

Abstract
Cytosolic phospholipase A2α (cPLA2α) catalyzes the hydrolysis of membrane glycerol-phospholipids to release arachidonic acid as the first step of the eicosanoid signaling pathway. This pathway contributes to proliferation in breast cancer, and numerous studies have demonstrated a crucial role of cyclooxygenase 2 and prostaglandin E2 release in breast cancer progression. The role of cPLA2α activation is less clear, and we recently showed that 17β-estradiol (E2) can rapidly activate cPLA2α in MCF-7 breast cancer cells. Overexpression or gene amplification of HER2 is found in approximately 30% of breast cancer patients and correlates with a poor clinical outcome and resistance to endocrine therapy. This study reports the first evidence for a correlation between cPLA2α enzymatic activity and overexpression of the HER2 receptor. The activation of cPLA2α in response to E2 treatment was biphasic with the first phase dependent on trans-activation through the matrix metalloproteinase-dependent release of heparin-bound epidermal growth factor. EGFR/HER2 heterodimerization resulted in downstream signaling through the ERK1/2 cascade to promote cPLA2α phosphorylation at Ser505. There was a correlation between HER2 and cPLA2α expression in six breast cancer cell lines examined, and inhibition of HER2 activation or expression in the SKBR3 cell line using herceptin or HER2-specific small interfering RNA, respectively, resulted in decreased activation and expression of cPLA2α. Pharmacological blockade of cPLA2α using a specific antagonist suppressed the growth of both MCF-7 and SKBR3 cells by reducing E2-induced proliferation and by stimulating cellular apoptosis and necrosis. This study highlights cPLAα2 as a potential target for therapeutic intervention in endocrine-dependent and endocrine-independent breast cancer.
Estrogen-induced activation of cytosolic phospholipase A2α is coupled to EGFR/HER2 heterodimerization and contributes to the proliferative effects of estrogen in breast carcinoma.
The phospholipase A2 (PLA2) enzymes catalyze the hydrolysis of the sn-2 linkage in membrane glycerol-phospholipids to release arachidonic acid (AA) and lysophospholipid secondary messengers. AA is then converted to bioactive eicosanoid lipid mediators such as prostaglandins, lipoxins, and leukotrienes, which play important regulatory roles in diverse cellular responses. There are three PLA2 isoform subfamilies: the Ca2+-dependent secretory PLA2, the Ca2+-independent intracellular PLA2, and the Ca2+-dependent cytosolic PLA2 (cPLA2). The cPLA2α isoform is constitutively expressed in most cells, and through the modulation of substrate availability, cPLA2α regulates the rate of AA metabolism to prostaglandins by cyclooxygenases (COX) and so indirectly regulates prostaglandin E2 (PGE2) production (1, 2). The AA-based eicosanoid signaling pathway plays an important role in normal cellular homeostasis, inflammation, and pathophysiological conditions. Specifically, eicosanoid signaling has been implicated in the development and progression of malignancy in different tissues including the lung (3), colon (4), prostate (5), and mammary gland (6). Overexpression of AA-metabolizing enzymes, principally COX-2, can be detected in many breast tumors and correlates with poor patient prognosis (7). COX inhibition decreases cell growth and promotes chemotherapy-induced apoptosis in breast cancer cells (8); epidemiological evidence also links the chronic use of COX-2 inhibitors with a reduced risk of breast cancer development (9, 10).
Recent data have suggested a link between eicosanoid signaling and estrogen-stimulated signaling events in breast cancer cells, at the level of both cPLA2α and COX-2 activity (11). The eicosanoid pathway has a potential role in estrogen-responsive breast cancer through a positive feedback loop, where COX-2 transcription is up-regulated by estrogen through epidermal growth factor receptor (EGFR) trans-activation (12), and COX-2 activity stimulates aromatase activity with important consequences for tumor cell proliferation (13). The mitogenicity of circulatory estrogens exerts a critical effect on the etiology and progression of breast cancer, where cumulative exposure of the mammary epithelium to estrogens is a significant risk factor (14, 15). The effects of estrogens, including the most biologically active 17β-estradiol (E2), are driven through the specific estrogen receptors (ERs) α and β (reviewed in Ref. 16). Antagonism of these receptors serves as the basis for therapeutic intervention in breast cancer using selective ER modulators (SERMs) such as tamoxifen and fulvestrant (17, 18, 19). ERs act by regulating gene transcription in the nucleus and by modulating the rapid activation of different signaling pathways from the plasma membrane (16). In particular, rapid activation of ERK1/2 MAPK by E2 through ERα and EGFR trans-activation has been reported in breast cancer cells (20).
Several studies have linked rapid estrogen-induced signaling to EGFR trans-activation: Filardo and Thomas (21) reported the involvement of the G protein-coupled receptor (GPR)-30 in the activation of matrix metalloproteinase (MMP), release of heparin-bound (HB)-EGF, and activation of EGFR in SKBR3 breast carcinoma cells. Razandi et al. (22, 23) demonstrated a direct interaction between ER and G proteins and also found that this interaction triggers a Gαq and Gβγ-dependent activation of MMPs leading to EGFR trans-activation and downstream signaling to ERK and phosphatidylinositol 3-kinase in breast cancer cells. These E2-induced indirect effects can potentiate the mitogenic action of estrogens and are also involved in the development of endocrine resistance by diverting the effects of E2 to alternative growth factor receptor signaling pathways that are insensitive to SERMs (24). The dependency of growth on estrogens can be circumvented by overexpression of EGFR/c-erbB1 and HER2/c-erbB2 (members of the EGFR family of receptor tyrosine kinases that also include c-erbB3 and c-erbB4), which is frequently found in invasive breast cancer and where it correlates with a decreased sensitivity to endocrine therapy and with poor patient prognosis (25).
Clinical, epidemiological, and molecular studies have investigated the role of eicosanoid signaling in breast cancer, focusing mainly on COX-2 and its metabolite PGE2 (11, 26). Animal models have shown that carcinogen-induced mammary tumor formation can be reduced by either treatment with COX inhibitors (27, 28, 29) or genetic ablation of Cox-2 (30). Conversely, COX-2 overexpression in mouse mammary gland increased tumor formation and potentiated angiogenesis (31, 32). As the major prostaglandin produced by COX-2 in breast cancer (33), PGE2 has been shown to play a key role in many aspects of COX-2-induced tumorigenesis. PGE2 levels are elevated in breast cancer (31), and in vitro studies have shown that PGE2 can stimulate both the proliferation (34) and migration (35) of mammary epithelial cells. PGE2 can stimulate the expression of growth-promoting genes such as c-fos and VEGF (36) and can also increase aromatase activity and consequent estrogen biosynthesis (13), indirectly contributing to cell proliferation.
Despite the body of data available on the role of COX-2 and PGE2 in breast cancer tumorigenesis, the role of cPLA2 in the cross talk between the estrogen and the eicosanoid signaling pathways in estrogen-responsive breast cancer remains unclear. cPLA2 is involved in the rapid estrogen-induced responses in the colon (37) and in embryonic membranes (38). Previous work from our laboratory showed that low concentrations of E2 rapidly promote the activation of cPLA2α in the MCF-7 breast cancer cell line, impacting on the rapid, estrogen-driven transient rise in intracellular Ca2+ concentration. cPLA2α was activated through ERK1/2 MAPK-dependent phosphorylation on Ser505 and intracellular translocation to perinuclear membranes (39). Here we have identified the receptors and characterized the molecular mechanisms involved in the rapid estrogen-induced activation of cPLA2α in both endocrine-sensitive and endocrine-resistant breast cancer cells.
Results
E2 rapidly and transiently stimulates cPLA2α phosphorylation through ER-dependent ERK1/2 activation in MCF-7 cells
We previously showed that E2 stimulated the phosphorylation of cPLA2α at residue Ser505 within 1 min of treatment in MCF-7 cells (39). To further characterize the the E2-induced cPLA2α response, we analyzed a time course ranging from 30 sec to 20 min. E2 (10 nm) induced a rapid, transient, and biphasic activation of cPLA2α, with a first peak of phosphorylation starting as early as 30 sec to 2 min after treatment and a second peak detectable from 4–15 min after treatment (Fig. 1A). Time points corresponding to the two maximal peaks of activation (1 and 10 min) were chosen for analysis in all subsequent experiments. The rapid E2-induced activation of signaling pathways is thought to be mediated by an ER localized at or near the plasma membrane. The nature of such a receptor has variously been reported to be either a truncated form of ERα, a lipid-modified form of ERα, or a GPR like GPR30 (40). We previously showed that the rapid activation of cPLA2α can be induced by both E2 and the membrane-impermeable E2-BSA (39), indicating the involvement of a membrane-localized receptor. Here we show that the specific ER antagonist ICI 182,780 (ICI) blocked the E2-induced phosphorylation of cPLA2 at both 1- and 10-min time points (Fig. 1B).
Fig. 1.
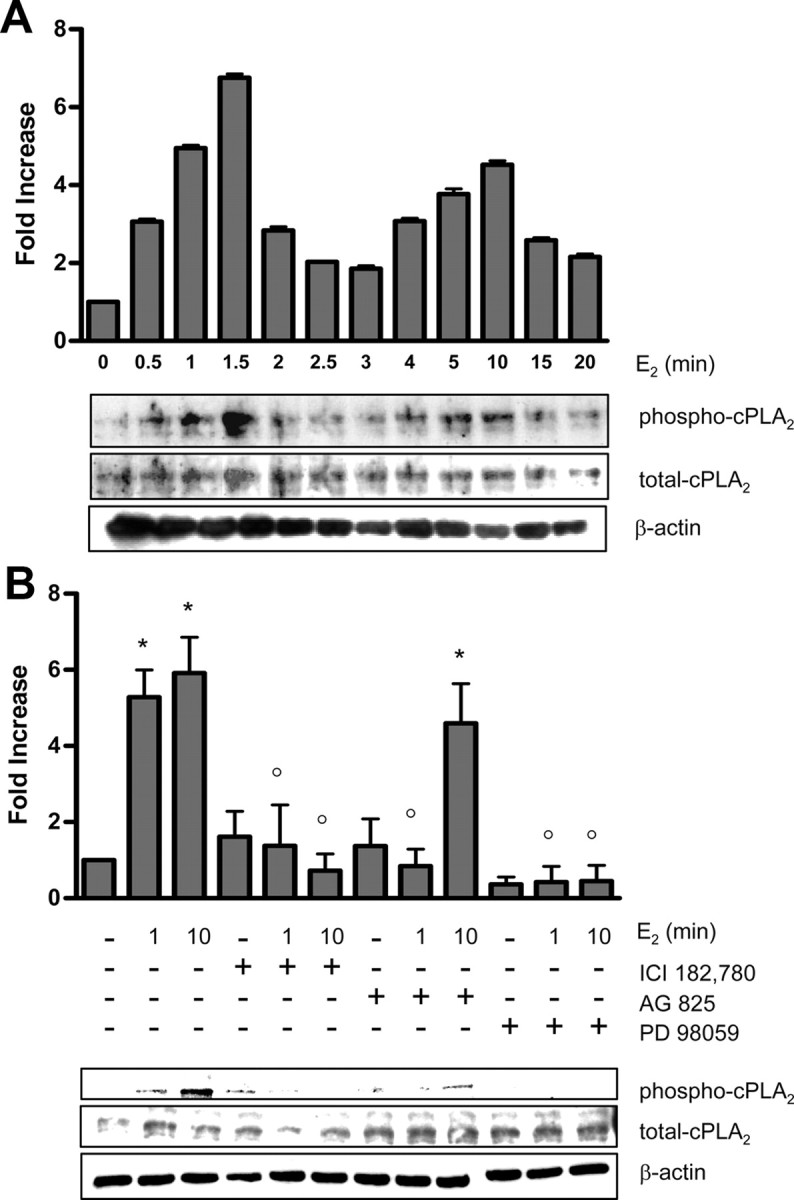
E2 induces transient phosphorylation of cPLA2α through ER- and EGFR-dependent MAPK activation. A, Western blot analysis of phospho-cPLA2α (Ser505) and total cPLA2α was performed on MCF-7 cells treated with either vehicle (0) or 10 nm E2 at the indicated time points. A representative blot is shown along with densitometric analysis of three independent experiments. B, Western blot analysis of phospho-cPLA2α (Ser505) and total cPLA2α was performed on MCF-7 cells treated for 1 and 10 min with either vehicle controls or E2 (10 nm) with or without the inhibitors ICI (10 μm), AG825 (5 μm), or PD98059 (20 μm). β-Actin was used for protein level normalization. Densitometric analysis of three independent experiments is shown with a representative blot. Data are mean values ± se. *, P < 0.01 compared with vehicle-treated control; °, P < 0.01 compared with E2-stimulated values at corresponding time points.
Phosphorylation of cPLA2α at Ser505 is mediated by members of the MAPK family (2), and in MCF-7 cells, the rapid effect of E2 on cPLA2α is specifically driven through ERK1/2 (39). The specific MAPK kinase-1 inhibitor PD98059 blocked the E2-induced phosphorylation of cPLA2α at 1 and 10 min (Fig. 1B), thus confirming the involvement of ERK1/2 MAPK upstream of cPLA2α. Interestingly, E2 promotes a transient and biphasic phosphorylation of ERK1/2 in MCF-7 cells that mirrors the time-course for cPLA2α activation (39). Because E2 can activate MAPK through trans-activation of EGFR (41, 42), we investigated the role of EGFR in mediating the stimulatory effect of E2 on cPLA2α. Pretreatment of MCF-7 cells with the specific EGFR/HER2 inhibitor AG825 blocked the first rapid peak of E2-induced cPLA2α phosphorylation but only partially suppressed the subsequent activation at 10 min (Fig. 1B). AG825 is an EGFR kinase inhibitor preferentially selective for HER2 over EGFR, suggesting that HER2 may play a greater role in the earlier phase of cPLA2α activation.
Rapid E2-induced cPLA2α activation is dependent on trans-activation of EGFR-HER2 heterodimers
Recent evidence demonstrates that in breast cancer cells, E2 promotes EGFR trans-activation and downstream signaling through the c-Src-mediated activation of the MMP cascade and the subsequent release of membrane-associated HB-EGF (23, 42). MCF-7 cells were pretreated with the diphtheria toxin mutant CRM197, which inhibits the mitogenic activity of HB-EGF by promoting its internalization from the cell membrane (43). CRM197 blocked E2-induced phosphorylation of cPLA2α after 1 min treatment but achieved only partial inhibition of E2-induced cPLA2 phosphorylation at 10 min (Fig. 2A). CRM197 also blocked the E2-induced activation of ERK1/2 upstream of cPLA2α at 1 min, but inhibition was not observed at 10 min. (Fig. 2B). Moreover, pretreating MCF-7 cells with the general MMP inhibitor GM6001 blocked the E2 effect on cPLA2α phosphorylation at 1 min (Fig. 2C). These data demonstrate that E2-induced cPLA2α activation at 1 and 10 min is differentially regulated, with the early phase of cPLA2α activation being dependent on the activation of a MMP cascade at the cell membrane leading to trans-activation of EGFR. The later phase of E2-induced cPLA2α activation at 10 min was largely independent of EGFR trans-activation. E2-bound ER binds to and activates the c-Src tyrosine kinase, leading to MAPK activation, through EGFR trans-activation or via direct activation of Ras (23, 42, 44). Pretreatment of MCF-7 cells with the specific c-Src inhibitor 4-amino-5-(4chorophenyl)-7-(t-butyl) pyrozolo[3,4-D]pyrimidine (PP2) blocked the E2-induced activation of cPLA2α at both 1 and 10 min (Fig. 2D), thus confirming the involvement of c-Src in the rapid activation of the MMP cascade leading to the EGFR-dependent early phase of cPLA2α activation at 1 min. Pretreatment of MCF-7 cells with PP2 also blocked the activation of ERK1/2 after 10 min E2 treatment (Fig. 2E). These data suggest that the later phase of cPLA2α activation at 10 min is largely driven by a c-Src-mediated direct activation of the MAPK cascade that augments the contribution of EGFR trans-activation.
Fig. 2.

E2 activates cPLA2 through MMP- and HB-EGF-dependent trans-activation of EGFR. A and B, Western blot analysis of cPLA2α phosphorylation at residue Ser505 (A) and ERK1/2 MAPK phosphorylation at residues Thr202/Tyr204 (B) was performed on MCF-7 cells treated with either vehicle (0) or 10 nm E2, with or without CRM197 (200 ng/ml, 2 h pretreatment) at the indicated time points. A line separates noncontiguous lanes on the same gel. C, Phosphorylation of cPLA2α at residue Ser505 was assessed by immunoblotting in MCF-7 cells treated with either vehicle (control) or 10 nm E2 for 1 min, with or without the MMP inhibitor GM6001 (10 μm). D, Phosphorylation of cPLA2α at residue Ser505 was assessed by immunoblotting in MCF-7 cells treated with either vehicle (0) or 10 nm E2 for 1 and 10 min, with or without the c-Src antagonist PP2 (100 nm). E, Phosphorylation of ERK1/2 at residues Thr202/Tyr204 was assessed by immunoblotting in MCF-7 cells treated with either vehicle (0) or 10 nm E2 for 10 min, with or without the c-Src antagonist PP2 (100 nm). All blots were stripped and reprobed with total cPLA2 or total ERK1/2 MAPK antibodies. Total cPLA2α or total ERK1/2 was used for protein level normalization as appropriate. Densitometric analysis of three independent experiments is shown with a representative blot. Data are mean values ± se. *, P < 0.01 compared with E2 stimulation.
EGFR and HER2 can form homodimers and heterodimers with each other and with the other two members of the EGFR family (45). Receptor dimerization and activation leads to trans-phosphorylation of specific tyrosine residues within the cytoplasmic tail of the receptors. Activation of EGFR/HER2 heterodimers has been observed in breast cancer cells, resulting in the activation of distinct signaling pathways (46). E2 treatment promoted tyrosine phosphorylation of EGFR in MCF-7 cells within 1 min of treatment (Fig. 3A). The effect was persistent for at least 10 min and abrogated by pretreatment with CRM197, confirming that in this cell line, E2 promotes EGFR trans-activation through release of HB-EGF. E2 treatment did not increase phosphorylation of HER2 above basal levels in MCF-7 cells (Fig. 3B), as compared with HCC38 (used here as HER2-negative control) and SKBR3 (HER2-overexpressing positive control). Coimmunoprecipitation studies showed that E2 treatment increased the basal level of association between EGFR and HER2 within a rapid time frame of 1–3 min, corresponding to the first peak of activation of cPLA2α (Fig. 3C). The effect was also blocked by pretreatment with CRM197. These data suggest that E2 induces a MMP-mediated release of HB-EGF acting in an autocrine fashion to promote trans-activation of EGFR through an increase in active EGFR/HER2 heterodimers.
Fig. 3.

E2 promotes activation of EGFR-HER2 heterodimers. MCF-7 cells were treated with either vehicle (0) or 10 nm E2 with or without CRM197 (200 ng/ml) at the indicated time points. A and C, Lysates were immunoprecipitated with 2 μg EGFR antibody and blotted for phosphotyrosines (A) or immunoprecipitated with 2 μg of either EGFR or HER2 antibody and blotted for HER2 and EGFR (C). Data are mean values ± se. *, P < 0.05 compared with E2 stimulation without CRM197 at corresponding time points. B, HCC38 (HER2-negative control breast cancer cell line), MCF-7, and SKBR3 cells were treated with either vehicle (0) or 10 nm E2 at indicated time points, and lysates were subjected to ELISA for phospho-HER2 (Tyr 1221/1222). The lower panel shows a representative Western blot of MCF-7 lysates immunoprecipitated with 2 μg HER2 antibody and blotted for phosphotyrosines. IP, Immunoprecipitation; WB, Western blot.
Increased cPLA2α expression and activity in HER2-overexpressing breast cancer cell lines
To determine whether there was a correlation between EGFR/HER2 heterodimerization and the activation of cPLA2α, we used the SKBR3 cell line, a breast cancer cell line that is ER negative but HER2 positive and is used as a model for endocrine-resistant, HER2-overexpressing ductal breast carcinoma. Semiquantitative RT-PCR analysis confirmed that SKBR3 cells expressed significantly higher levels of HER2 mRNA compared with MCF-7 cells (53 ± 11% increase, P < 0.001). cPLA2α mRNA levels were also greater (30 ± 4.9%, P < 0.01) in SKBR3 cells compared with MCF-7, whereas no significant difference was measured in EGFR (17.5 ± 2.8%) and COX-2 (9.5 ± 1.2%) mRNA levels (Fig. 4A). Western blot analysis confirmed that protein expression levels for EGFR (6.3-fold, P < 0.001), HER2 (4.2-fold, P < 0.01), and cPLA2α (12.9-fold, P < 0.001) were also greater in SKBR3 cells compared with MCF-7 cells. COX-2 expression was also slightly higher (1.4-fold) but was not statistically significant (Fig. 4B). Quantitative real-time PCR confirmed mRNA expression levels of both HER2 and cPLA2α were significantly higher in SKBR3 cells when compared with MCF-7 cells (Fig. 4C). To address the question of whether the increased expression of cPLA2α in SKBR3 cells was coupled to an increased enzymatic activity, we measured hydrolysis of the substrate arachidonoyl thio-phosphatidylcholine in vitro (Fig. 4D). SKBR3 cells showed a 2-fold greater cPLA2α catalytic activity when compared with MCF-7 cells (9.9 ± 0.2 vs. 5.8 ± 0.3 nmol/min·ml, respectively), confirming that the greater expression of cPLA2α in SKBR3 cells translates into a higher enzymatic activity to drive production of AA. To investigate whether the correlation between HER2 and cPLA2α is a peculiar characteristic of the SKBR3 cell line, we compared a panel of five breast cancer cell lines that differentially expressed HER2 with MCF-7 cells for both HER2 and cPLA2α mRNA (Fig. 5A) and protein (Fig. 5B) abundance. All cell lines reported to be HER2 overexpressing (BT474, SKBR3, and UACC893) (47) showed higher levels of HER2 expression when compared with MCF-7 cells, at both the mRNA and protein level. SKBR3 and UACC893 also showed higher levels of cPLA2α mRNA and protein compared with MCF-7 cells. BT474 cells had less mRNA but the same amount of cPLA2α protein as compared with MCF-7 cells. The two cell lines reported to be nonoverexpressing (HCC38 and MDA-MB-231) (47) expressed HER2 mRNA at comparable levels to MCF-7 cells; both of these cell lines also expressed higher levels of cPLA2α mRNA and protein compared with MCF-7 cells (Fig. 5).
Fig. 4.

The endocrine-resistant breast cancer cell line SKBR3 overexpresses EGFR/HER2 and shows increased expression and activity of cPLA2. A and C, Total mRNA was extracted from untreated MCF-7 and SKBR3 cells, reverse transcribed into cDNA, and either subjected to semiquantitative PCR using specific primers for EGFR, HER2, cPLA2α and COX-2 (expression levels were normalized for GAPDH (a representative agarose gel is shown along with densitometric analysis of six experiments) (A) or subjected to real-time quantitative PCR with specific primers for HER2 and cPLA2 (C). mRNA expression levels were normalized to 18S and expressed as fold difference in relative quantity relative to MCF-7. Data are mean values ± se. *, P < 0.001; **, P < 0.01 compared with MCF-7 values. B, Western blot analysis of total EGFR, HER2, cPLA2, and COX-2 was performed on unstimulated MCF-7 and SKBR3 cells. β-Actin was used for protein level normalization. Densitometric analysis of three different experiments is shown with a representative blot. Data are mean values ± se. *, P < 0.001; **, P < 0.01 compared with MCF-7 values. D, cPLA2 enzymatic activity was measured in total lysates from MCF-7 and SKBR3 cells. *, P < 0.01 compared with MCF-7 values.
Fig. 5.

Analysis of HER2 and cPLA2 expression in a panel of breast cancer cell lines. A, Total mRNA was extracted from untreated HCC38, MDA-MB-231, MCF-7, BT474, SKBR3, and UACC893 cells, reverse-transcribed into cDNA, and subjected to real-time quantitative PCR with specific primers for HER2 and cPLA2. mRNA expression levels were normalized to 18S and expressed as fold difference in relative quantity relative to MCF-7. B, Western blot analysis of total HER2 and cPLA2 was performed on unstimulated HCC38, MDA-MB-231, MCF-7, BT474, SKBR3, and UACC893 cells. α-Tubulin was used for protein level normalization.
Inhibition of HER2 impacts on cPLA2α activation in SKBR3 cells
The coupling of HER2 to cPLA2α activation in SKBR3 cells was investigated by studying the effect of HER2 inhibition on the expression and activation of cPLA2α. To do so, we used both a pharmacological inhibition approach using herceptin and a gene silencing approach using small interfering RNA (siRNA). Herceptin (Trastuzumab) is a recombinant humanized monoclonal antibody directed against the extracellular domain of HER2 that is extensively used in the clinical setting to treat HER2-positive metastatic breast cancer (48). The mechanism of action of herceptin is still not completely clear, but several reports suggest that its action could be explained by an induced impairment of HER2 heterodimerization and consequent blockade of downstream signaling events (48). Treatment of SKBR3 cells with 20 μg/ml herceptin in the presence of serum for 48 h resulted in inhibition of HER2 phosphorylation, with no change in total HER2 protein expression (49). We found that treating cells with herceptin (20 μg/ml) in the presence of E2 (10 nm) resulted in a similar inhibition of HER2 phosphorylation to that found for herceptin in the presence of serum, with no change in total HER2 protein expression levels (Fig. 6A). The reduction in HER2 phosphorylation upon treatment with herceptin in conjunction with E2 was coupled to a reduction in cPLA2α phosphorylation, with no change in total cPLA2α protein abundance (Fig. 6A). Treatment with either herceptin alone or E2 alone did not change total expression or the phosphorylation states of either HER2 or cPLA2α. When SKBR3 cells were transfected with a pool of four different siRNA species specific for HER2, expression of the receptor was partially silenced, with protein expression levels reduced to 30% of nontransfected control. Silencing of HER2 reduced cPLA2α protein expression to 58% of nontransfected control and cPLA2α phosphorylation levels to 43% of nontransfected control (Fig. 6B), confirming the positive correlation between HER2 overexpression and the abundance of activated cPLA2α in SKBR3 cells. To rule out any non-sequence-specific effects of gene silencing, a negative nontargeting siRNA control was used that is designed to have at least four mismatches with all known human genes. This negative siRNA had no effect on the expression levels of HER2 or cPLA2α, confirming the specificity of cPLA2α down-regulation after selective HER2 silencing (Fig. 6B). The HER2 siRNA did not change cPLA2α expression at the mRNA level as compared with nontransfected or negative siRNA-transfected controls (Fig. 6C), indicating that HER2 exerts a posttranslational control of cPLA2α protein expression.
Fig. 6.

Inhibition of HER2 in SKBR3 cells decreases cPLA2 activation and expression. Panel A, SKBR3 cells were treated with 20 μg/ml herceptin for 48 h with or without 10 nm E2 as indicated. Western blot analysis of phospho-cPLA2α, total cPLA2α, phospho-HER2, and total HER2 was performed. β-Actin was used for protein level normalization, and densitometric values are expressed as fold difference vs. vehicle-treated controls. Data are mean values ± se. *, P < 0.01 compared with control values. Panels B and C, SKBR3 cells were transfected with 100 nm of a pool of four different siRNA targeting HER2. Negative control cells were transfected with 100 nm nontargeting siRNA. At 72 h after transfection, total proteins and mRNA were extracted and subjected to either Western blot analysis for total HER2, phospho-cPLA2 (Ser505) and total cPLA2α, (total cPLA2α was used for protein level normalization (panel B) (data are mean values ± se; *, P < 0.001 compared with vehicle-treated control) or semiquantitative RT-PCR using specific primers for cPLA2 (panel C) (expression levels were normalized for GAPDH, and a representative agarose gel is shown along with densitometric analysis of two experiments). C, Control.
E2 rapidly promotes cPLA2 phosphorylation in SKBR3 cells through GPR30-dependent EGFR trans-activation
In the HER2-positive SKBR3 cells, cPLA2α was overexpressed and was also constitutively activated (Figs. 4 and 6). However, E2 still promoted cPLA2α activation above basal levels of phosphorylation, at both 1 and 10 min (Fig. 7A). This effect was blocked by pretreatment with CRM197, demonstrating that the effect of E2 is driven by EGFR trans-activation in both ER-positive (MCF-7) and ER-negative (SKBR3) cell lines (Fig. 7A). Unlike in MCF-7 cells, CRM197 treatment fully blocked E2-induced phosphorylation of cPLA2α in SKBR3 cells at both 1 and 10 min. Pretreatment with the selective EGFR/HER2 inhibitor AG825 also blocked the E2-induced activation of cPLA2α at 10 min (Fig. 7B). The effect of E2 on ERK1/2 activation showed a different temporal activation profile in SKBR3 to that in MCF-7 cells. In SKBR3 cells, ERK activation started 1 min after E2 stimulation and increased to a maximum at 5 min, remaining constant for at least 10 min, in contrast to the biphasic response observed in MCF-7 cells. CRM197 blocked the stimulation of ERK1/2 activation by E2 in SKBR3 cells over the entire duration of a 10-min time course (Fig. 7D), whereas CRM197 completely blocked only the first transient phase of ERK activation in MCF-7 cells (Fig. 2A). SKBR3 cells are described as ER negative as well as being HER2 positive, but they do express GPR30, which binds E2 to activate MAPK through MMP-mediated EGFR trans-activation (42). The rapid effect of E2 on cPLA2α activation in SKBR3 cells was mimicked by the selective GPR30 agonist G1 and by ICI, which also acts as a GPR30 agonist (21). The effects of E2, G1, and ICI were nonadditive, indicating that E2 and GPR30 agonists may act through a common receptor and signaling pathway in SKBR3 cells (Fig. 7C). The physiological role of GPR30 and its capacity to bind to and mediate the effects of E2 are still controversial.
Fig. 7.

E2 increases the basal activation of cPLA2 in SKBR3 cells, acting through GPR30-mediated EGFR trans-activation. A–C, Western blot analysis of phospho-cPLA2α (Ser505) and total cPLA2 was performed on SKBR3 cells treated with either vehicle (0) or E2 (10 nm), with or without CRM197 (200 ng/ml) at the indicated time points (A); treated with vehicle (0) or E2 (10 nm), with or without AG825 (5 μm) for 10 min (B); or treated with vehicle (C) or E2 (10 nm) or ICI (10 mm) or the GPR30-selective agonist G1 (100 nm) for 10 min (C). A line separates noncontiguous lanes on the same gel. Data are mean values ± se. **, P < 0.01 compared with vehicle-treated control; *, P < 0.01 compared with E2-stimulated values at corresponding time point. D, Western blot analysis of phospho-ERK1/2 MAPK (Thr202/Tyr204) and total ERK1/2 MAPK was performed on SKBR3 cells treated with either vehicle (0) or E2 (10 nm), with or without CRM197 (200 ng/ml) at the indicated time points. Total cPLA2α or ERK1/2 was used for protein level normalization as appropriate.
Researchers have reported the expression of differentially spliced ERα isoforms, namely ERα46 (50) and ERα36 (51), which inhibit the transcriptional activity of wild-type ERα and which could mediate the transduction of estrogen- and antiestrogen-mediated mitogenic signaling from the plasma membrane of endothelial and breast cancer cells (52, 53, 54). Western blotting with a specific ERα antibody showed that SKBR3 cells do not express the 66-kDa wild-type ERα, but two bands of approximately 36 and 46 kDa were present that were also detected in MCF-7 cells (Fig. 8A). These bands could represent degradation products, or alternatively spliced receptor isoforms with a similar molecular mass. However, the antibody that was used in this study is directed against an epitope surrounding Ser118 in the A/B domain of ERα, a region that is completely deleted in the ERα36 and ERα46 isoforms (Fig. 8B). Furthermore, when SKBR3 cells were grown in the presence of E2 for 24 h, the 46-kDa band was not present, and the 36-kDa band was reduced compared with cells grown in the absence of E2 (Fig. 8A). To establish whether the mitogenic effects of E2 (namely, activation of ERK1/2 MAPK and subsequently cPLA2α) in the SKBR3 cells were mediated by GPR30, we performed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays to study cell growth (Fig. 8C). Treatment with the antiestrogens ICI and tamoxifen, both of which have been shown to act as GPR30 agonists (21), mimicked the E2-induced increase in cell growth with no additive effect. The selective GPR30 agonist G1 also increased cell growth with no additive effects with E2 (Fig. 8C), suggesting that G1 and E2 were acting through a common receptor to promote cell growth.
Fig. 8.

Involvement of ER isoforms and GPR30 in mediating the effects of E2 in SKBR3 cells. Panel A, Western blot analysis of total ERα was performed on SKBR3 cells treated with either vehicle (0) or E2 (10 nm) for 24 h and on untreated MCF-7 cells. The different isoforms are indicated at their respective molecular weight. Samples were also probed for c-Fos as a positive control for E2 treatment because E2 was reported to up-regulate c-Fos in the SKBR3 cell line (71 ). β-Actin was used for protein level normalization. Panel B, Structure of the 66-kDa wild-type ERα and the two splice variants ERα36 and ERα46 lacking the N-terminal A/B domain. The different domains (A–F) and amino acid sequence numbers are indicated. An arrow indicates the epitope recognized by the ERα antibody used in this study (adapted from Ref. 51 ). Panel C, MTT cell growth assay was performed on SKBR3 cells treated with either vehicle or E2 (10 nm) with or without ICI (10 μm), tamoxifen (10 μm), or G1 (100 nm) for 48 h. Data are mean values ± se of three independent experiments.
Antagonism of cPLA2α inhibits E2-induced cell proliferation in MCF-7 and SKBR3 cells
The proliferative effect of E2 and EGFR ligands on breast cancer cells is well characterized (16, 25); to investigate whether the eicosanoid signaling pathway and specifically cPLA2 activation is involved in these proliferative events, we measured E2-induced cell growth using the MTT cell growth assay in MCF-7 and SKBR3 cells pretreated with a specific cPLA2 inhibitor (Fig. 9). Treatment of MCF-7 cells with 10 nm E2 resulted in an increased cell growth (24% increase compared with vehicle control). Pharmacological inhibition of cPLA2α completely abolished the E2-stimulated cell growth. This effect of cPLA2 inhibition on cell growth was comparable to growth inhibition after ICI treatment, which blocked the effect of E2, reducing growth levels (Fig. 9A). In SKBR3 cells, E2 (10 nm) induced a 35% increase in cell growth compared with vehicle-treated control. Pharmacological inhibition of cPLA2α blocked the cell growth effect of E2 and restored growth levels to control levels. Inhibition of HER2 with herceptin also down-regulated E2-stimulated cell growth to control levels (Fig. 9B). In addition to inhibiting the E2-induced cell growth, the cPLA2α inhibitor also reduced MCF-7 cell numbers below control basal levels, which implied a homeostatic role for cPLA2α in regulating cell viability. Treatment of both MCF-7 and SKBR3 cell lines with the specific cPLA2α inhibitor increased the incidence of both apoptotic and necrotic cell death compared with vehicle-treated controls (Fig. 9C). In MCF-7 cells, the cPLA2α inhibitor caused an 85% increase in apoptosis compared with control and a 2.4-fold increase in necrosis. Tamoxifen was used as a positive control, because its effect on cell death in MCF-7 has been previously reported (55). In SKBR3 cells, the inhibition of cPLA2 caused a 2.6-fold increase in apoptosis and a 2.4-fold increase in necrosis, compared with control. Inhibition of HER2 with herceptin, which has been shown to induce cell death (48), also increased cell necrosis and apoptosis in SKBR3 cells, and this response was similar to that observed with the cPLA2 inhibitor (Fig. 9C).
Fig. 9.

Pharmacological inhibition of cPLA2 decreases E2-induced cell growth and increases apoptosis and necrosis in both MCF-7 and SKBR3. Panel A, MTT cell growth assay was performed on MCF-7 treated with either vehicle [control (C)] or E2 (10 nm) with or without ICI (10 μm) or the specific cPLA2α inhibitor (50 nm), and (panel B) on SKBR3 treated with either vehicle (C) or E2 (10 nm) with or without the monoclonal HER2 antibody herceptin (20 mg/ml) or the cPLA2α inhibitor (50 nm). Data are mean values ± se. *, P < 0.01 compared with vehicle-treated control; **, P < 0.01 compared with E2 stimulation. Panel C, An ELISA to detect oligonucleosomes in the cytoplasm or in the cell culture medium was performed on both MCF-7 and SKBR3 treated with the cPLA2α inhibitor (50 nm) or ICI (10 mm) or herceptin (20 μg/ml) for 48 h. Data are expressed as percent increase of both apoptosis and necrosis compared with vehicle-treated controls.
Discussion
The activation of cPLA2α is the rate-limiting step in the physiological production of AA, which is rapidly metabolized by COX enzymes to produce PGE2 (1). Prostaglandins regulate many physiological processes through GPR activation leading to the production of second messengers that induce proliferation, migration, apoptosis, and angiogenesis (56). In addition, cPLA2α can also promote carcinogenesis by liberating membrane lysophospholipids that can induce cell growth through their metabolism to lysophosphatidic acid (57). Consequently, cPLA2α activity is tightly controlled to maintain low intracellular concentrations of AA in resting cells. However, dysregulation of cPLA2α activity is detected in many human malignancies, including mammary adenocarcinoma (6). Increased cPLA2α activity, coupled to increased activity of AA-metabolizing enzymes such as COX-2, leads to high levels of proliferative eicosanoids (31, 33). Recent studies have focused on the regulatory mechanisms controlling the activity of COX-2 during carcinogenesis. These studies have provided the rationale for the use of nonsteroidal antiinflammatory drugs (such as indomethacin and flurbiprofen) and specific COX inhibitors (such as celecoxib and nimesulide) as chemotherapeutic agents. Despite their efficacy in slowing the progression of malignancy (58), these therapies are often associated with detrimental side effects including gastrointestinal bleeding and cardiovascular toxicity. Other components of the AA-based signaling pathway have been proposed as potential targets for chemoprevention and therapy, including cPLA2α, and therefore, a better understanding of the precise mechanism underlying the activation of cPLA2α in breast cancer and its role in proliferation would enhance the development of specific pharmacological strategies for the treatment of breast carcinoma and also other malignancies.
We have previously shown that cPLA2α is expressed in the MCF-7 breast carcinoma cell line and is rapidly activated after treatment with physiological concentrations of E2 (39). In this present study, we investigated the molecular mechanism of E2-induced cPLA2α activation in breast cancer cell lines that differentially express ERα and HER2. In ERα-positive, HER2-negative MCF-7 cells, E2 elicited a biphasic activation of cPLA2α that was driven by trans-activation of EGFR resulting in activation of the ERK1/2 MAPK cascade. Evidence of a synergism between EGFR and eicosanoid signaling has been described in other experimental systems. EGFR is required for the phosphorylation of cPLA2 induced by neurotensin and EGF in prostate cancer cells (59), and a correlation has been found between COX-2 activity and EGFR activity in breast cancer (12, 60). We found that the initial ERK1/2 activation and downstream phosphorylation of cPLA2α in response to E2 was dependent on EGFR trans-activation in MCF-7 cells, through MMP-dependent release of HB-EGF and the formation of EGFR/HER2 heterodimers. The MMP inhibitor GM6001, the HB-EGF inhibitor CRM197, and the EGFR/HER2 inhibitor AG825 also blocked the phosphorylation of cPLA2α induced by E2 in this experimental model. E2-induced ERK1/2 activation in breast cancer cells can be mediated by direct interaction of ER with the nonreceptor tyrosine kinase c-Src to activate Ras. E2 also down-regulates MAPK phosphatase 1 (MKP-1) leading to up-regulation of ERK1/2 activity within 10 min of treatment (61). This present study suggests that E2 signals through c-Src-dependent, EGFR trans-activation to promote the early phase of ERK1/2 phosphorylation and subsequent cPLA2α activation within 1 min, whereas the later phase of cPLA2α activation after 5–10 min is largely driven by EGFR-independent mechanisms through ER-mediated ERK1/2 phosphorylation but which is still c-Src dependent.
Approximately 25–30% of human breast cancers display overexpression or gene amplification of HER2, and its increased expression correlates with poor clinical outcome and with resistance to endocrine therapy (25, 62). EGFR is also overexpressed in 50% of breast tumors and correlates with resistance to hormonal therapy (25). In these tumors, the cross talk between ER and EGFR/HER2 signaling pathways results in a positive feedback cycle of cell survival stimuli. HER2 has the strongest catalytic activity of the four members of the EGFR family, and HER2-containing heterodimers have the greatest capacity for inducing intracellular signaling (46). HER2 is also less sensitive to inactivating signals, and its recruitment into heterodimeric signaling complexes leads to more sustained signaling responses. In the MCF-7 cell line, E2 rapidly promoted an increased dimerization of EGFR with HER2, which was coupled to increased phosphorylation of EGFR but not of HER2. A correlation between overexpression of COX-2 and HER2 gene amplification in breast cancer was previously reported by Ristimäki and colleagues (7). This was subsequently confirmed by the finding that HER2 abundance and activity determines Cox-2 gene expression (12). This present study is the first report of a correlation between cPLA2α and HER2 overexpression in a breast cancer cell line. Western blot analysis of phosphorylated cPLA2 showed a constitutive basal activation of cPLA2 in SKBR3 cells that was further increased after E2 treatment. In contrast to MCF-7 cells, the E2-induced activation of cPLA2 in SKBR3 cells was entirely dependent on EGFR trans-activation signaling to ERK1/2. SKBR3 cells are ER negative but do express GPR30, which binds to E2 and activates MAPK through MMP-mediated EGFR trans-activation (42). The selective GPR30 agonist G1 and ICI both mimicked the effect of E2 and rapidly stimulated the phosphorylation of cPLA2α. Inhibition of EGFR trans-activation by CRM197 and AG825 blocked the E2-induced activation of ERK and cPLA2 at all time points analyzed.
If constitutive overexpression of HER2 were the driver for the increased expression and activation of cPLA2α, then inhibition or down-regulation of HER2 would also suppress cPLA2α. Treatment of SKBR3 cells with the anti-HER2 monoclonal antibody herceptin in combination with E2 treatment down-regulated the phosphorylation of both HER2 and cPLA2α, without affecting HER2 or cPLA2α protein expression levels. Treatment with either herceptin or E2 alone did not elicit any change in the phosphorylation state of either HER2 or cPLA2α, suggesting a synergism between herceptin action and the presence of estrogen. When HER2 protein expression was down-regulated using siRNA, cPLA2α protein basal expression and phosphorylation were also diminished. This supports the hypothesis that HER2 overexpression drives constitutive cPLA2α expression and activation in ER-negative breast carcinoma cells. cPLA2α controls cell proliferation in both normal and malignant thyroid epithelial cells (63, 64), and other reports indicate that cPLA2α can mediate proliferation in human umbilical vein endothelial cells (65) and also in prostate cancer cells (5). The general PLA2 inhibitor quinacrine reduced both basal and E2-induced cell growth in MCF-7 cells (66), whereas this present study demonstrates that more specific pharmacological inhibition of cPLA2 reduced E2-induced cell proliferation of both ER-positive (MCF-7) and ER-negative (SKBR3) breast cancer cells. cPLA2 antagonism in the absence of E2 inhibited MCF-7 but not SKBR3 cell growth. This may reflect the lower basal levels of cPLA2α activity in the HER2-negative MCF-7 cells, which makes them more sensitive to antagonism of both the homeostatic function of cPLA2 as well as its contribution to E2-induced cell proliferation. For both cell lines, the reduced cell growth was at least in part due to an increase in cell death, because the cPLA2 inhibitor induced both apoptosis and necrosis. The increase in apoptosis could explain why the levels of cell growth in MCF-7 cells fell below the basal level of control when cells are treated with the inhibitor, either alone or in combination with E2.
This study demonstrates a novel role for the rapid, E2-induced trans-activation of EGFR/HER2 heterodimers in promoting ERK1/2-induced phosphorylation and activation of cPLA2α in breast cancer cells that differentially express ER and EGFR/HER2 receptors. HER2 overexpression is a well characterized prognostic marker for invasive breast cancer that is associated with loss of ER expression and resistance to antiestrogen therapy. Our data suggest that HER2 overexpression drives increased cPLA2α expression and constitutive activation, although loss or inhibition of HER2 can reduce the expression and activation of cPLA2α. In breast cancer cells lacking ER, E2 can increase basal activation of cPLA2α by trans-activating EGFR/HER2, possibly via GPR30. As a consequence, cPLA2α may contribute to proliferative E2 signaling in tumors that are ER negative and resistant to endocrine therapy. E2 exerts a proliferative effect in breast cancer cells through ER and via EGFR/HER2 in ER-negative tumors. Lipid mediators produced through cPLA2α activation could play an important role in mediating proliferation of both endocrine-sensitive and endocrine-resistant breast cancer cells. Our data show that pharmacological inhibition of cPLA2α reduced cell growth in vitro through increases in apoptotic and necrotic cell death in both ER-positive and ER-negative cells. Therapeutic strategies to target the eicosanoid signaling pathway have focused mainly on COX-2 inhibition, which results in adverse side effects on the cardiovascular system. This study identifies cPLA2α as a potential, alternative target for therapeutic intervention in breast cancer.
Materials and Methods
Cell culture
MCF-7, UACC 893, and HCC38 breast carcinoma cells (American Type Culture Collection, Teddington, UK) were routinely grown in Eagle’s MEM, Leibovitz L-15, or RPMI 1640 (Sigma-Aldrich, Tallaght, Ireland) culture medium, respectively, supplemented with 2 mm l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin and 10% fetal bovine serum (FBS) (GIBCO, Paisley, UK). SKBR3 (American Type Culture Collection), MDA-MB-231, and BT474 breast carcinoma cell lines (Dr. R. J. Santen, University of Virginia School of Medicine, Charlottesville, VA) were maintained in DMEM/F12 (Sigma-Aldrich) supplemented with nonessential amino acids, 2 mm l-glutamine, 100 μg/ml gentamicin, and 10% FBS. All cell lines were incubated in a humidified atmosphere of 5% CO2 at 37 C with the exception of the UACC 893 cell line, which was maintained at atmospheric CO2 concentration. For the purpose of experiments, cells were seeded in six-well plates or 10-cm-diameter dishes at 80% confluency and then serum starved for 48 h before treatment at 100% confluency as indicated.
Reagents and antibodies
E2 was purchased from Sigma-Aldrich and dissolved in ethanol before being diluted in cell culture medium to a final concentration of 10 nm. The GPR30-specific agonist G1, the MEK inhibitor PD98059, the c-Src inhibitor PP2, the matrix metalloproteinase inhibitor GM6001, and the specific cPLA2α inhibitor N-c-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl] acrylamide, HCl (67) were obtained from Calbiochem (Nottingham, UK) and dissolved in dimethylsulfoxide (DMSO) (the cPLA2α inhibitor was dissolved in 75% acetic acid). The HER2/EGFR inhibitor AG825 and the ER inhibitors ICI and tamoxifen were purchased from Tocris (Avonmouth, UK) and dissolved in DMSO or methanol, respectively. The D2189 [Glu52] diphtheria toxin CRM197 was obtained from Sigma-Aldrich and diluted in distilled water to 1 mg/ml. Herceptin (Roche, Clairecastle, Ireland) was diluted in PBS to 10 μg/ml. The bicinchoninic acid protein assay was purchased from Pierce (Northumberland, UK). The Rainbow molecular weight marker, the ECL chemiluminescence reagents, and hyperfilm were from Amersham Bioscience (Little Chalfont, UK). The MTT cell growth assay was from Promega (Southampton, UK). The apoptosis assay was from Roche Applied Science (Burgess Hill, UK). The anti-ERα, anti-cPLA2, anti-HER2, anti-phospho-HER2 (Tyr1221/1222), anti-EGFR, anti-phospho-EGFR (Tyr845), anti-p44/42 MAPK, anti-phospho-p44/42 MAPK (Thr202/Tyr204), the anti-c-Fos, the antimouse IgG horseradish peroxidase conjugate antibodies, and the phospho-HER2 (Tyr1221/1222) ELISA kit were from Cell Signaling Technology (Hitchin, UK). The anti-phospho-cPLA2 antibody was from Santa Cruz Biotechnology (Heidelberg, Germany). The antirabbit IgG horseradish peroxidase conjugate and the anti-β-actin antibodies were from Sigma-Aldrich. All other chemical reagents used were purchased from Sigma-Aldrich, unless otherwise specified.
Immunoprecipitation and Western blotting
Cells were treated with 10 nm E2 or vehicle control for the indicated times. Preincubation with the indicated inhibitors was performed as described. Cells were then transferred onto ice, washed twice with ice-cold PBS, and then ultrasonicated in lysis buffer [20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, complete mini EDTA-free protease inhibitor mixture tablets (one tablet per 10 ml lysis buffer; Roche) and phosphatase inhibitors]. Samples were clarified by centrifugation at 13,000 rpm for 15 min, and supernatants were collected and stored at −80 C for subsequent analysis. Total protein concentration was quantified using the bicinchoninic acid assay (68). For immunoprecipitation, equal amounts of soluble cell extracts were incubated with 2 μg of either anti-EGFR or anti-HER2 antibody for 16 h at 4 C with rotation. Washed EZ-view Red Protein A Beads (Sigma-Aldrich) were combined with samples and incubated for 1 h at 4 C on a rotor. Complexes were centrifuged at 13,000 rpm for 3 min, the supernatants were removed, and pelleted beads were washed five times in lysis buffer. Finally, samples were resuspended in 20 μl 2× Laemmli sample buffer (Sigma-Aldrich) and boiled for 5 min at 95 C. For nonimmunoprecipitated samples, total cell extracts were combined with equal amounts of 2× Laemmli sample buffer and heated for 5 min at 95 C.
Solubilized proteins (40 μg) were resolved by SDS-PAGE on 6% (EGFR, HER2, cPLA2, and COX-2) or 10% (ERK1/2) gels (100 V, 90 min). Proteins were then transferred to nitrocellulose membranes (15 V, 45 min to 2 h) with a Trans-Blot SD system (Bio-Rad, Hemel Hempstead, UK). Membranes were blocked in TBS with 0.1% Tween 20 and 5% nonfat dry milk for 1 h at 25 C, incubated with the indicated primary antibody for 16 h at 4 C, and probed with the appropriate secondary antibody for 1 h at 25 C. Membranes were washed three times in TBS with 0.1% Tween 20 at 25 C, and antibody reaction was visualized by enhanced chemiluminescence on an autoradiographic film. Membranes were stripped with Restore Western blot stripping buffer (Pierce Chemical Co., Rockford, IL) for 10 min at 25 C and reprobed with the anti-β-actin or total target protein antibody as indicated to normalize densitometry data for gel loading. Tubulin was used in normalization of the cell line comparison (Fig. 6B) due to large variation in β-actin expression between the cell lines.
RNA isolation and RT-PCR analysis
Total RNA was extracted from the cell lines indicated using the RNeasy mini kit (QIAGEN, Crawley, UK) according to the manufacturer’s instructions. RNA was finally eluted in diethylpyrocarbonate-treated water (30 μl) and stored at −80 C. The quantity and quality of the extracted RNA was confirmed by absorption measurements at 260 and 280 nm. Single-strand cDNA was synthesized using the ImProm II reverse transcriptase kit (Promega). cDNA was quantified and corrected for loading into RT-PCR mixes. GoTaq polymerase mix (Promega) was used in the PCR amplification. Touchdown PCR was used to amplify cDNA for the indicated number of cycles and annealing temperature range for each primer set used. The RT-PCR product was analyzed on a 1% Tris acetate-EDTA agarose gel and visualized using a UV light source. The abundance of target mRNA detected was normalized in comparison with the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) internal control. The sequences for gene-specific forward and reverse primers were designed using the OligoPerfect Designer software program (Invitrogen), unless a different source is specified. Sequences were as follows: for ErbB2/HER2 (GeneID 2064), 5′-CCATAACACCCACCTCTGCT-3′ (forward) and 5′-ACTGGCTGCAGTTGACACAC-3′ (reverse), 20 cycles at 58–68 C; for EGFR (GeneID 1956), 5′-ATGTCCGGGAACACAAAGAC-3′ (forward) and 5′-TTCCGTCATATGGCTTGGAT-3′ (reverse), 40 cycles at 56 C (69); for cPLA2α (PLA2G4A, GeneID 5321), 5′-ACGTTTCAGAGCTGATGTTT-3′ (forward) and 5′-CTTCCAGCATCTTCATTTTC-3′ (reverse), 30 cycles at 52–62 C; for COX-2 (PTGS2, GeneID 5743), 5′-TGAAACCCACTCCAAACACA-3′ (forward) and 5′-GAGAAGGCTTCCCAGCTTTT-3′ (reverse), 40 cycles at 58–63 C; and for GAPDH (GeneID 2597), 5′-GTCATCATCTCTGCCCCCTCTGC-3′ (forward) and 5′-CGACGGCTGCTTCACCACCTTCT-3′ (reverse), 14 cycles at 52 C (Table 1).
Table 1.
Primer sequences
| Gene name | Primer sequences (5′–3′) | Product size (bp) | Number of cycles | Annealing temperature (°C) |
|---|---|---|---|---|
| ErbB2-HER2 | ||||
| Forward | CCATAACACCCACCTCTGCT | 194 | 20 | 58–68 |
| Reverse | ACTGGCTGCAGTTGACACAC | |||
| EGFR (80) | ||||
| Forward | ATGTCCGGGAACACAAAGAC | 351 | 40 | 56 |
| Reverse | TTCCGTCATATGGCTTGGAT | |||
| cPLA2 | ||||
| Forward | ACGTTTCAGAGCTGATGTTT | 352 | 30 | 52–62 |
| Reverse | CTTCCAGCATCTTCATTTTC | |||
| COX-2 | ||||
| Forward | TGAAACCCACTCCAAACACA | 187 | 40 | 58–63 |
| Reverse | GAGAAGGCTTCCCAGCTTTT | |||
| GAPDH | ||||
| Forward | GTCATCATCTCTGCCCCCTCTGC | 444 | 14 | 52 |
| Reverse | CGACGGCTGCTTCACCACCTTCT |
For quantitative real-time PCR, 2 μl cDNA was loaded in a 96-well plate with SYBR Green I Master mix (Roche), and amplification was carried out in a LightCycler 480 (Roche) as follows: a preincubation step at 95 C for 10 min was followed by 45 cycles of denaturation at 95 C for 10 sec, annealing at 60 C for 10 sec, and elongation at 72 C for 10 sec. Efficiencies for each primer set were calculated from the PCR kinetic curve using the linear regression method with LinRegPCR software (70) and used to measure relative quantification of gene expression with the comparative cycle threshold method. All samples were normalized for 18S rRNA expression levels. The sequences for 18S primers were 5′-GTCCCCCAACTTCTTAGAG-3′ (forward) and 5′-CACCTACGGAAACCTTGTTAC-3′ (reverse).
Cell growth and apoptosis assays
Reduction of MTT by mitochondrial respiration was used to measure cell growth. Cells were harvested, counted in a Neubauer chamber, and seeded in a 96-well plate at 105 cells per well in medium containing 2% charcoal-stripped FBS. After 16 h, cells were stimulated with 10 nm E2 or vehicle control, with or without the indicated inhibitors. Stimulation was repeated after 48 h and carried out for a total 96 h before performing the growth assay, in which cells were incubated with 1 mg/ml MTT for 4 h at 37 C in a humidified atmosphere containing 5% CO2. The reaction was stopped by the addition of a DMSO solution and solubilization of formazan crystals was allowed for 2 h at 37 C. Absorbance was measured at 570 nm using a Multiskan EX plate reader (Thermo Scientific, Northumberland, UK). A photometric enzyme immunoassay (Roche) was used for the quantitative determination of cytoplasmic histone-associated DNA fragments to measure cell death. Cells were seeded on a 96-well plate at 105 cells per well in medium containing vehicle or the indicated inhibitors and incubated for 48 h. Medium was collected and cells lysed for 30 min at 25 C; then both cell lysates and medium supernatants were used for the ELISA following the manufacturer’s instructions, and absorbance was measured at 405 nm.
cPLA2 enzymatic activity
MCF-7 and SKBR3 cells were lysed as previously described, and lysates were incubated with 5 μm bromoenol lactone and 200 μm thioetheramide-phosphatidylcholine (Cayman Europe, Tallinn, Estonia) for 15 min at 25 C to inhibit either Ca2+-independent intracellular PLA2 or Ca2+-dependent secretory PLA2, respectively. Samples were then incubated with arachidonoyl thio-phosphatidylcholine using a cPLA2 assay kit (Cayman) according to the manufacturer’s directions. Briefly, 60 min after incubation, samples were mixed with a solution of 5,5′-dithio-bis2-nitrobenzoic acid/EGTA to detect free thiols released by hydrolysis of arachidonoyl thioester bonds by cPLA2. Absorbances were measured at 405 nm using a Multiskan EX iplate reader (Thermo Scientific). Enzymatic activity was calculated using the 5,5′-dithio-bis2-nitrobenzoic acid extinction coefficient of 10 mm−1.
RNA silencing
A pool of four different siRNA specific for ErbB2 (NCBI gene ID 2064) was purchased from Dharmacon (Lafayette, CO). Sequences were as follow: siRNA 1, GGACGAAUUCUGCACAAUG; siRNA 2, GACGAAUUCUGCACAAUGG; siRNA 3, CUACAACACAGACACGUUU; and siRNA 4, AGACGAAGCAUACGUGAUG. A nontargeting siRNA with at least four mismatches with all known human genes (Dharmacon D-001210-01) was used as negative control. All siRNAs were resuspended to a 20 μm concentration in a buffer containing 60 mm KCl, 6 mm HEPES (pH 7.5), and 0.2 mm MgCl2. SKBR3 cells were transfected with 100 nm siRNA using DharmaFECT (Dharmacon) and silencing of HER2 expression was assessed by Western blotting over a time course of 24–96 h. Maximal silencing (∼65%) was obtained 72 h after transfection; cells were then lysed, and Western blotting for HER2 and cPLA2 was performed as previously described.
Statistical analysis
Densitometric analysis of polyacrylamide and agarose gels was performed using GeneTools software (Syngene, Cambridge, UK). Statistical analysis of the data was performed using paired Student’s t test for analysis between two groups. One-way ANOVA was used for multiple analyses of more than two groups. P values < 0.05 were considered statistically significant. Data are expressed as mean ± se of the indicated number of experiments (at least three different experiments performed in duplicate).
Acknowledgments
Herceptin was kindly provided by Dr. P. Jacob, Beaumont Hospital pharmacy (Dublin, Ireland). The BT474 and MDA-MB-231 cells were a kind gift from Dr. R. J. Santen (University of Virginia School of Medicine, Charlottesville, VA).
NURSA Molecule Pages:
Ligands: 17β-estradiol | Fulvestrant.
Footnotes
This work was supported by the Higher Education Authority of Ireland (PRTLI Cycle 4) through the National Bio-Photonics and Imaging Platform, and by a Ph.D. scholarship to F.C. from The Royal College of Surgeons in Ireland.
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 8, 2010
Abbreviations: AA, Arachidonic acid; COX, cyclooxygenase; PLA2, cytosolic PLA2; DMSO, dimethylsulfoxide; E2, 17β-estradiol; EGFR, epidermal growth factor receptor; ER, estrogen receptor; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GPR, G protein-coupled receptor; HB, heparin-bound; ICI, ICI 182,780; MMP, matrix metalloproteinase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PGE2, prostaglandin E2; PLA2, phospholipase A2; PP2, 4-amino-5-(4chorophenyl)-7-(t-butyl)pyrozolo[3,4-D]pyrimidine; SERM, selective ER modulator; siRNA, small interfering RNA.
References
- 1.Park JY, Pillinger MH, Abramson SB2006. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol 119:229–240 [DOI] [PubMed] [Google Scholar]
- 2.Leslie CC1997. Properties and regulation of cytosolic phospholipase A2. J Biol Chem 272:16709–16712 [DOI] [PubMed] [Google Scholar]
- 3.Heasley LE, Thaler S, Nicks M, Price B, Skorecki K, Nemenoff RA1997. Induction of cytosolic phospholipase A2 by oncogenic Ras in human non-small cell lung cancer. J Biol Chem 272:14501–14504 [DOI] [PubMed] [Google Scholar]
- 4.Panel V, Boëlle PY, Ayala-Sanmartin J, Jouniaux AM, Hamelin R, Masliah J, Trugnan G, Fléjou JF, Wendum D2006. Cytoplasmic phospholipase A2 expression in human colon adenocarcinoma is correlated with cyclooxygenase-2 expression and contributes to prostaglandin E2 production. Cancer Lett 243:255–263 [DOI] [PubMed] [Google Scholar]
- 5.Patel MI, Singh J, Niknami M, Kurek C, Yao M, Lu S, Maclean F, King NJ, Gelb MH, Scott KF, Russell PJ, Boulas J, Dong Q2008. Cytosolic phospholipase A2-α: a potential therapeutic target for prostate cancer. Clin Cancer Res 14:8070–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakanishi M, Rosenberg DW2006. Roles of cPLA2α and arachidonic acid in cancer. Biochim Biophys Acta 1761:1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H, Isola J2002. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res 62:632–635 [PubMed] [Google Scholar]
- 8.Basu GD, Pathangey LB, Tinder TL, Lagioia M, Gendler SJ, Mukherjee P2004. Cyclooxygenase-2 inhibitor induces apoptosis in breast cancer cells in an in vivo model of spontaneous metastatic breast cancer. Mol Cancer Res 2:632–642 [PubMed] [Google Scholar]
- 9.Harris RE, Beebe-Donk J, Alshafie GA2007. Cancer chemoprevention by cyclooxygenase 2 (COX-2) blockade: results of case control studies. Subcell Biochem 42:193–212 [DOI] [PubMed] [Google Scholar]
- 10.Khuder SA, Mutgi AB2001. Breast cancer and NSAID use: a meta-analysis. Br J Cancer 84:1188–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas W, Caiazza F, Harvey BJ2008. Estrogen, phospholipase A and breast cancer. Front Biosci 13:2604–2613 [DOI] [PubMed] [Google Scholar]
- 12.Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, Ali-Seyed M, Lee DF, Bartholomeusz G, Ou-Yang F, Giri DK, Hung MC2004. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 6:251–261 [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y, Agarwal VR, Mendelson CR, Simpson ER1996. Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology 137:5739–5742 [DOI] [PubMed] [Google Scholar]
- 14.Henderson BE, Feigelson HS2000. Hormonal carcinogenesis. Carcinogenesis 21:427–433 [DOI] [PubMed] [Google Scholar]
- 15.Russo J, Russo IH2006. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol 102:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ascenzi P, Bocedi A, Marino M2006. Structure-function relationship of estrogen receptor α and β: impact on human health. Mol Aspects Med 27:299–402 [DOI] [PubMed] [Google Scholar]
- 17.Jordan VC, Brodie AM2007. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids 72:7–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howell A2006. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr Relat Cancer 13:689–706 [DOI] [PubMed] [Google Scholar]
- 19.Katzenellenbogen BS, Katzenellenbogen JA2000. Estrogen receptor transcription and transactivation: estrogen receptor α and estrogen receptor β: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res 2:335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F1996. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 15:1292–1300 [PMC free article] [PubMed] [Google Scholar]
- 21.Filardo EJ, Thomas P2005. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab 16:362–367 [DOI] [PubMed] [Google Scholar]
- 22.Razandi M, Pedram A, Greene GL, Levin ER1999. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol 13:307–319 [DOI] [PubMed] [Google Scholar]
- 23.Razandi M, Pedram A, Park ST, Levin ER2003. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem 278:2701–2712 [DOI] [PubMed] [Google Scholar]
- 24.Levin ER2003. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol 17:309–317 [DOI] [PubMed] [Google Scholar]
- 25.Pietras RJ2003. Interactions between estrogen and growth factor receptors in human breast cancers and the tumor-associated vasculature. Breast J 9:361–373 [DOI] [PubMed] [Google Scholar]
- 26.Howe LR2007. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res 9:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakatsugi S, Ohta T, Kawamori T, Mutoh M, Tanigawa T, Watanabe K, Sugie S, Sugimura T, Wakabayashi K2000. Chemoprevention by nimesulide, a selective cyclooxygenase-2 inhibitor, of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mammary gland carcinogenesis in rats. Jpn J Cancer Res 91:886–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris RE, Alshafie GA, Abou-Issa H, Seibert K2000. Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Res 60:2101–2103 [PubMed] [Google Scholar]
- 29.Lanza-Jacoby S, Miller S, Flynn J, Gallatig K, Daskalakis C, Masferrer JL, Zweifel BS, Sembhi H, Russo IH2003. The cyclooxygenase-2 inhibitor, celecoxib, prevents the development of mammary tumors in Her-2/neu mice. Cancer Epidemiol Biomarkers Prev 12:1486–1491 [PubMed] [Google Scholar]
- 30.Howe LR, Chang SH, Tolle KC, Dillon R, Young LJ, Cardiff RD, Newman RA, Yang P, Thaler HT, Muller WJ, Hudis C, Brown AM, Hla T, Subbaramaiah K, Dannenberg AJ2005. HER2/neu-induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase-2 knockout mice. Cancer Res 65:10113–10119 [DOI] [PubMed] [Google Scholar]
- 31.Chang SH, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, Lane TF, Hla T2004. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA 101:591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T2001. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem 276:18563–18569 [DOI] [PubMed] [Google Scholar]
- 33.Rolland PH, Martin PM, Jacquemier J, Rolland AM, Toga M1980. Prostaglandin in human breast cancer: evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J Natl Cancer Inst 64:1061–1070 [PubMed] [Google Scholar]
- 34.Bandyopadhyay GK, Imagawa W, Wallace D, Nandi S1987. Linoleate metabolites enhance the in vitro proliferative response of mouse mammary epithelial cells to epidermal growth factor. J Biol Chem 262:2750–2756 [PubMed] [Google Scholar]
- 35.Ma X, Kundu N, Rifat S, Walser T, Fulton AM2006. Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res 66:2923–2927 [DOI] [PubMed] [Google Scholar]
- 36.Mauritz I, Westermayer S, Marian B, Erlach N, Grusch M, Holzmann K2006. Prostaglandin E2 stimulates progression-related gene expression in early colorectal adenoma cells. Br J Cancer 94:1718–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harvey BJ, Alzamora R, Healy V, Renard C, Doolan CM2002. Rapid responses to steroid hormones: from frog skin to human colon. A homage to Hans Ussing. Biochim Biophys Acta 1566:116–128 [DOI] [PubMed] [Google Scholar]
- 38.Fiorini S, Ferretti ME, Biondi C, Pavan B, Lunghi L, Paganetto G, Abelli L2003. 17β-Estradiol stimulates arachidonate release from human amnion-like WISH cells through a rapid mechanism involving a membrane receptor. Endocrinology 144:3359–3367 [DOI] [PubMed] [Google Scholar]
- 39.Thomas W, Coen N, Faherty S, Flatharta CO, Harvey BJ2006. Estrogen induces phospholipase A2 activation through ERK1/2 to mobilize intracellular calcium in MCF-7 cells. Steroids 71:256–265 [DOI] [PubMed] [Google Scholar]
- 40.Marino M, Caiazza F2007. Estrogen signal transduction pathways from plasma membrane to the nucleus. In: Grachevsky NO, ed. Signal transduction research trends. New York: Nova Science Publishers; 17–44
- 41.Song RX, Zhang Z, Chen Y, Bao Y, Santen RJ2007. Estrogen signaling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells. Endocrinology 148:4091–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Filardo EJ, Quinn JA, Bland KI, Frackelton Jr AR2000. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649–1660 [DOI] [PubMed] [Google Scholar]
- 43.Mitamura T, Higashiyama S, Taniguchi N, Klagsbrun M, Mekada E1995. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J Biol Chem 270:1015–1019 [DOI] [PubMed] [Google Scholar]
- 44.Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, Fan P, Santen RJ 6 October 2009. Estrogen utilization of IGF-1-R and EGF-R to signal in breast cancer cells. J Steroid Biochem Mol Biol 10.1016/j.jsbmb.2009.09.018 [DOI] [PMC free article] [PubMed]
- 45.Landgraf R2007. HER2 therapy. HER2 (ERBB2): functional diversity from structurally conserved building blocks. Breast Cancer Res 9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moasser MM2007. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26:6469–6487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD, Pollack JR2009. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One 4:e6146 [DOI] [PMC free article] [PubMed]
- 48.Nahta R, Esteva FJ2006. Herceptin: mechanisms of action and resistance. Cancer Lett 232:123–138 [DOI] [PubMed] [Google Scholar]
- 49.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, Miele L2008. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a γ-secretase inhibitor. Oncogene 27:5019–5032 [DOI] [PubMed] [Google Scholar]
- 50.Flouriot G, Brand H, Denger S, Metivier R, Kos M, Reid G, Sonntag-Buck V, Gannon F2000. Identification of a new isoform of the human estrogen receptor-α (hER-α) that is encoded by distinct transcripts and that is able to repress hER-α activation function 1. EMBO J 19:4688–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF2005. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α66. Biochem Biophys Res Commun 336:1023–1027 [DOI] [PubMed] [Google Scholar]
- 52.Li L, Haynes MP, Bender JR2003. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA 100:4807–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penot G, Le Péron C, Mérot Y, Grimaud-Fanouillère E, Ferrière F, Boujrad N, Kah O, Saligaut C, Ducouret B, Métivier R, Flouriot G2005. The human estrogen receptor-α isoform hERα46 antagonizes the proliferative influence of hERα66 in MCF7 breast cancer cells. Endocrinology 146:5474–5484 [DOI] [PubMed] [Google Scholar]
- 54.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF2006. A variant of estrogen receptor-α, hER-α36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA 103:9063–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng A, Kallio A, Härkönen P2007. Tamoxifen-induced rapid death of MCF-7 breast cancer cells is mediated via extracellularly signal-regulated kinase signaling and can be abrogated by estrogen. Endocrinology 148:2764–2777 [DOI] [PubMed] [Google Scholar]
- 56.Cuendet M, Pezzuto JM2000. The role of cyclooxygenase and lipoxygenase in cancer chemoprevention. Drug Metabol Drug Interact 17:109–157 [DOI] [PubMed] [Google Scholar]
- 57.Aoki J2004. Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol 15:477–489 [DOI] [PubMed] [Google Scholar]
- 58.Abir F, Alva S, Kaminski DL, Longo WE2005. The role of arachidonic acid regulatory enzymes in colorectal disease. Dis Colon Rectum 48:1471–1483 [DOI] [PubMed] [Google Scholar]
- 59.Hassan S, Carraway RE2006. Involvement of arachidonic acid metabolism and EGF receptor in neurotensin-induced prostate cancer PC3 cell growth. Regul Pept 133:105–114 [DOI] [PubMed] [Google Scholar]
- 60.Lanza-Jacoby S, Burd R, Rosato Jr FE, McGuire K, Little J, Nougbilly N, Miller S2006. Effect of simultaneous inhibition of epidermal growth factor receptor and cyclooxygenase-2 in HER-2/neu-positive breast cancer. Clin Cancer Res 12:6161–6169 [DOI] [PubMed] [Google Scholar]
- 61.Levin ER, Pietras RJ2008. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat 108:351–361 [DOI] [PubMed] [Google Scholar]
- 62.Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ1995. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene 10:2435–2446 [PubMed] [Google Scholar]
- 63.Mariggiò S, Filippi BM, Iurisci C, Dragani LK, De Falco V, Santoro M, Corda D2007. Cytosolic phospholipase A2α regulates cell growth in RET/PTC-transformed thyroid cells. Cancer Res 67: 11769–11778 [DOI] [PubMed]
- 64.Mariggiò S, Sebastià J, Filippi BM, Iurisci C, Volonté C, Amadio S, De Falco V, Santoro M, Corda D2006. A novel pathway of cell growth regulation mediated by a PLA2alpha-derived phosphoinositide metabolite. FASEB J 20:2567–2569 [DOI] [PubMed] [Google Scholar]
- 65.Herbert SP, Ponnambalam S, Walker JH2005. Cytosolic phospholipase A2-α mediates endothelial cell proliferation and is inactivated by association with the Golgi apparatus. Mol Biol Cell 16:3800–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Markaverich BM, Crowley J, Rodriquez M, Shoulars K, Thompson T2007. Tetrahydrofurandiol stimulation of phospholipase A2, lipoxygenase, and cyclooxygenase gene expression and MCF-7 human breast cancer cell proliferation. Environ Health Perspect 115:1727–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M2000. Pyrrolidine inhibitors of human cytosolic phospholipase A2 J Med Chem 43:1041–1044 [DOI] [PubMed] [Google Scholar]
- 68.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC1985. Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85 [DOI] [PubMed] [Google Scholar]
- 69.Peghini PL, Iwamoto M, Raffeld M, Chen YJ, Goebel SU, Serrano J, Jensen RT2002. Overexpression of epidermal growth factor and hepatocyte growth factor receptors in a proportion of gastrinomas correlates with aggressive growth and lower curability. Clin Cancer Res 8:2273–2285 [PubMed] [Google Scholar]
- 70.Ramakers C, Ruijter JM, Deprez RH, Moorman AF2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66 [DOI] [PubMed] [Google Scholar]
- 71.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, Andò S2004. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-estradiol and phytoestrogens in breast cancer cells. J Biol Chem 279: 27008–27016 [DOI] [PubMed]