany), and Dr. Anna Bigas (Centre Oncologia Molecular, IDIBELL-Institut de Recerca Oncologica, Barcelona, Spain) for the NF-κB (c-Rel, RelB, p52, and p50) and human p65 expression plasmids, respectively. We also thank Dr. Cecilia Alvarez (Centro de Investigaciones en Bioquímica Clínica e Inmunología-Consejo Nacional de Investigaciones Científicas y Técnicas, Córdoba, Argentina) for pGEX-4T plasmid, Liliana Franchioni (Hospital de Niños de la Santísima Trinidad de Córdoba, Córdoba, Argentina) for hormonal measurements, and the members of our laboratory for providing technical assistance and critical insights, especially to Sebastián Susperreguy for his help and advice in qPCR reactions and cloning procedures.
Footnotes
This work was supported by grants from Fondo Nacional de Ciencia y Tecnología (FONCYT), CONICET, Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba (SeCyT), and Agencia Córdoba Ciencia. J.P.N. and I.D.M. are doctoral and postdoctoral fellows at CONICET, respectively; M.N is a doctoral fellow at FONCYT; C.G.P. is member of the research career of CONICET; C.G.P. and A.M.M-R. are established researchers at Departamento de Bioquímica Clínica, (CIBICI-CONICET), Facultad de Ciencias Químicas, Universidad Nacional de Córdoba.
Disclosure Summary: The authors have nothing to disclose.
First Published Online July 28, 2010
Abbreviations: aa, Amino acids; ChIP, chromatin immunoprecipitation; CRE-L, degenerate cAMP-response element; D1, type 1 deiodinase; FRTL-5, Fisher rat thyroid cell line 5; GSBs, glutathione-Sepharose beads; GST, glutathione-S-transferase; IκB-α, NF-κB inhibitor-α; IP, immunoprecipitation; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; NIS, Na+/I− symporter; NTI, nonthyroidal illness; NUE, NIS upstream enhancer; Pax8, paired box gene 8; qPCR, quantitative PCR; RHD, Rel homology domain; SDS, sodium dodecyl sulfate; siRNA, small interfering RNA; SZ, sulfasalazine; TAD, transactivation domain; TLR, Toll-like receptor; TTF-1, thyroid transcription factor-1.
References
- 1.Dohán O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, Ginter CS, Carrasco N2003. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 24:48–77 [DOI] [PubMed] [Google Scholar]
- 2.Kogai T, Taki K, Brent GA2006. Enhancement of sodium/iodide symporter expression in thyroid and breast cancer. Endocr Relat Cancer 13:797–826 [DOI] [PubMed] [Google Scholar]
- 3.Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N2009. The Na+/I− symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol 296:C654–C662 [DOI] [PMC free article] [PubMed]
- 4.Bizhanova A, Kopp P2009. Minireview: The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology 150:1084–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spitzweg C, Dutton CM, Castro MR, Bergert ER, Goellner JR, Heufelder AE, Morris JC2001. Expression of the sodium iodide symporter in human kidney. Kidney Int 59:1013–1023 [DOI] [PubMed] [Google Scholar]
- 6.Chun JT, Di Dato V, D'Andrea B, Zannini M, Di Lauro R2004. The CRE-like element inside the 5′-upstream region of the rat sodium/iodide symporter gene interacts with diverse classes of b-Zip molecules that regulate transcriptional activities through strong synergy with Pax-8. Mol Endocrinol 18:2817–2829 [DOI] [PubMed] [Google Scholar]
- 7.Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R1999. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol 19:2051–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S2008. Shared principles in NF-κB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 9.Ghosh S, Karin M2002. Missing pieces in the NF-κB puzzle. Cell 109(Suppl):S81–S96 [DOI] [PubMed]
- 10.Karin M, Ben-Neriah Y2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18:621–663 [DOI] [PubMed] [Google Scholar]
- 11.Vallabhapurapu S, Karin M2009. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol 27:693–733 [DOI] [PubMed] [Google Scholar]
- 12.Perkins ND2007. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol 8:49–62 [DOI] [PubMed] [Google Scholar]
- 13.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST2000. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci USA 97:12705–12710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenner RG, Young RA2005. Insights into host responses against pathogens from transcriptional profiling. Nat Rev Microbiol 3:281–294 [DOI] [PubMed] [Google Scholar]
- 15.Miggin SM, O'Neill LA2006. New insights into the regulation of TLR signaling. J Leukoc Biol 80:220–226 [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Akira S2007. Signaling to NF-κB by Toll-like receptors. Trends Mol Med 13:460–469 [DOI] [PubMed] [Google Scholar]
- 17.Krappmann D, Wegener E, Sunami Y, Esen M, Thiel A, Mordmuller B, Scheidereit C2004. The IκB kinase complex and NF-κB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol Cell Biol 24:6488–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bottero V, Withoff S, Verma IM2006. NF-κB and the regulation of hematopoiesis. Cell Death Differ 13:785–797 [DOI] [PubMed] [Google Scholar]
- 19.Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, Sung WK, Lipovich L, Vega VB, Chen J, Shahab A, Zhao XD, Hibberd M, Wei CL, Lim B, Ng HH, Ruan Y, Chin KC2007. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol Cell 27:622–635 [DOI] [PubMed] [Google Scholar]
- 20.Raetz CR, Whitfield C2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guha M, Mackman N2001. LPS induction of gene expression in human monocytes. Cell Signal 13:85–94 [DOI] [PubMed] [Google Scholar]
- 22.Vakharia K, Hinson JP2005. Lipopolysaccharide directly stimulates cortisol secretion by human adrenal cells by a cyclooxygenase-dependent mechanism. Endocrinology 146:1398–1402 [DOI] [PubMed] [Google Scholar]
- 23.Vives-Pi M, Somoza N, Fernández-Alvarez J, Vargas F, Caro P, Alba A, Gomis R, Labeta MO, Pujol-Borrell R2003. Evidence of expression of endotoxin receptors CD14, toll-like receptors TLR4 and TLR2 and associated molecule MD-2 and of sensitivity to endotoxin (LPS) in islet β cells. Clin Exp Immunol 133:208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batra A, Pietsch J, Fedke I, Glauben R, Okur B, Stroh T, Zeitz M, Siegmund B2007. Leptin-dependent toll-like receptor expression and responsiveness in preadipocytes and adipocytes. Am J Pathol 170:1931–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicola JP, Vélez ML, Lucero AM, Fozzatti L, Pellizas CG, Masini-Repiso AM2009. Functional toll-like receptor 4 conferring lipopolysaccharide responsiveness is expressed in thyroid cells. Endocrinology 150:500–508 [DOI] [PubMed] [Google Scholar]
- 26.Vélez ML, Costamagna E, Kimura ET, Fozzatti L, Pellizas CG, Montesinos MM, Lucero AM, Coleoni AH, Santisteban P, Masini-Repiso AM2006. Bacterial lipopolysaccharide stimulates the thyrotropin-dependent thyroglobulin gene expression at the transcriptional level by involving the transcription factors thyroid transcription factor-1 and paired box domain transcription factor 8. Endocrinology 147:3260–3275 [DOI] [PubMed] [Google Scholar]
- 27.Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A, Onaya T1997. Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology 138:2227–2232 [DOI] [PubMed] [Google Scholar]
- 28.Costamagna E, García B, Santisteban P2004. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-β repression of the sodium/iodide symporter gene. J Biol Chem 279:3439–3446 [DOI] [PubMed] [Google Scholar]
- 29.García B, Santisteban P2002. PI3K is involved in the IGF-I inhibition of TSH-induced sodium/iodide symporter gene expression. Mol Endocrinol 16:342–352 [DOI] [PubMed] [Google Scholar]
- 30.Firestone GL, Giampaolo JR, O'Keeffe BA2003. Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cell Physiol Biochem 13:1–12 [DOI] [PubMed] [Google Scholar]
- 31.Alotaibi H, Yaman E, Salvatore D, Di Dato V, Telkoparan P, Di Lauro R, Tazebay UH2010. Intronic elements in the Na+/I− symporter gene (NIS) interact with retinoic acid receptors and mediate initiation of transcription. Nucleic Acids Res 38:3172–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin X, Ryu KY, Jhiang SM2004. Cloning of the 5′-flanking region of mouse sodium/iodide symporter and identification of a thyroid-specific and TSH-responsive enhancer. Thyroid 14:19–27 [DOI] [PubMed] [Google Scholar]
- 33.Taki K, Kogai T, Kanamoto Y, Hershman JM, Brent GA2002. A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3′,5′-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol 16:2266–2282 [DOI] [PubMed] [Google Scholar]
- 34.Endo T, Kaneshige M, Nakazato M, Ohmori M, Harii N, Onaya T1997. Thyroid transcription factor-1 activates the promoter activity of rat thyroid Na+/I− symporter gene. Mol Endocrinol 11:1747–1755 [DOI] [PubMed] [Google Scholar]
- 35.Ohmori M, Endo T, Harii N, Onaya T1998. A novel thyroid transcription factor is essential for thyrotropin-induced up-regulation of Na+/I− symporter gene expression. Mol Endocrinol 12:727–736 [DOI] [PubMed] [Google Scholar]
- 36.Plevy SE, Gemberling JH, Hsu S, Dorner AJ, Smale ST1997. Multiple control elements mediate activation of the murine and human interleukin 12 p40 promoters: evidence of functional synergy between C/EBP and Rel proteins. Mol Cell Biol 17:4572–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T1997. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 94:2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pasca di Magliano M, Di Lauro R, Zannini M2000. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci USA 97:13144–13149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riesco-Eizaguirre G, Santisteban P2007. New insights in thyroid follicular cell biology and its impact in thyroid cancer therapy. Endocr Relat Cancer 14:957–977 [DOI] [PubMed] [Google Scholar]
- 40.De Leo R, Miccadei S, Zammarchi E, Civitareale D2000. Role for p300 in Pax 8 induction of thyroperoxidase gene expression. J Biol Chem 275:34100–34105 [DOI] [PubMed] [Google Scholar]
- 41.Di Palma T, Nitsch R, Mascia A, Nitsch L, Di Lauro R, Zannini M2003. The paired domain-containing factor Pax8 and the homeodomain-containing factor TTF-1 directly interact and synergistically activate transcription. J Biol Chem 278:3395–3402 [DOI] [PubMed] [Google Scholar]
- 42.Beutler B2004. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430:257–263 [DOI] [PubMed] [Google Scholar]
- 43.Egan LJ, Sandborn WJ1998. Inhibition of nuclear factor κB by sulfasalazine: a new target for inflammatory bowel disease therapy? Gastroenterology 115:1295–1296 [DOI] [PubMed] [Google Scholar]
- 44.Morshed SA, Latif R, Davies TF2009. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology 150:519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voigt C, Prodinger C, Paschke R2007. The TSH receptor is linked with AP1 and NFκB signaling in COS7 cells. Exp Clin Endocrinol Diabetes 115:590–593 [DOI] [PubMed] [Google Scholar]
- 46.De Groot LJ2006. Non-thyroidal illness syndrome is a manifestation of hypothalamic-pituitary dysfunction, and in view of current evidence, should be treated with appropriate replacement therapies. Crit Care Clin 22:57–86, vi [DOI] [PubMed] [Google Scholar]
- 47.Renner F, Schmitz ML2009. Autoregulatory feedback loops terminating the NF-κB response. Trends Biochem Sci 34:128–135 [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, Ouaaz F, Beg AA2007. Distinct roles of different NF-κ B subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. J Immunol 178:6777–6788 [DOI] [PubMed] [Google Scholar]
- 49.Boelen A, Kwakkel J, Thijssen-Timmer DC, Alkemade A, Fliers E, Wiersinga WM2004. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol 182:315–323 [DOI] [PubMed] [Google Scholar]
- 50.Boelen A, Platvoet-ter Schiphorst MC, Bakker O, Wiersinga WM1995. The role of cytokines in the lipopolysaccharide-induced sick euthyroid syndrome in mice. J Endocrinol 146:475–483 [DOI] [PubMed] [Google Scholar]
- 51.Riesco-Eizaguirre G, Santisteban P2006. A perspective view of sodium iodide symporter research and its clinical implications. Eur J Endocrinol 155:495–512 [DOI] [PubMed] [Google Scholar]
- 52.Fenton MS, Marion KM, Hershman JM2008. Identification of cyclic adenosine 3′,5′-monophosphate response element modulator as an activator of the human sodium/iodide symporter upstream enhancer. Endocrinology 149:2592–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dubchak I, Brudno M, Loots GG, Pachter L, Mayor C, Rubin EM, Frazer KA2000. Active conservation of noncoding sequences revealed by three-way species comparisons. Genome Res 10:1304–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mascia A, Nitsch L, Di Lauro R, Zannini M2002. Hormonal control of the transcription factor Pax8 and its role in the regulation of thyroglobulin gene expression in thyroid cells. J Endocrinol 172:163–176 [DOI] [PubMed] [Google Scholar]
- 55.Zaballos MA, Garcia B, Santisteban P2008. Gbetagamma dimers released in response to thyrotropin activate phosphoinositide 3-kinase and regulate gene expression in thyroid cells. Mol Endocrinol 22:1183–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong H, Voll RE, Ghosh S1998. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1:661–671 [DOI] [PubMed] [Google Scholar]
- 57.Yoon C, Korade Z, Carter BD2008. Protein kinase A-induced phosphorylation of the p65 subunit of nuclear factor-κB promotes Schwann cell differentiation into a myelinating phenotype. J Neurosci 28:3738–3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okazaki T, Sakon S, Sasazuki T, Sakurai H, Doi T, Yagita H, Okumura K, Nakano H2003. Phosphorylation of serine 276 is essential for p65 NF-κB subunit-dependent cellular responses. Biochem Biophys Res Commun 300:807–812 [DOI] [PubMed] [Google Scholar]
- 59.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S1997. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89:413–424 [DOI] [PubMed] [Google Scholar]
- 60.Zhong H, May MJ, Jimi E, Ghosh S2002. The phosphorylation status of nuclear NF-κ B determines its association with CBP/p300 or HDAC-1. Mol Cell 9:625–636 [DOI] [PubMed] [Google Scholar]
- 61.Benezra M, Chevallier N, Morrison DJ, MacLachlan TK, El-Deiry WS, Licht JD2003. BRCA1 augments transcription by the NF-κB transcription factor by binding to the Rel domain of the p65/RelA subunit. J Biol Chem 278:26333–26341 [DOI] [PubMed] [Google Scholar]
- 62.Anrather J, Racchumi G, Iadecola C2006. NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem 281:5657–5667 [DOI] [PubMed] [Google Scholar]
- 63.Sasaki CY, Barberi TJ, Ghosh P, Longo DL2005. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J Biol Chem 280:34538–34547 [DOI] [PubMed] [Google Scholar]
- 64.Mascanfroni ID, Montesinos MD, Alamino VA, Susperreguy S, Nicola JP, Ilarregui JM, Masini-Repiso AM, Rabinovich GA, Pellizas CG2010. Nuclear factor (NF)-κB-dependent thyroid hormone receptor β1-expression controls dendritic cell function via AKT signaling. J Biol Chem 285:9569–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, Ringel MD, Saji M, Giuliani C, Napolitano G, Goetz DJ, Kohn LD2005. Thyrocytes express a functional toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. Mol Endocrinol 19:1231–1250 [DOI] [PubMed] [Google Scholar]
- 66.Boelaert K, Smith VE, Stratford AL, Kogai T, Tannahill LA, Watkinson JC, Eggo MC, Franklyn JA, McCabe CJ2007. PTTG and PBF repress the human sodium iodide symporter. Oncogene 26:4344–4356 [DOI] [PubMed] [Google Scholar]
- 67.Smale ST2010. Selective transcription in response to an inflammatory stimulus. Cell 140:833–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamazaki K, Suzuki K, Yamada E, Yamada T, Takeshita F, Matsumoto M, Mitsuhashi T, Obara T, Takano K, Sato K2007. Suppression of iodide uptake and thyroid hormone synthesis with stimulation of the type I interferon system by double-stranded ribonucleic acid in cultured human thyroid follicles. Endocrinology 148:3226–3235 [DOI] [PubMed] [Google Scholar]
- 69.Maier HJ, Marienfeld R, Wirth T, Baumann B2003. Critical role of RelB serine 368 for dimerization and p100 stabilization. J Biol Chem 278:39242–39250 [DOI] [PubMed] [Google Scholar]
- 70.Espinosa L, Santos S, Inglés-Esteve J, Muñoz-Canoves P, Bigas A2002. p65-NFκB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. J Cell Sci 115:1295–1303 [DOI] [PubMed] [Google Scholar]
- 71.Fozzatti L, Vélez ML, Lucero AM, Nicola JP, Mascanfroni ID, Macció DR, Pellizas CG, Roth GA, Masini-Repiso AM2007. Endogenous thyrocyte-produced nitric oxide inhibits iodide uptake and thyroid-specific gene expression in FRTL-5 thyroid cells. J Endocrinol 192:627–637 [DOI] [PubMed] [Google Scholar]
- 72.Mori K, Yoshida K, Tani J, Nakagawa Y, Hoshikawa S, Ito S2001. Double-stranded RNA-induced interferon regulatory factor-1 gene expression in FRTL-5 rat thyroid cells. Mol Cell Endocrinol 184:77–86 [DOI] [PubMed] [Google Scholar]
- 73.Livak KJ, Schmittgen TD2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 74.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M2003. Distribution of NF-κB-binding sites across human chromosome 22. Proc Natl Acad Sci USA 100:12247–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nelson JD, Denisenko O, Bomsztyk K2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1:179–185 [DOI] [PubMed] [Google Scholar]
- 76.Wu S, Fadoju D, Rezvani G, De Luca F2008. Stimulatory effects of insulin-like growth factor-I on growth plate chondrogenesis are mediated by nuclear factor-κB p65. J Biol Chem 283:34037–34044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmitt TL, Espinoza CR, Loos U2002. Characterization of a thyroid-specific and cyclic adenosine monophosphate-responsive enhancer far upstream from the human sodium iodide symporter gene. Thyroid 12:273–279 [DOI] [PubMed] [Google Scholar]