

Abstract
Doxorubicin (Dox) has been used as a potent anticancer agent, but serious cardiotoxicity precludes its use in a wide range of patients. We have reported that the androgen-androgen receptor (AR) system plays important roles in cardiac growth and protection from angiotensin II-induced cardiac remodeling. The present study was undertaken to clarify whether the androgen-AR system exerts a cardioprotective effect against Dox-induced cardiotoxicity. Male AR knockout (ARKO) and age-matched littermate male wild-type (WT) mice at 25 wk of age were given ip injections of Dox (20 mg/kg) or a vehicle. The survival rate and left ventricular function in Dox-treated male ARKO mice were reduced compared with those in Dox-treated male WT mice. Electron microscopic study showed prominent vacuole formation of myocardial mitochondria in Dox-treated male ARKO mice. Cardiac oxidative stress and apoptosis of cardiomyocytes were increased more prominently by Dox treatment in male ARKO mice than in male WT mice. In addition, Dox-induced reduction in the expression of cardiac mitochondria transcription factor A (Tfam) and phosphorylation of serine-threonine kinase (Akt) was more pronounced in male ARKO mice than in male WT mice. In cardiac myoblast cells, testosterone up-regulated Akt phosphorylation and Tfam expression and exerted an antiapoptotic effect against Dox-induced cardiotoxicity. Collectively, the results demonstrate that Dox-induced cardiotoxicity is aggravated in male ARKO mice via exacerbation of mitochondrial damage and superoxide generation, leading to enhanced apoptosis of cardiomyocytes. Thus, the androgen-AR system is thought to counteract Dox-induced cardiotoxicity partly through activation of the Akt pathway and up-regulation of Tfam to protect cardiomyocytes from mitochondrial damage and apoptosis.
Doxorubicin, an anticancer agent, causes acceleration of cardiac oxidative stress and cell apoptosis leading to reduced survival rate in male mice lacking androgen receptor.
Doxorubicin (Dox), an antitumor agent, is widely used for treatment of hematological malignancies and solid tumors. However, its clinical application is often limited due to its dose-dependent cardiotoxicity, which leads to congestive heart failure ( 1, 2, 3). A major cause of Dox-induced cardiotoxicity is thought to be increased oxidative stress ( 4, 5), by which mitochondrial dysfunction ( 6) and apoptosis of cardiomyocytes ( 7, 8) are elicited.
Androgens, male sex hormones, exert their biological effects in various target organs, and most of the biological actions of androgens are mediated through transcriptional regulation by activation of the nuclear androgen receptor (AR). To analyze the physiological function of the androgen-AR system in vivo, we have generated AR knockout (ARKO) mice and have shown that male ARKO mice manifest late-onset obesity ( 9), high-turnover osteopenia ( 10), impaired brain masculinization (11), and aberrant adiponectin expression ( 12). In addition, AR exists in cardiomyocytes and promotes physiological cardiac hypertrophy by a direct receptor-specific mechanism in vitro ( 13). Recently, we have demonstrated, by using male ARKO mice, that the androgen-AR system plays a pivotal role in not only physiological cardiac growth but also protection from pathological angiotensin II-induced cardiovascular remodeling ( 14, 15). Based on these observations, we hypothesized that androgens play a protective role against the development of Dox-induced cardiotoxicity. In this study, we examined whether the androgen-AR system has a protective effect against Dox-induced cardiotoxicity using male ARKO mice.
Results
Reduced survival rate of Dox-treated male ARKO mice
The survival rates of vehicle-treated male wild-type (WT) and male ARKO mice were consistently 100% in all series of experiments. Although Dox treatment caused a reduction in the survival rates of both male WT and male ARKO mice at d 7, male ARKO mice showed a lower survival rate than that of male WT mice (28 vs. 68%, P < 0.05) (Fig. 1A). The reduced survival rate in male ARKO mice was not associated with carrying cytomegalovirus/Cre-recombinase transgene (Fig. 1B). In addition, there was no significant difference in mortality among littermate female WT mice, female ARKO mice, and male WT mice by Dox treatment (75 vs. 68%, not significant) (Fig. 1C). Thus, AR deficiency aggravates Dox-induced cardiotoxicity in male but not female mice. Furthermore, the survival rate of orchidectomized (castration performed at age of 20 wk) male WT mice with Dox treatment was almost the same as that of male WT mice with Dox treatment (75 vs. 68%, not significant) (Fig. 1D). The results suggest that male ARKO mice have preexisting cardiac impairment in the process of growth and maturation of the cardiovascular system.
Fig. 1.

Survival rates of male and female WT mice, male and female ARKO mice, and castrated male WT mice after Dox administration. Survival rates were calculated by the Kaplan-Meier method and compared by the log-lank test. n = 50 in each group. *, P < 0.05. NS, Not significant.
Early-phase left ventricular dysfunction in Dox-treated male ARKO mice
Beginning of deaths in these experimental mice was observed at the fifth day after Dox injection (Fig. 1A). Therefore, we performed echocardiographical studies at the fifth day after Dox administration to detect early change of cardiac dysfunction. We found that left ventricular function, evaluated by fractional shortening (%FS), was similar in male WT and male ARKO mice after vehicle injection. On d 5 after Dox administration, slight but significant reduction of %FS was found in male WT mice, whereas more pronounced reduction in %FS was observed in male ARKO mice with Dox treatment (Table 1 and Fig. 2A).
Table 1.
Echocardiographic measurement
| Parameter | WT | ARKO | ||||
|---|---|---|---|---|---|---|
| Vehicle | Dox | Vehicle | Dox | |||
| LVDd (mm) | 3.98 ± 0.09 | 3.78 ± 0.10 | 3.21 ± 0.0424 | 3.72 ± 0.095 | ||
| LVDs (mm) | 2.44 ± 0.06 | 2.46 ± 0.08 | 1.91 ± 0.0424 | 2.66 ± 0.09††5 | ||
| IVS (mm) | 0.80 ± 0.02 | 0.81 ± 0.03 | 0.62 ± 0.0224 | 0.58 ± 0.0224 | ||
| PW (mm) | 0.84 ± 0.03 | 0.80 ± 0.03 | 0.68 ± 0.0223 | 0.64 ± 0.0224 | ||
| FS (%) | 39.4 ± 0.9 | 35.7 ± 0.71 | 40.7 ± 0.84 | 28.1 ± 1.2245 | ||
LVDd, Left ventricular end-diastolic dimension; LVDs, left ventricular end-systolic dimension; IVS, interventricular septal thickness; PW, posterior wall. Values are means ± sem, each group n = 20.
P < 0.05 vs. WT-vehicle.
P < 0.01 vs. WT-vehicle.
P < 0.05 vs. WT-Dox.
P < 0.01 vs. WT-Dox.
P < 0.01 vs. KO-vehicle.
Fig. 2.
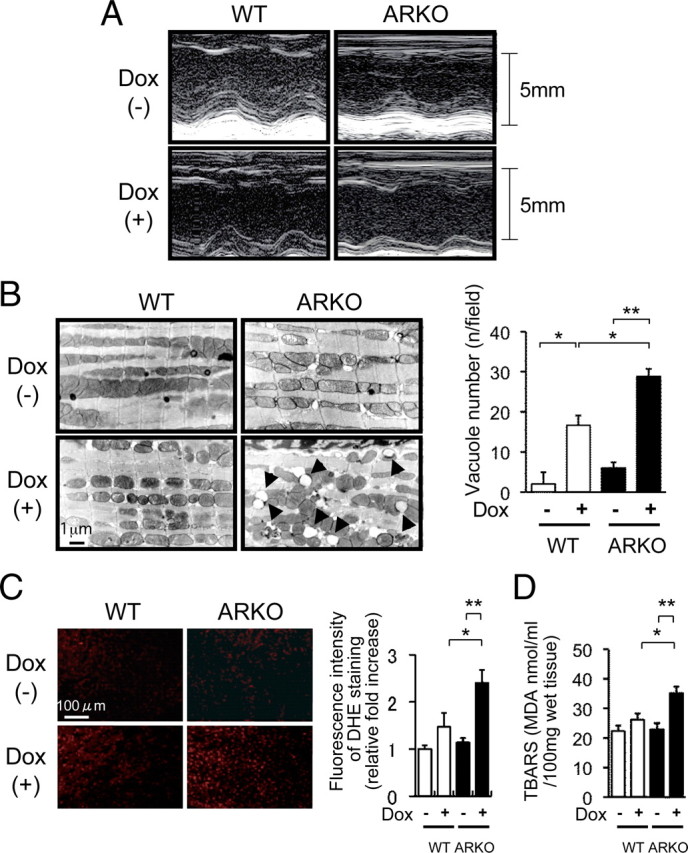
Left ventricular function, electron microscopic findings, superoxide generation, and lipid peroxidation after Dox administration in male WT mice and male ARKO mice. A, Representative M-mode echocardiogram of left ventricular wall motion. M-mode echocardiogram of the left ventriculum at 5 d after administration of the vehicle or Dox in male WT and male ARKO mice. Although there were slight extensional changes in left ventricular end-diastolic dimension (LVDd) and left ventricular end-systolic dimension (LVDs) of Dox-treated male WT mice, acute dilatation of LVDd and LVDs was markedly apparent in Dox-treated male ARKO mice. B, left panel, Electron microscopic findings of the left ventricular myocardium after administration of the vehicle or Dox in male WT and male ARKO mice. Accelerated vacuole formation of myocardial mitochondria was observed in Dox-treated male ARKO mice. Right panel, Quantitative analysis of vacuole number after administration of the vehicle or Dox in male WT (white bars) and male ARKO (black bars) mice. Values are expressed as means ± sem. *, P < 0.05; **, P < 0.01, n = 4–6 in each group. C, DHE bromide staining analysis after administration of the vehicle or Dox in male WT and male ARKO mice. D, Myocardial TBARS assay after administration of the vehicle or Dox in male WT (white bars) and male ARKO (black bars) mice. Values are expressed as means ± sem. *, P < 0.05; **, P < 0.01, n = 6–8 in each group.
Accelerated mitochondrial damage of cardiomyocytes in Dox-treated male ARKO mice
Electron microscopic examination of the heart revealed that Dox treatment caused prominent mitochondrial vacuolization in ARKO mice compared with that in Dox-treated WT mice (Fig. 2B). In addition, vehicle-treated male ARKO mice also manifested several small mitochondrial vacuolizations in the myocardium. These findings suggest that there is a basal mitochondrial dysfunction in the male ARKO mouse heart and that the lack of AR-mediated signals affects cardiac oxidative stress even in the absence of Dox treatment.
Enhanced cardiac superoxide production in Dox-treated male ARKO mice
Fluorescence microscopic examination of ventricular tissue with dihydroethidium (DHE) bromide staining showed very weak signals in both vehicle-treated male WT mice and vehicle-treated male ARKO mice. On the other hand, superoxide production after Dox treatment was markedly enhanced in male ARKO mice compared with that in male WT mice (Fig. 2C).
Increased cardiac lipid peroxidation in Dox-treated male ARKO mice
To quantify cardiac lipid peroxidation for evaluation of in vivo oxidative stress, a thiobarbital reactive substances (TBARS) assay of cardiac tissues was performed. Dox-treated male ARKO mice showed higher levels of cardiac lipid peroxidation than did Dox-treated male WT mice (Fig. 2D). Superoxide dismutase (SOD) components such as Cu/ZnSOD and MnSOD are recognized as endogenous and crucial radical scavengers. However, neither Cu/ZnSOD nor MnSOD protein expression was significantly different in the myocardium among the experimental groups of mice (data not shown).
Accelerated apoptosis of cardiac cells in Dox-treated male ARKO mice
To evaluate Dox-induced apoptosis of cardiac cells in male ARKO and male WT mice, we quantified terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) values as the ratio of TUNEL-positive cells to total cell numbers in ventricular tissues. Without Dox treatment, the number of TUNEL-positive cells was small and there was no difference between the numbers of apoptotic cells in male ARKO and male WT mice. Dox treatment increased TUNEL-positive cells in both male ARKO and male WT mice, but the number of apoptotic cells in the ventricular tissues was significantly larger in male ARKO mice than in male WT mice (Fig. 3A). To further examine apoptotic changes of cardiomyocytes, cardiac Bcl-2 and Bax proteins were quantified by Western blot analysis using myocardial tissues. Although Dox treatment caused a reduction in the Bcl-2-to-Bax ratio in the myocardium of both male WT and male ARKO mice, the Bcl-2-to-Bax ratio after Dox treatment was significantly lower in male ARKO mice than in male WT mice (Fig. 3B).
Fig. 3.

Apoptosis of cardiac cells, cardiac Akt activation, and Tfam expression after Dox treatment in male WT and male ARKO mice. A, Numbers of TUNEL-positive cells in three separate left ventricular sections per mouse after administration of the vehicle or Dox in male WT (white bars) and male ARKO (black bars) mice. Values are expressed as means ± sem. *, P < 0.05; **, P < 0.01, n = 6 in each group. B, Cardiac expression of Bcl-2 and Bax. Upper panel, Representative blots of Bcl-2, Bax, and β-actin. Lower panel, Results of densitometric analysis of Bcl-2-to-Bax ratio after administration of the vehicle or Dox in male WT (white bars) and male ARKO (black bars) mice. Values are expressed as means ± sem. *, P < 0.05, n = 8 in each group. C, Cardiac Akt phosphorylation in male WT mice and male ARKO mice with or without Dox. Upper panels, Representative blots of phosphorylated forms of Akt and total Akt. Lower panels, Results of densitometric analysis of phosphorylated Akt (p-Akt) after administration of the vehicle or Dox in male WT (white bars) and male ARKO (black bars) mice. Values are expressed as means ± sem. *, P < 0.05, n = 8 in each group. D, Cardiac expression of Tfam with or without Dox treatment. Right panel, Representative blots of Tfam protein with results of densitometric analysis of Tfam expression after administration of the vehicle or Dox in male WT (white bars) and male ARKO (black bars) mice. Values are expressed as means ± sem. *, P < 0.05, n = 6 in each group.
Suppressed cardiac serine-threonine kinase (Akt) phosphorylation in Dox-treated male ARKO mice
The Akt pathway plays an important role in the intracellular transduction of antiapoptotic signals, and elevation of Akt signaling ameliorates Dox-induced cardiac toxicity ( 16). To clarify whether the androgen-AR system acts via the Akt pathway to alleviate apoptosis signals, we examined phosphorylation of Akt protein in cardiomyocytes in male ARKO and male WT mice with or without Dox treatment. Without Dox treatment, Akt phosphorylation was lower by about 10% in male ARKO mice than in male WT mice, although the difference was not statistically significant. Similarly, Akt phosphorylation was slightly but not significantly reduced by Dox treatment in male WT mice. However, Dox treatment caused a significant reduction in Akt phosphorylation in cardiomyocytes of male ARKO mice (Fig. 3C).
Reduced expression of mitochondria transcription factor A (Tfam) in the myocardium of Dox-treated male ARKO mice
Tfam regulates mitochondrial DNA (mtDNA) transcription and replication. Because mitochondria play a pivotal role in the regulation of energy production, superoxide generation, and apoptosis of cardiomyocytes, we examined Tfam mRNA and protein expression in cardiac tissues. Although Dox treatment did not significantly affect the expression of Tfam protein in cardiac tissues of male WT mice, Tfam expression level was reduced to almost half by Dox treatment in male ARKO mice (Fig. 3D).
Androgen enhances Akt phosphorylation and Tfam expression through an AR-and phosphatidyl inositol 3 phosphatase kinase (PI3K)-dependent pathway in cardiac myoblast cells
To clarify the mechanism of the protective action of the androgen-AR system in Dox-induced cardiotoxicity, H9c2 cardiac myoblast cells were treated with testosterone as an AR agonist, and Akt phosphorylation status was assessed. First, we confirmed that the H9c2 cell line includes cells derived from male rats. In the present study, we found mRNA expression of both AR and sex-determining region Y gene in H9c2 cells by conventional RT-PCR analysis in this cell line (data not shown). Testosterone significantly increased Akt phosphorylation, and the level of Akt phosphorylation peaked at 30 min after testosterone treatment (Fig. 4A). Pretreatment with flutamide, an AR antagonist, or LY294002, a PI3K inhibitor, before testosterone stimulation significantly reduced Akt phosphorylation levels (Fig. 4B).
Fig. 4.

Acute effect of androgen action on Akt phosphorylation (p-Akt) and Tfam expression in H9c2 cells. A, upper panel, Representative blots of phosphorylated Akt and total Akt. Lower panel, Results of densitometric analysis of phosphorylated Akt. Values are expressed as means ± sem. *, P < 0.05 vs. with control, n = 4 in each group. B, Akt phosphorylation in H9c2 cells with or without flutamide or LY294002. Upper panels, Representative blots of phosphorylated forms of Akt and total Akt. Lower panels, Results of densitometric analysis of phosphorylation of Akt pretreated with or without flutamide or LY294002 for 1 h before testosterone administration in H9c2 cells. Values are expressed as means ± sem. *, P < 0.05; **, P < 0.01 n = 6 in each group. C, Tfam expression in H9c2 cells with or without flutamide or LY294002. Upper panels, Representative blots of Tfam expression and β-actin. Lower panels, Results of densitometric analysis of phosphorylation of Akt pretreated with or without flutamide or LY294002 for 1 h before testosterone (T) administration for 24 h in H9c2 cells. Values are expressed as means ± sem. *, P < 0.05; **, P < 0.01, n = 4 in each group.
We also examined whether androgen affects Tfam expression in vitro. The expression of Tfam protein was significantly increased by testosterone treatment in H9c2 cells. Moreover, we tested whether testosterone-induced up-regulation of Tfam expression is dependent on AR-dependent or PI3K-Akt-dependent pathways. Testosterone-induced Tfam expression was blocked by pretreatment with flutamide or LY294002 for 1 h (Fig. 4C). We also investigated whether androgen-mediated Akt phosphorylation and Tfam expression are directly involved in AR by using small interfering RNA (siRNA). Because testosterone-induced Akt phosphorylation and Tfam expression were abolished by AR siRNA, testosterone preserved Akt phosphorylation and Tfam expression through AR activation even in the Dox-treated condition (Fig. 5, A and B).
Fig. 5.

Effect of AR deficiency on Akt phosphorylation, Tfam expression, and interaction of phosphatidylinositol 3-kinase regulatory subunit α in H9c2 Cells. A, Akt phosporylation after AR siRNA transfection with or without Dox treatment in H9c2 cells. Representative blots of phosphorylated Akt and total Akt are presented. B, Tfam expression after AR siRNA transfection with or without Dox treatment in H9c2 cells. Representative blots of Tfam and β-actin are presented. C, Action of androgen on AR interaction with p85α in H9c2 cells. Left panel, Representative blots of p85α and AR expression. Right panel, A result of densitometric analysis of p85α expression corrected by AR. IP, Immunoprecipitation: WB, Western blot analysis. Values are expressed as means ± sem. *, P < 0.05 vs. with control, n = 4 in each group.
Effects of androgen action are associated with interaction of AR with p85α
Because it has been shown that androgen promotes Akt phosphorylation thorough AR interaction with p85α, a regulatory subunit of PI3K, we examined the interaction of AR with p85α by immunoprecipitation ( 17) with or without Dox. As shown in Fig. 5C, the absence of testosterone did not promote interaction of AR with p85α in H9c2 cells. When H9c2 cells were treated with testosterone, accelerated interaction of AR with p85α was observed. However, Dox treatment abolished the testosterone-induced interactive change (Fig. 5C).
Androgen protects H9c2 cells against Dox-induced cell damage
To clarify whether the up-regulation of Akt phosphorylation and/or Tfam induction by androgen exert a protective effect against Dox cardiotoxicity, we investigated the effect of androgen on H9c2 cell damage induced by Dox. There was no difference in cell viability between control and testosterone treatment in H9C2 cells under basal conditions. Treatment with Dox for 24 h significantly decreased cell viability to 73%. However, cotreatment with testosterone reversed the Dox-induced toxicity. The cellular-protective effects of testosterone were inhibited by treatment with flutamide or LY294002 (Fig. 6A). We also evaluated the antiapoptotic action of testosterone by using TUNEL staining. Treatment of H9c2 cells with Dox increased the percentage of TUNEL-positive cells from 0.9 to 45% after 24 h. Cotreatment with testosterone significantly reduced TUNEL-positive cells to 25%, and the antiapoptotic effect of testosterone was abolished by flutamide or LY294002 (Fig. 6B). These results indicate that androgen action through AR increases cell viability and prevents apoptosis by promoting Akt phosphorylation and Tfam expression.
Fig. 6.
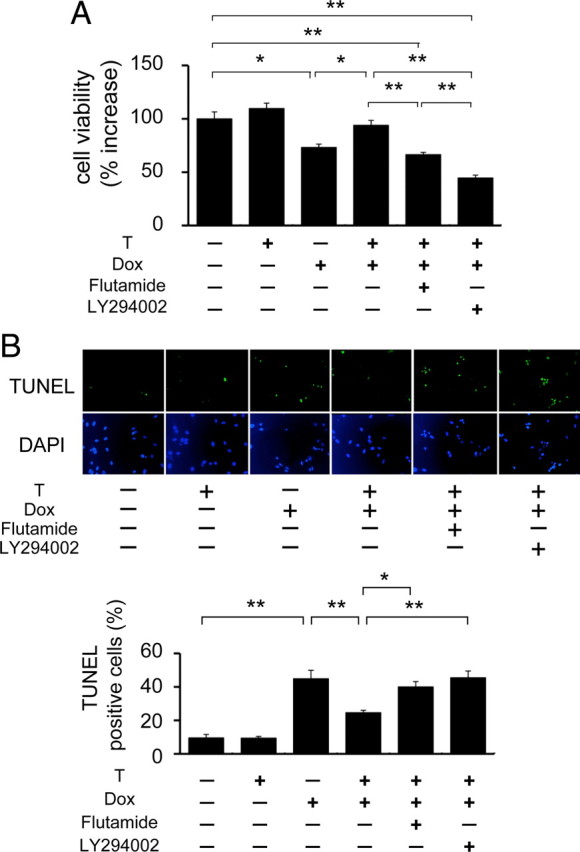
Effect of androgen action on cell viability and apoptosis with or without Dox in H9c2 Cells. H9c2 cells were pretreated with or without flutamide or LY294002 for 1 h before testosterone (T) administration. After 24 h, cells were coincubated with Dox for 24 h and then analyzed. A, Cell viability after Dox treatment for 24 h in H9c2 cells. Cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt assay. Values are relative change to control value and expressed as means ± sem. *, P < 0.05; **, P < 0.01, n = 6 in each group. B, Apoptotic cells after Dox treatment of H9c2 cells. Upper panel, Representative photomicrographs of TUNEL-positive cells. The nuclei of apoptotic cells were determined by TUNEL staining (green), and total nuclei were demonstrated by DAPI staining (blue). Lower panel, The percentage of TUNEL-positive cells in three random fields. Values are relative change to control value and expressed as means ± sem. *, P < 0.05; **, P < 0.01, n = 4 in each group.
Discussion
Although Dox is one of the most potent anticancer agents against a variety of malignant tumors ( 18), wide clinical use of Dox has been hampered because of its dose-dependent cardiotoxicity leading to irreversible heart failure and cardiac death ( 18, 19).
We have reported that male ARKO mice manifest impaired cardiac growth and maladaptation in angiotensin II-induced cardiovascular remodeling ( 14, 15). Those findings suggest that endogenous and physiological androgen actions have a pivotal role in morphological and functional homeostasis on male heart. The present study using male ARKO mice was undertaken to clarify whether physiological androgen action plays a cardioprotective role in the male heart against Dox-induced cardiotoxicity. The results showed that there were significant reductions in survival rate and left ventricular contractility in Dox-treated male ARKO mice. In addition, electron microscopic examination demonstrated severe vacuolization of cardiac mitochondria, suggesting increased oxidative stress in Dox-treated male ARKO mice.
Accelerated apoptosis has been shown to be involved in cardiac dysfunction under some experimental and clinical conditions ( 20, 21, 22, 23, 24). Previous studies also demonstrated that Dox treatment results in cytochrome c release, leading to caspase-3 activation and apoptosis ( 25), and that the increased apoptosis in cardiac myocytes contributes to heart failure. In the present study, Dox-treated male ARKO mice showed prominent apoptosis as evidenced by an increased number of TUNEL-positive cells and a reduced cardiac Bcl-2-to-Bax expression ratio. We also demonstrated that androgen treatment restored the reduced cell viability and prevented apoptosis in Dox-treated cardiac myoblast cells (Fig. 5A). In addition to the regulation of cellular apoptosis in the prostate, androgen also suppresses granulosa cell apoptosis in overies of female mice ( 26, 27). These findings suggest that androgen regulates apoptosis in various tissues and cell types, including cardiomyocytes.
Akt is involved in multiple cellular signaling pathways, including regulation of glucose metabolism, gene transcription, protein synthesis, cell cycle, and cell survival ( 28). Phosphorylation and activation of Akt are evoked by activation of PI3K, which is caused by binding of ligands to membrane receptors such as insulin and IGF-I ( 29, 30). Negoro et al. ( 31) showed that Dox inhibits PI3K-Akt activation and enhances apoptosis of cardiomyocytes, and Taniyama and Walsh ( 16) showed that Akt activation protects hearts from Dox-induced toxicity. The present study also revealed that Dox treatment caused a more severe impairment of cardiac Akt phosphorylation in male ARKO mice than in male WT mice. The PI3K-Akt signaling pathway has been reported to participate in cardiovascular protection ( 32, 33, 34), and Bai et al. ( 35) reported that androgen activates the PI3K-Akt pathway in cardiomyocytes via AR activation. These results of previous studies and our present observations are consistent with the notion that the androgen-AR system suppresses apoptosis of cardiomyocytes and contributes to cardiac protection at least in part via activation of the PI3K-Akt pathway.
Dox is known to produce free radicals ( 36, 37, 38), and superoxide radicals can contribute to the formation of hydroxyl radicals by reducing oxidized transition metals. The reduced transition metals then react with hydrogen peroxide by the Haber-Weiss reaction to produce hydroxyl radicals ( 39). The hydroxyl radicals cause protein and DNA damage and promote lipid peroxidation. In several experimental studies, increased lipid peroxidation and enhanced free radical generation were observed in Dox-treated mammalian hearts ( 37, 40, 41) and are thought to play a pivotal role in the development of Dox-induced cardiotoxicity. In the present study, Dox treatment caused higher levels of cardiac superoxide generation and lipid peroxidation in male ARKO mice than in male WT mice, and such changes were in parallel with the lower survival rate and more severe left ventricular dysfunction of male ARKO mice. These results indicated that the cardioprotective action of androgen in Dox toxicity is closely associated with its antioxidant potency.
Mitochondrial injury causes mtDNA damage, as well as a reduction in mitochondrial function ( 42). Tfam is an essential factor for mtDNA transcription and replication ( 43, 44, 45, 46), and Tfam plays an important role in maintaining mitochondrial function under the condition of mitochondrial injury. The expression level of Tfam is reduced in various animal models of cardiac failure involving myocardial infarction ( 42) and pressure overload ( 47). Dox administration is known to cause cardiac mitochondrial injury, including mitochondrial vacuolization ( 48, 49, 50), and there is a possibility that one of the primary targets of Dox-induced cardiotoxicity is mitochondria of the myocardium. In the present study, Dox treatment resulted in more prominent mitochondrial vacuolization in male ARKO mice than in male WT mice, and Tfam expression was reduced in Dox-treated male ARKO mice. In addition, we showed that androgen up-regulated Tfam expression through a PI3K-dependent pathway in cardiac myoblast cells. It has been reported that Tfam expression is regulated by peroxisome proliferators-activated receptor γ coactivator-1α and that the levels of peroxisome proliferators-activated receptor γ coactivator-1α mRNA in skeletal muscle and the liver are decreased in male ARKO mice ( 12). These observations together suggest that Tfam is required for alleviation of Dox-induced mitochondrial damage and protection of cardiomyocytes from oxidative stress and that the androgen-AR system plays an important role in maintenance of Tfam expression. Although our DHE staining and TBARS assay did not show a significant increase in cardiac superoxide generation in vehicle-treated male ARKO mice (Fig. 2, C and D), several vacuole formations of cardiac mitochondria were observed even in vehicle-treated ARKO mice (Fig. 2B). We speculate that DHE staining and TBARS assay are not sensitive enough to detect the small changes in oxidative stress in vehicle-treated ARKO mice. Finally, there is a possibility that androgen acts via AR to preserve cardiac mitochondrial function with or without cardiac stress, such as Dox treatment.
In the present study, no difference in mortality was seen in castrated and noncastrated male WT mice after Dox injection. ARKO mice lacked androgen action from the embryonic stage throughout their life, whereas the castrated male WT mice had normal androgen level until castration. Therefore, it is plausible to assume that androgen action in cardiomyocytes during periods of growth is important for the development of tolerability against cardiac stress. In addition, adrenal androgen can exert low levels of androgen action even after castration of WT mice. The observation that mitochondrial vacuolization was detected in the myocardium of saline-treated ARKO mice further supports the notion that ARKO mice are much more vulnerable to Dox treatment compared with castrated WT mice. Differences in body fat may also influence drug kinetics of Dox. Because Dox does not reach a high concentration in fat and because male ARKO mice exhibit late-onset obesity, the increase in male ARKO mice may be associated with increased vulnerability to Dox.
Excessive single administration of Dox is effective for analyzing the direct impact of Dox in the heart, and this method has been widely used to analyze Dox-induced cardiomyopathy in a number of animal models ( 51, 52, 53). However, the dosage and administration strategy of Dox is different from clinical usage, and the results of this study do not emulate clinical conditions. Therefore, further examination is required to determine the pathophysiological role of androgen in protection in human against Dox-induced cardiac injury.
In conclusion, the present study demonstrated that Dox-induced cardiotoxicity is accelerated in ARKO mice and that Dox treatment causes aggravated mitochondrial damage and superoxide generation, leading to enhanced apoptosis of cardiomyocytes in male ARKO mice. Because the androgen-AR system counteracts Dox to activate PI3K-Akt signaling and because androgen preserves Tfam expression, the present observations are consistent with the assumption that the androgen-AR system protects cardiomyocytes from Dox-induced mitochondrial damage and apoptosis via activation of PI3K-Akt signaling and up-regulation of Tfam expression.
Materials and Methods
Animal preparation
We used 25-wk-old male ARKO mice and age-matched littermate male WT mice after backcrossing for 10 generations with the C57BL/6J strain. These male ARKO mice were generated by targeted disruption of the AR gene by using the Cre-loxP system ( 9, 10, 54). Testicular androgen production seems to be severely impaired, leading to a reduction in serum gonadal androgen levels, whereas serum adrenal androgen and estrogen levels remain normal in male ARKO mice. The methods of generation and maintenance of male ARKO mice and the characteristics of male ARKO mice were previously reported in detail ( 9, 10, 54). In this study, littermate male WT mice were used as controls for male ARKO mice. Both WT and ARKO mice were assigned to a vehicle-treated group or a Dox-treated group. Dox (Dox hydrochloride; Kyowa Hakko, Tokyo, Japan) was dissolved in saline and administered by ip injection at a single dose of 20 mg/kg. The vehicle-treated group was given ip injection of saline. We finally divided these mice into four groups: male WT and male ARKO mice with and without Dox administration. All experimental procedures were performed in accordance with the guidelines of the Animal Research Committee of The University of Tokushima Graduate School.
Echocardiographic study
Transthoracic echocardiography was performed 5 d after Dox or vehicle injection using of an Aplio 80 (Toshiba Medical Systems Corp., Ltd, Tochigi, Japan) with a 15-MHz imaging transducer. Mice from each group were anesthetized by peritoneal injection with 20 mg/kg of pentobarbital, and heart rates of all mice were kept at 450–500 beats per minute. M-mode measurements, including left ventricular %FS calculation, were described previously ( 14).
Electron microscopy
Left ventricular tissue was trimmed into 1- to 2-mm blocks and was used for electron microscopic analysis after fixing with 3% glutaraldehyde solution in 0.1 mol/liter sodium cacodylate-HCl buffer (pH 7.3) containing 0.05 mol/liter sucrose. The methods used for electron microscopic analysis were described in detail previously ( 55). In brief, the specimens were further fixed for 2 h at 4 C with 2% osmium tetoxide in 0.1 mol/liter cacodylate buffer (pH 7.3) containing 0.5% potassium ferrocyanate, dehydrated in graded ethanol (70–100%), stained with 2% uranyl acetate in ethanol, and embedded in Spurr low viscosity epoxy resin. The sections were cut with a diamond knife on an LKB Ultratome IV, selected on carbon-coated films, stained with alkaline lead citrate, examined using a JOEL 100CX electron microscope at 60 kV, and printed on the film (Kodak MN film 4489; Eastman Kodak Co., Rochester, NY). Semiquantitative analysis of vacuole formation was performed in five randomly selected power fields in each mouse.
Superoxide detection in myocardial tissue
We evaluated myocardial superoxide production with in situ DHE bromide staining ( 56, 57). In brief, hearts from mice were excised, rinsed in physiological saline, and frozen in optimal cutting temperature compound (Tissue-Tek; Sakura Fine Chemical, Tokyo, Japan) until use. Transverse sections (10 μm in thickness) were cut with a cryostat and placed on silane-coated glass slides. The slide-glasses with heart tissue were incubated with DHE in PBS (10 μmol/liter) in a dark, humidified container at room temperature for 30 min. DHE is oxidized upon reaction with superoxide to ethidium bromide, which binds to DNA in the nucleus and fluoresces red. After placing a cover glass over the heart tissue, the heart tissue was observed using a laser scanning confocal microscope (Leica TCS-NT, mounted on a Leica DMRB light microscope; Leica, Mannheim, Germany). The excitation wavelength was 488 nm, and emission fluorescence was detected with a −568-nm long-pass filter.
Measurement of TBARS in the myocardium
Sample preparation was performed as described above, and cardiac ventricular tissues were homogenized in ice-cooled physiological saline. The uncentrifuged whole homogenate suspension was used for analysis. The value of TBARS in the myocardium was determined by using a commercially available assay kit (TBARS assay kit; ZeptoMetrix Corp., Buffalo, NY) according to the manufacturer’s instructions. In brief, homogenized and uncentrifuged samples were used. After adding an equal volume of sodium dodecyl sulfate solution, the mixture was incubated at 95 C for 60 min and then cooled to room temperature. After centrifuging the sample, the supernatant was used for analysis with absorbance read at 532 nm.
TUNEL staining in vivo
Five days after vehicle or Dox injection, mice were killed with an overdose injection of pentobarbital, and hearts were excised. The hearts were fixed in 5% neutral buffered formalin overnight. After fixation, the hearts were embedded in paraffin and were serially cut into 3-μm-thick slices. Sections were used for TUNEL staining for detecting apoptotic cardiomyocytes. After being deparaffinized and hydrated, the sections were washed in deionized water, and endogenous peroxidase was blocked with 0.3% hydrogen peroxide for 30 min. Next, the sections were treated with 20 μg/ml proteinase K (DAKO Japan, Inc., Kyoto, Japan) for 10 min at room temperature and rinsed in PBS. Then the sections were incubated with a reaction reagent (terminal transferase, biotin-16–2′-deoxyuridine 5′-triphosphate, TUNEL dilution buffer, all supplied by Roche Diagnostic Corp., Indianapolis, IN) for 60 min at 37 C and with 1:500-diluted peroxidase-conjugated streptavidin (DAKO Japan, Inc.) for 30 min at room temperature, rinsed in PBS, incubated with 3.3′-diaminobenzidine tetrahydrochloride for 10 min at room temperature, washed in water, and finally counterstained with methyl green for 30 min. To determine the percentage of apoptotic cells, TUNEL-positive and TUNEL-negative cells were counted. Results are expressed as number of TUNEL-positive cells/total cells × 100%.
Western blot analysis
Protein extraction from cardiac tissue and Western blot analysis were performed as described previously ( 14, 58). In brief, protein extracts from the hearts were boiled for 5 min in Laemmli sample buffer and then run on SDS-PAGE. The protein extracts were then transferred to a nitrocellulose membrane [Hybond-enhanced chemiluminescence (ECL); Amersham Pharmacia Biotech (Buckinghamshire, UK)]. The membrane was blocked for 1 h at room temperature with 5% nonfat milk in PBS-Tween 20. The blots were incubated overnight at 4 C with each antibody, followed by incubation for 1 h with a secondary antibody. Immunoreactive bands were visualized using ECL-PLUS chemiluminescence reagent (Amersham Pharmacia Biotech, Piscataway, NJ) treatment and exposure to Hyperfilm-ECL. The signals were quantified by densitometry using Image J version 1.29. We used antibodies against Cu/ZnSOD, MnSOD, Bcl-2, Bax, and Tfam (all from Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Activation of Akt was assessed with antibodies against Akt and phospho-Akt antibody (Cell Signaling Technology, Beverly, MA). An antibody against β-actin (Biolegend, San Diego, CA) served as a loading control.
Cell preparation
Rat H9c2 cardiac myoblast cells (European Collection Cell Cultures, Porton Down, UK) were maintained in DMEM supplemented with 10% fetal bovine serum and 2 mm l-glutamine in a humidified atmosphere of 95% air and 5% CO2 at 37 C. Cells were grown to confluence and then changed to a serum-free medium before exposure to all experiments, and they were then incubated with or without 10 μm flutamide, 10 μm LY294002 (all obtained from Wako Pure Chemical Industries Ltd., Osaka, Japan), 1 μm testosterone, and 1 μm Dox (Kyowa Hakko). Cultured cells were harvested after rinsing in PBS and lysed for 30 min at 4 C in lysis buffer (Cell Signaling Technology). Supernatants obtained by centrifugation were used for Western blot analysis. The procedure for Western blot analysis was a described previously ( 14, 58).
Cell viability assay by using 3-(4,5-dimethylthiazol-2-yl)-2,5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt dye
To assess cell viability based on mitochondrial activity in H9c2 cells, a tetrazolium assay was performed by using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI). In brief, H9c2 cells were cultured at a density of 1 × 104 per well in 100 μl medium on a 96-well plate. The cells were incubated in serum-free media for 24 h before experiments. They were pretreated with or without flutamide or LY294002 for 1 h before testosterone treatment and incubated for 24 h. Consequently, cells were cocultured with Dox for 24 h. After treating the cells, 20 μl Cell Titer 96 AQueous One Solution was added to each well. Absorbance at 490 nm was determined with a plate reader after 1 h at 37 C in a humidified 5% CO2 incubator.
Immunoprecipitation
Cell lysates, including 500 μg protein, were precleaned with protein A/G agarose beads (Santa Cruz Biotechnology, Inc.) and rabbit IgG antibody at 4 C for 30 min, and then supernatants were collected and incubated with anti-AR antibody (Santa Cruz Biotechnology, Inc.) at 4 C overnight. The lysates were then gently agitated with agarose beads for 1 h and boiled in sample buffer for 5 min. The immunoprecipitated proteins were subjected to Western blot analysis. Antibody against anti-p85 was purchased from Santa Cruz Biotechnology, Inc.
Small interfering RNA experiments
Small interfering RNA targeting rat AR and nontargeting siRNA control were purchased from Sigma-Aldrich (MISSION siRNA and MISSION siRNA Universal Negative Control; Sigma-Aldrich, St. Louis, MO), and transfection with this agent was performed as previously reported ( 59). Testosterone stimulation was performed 48 h after siRNA transfection.
TUNEL staining in vitro
Apoptotic cell frequency in vitro was analyzed by TUNEL staining using a In Situ Cell Death Detection kit, Fluorescein (Roche Diagnostic Corp., Indianapolis, IN). Briefly, H9c2 cells were prepared and cultured in a chamber slide (Lab-teckII; Nalge Nunc International, Rochester, NY). The samples were washed with PBS two times and then fixed in 4% paraformaldehyde for 30 min at room temperature. After incubating in permeabilization solution for 5 min and washing with PBS, the samples were incubated with TUNEL reaction mixture at 37 C in a humid condition for 1 h and then encapsulated by mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). We evaluated the number of apoptotic cells using fluorescence microscopy. Several fields were randomly chosen, and the number of TUNEL-positive cells in each plate was counted. TUNEL-positive cells were expressed as percentage of total number of DAPI-positive cells.
Statistical analysis
Values for each parameter within a group are expressed as means ± sem. For comparisons among groups, statistical significance was assessed using one-way ANOVA, and the significance of each difference was determined by post hoc testing using Tukey-Kramer’s method. Survival curves after Dox administration were created by the Kaplan-Meier method and tested by a log-rank test. These analyses were performed on an Apple Macintosh computer with the use of Excel (Microsoft X) and Stat View statistical package (Stat View 5.0; SAS Institute, Inc.) ( 60). Statistical significance was considered at P < 0.05.
Acknowledgments
We thank Linda Whittaker for assistance in preparations of the manuscript.
NURSA Molecule Pages:
Ligands: Flutamide | Testosterone;
Nuclear Receptors: AR.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research and a grant for a Study Group on Aseptic Femoral Neck Necrosis from the Ministry of Health, Labor, and Welfare of Japan, and grants for Clinical Vascular Function from the Kimura Memorial Cardiovascular Foundation.
Disclosure Summary: The authors have nothing to disclose.
First Published Online May 25, 2010
Abbreviations: Akt, Serine-threonine kinase; AR, androgen receptor; ARKO, AR knockout; DAPI, 4′,6-diamidino-2-phenylindole; DHE, dihydroethidium; Dox, doxorubicin; ECL, enhanced chemiluminescence; %FS, fractional shortening; mtDNA, mitochondrial DNA; PI3K, phosphatidyl inositol 3 phosphatase kinase; siRNA, small interfering RNA; SOD, superoxide dismutase; TBARS, thiobarbital reactive substances; Tfam, mitochondria transcription factor A; TUNEL, terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling; WT, wild type.
References
- 1.Von Hoff DD, Layard MW, Basa P, Davis Jr HL, Von Hoff AL, Rozencweig M, Muggia FM1979. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 91:710–717 [DOI] [PubMed] [Google Scholar]
- 2.Billingham ME, Mason JW, Bristow MR, Daniels JR1978. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer Treat Rep 62:865–872 [PubMed] [Google Scholar]
- 3.Bristow MR, Thompson PD, Martin RP, Mason JW, Billingham ME, Harrison DC1978. Early anthracycline cardiotoxicity. Am J Med 65:823–832 [DOI] [PubMed] [Google Scholar]
- 4.Rajagopalan S, Politi PM, Sinha BK, Myers CE1988. Adriamycin-induced free radical formation in the perfused rat heart: implications for cardiotoxicity. Cancer Res 48:4766–4769 [PubMed] [Google Scholar]
- 5.Gupta M, Singal PK1987. Oxygen radical injury in the presence of desferal, a specific iron-chelating agent. Biochem Pharmacol 36:3774–3777 [DOI] [PubMed] [Google Scholar]
- 6.Fleury C, Mignotte B, Vayssière JL2002. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84:131–141 [DOI] [PubMed] [Google Scholar]
- 7.Cook SA, Sugden PH, Clerk A1999. Regulation of bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: association with changes in mitochondrial membrane potential. Circ Res 85:940–949 [DOI] [PubMed] [Google Scholar]
- 8.von Harsdorf R, Li PF, Dietz R1999. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 99:2934–2941 [DOI] [PubMed] [Google Scholar]
- 9.Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S2003. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun 300:167–171 [DOI] [PubMed] [Google Scholar]
- 10.Kawano H, Sato T, Yamada T, Matsumoto T, Sekine K, Watanabe T, Nakamura T, Fukuda T, Yoshimura K, Yoshizawa T, Aihara K, Yamamoto Y, Nakamichi Y, Metzger D, Chambon P, Nakamura K, Kawaguchi H, Kato S2003. Suppressive function of androgen receptor in bone resorption. Proc Natl Acad Sci USA 100:9416–9421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato T, Matsumoto T, Kawano H, Watanabe T, Uematsu Y, Sekine K, Fukuda T, Aihara K, Krust A, Yamada T, Nakamichi Y, Yamamoto Y, Nakamura T, Yoshimura K, Yoshizawa T, Metzger D, Chambon P, Kato S2004. Brain masculinization requires androgen receptor function. Proc Natl Acad Sci USA 101:1673–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan W, Yanase T, Nomura M, Okabe T, Goto K, Sato T, Kawano H, Kato S, Nawata H2005. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes 54:1000–1008 [DOI] [PubMed] [Google Scholar]
- 13.Marsh JD, Lehmann MH, Ritchie RH, Gwathmey JK, Green GE, Schiebinger RJ1998. Androgen receptors mediate hypertrophy in cardiac myocytes. Circulation 98:256–261 [DOI] [PubMed] [Google Scholar]
- 14.Ikeda Y, Aihara K, Sato T, Akaike M, Yoshizumi M, Suzaki Y, Izawa Y, Fujimura M, Hashizume S, Kato M, Yagi S, Tamaki T, Kawano H, Matsumoto T, Azuma H, Kato S, Matsumoto T2005. Androgen receptor gene knockout male mice exhibit impaired cardiac growth and exacerbation of angiotensin II-induced cardiac fibrosis. J Biol Chem 280:29661–29666 [DOI] [PubMed] [Google Scholar]
- 15.Ikeda Y, Aihara K, Yoshida S, Sato T, Yagi S, Iwase T, Sumitomo Y, Ise T, Ishikawa K, Azuma H, Akaike M, Kato S, Matsumoto T, Matsumoto T2009. Androgen-androgen receptor system protects against angiotensin II-induced vascular remodeling. Endocrinology 150:2857–2864 [DOI] [PubMed] [Google Scholar]
- 16.Taniyama Y, Walsh K2002. Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol 34:1241–1247 [DOI] [PubMed] [Google Scholar]
- 17.Sun M, Yang L, Feldman RI, Sun XM, Bhalla KN, Jove R, Nicosia SV, Cheng JQ2003. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85α, androgen receptor, and Src. J Biol Chem 278:42992–43000 [DOI] [PubMed] [Google Scholar]
- 18.Singal PK, Iliskovic N1998. Doxorubicin-induced cardiomyopathy. N Engl J Med 339:900–905 [DOI] [PubMed] [Google Scholar]
- 19.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA1973. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 32:302–314 [DOI] [PubMed] [Google Scholar]
- 20.Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A, Dec GW, Israels S, Ballester M, Virmani R, Saxena S, Kharbanda S1999. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA 96:8144–8149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA1996. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 335:1182–1189 [DOI] [PubMed] [Google Scholar]
- 22.Sharov VG, Sabbah HN, Shimoyama H, Goussev AV, Lesch M, Goldstein S1996. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am J Pathol 148:141–149 [PMC free article] [PubMed] [Google Scholar]
- 23.Saraste A, Voipio-Pulkki LM, Parvinen M, Pulkki K1997. Apoptosis in the heart. N Engl J Med 336:1025–1026; author reply 1026 [DOI] [PubMed] [Google Scholar]
- 24.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM1997. Apoptosis in human acute myocardial infarction. Circulation 95:320–323 [DOI] [PubMed] [Google Scholar]
- 25.Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C2002. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res 62:4592–4598 [PubMed] [Google Scholar]
- 26.Hu YC, Wang PH, Yeh S, Wang RS, Xie C, Xu Q, Zhou X, Chao HT, Tsai MY, Chang C2004. Subfertility and defective folliculogenesis in female mice lacking androgen receptor. Proc Natl Acad Sci USA 101:11209–11214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiina H, Matsumoto T, Sato T, Igarashi K, Miyamoto J, Takemasa S, Sakari M, Takada I, Nakamura T, Metzger D, Chambon P, Kanno J, Yoshikawa H, Kato S2006. Premature ovarian failure in androgen receptor-deficient mice. Proc Natl Acad Sci USA 103:224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandel ES, Hay N1999. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res 253:210–229 [DOI] [PubMed] [Google Scholar]
- 29.Burgering BM, Coffer PJ1995. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376:599–602 [DOI] [PubMed] [Google Scholar]
- 30.Franke TF, Kaplan DR, Cantley LC, Toker A1997. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275:665–668 [DOI] [PubMed] [Google Scholar]
- 31.Negoro S, Oh H, Tone E, Kunisada K, Fujio Y, Walsh K, Kishimoto T, Yamauchi-Takihara K2001. Glycoprotein 130 regulates cardiac myocyte survival in doxorubicin-induced apoptosis through phosphatidylinositol 3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction. Circulation 103:555–561 [DOI] [PubMed] [Google Scholar]
- 32.Matsui T, Nagoshi T, Rosenzweig A2003. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle 2:220–223 [PubMed] [Google Scholar]
- 33.Latronico MV, Costinean S, Lavitrano ML, Peschle C, Condorelli G2004. Regulation of cell size and contractile function by AKT in cardiomyocytes. Ann NY Acad Sci 1015:250–260 [DOI] [PubMed] [Google Scholar]
- 34.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K2005. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115:2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai CX, Kurokawa J, Tamagawa M, Nakaya H, Furukawa T2005. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation 112:1701–1710 [DOI] [PubMed] [Google Scholar]
- 36.Kalyanaraman B, Perez-Reyes E, Mason RP1980. Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs. Biochim Biophys Acta 630:119–130 [DOI] [PubMed] [Google Scholar]
- 37.Doroshow JH1983. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res 43:460–472 [PubMed] [Google Scholar]
- 38.Iliskovic N, Hasinoff BB, Malisza KL, Li T, Danelisen I, Singal PK1999. Mechanisms of beneficial effects of probucol in adriamycin cardiomyopathy. Mol Cell Biochem 196:43–49 [PubMed] [Google Scholar]
- 39.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK1996. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest 98:1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singal PK, Deally CM, Weinberg LE1987. Subcellular effects of adriamycin in the heart: a concise review. J Mol Cell Cardiol 19:817–828 [DOI] [PubMed] [Google Scholar]
- 41.Olson RD, Mushlin PS1990. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. FASEB J 4:3076–3086 [PubMed] [Google Scholar]
- 42.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A2001. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88:529–535 [DOI] [PubMed] [Google Scholar]
- 43.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA1998. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18:231–236 [DOI] [PubMed] [Google Scholar]
- 44.Garesse R, Vallejo CG2001. Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene 263:1–16 [DOI] [PubMed] [Google Scholar]
- 45.Davis AF, Ropp PA, Clayton DA, Copeland WC1996. Mitochondrial DNA polymerase γ is expressed and translated in the absence of mitochondrial DNA maintenance and replication. Nucleic Acids Res 24:2753–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larsson NG, Oldfors A, Holme E, Clayton DA1994. Low levels of mitochondrial transcription factor A in mitochondrial DNA depletion. Biochem Biophys Res Commun 200:1374–1381 [DOI] [PubMed] [Google Scholar]
- 47.Garnier A, Fortin D, Deloménie C, Momken I, Veksler V, Ventura-Clapier R2003. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551:491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaiswing L, Cole MP, St Clair DK, Ittarat W, Szweda LI, Oberley TD2004. Oxidative damage precedes nitrative damage in adriamycin-induced cardiac mitochondrial injury. Toxicol Pathol 32:536–547 [DOI] [PubMed] [Google Scholar]
- 49.Lebrecht D, Kokkori A, Ketelsen UP, Setzer B, Walker UA2005. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J Pathol 207:436–444 [DOI] [PubMed] [Google Scholar]
- 50.Santos DL, Moreno AJ, Leino RL, Froberg MK, Wallace KB2002. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicol Appl Pharmacol 185:218–227 [DOI] [PubMed] [Google Scholar]
- 51.Nozaki N, Shishido T, Takeishi Y, Kubota I2004. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation 110:2869–2874 [DOI] [PubMed] [Google Scholar]
- 52.Fan GC, Zhou X, Wang X, Song G, Qian J, Nicolaou P, Chen G, Ren X, Kranias EG2008. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ Res 103:1270–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu L, Zhang X, Qian B, Min X, Gao X, Li C, Cheng Y, Huang J2007. Over-expression of heat shock protein 27 attenuates doxorubicin-induced cardiac dysfunction in mice. Eur J Heart Fail 9:762–769 [DOI] [PubMed] [Google Scholar]
- 54.Matsumoto T, Takeyama K, Sato T, Kato S2003. Androgen receptor functions from reverse genetic models. J Steroid Biochem Mol Biol 85:95–99 [DOI] [PubMed] [Google Scholar]
- 55.Yoshida T, Azuma H, Aihara K, Fujimura M, Akaike M, Mitsui T, Matsumoto T2005. Vascular smooth muscle cell proliferation is dependent upon upregulation of mitochondrial transcription factor A (mtTFA) expression in injured rat carotid artery. Atherosclerosis 178:39–47 [DOI] [PubMed] [Google Scholar]
- 56.Fink B, Laude K, McCann L, Doughan A, Harrison DG, Dikalov S2004. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am J Physiol Cell Physiol 287:C895–902 [DOI] [PubMed]
- 57.Aihara K, Azuma H, Akaike M, Ikeda Y, Sata M, Takamori N, Yagi S, Iwase T, Sumitomo Y, Kawano H, Yamada T, Fukuda T, Matsumoto T, Sekine K, Sato T, Nakamichi Y, Yamamoto Y, Yoshimura K, Watanabe T, Nakamura T, Oomizu A, Tsukada M, Hayashi H, Sudo T, Kato S, Matsumoto T2007. Strain-dependent embryonic lethality and exaggerated vascular remodeling in heparin cofactor II-deficient mice. J Clin Invest 117:1514–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aihara K, Azuma H, Akaike M, Ikeda Y, Yamashita M, Sudo T, Hayashi H, Yamada Y, Endoh F, Fujimura M, Yoshida T, Yamaguchi H, Hashizume S, Kato M, Yoshimura K, Yamamoto Y, Kato S, Matsumoto T2004. Disruption of nuclear vitamin D receptor gene causes enhanced thrombogenicity in mice. J Biol Chem 279:35798–35802 [DOI] [PubMed] [Google Scholar]
- 59.Ikeda Y, Sato K, Pimentel DR, Sam F, Shaw RJ, Dyck JR, Walsh K2009. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem 284:35839–35849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aihara K, Azuma H, Takamori N, Kanagawa Y, Akaike M, Fujimura M, Yoshida T, Hashizume S, Kato M, Yamaguchi H, Kato S, Ikeda Y, Arase T, Kondo A, Matsumoto T2004. Heparin cofactor II is a novel protective factor against carotid atherosclerosis in elderly individuals. Circulation 109:2761–2765 [DOI] [PubMed] [Google Scholar]