

Abstract
Cytosolic phospholipase A (cPLA2α) catalyzes the formation of arachidonic acid in prostaglandin synthesis. In contrast to the well-described down-regulation of cPLA2α, up-regulation of cPLA2α by glucocorticoids has been reported in human amnion fibroblasts, which may play a key role in parturition. The mechanisms underlying this paradoxical induction of cPLA2α by glucocorticoids remain largely unknown. Using cultured human amnion fibroblasts, we found that the induction of cPLA2α by cortisol required ongoing transcription and synthesis of at least one other protein. The induction of cPLA2α by cortisol was abolished by mutagenesis of a glucocorticoid response element (GRE) in the promoter. The same GRE was found mediating the classical inhibition of cPLA2α expression by cortisol in human fetal lung fibroblasts (HFL-1). Cortisol increased Gαs expression in amnion fibroblasts but not in HFL-1 cells. Inhibition of Gαs with NF449 attenuated the phosphorylation of cAMP response element-binding protein-1 (CREB-1) and the induction of cPLA2α by cortisol in amnion fibroblasts. Both glucocorticoid receptor (GR) and CREB-1 were found bound to the GRE upon cortisol stimulation of amnion fibroblasts. The induction of cPLA2α by cortisol was blocked by GR antagonist RU486 or protein kinase A inhibitor H89 or dominant-negative CREB-1. In conclusion, cortisol activates the cAMP/protein kinase A/CREB-1 pathway via Gαs induction, and the phosphorylated CREB-1 interacts with GR at the GRE to promote cPLA2α expression in amnion fibroblasts.
Glucocorticoids activate cAMP/PKA/CREB pathway via induction of Gαs, and the phosphorylated CREB interacts with GR at target glucocorticoid response elements to increase cPLA2α expression in human amnion.
Preterm delivery (<37 completed weeks of gestation) occurs in approximately 10% of all pregnancies but accounts for more than 75% of the perinatal mortality and morbidity rate (1, 2). Despite progress in this field, the lack of identification of the mechanisms of human parturition has limited the specific and effective diagnosis and treatment of preterm labor and subsequent birth. Activation of prostaglandin (PG) synthesis, especially of PGE2 and PGF2α, in the intrauterine tissues toward term, has been identified as one of the final events leading to parturition (3, 4). PGs induce cervical ripening, fetal membrane rupture, and myometrial contraction at term (3, 4). Among the intrauterine tissues, the amnion, amnion fibroblasts in particular, produces most of the PGs toward term and during labor (5, 6, 7). In most mammalian species including humans, there is an increase in glucocorticoid concentration in the maternal and fetal circulations as well as in the amniotic fluid toward the end of gestation and at the onset of labor (8, 9). This surge of glucocorticoid is believed to be crucial to the maturation of fetal organs as well as to be integral to the cascade of events in the initiation and maintenance of labor (8). This concurrent increase in glucocorticoids and PGs toward the end of gestation displays a paradoxical phenomenon contradicting the classical relationship between these two classes of hormones involved in inflammation. The cascade of reactions for the synthesis of PGs involves a number of enzymes. The initial reaction catalyzed by cytosolic phospholipase A2α (cPLA2α) provides the rate-limiting substrate, arachidonic acid (10), for conversion to PG endoperoxide by the inducible cyclooxygenase-2 (COX-2). As a well-described antiinflammatory hormone, glucocorticoid exerts potent inhibition on PG synthesis by inhibiting the expression of cPLA2α and COX-2 at most sites in the body (11, 12, 13). However, we and others have shown that glucocorticoids can potently stimulate PG output by inducing the expression of cPLA2α and COX-2 in amnion fibroblasts (6, 7, 14, 15). The mechanisms underlying the paradoxical induction of cPLA2α and COX-2 by glucocorticoids remain, however, largely unknown. Recently, we found that glucocorticoids stimulated COX-2 expression via activation of the cAMP/ protein kinase A (PKA)/cAMP response element (CRE)-binding protein-1 (CREB-1) pathway by inducing an unknown protein in human amnion fibroblasts (16).
Consistent with the capability of PG synthesis, cPLA2α is expressed in greater abundance in the fetal membranes among the intrauterine tissues (17). In the amnion, cPLA2α activity has been shown to increase with gestation age, being highest at term before labor but then depleted during labor (18). Moreover, cPLA2α knockout female mice fail to labor at term (19). All these findings point to the crucial role of cPLA2α in parturition. Thus, understanding the molecular mechanisms underlying the paradoxical induction of cPLA2α by cortisol may help us better understand the events leading to parturition. In this study, we investigated the cis element, trans-acting factor mediating the induction of cPLA2α expression by cortisol and identified the protein induced by cortisol that is involved in the induction of both cPLA2α and COX-2 expression.
Results
Glucocorticoid receptor (GR)-mediated induction of cPLA2α expression by cortisol via induction of mRNA transcription and de novo protein synthesis in human amnion fibroblasts
Analysis with quantitative real-time PCR (qRT-PCR) and Western blotting showed that treatment of cultured human amnion fibroblasts with cortisol (0.01–1 μm, 24 h) caused a concentration-dependent increase in cPLA2α mRNA and protein levels, which was blocked by GR antagonist RU486 (1 μm) (Fig. 1, A and B). Treatment of the cells with mRNA transcription inhibitor 5,6- dichlorobenzimidazole riboside (DRB, 75 μm) or with protein synthesis inhibitor cycloheximide (CHX, 10 μm) attenuated significantly the induction of cPLA2α mRNA expression by cortisol (1 μm, 24 h) (Fig. 1, C and D). These results suggest that the induction of cPLA2α mRNA transcription is a GR-mediated process via the induction of the synthesis of at least one other protein. To verify the paradoxical induction of cPLA2α expression by cortisol specific to human amnion fibroblasts, we also carried out a control study in human fetal lung fibroblasts (HFL-1), an immortalized cell line. In contrast to the amnion fibroblasts, cortisol (1 μm) treatment of the HFL-1 cells caused a significant reduction in cPLA2α mRNA and protein levels (Fig. 2, A and B).
Fig. 1.

A and B, Concentration-dependent stimulation of cPLA2α mRNA and protein expression by cortisol, which could be blocked by GR antagonist RU486 in human amnion fibroblasts. The top panel of B is a representative immunoblot; bottom panel shows the mean data. C and D, The stimulation of cPLA2α mRNA expression by cortisol (1 μm) was blocked by mRNA transcription inhibitor DRB (75 μm) and protein synthesis inhibitor CHX (10 μm) in human amnion fibroblasts. n = 3–7. *, P < 0.05; **, P < 0.01 vs. vehicle control; #, P < 0.05 vs. cortisol without RU486.
Fig. 2.
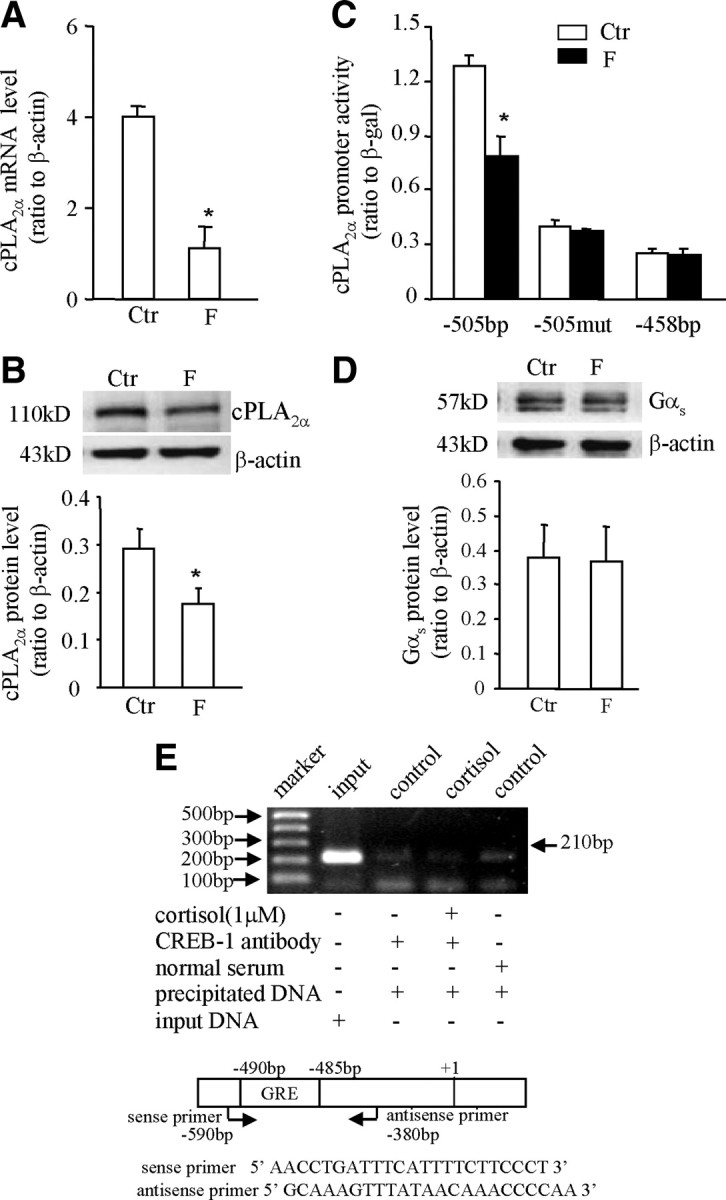
A and B, Inhibition of cPLA2α mRNA and protein expression by cortisol (F, 1 μm) in HFL-1 cells. The top panel of B is the representative immunoblot; bottom panel shows the mean data. C, Inhibition of cPLA2α promoter (−505 bp) activity by cortisol (F, 1 μm) could be diminished either by introduction of triple-nucleotide mutations (−505mut) into the putative GRE at −485 to −490 bp or by complete removal of the GRE as −458 bp in HFL-1 cells. D, Cortisol (F, 1 μm) treatment did not affect the expression of Gαs protein in HFL-1 cells. The top panel of D is the representative immunoblot; bottom panel shows the mean data. n = 3–4. *, P < 0.05 vs. control (Ctr). E, ChIP demonstrated that cortisol (1 μm) caused no obvious binding of CREB to the GRE in the cPLA2α promoter as revealed by gel electrophoresis of the PCR products amplified from the DNA fragments precipitated by CREB antibody in HFL-1 cells. The top panel is a representative gel of three individual experiments; bottom panel illustrates the positions and sequences of the primers used for PCR.
Induction of cPLA2α expression by cortisol via a glucocorticoid response element (GRE) in the promoter in human amnion fibroblasts
Bioinformatic analysis of the nucleotide sequences of the cPLA2α promoter (−595 bp upstream to the transcription start site) with Transcription Element Search System (TESS) revealed several putative GREs (TGTTCT or AGAAGA), but no CRE or nuclear factor-κB binding site within this region (Fig. 3). In addition, there exist tandem repeats of CA nucleotides from −221 to −257 bp. Treatment of the amnion cells transfected with pGL3-enhancer plasmid carrying −595 bp cPLA2α promoter and luciferase reporter gene with cortisol (1 μm, 24 h) significantly increased the promoter activity, which was blocked by RU486 (1 μm) (Fig. 4A). Deletion of the first 90 bases from the 5′ end did not affect cortisol-induced cPLA2α promoter activity. However, cortisol-induced cPLA2α promoter activity was abolished when the deletion progressed to −458 bp (Fig. 4B), suggesting a crucial element responsible for the induction by cortisol existing between −505 and −458 bp. Within this region, there is a typical GR binding sequence (TGTTCT) (20) from −485 to −490 bp (Fig. 3). Introduction of triple-nucleotide mutations (from TGTTCT to TGCCTT; mutation underlined) into this putative GRE abolished the stimulation of cPLA2α promoter activity by cortisol (1 μm) (Fig. 4C). Chromatin immunoprecipitation assay (ChIP) demonstrated that cortisol treatment (1 μm, 24 h) enhanced the binding of GR to the DNA fragment containing this putative GRE in human amnion fibroblasts (Fig. 5). In contrast to the findings in human amnion fibroblasts, the same GRE was found to mediate the inhibition of the cPLA2α promoter activity by cortisol (1 μm) in HFL-1 cells. This inhibition of the cPLA2α promoter activity (−505 bp) by cortisol was abolished by the same triple-nucleotide mutations in the GRE as described above or by complete removable of this GRE (−458 bp) in HFL-1 cells (Fig. 2C). These findings suggest a cofactor interacting with GR might be involved in the paradoxical induction of cPLA2α expression by cortisol via this GRE in human amnion fibroblasts.
Fig. 3.

Bioinformatic analysis of the putative GRE in cPLA2α promoter (−595 bp). Boxed nucleotides indicate the positions of the putative GREs, and the arrows indicate the sites of antisense (pointing to the right) and sense (pointing to the left) primers used for PCR subcloning of 5′-end truncated cPLA2α promoters.
Fig. 4.
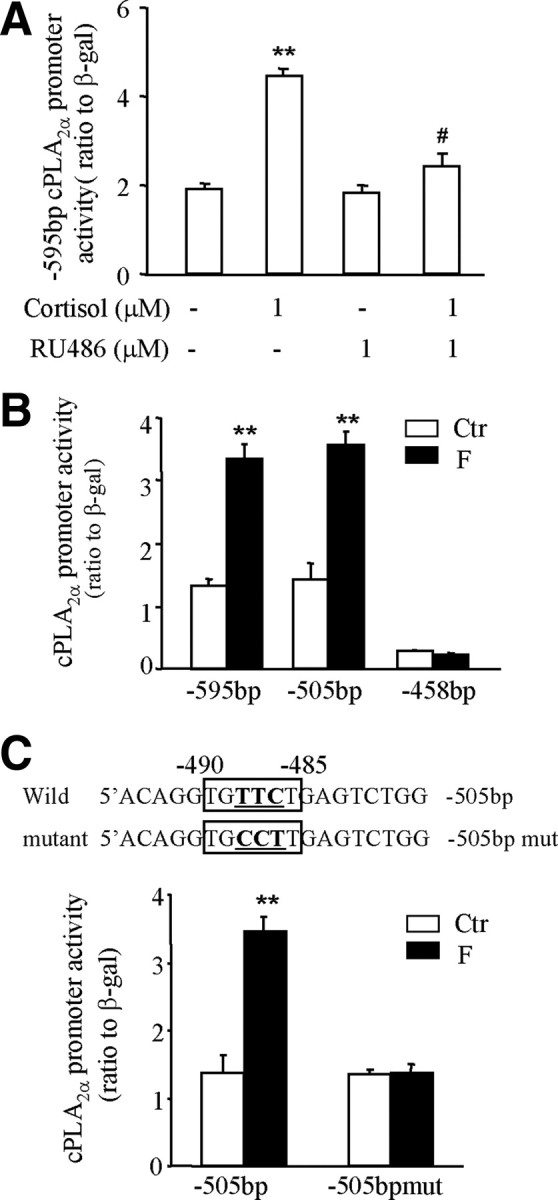
A, Stimulation of cPLA2α promoter (−595 bp) activity by cortisol could be blocked by GR antagonist RU486 in human amnion fibroblasts. B, Deletion of the first 90 nucleotides (as −505 bp) from the 5′ end of −595-bp cPLA2α promoter did not affect the stimulation of cPLA2α promoter activity by cortisol (F, 1 μm). Further deletion of the putative GRE at −485 to −490 bp (as −458 bp) diminished the stimulation of cPLA2α promoter activity by cortisol (F, 1 μm). C, The stimulation of of cPLA2α promoter activity by cortisol (F, 1 μm) could be abolished by introducing triple-nucleotide mutations (underlined bold letters) into the putative GRE at −485 to −490 bp (boxed letters). n = 3. **, P < 0.01 vs. control (Ctr); #, P < 0.05 vs. cortisol (F) without RU486.
Fig. 5.

ChIP demonstrated that cortisol (1 μm) stimulated the binding of GR to the GRE in the cPLA2α promoter as revealed by gel electrophoresis of the PCR products amplified from the DNA fragments precipitated by GR antibody in human amnion fibroblasts. The top panel is a representative gel of three individual experiments; bottom panel illustrates the positions and sequences of the primers used for PCR.
Interaction of GR and CREB-1 in the induction of cPLA2α expression in human amnion fibroblasts
In our recent study, we found that cortisol induced COX-2 expression in human amnion fibroblasts by stimulation of the cAMP/PKA pathway resulting in the phosphorylation of CREB-1 and its subsequent binding to the CRE in the COX-2 promoter (16). In the present study, we found that stimulation of adenylyl cyclase with forskolin (100 μm, 24 h) increased not only COX-2 mRNA expression but also cPLA2α mRNA expression in human amnion fibroblasts (Fig. 6A). Furthermore, inhibition of PKA by H89 (20 μm) (Fig. 6, B and C) or inhibition of CREB-1 by transfecting the plasmid expressing the dominant-negative (dn)-CREB-1 (Fig. 7, A and B) into the cells significantly attenuated the induction of cPLA2α mRNA and protein expression by cortisol (1 μm, 24 h), suggesting that activation of the cAMP/PKA/CREB-1 pathway also plays a significant role in the induction of cPLA2α by cortisol in human amnion fibroblasts. Coimmunoprecipitation assay revealed that GR was detectable in the protein complex precipitated by either GR antibody or CREB-1 antibody but not in the control sample precipitated with normal serum (Fig. 8A). ChIP assay demonstrated that cortisol treatment (1 μm, 24 h) enhanced the binding of CREB-1 to the same DNA fragment that binds GR in the amnion cells (Fig. 8B) but not in the HFL-1 cells (Fig. 2E). These results suggest that interaction of GR and CREB-1 occurs upon cortisol stimulation of the cells.
Fig. 6.
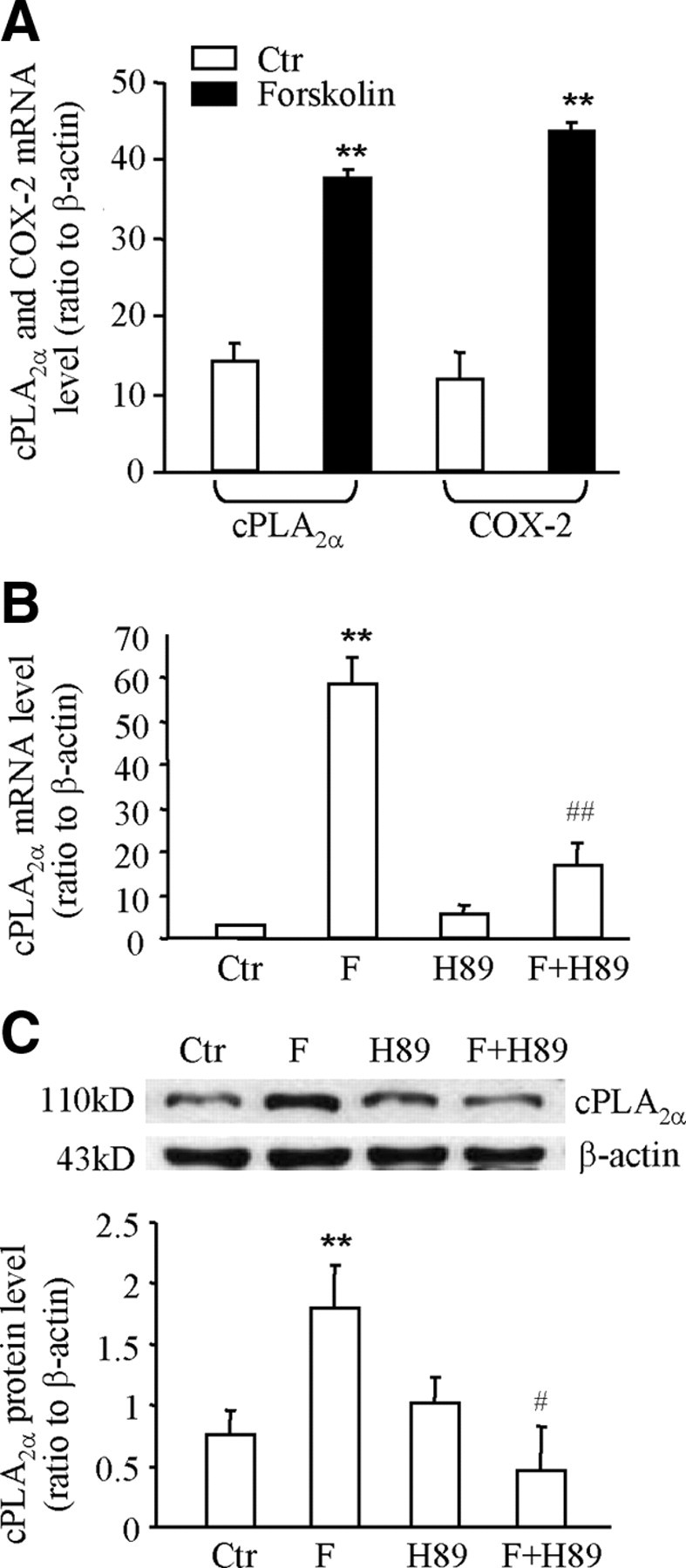
A, Forskolin (100 μm), an adenylyl cyclase stimulator, increased the level of cPLA2α and COX-2 mRNA in human amnion fibroblasts. B and C, Inhibition of PKA with H89 (20 μm) attenuated the stimulation of cPLA2α mRNA and protein expression by cortisol (F, 1 μm) in human amnion fibroblasts. The top panel of C is the representative immunoblot; bottom panel shows the mean data (n = 3). **, P < 0.01 vs. control (Ctr); #, P < 0.05; ##, P < 0.01 vs. cortisol (F) without H89.
Fig. 7.

Stimulation of cPLA2α mRNA (A, n = 3) and protein (B) expression by cortisol (1 μm) was attenuated by transfection of a vector expressing dn-CREB-1 protein in human amnion fibroblasts. The top panel of B is the representative immunoblot; bottom panel shows the mean data of three individual experiments. *, P < 0.05 vs. the fold induction in cells transfected with negative control vector (NC).
Fig. 8.

A, Representative immunoblot showing GR in the protein complex immunoprecipitated (IP) by GR antibody (Ab) or CREB-1 antibody in human amnion fibroblasts stimulated by cortisol (1 μm). IgG(H), IgG heavy chain; IgG(L), IgG light chain. B, ChIP demonstrated that cortisol (1 μm) stimulated the binding of CREB-1 to the GRE in the cPLA2α promoter as revealed by gel electrophoresis of the PCR products amplified from the DNA fragments precipitated by CREB-1 antibody in human amnion fibroblasts. The top panel is the representative gel image of three individual experiments; bottom panel illustrates the positions and sequences of the primers used for PCR.
Induction of Gαs expression plays a role in the stimulation of cPLA2α and COX-2 expression by cortisol in human amnion fibroblasts
As illustrated in Fig. 1D, CHX treatment of human amnion fibroblasts significantly attenuated the induction of cPLA2α mRNA expression by cortisol as in the case of COX-2 mRNA expression (16), suggesting that de novo protein synthesis participates in the paradoxical stimulation of cPLA2α and COX-2 mRNA expression by cortisol. Analysis with Western blotting revealed that cortisol treatment (1 μm, 12 h) significantly increased the levels of Gαs protein in human amnion fibroblasts (Fig. 9A) but not in HFL-1 cells (Fig. 2D). Consistent with GR-mediated induction of cPLA2α and COX-2 expression (16) by cortisol, the induction of Gαs expression by cortisol could also be blocked by the GR antagonist RU486 (1 μm) (Fig 9A). Cotreatment of the cells with cortisol (1 μm, 24 h) and NF449 (20 μm), a selective inhibitor of GTP binding with Gαs (21), significantly attenuated cortisol-stimulated CREB-1 phosphorylation (Fig. 9B) as well as cPLA2α and COX-2 expression (Fig. 9C).
Fig. 9.

A, Induction of Gαs protein expression by cortisol (1 μm) was blocked by GR antagonist RU486 (RU, 1 μm) in human amnion fibroblasts. B, Stimulation of CREB-1 phosphorylation by cortisol (1 μm) was attenuated by Gαs protein inhibitor NF449 (20 μm) in human amnion fibroblasts. C, Stimulation of cPLA2α and COX-2 protein expression by cortisol (1 μm) was attenuated by Gαs protein inhibitor NF449 (20 μm). The top panels of A–C are the representative immunoblots; bottom panel shows the mean data. n = 3–4. *, P < 0.05 vs. control (Ctr); #, P < 0.05 vs. cortisol without RU486 or NF449.
Discussion
Because the availability of free arachidonic acid is particularly important for the synthesis of PGs (10, 22), cPLA2α, the enzyme catalyzing its release from phospholipids, is crucial in the initiation of parturition. This is exemplified by the findings that the expression of cPLA2α is increased in the amnion, the major PG source in pregnancy, toward the end of normal pregnancy as well as in preterm labor (18, 23), and cPLA2α-null mice failed to labor at term (19). Glucocorticoids are well-known antiinflammatory drugs. One aspect of their effects is either inhibition of basal cPLA2α gene expression by itself or inhibition of proinflammatory cytokine-induced cPLA2α gene expression (12, 13, 24, 25, 26). Because multiple putative GREs exist in the cPLA2α promoter, the inhibition of cPLA2α expression by glucocorticoids has been suggested to be mediated by these GREs (27, 28). In this study, we proved the GRE at −485 to −490 bp was particularly important in mediating the classical inhibitory effect of cortisol on cPLA2α expression in HFL-1 cells. By contrast, we confirmed our previous findings that cortisol, like the synthetic dexamethasone, is a potent stimulator of cPLA2α expression in human amnion fibroblasts (7). Of interest, this kind of paradoxical stimulation of PG synthesis by glucocorticoids has also been recently observed in other cell types such as cardiomyocytes (29, 30), which is believed to provide a survival pathway against ischemia/reperfusion injury. In this study, we found that the stimulation of cPLA2α expression by glucocorticoids in human amnion fibroblasts required ongoing transcription and was achieved through stimulation of the cPLA2α promoter activity via the same cis-acting GRE that mediates the classical inhibitory effect of cortisol in HFL-1 cells. Although there exist multiple putative GREs in the cPLA2α promoter (31, 32), this distal GRE appears to be more crucial in mediating the stimulatory effect of glucocorticoids. The function of the proximal GREs might be interfered with by the neighboring tandem CA repeats, which have been shown to exert a strong negative regulation of cPLA2α promoter activity (32). However, the GREs found in the cPLA2α promoter appear to be half-sites, which may not allow the discrimination of repression and stimulation by the sequence alone (27, 33). Thus, it is very likely that the stimulation of cPLA2α expression by cortisol is determined by the interaction of GR with other transcription factors induced by glucocorticoids in human amnion fibroblasts. We found that recruitment of CREB-1 to the GRE is crucial for the stimulation of cPLA2α expression to occur. The CREB-1 protein was found not only in the protein complex containing GR but also bound to the distal GRE with GR by ChIP assay. Additionally, inhibiting PKA-induced CREB-1 phosphorylation with H89 or blocking the function of CREB-1 with dn-CREB-1 greatly attenuated the induction of cPLA2α by cortisol. All these findings resemble the mechanism of cortisol-induced COX-2 expression in human amnion fibroblasts (16). These results suggest that the stimulation of cPLA2α and COX-2 by cortisol shares one common pathway, namely cAMP/PKA/CREB-1, which is further supported by the findings that stimulation of adenylyl cyclase with forskolin increased both cPLA2α and COX-2 expression in human amnion fibroblasts. Of interest, different cis elements, respectively GRE and CRE, were used by this pathway in mediating the paradoxical induction of cPLA2α and COX-2 expression by cortisol in human amnion fibroblasts. This interaction of GR and CREB-1 has also been reported to be a unique mechanism for the up-regulation of the expression of a number of other genes such as phosphoenolpyruvate carboxykinase and somatostatin by glucocorticoids (34, 35), which might be one of the key rules to the cell- and stimulus-specific effects of glucocorticoids.
Our present and previous studies (16) showed that the stimulation of both cPLA2α and COX-2 mRNA expression by cortisol was blocked when the de novo protein synthesis was inhibited by CHX in human amnion fibroblasts, suggesting that cortisol-induced protein synthesis is required for the stimulation of cPLA2α and COX-2 transcription to occur. It is generally accepted that the activation of cAMP/PKA/CREB pathway is initiated by the activation of G protein-coupled receptors (36). The heterotrimeric G protein consists of an α-subunit that binds and hydrolyzes GTP as well as β- and γ-subunits that form an undissociable complex. Coupling of the activated receptor to the heterotrimeric G protein promotes the exchange of GDP for GTP on the α-subunit resulting in the dissociation of the α-subunit from the βγ-complex. It is the α-subunit that defines the basic properties of the G protein. There are at least four families of the α-subunit: Gαs, Gαi/Gα0, Gαq/Gα11, and Gα12/Gα13 (36). The Gαs couples many receptors to adenylyl cyclase and mediates receptor-dependent adenylyl cyclase activation resulting in increases in the intracellular cAMP level. The Gαs is encoded by GNAS, a complex imprinted gene. Although it is generally believed that Gαs is ubiquitously expressed, its expression has been reported to be inducible by glucocorticoid treatment, thus enhancing the efficacy of receptors coupled to Gαs (37, 38). In the present study, we found that the expression of Gαs was greatly increased by cortisol treatment of the human amnion fibroblasts. We believe that this induction of Gαs is crucial to the induction of cPLA2α and COX-2 expression by cortisol in human amnion fibroblasts because inhibiting Gαs function with NF449 greatly attenuated the induction of both cPLA2α and COX-2 expression as well as the phosphorylation of CREB-1 by cortisol. These findings lead us to speculate that the induction of Gαs may facilitate the actions of the paracrine or intracrine hormones secreted by the amnion fibroblasts, whose receptors coupled to Gαs, or simply activate the adenylyl cyclase directly to promote PG synthesis. As we know, human amnion fibroblasts synthesize and secrete a large amount of PGs as well as express virtually all the subtypes of PG receptors including EP2, EP4, and IP, which are coupled to Gαs protein (39, 40). Thus, we speculate that this paradoxical stimulation of PG synthesis by cortisol may form a feed-forward mechanism of PG synthesis via induction of Gαs protein toward the end of pregnancy and during parturition.
In conclusion, we demonstrated that cortisol, by inducing the expression of Gαs protein, activated the cAMP/PKA/CREB-1 pathway, resulting in the increased level of phosphorylated CREB-1, which, in turn, interacts with GR on the cis-acting GRE in cPLA2α promoter to promote its expressions in human amnion fibroblasts (Fig. 10).
Fig. 10.

The hypothesized mechanism underlying the induction of cPLA2α and COX-2 expression by cortisol in human amnion fibroblasts.
Materials and Methods
Human amnion fibroblast and fetal lung fibroblast cell culture
Human amnion fibroblasts were prepared from the fetal membranes collected at term from women undergoing elective cesarean section under a protocol approved by the ethics committee of the School of Life Sciences, Fudan University. Patients treated with steroids or other antiinflammatory agents or with a clinical indication of inflammation were excluded from this study. Amnion was digested with 0.125% trypsin (Sigma, St. Louis, MO) and washed to get rid of epithelial cells. The remaining amnion tissue was digested with 0.1% collagenase (Roche, Indianapolis, IN), and the cells were collected for purification with Percoll (GE Healthcare, Uppsala, Sweden) gradients (5, 20, 40, and 60%). The cells were cultured in DMEM (GIBCO, Grand Island, NY) containing 10% newborn calf serum (NCS) (GIBCO) and antibiotic-antimycotic (GIBCO). The identity of cells has been previously verified (7). To identify the role of the same putative GRE in the classical inhibitory effect of glucocorticoids on cPLA2α expression, HFL-1 cells (American Type Culture Collection, Rockville, MD) were cultured in MEMα (Invitrogen, Grand Island, NY) supplemented with 10% NCS and antibiotic-antimycotic.
Treatment of human amnion fibroblasts and HFL-1 cells
On the third day of amnion fibroblast culture, the cells were washed with PBS, and the culture medium was changed to NCS-free medium. The cells were then treated with different concentrations of cortisol (0.01–1 μm; Sigma). To examine whether the effect of cortisol on cPLA2α mRNA expression is dependent on ongoing transcription and requires de novo protein synthesis, the cells were pretreated with the mRNA transcription inhibitor DRB (75 μm; Sigma) or the protein synthesis inhibitor CHX (10 μm; Sigma) for 0.5 h before cortisol treatment and the DRB or CHX treatment was continued together with cortisol treatment for 24 h. To explore whether the effect of cortisol on cPLA2α expression is dependent on GR, the cells were treated with cortisol (1 μm, 24 h) in the presence or absence of GR antagonist RU486 (1 μm; Sigma). To study the role of cAMP, PKA, and CREB-1 in the regulation of cPLA2α expression by cortisol, the cells were treated with forskolin (100 μm, a stimulator of adenylyl cyclase; Sigma) or cortisol (1 μm, 24 h) in the presence and absence of H89 (20 μm, an inhibitor of PKA; Sigma) or Gαs inhibitor NF449 (20 μm) (Calbiochem, La Jolla, CA) or transfected with a RSV-SG plasmid carrying a dominant-negative mutant of CREB-1 (dn-CREB-1, 1 μg/well) in which Ser133 is replaced with Ala (kindly provided by Dr. M. R. Montminy, The Salk Institute, La Jolla, CA). The transfection method is described below. Upon 70–90% confluence, cortisol (1 μm) treatment of the HFL-1 cells was carried out for the measurement of cPLA2α mRNA and protein, Gαs protein, or cPLA2α promoter-driven reporter gene activity as described below.
Determination of cPLA2α mRNA levels with qRT-PCR
After removal of culture medium, cells were scraped into cell lysis buffer for subsequent extraction of total RNA for analysis with PCR using UNIQ-10 RNA extraction kit (Sangon Biotech, Shanghai, China). One microgram of RNA was reverse-transcribed to cDNA with oligo(deoxythymidine)12-18 primers using Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI), and the cDNA was used for subsequent measurement of cPLA2α mRNA level with qRT-PCR. The reaction solution of qRT-PCR consisted of 2.0 μl diluted cDNA, 0.2 μm of each paired primer, and power SYBR green PCR master mix (Toyobo, Osaka, Japan). The annealing temperature was set at 61 C. The absolute mRNA levels in each sample were calculated according to a standard curve set up using serial dilutions of known amounts of specific templates against corresponding cycle threshold values. To control for sampling errors, qRT-PCR for the housekeeping gene β-actin was routinely performed on each sample. The ratio of the target genes over β-actin in each sample was obtained to normalize the expression of the target gene. The specificity of the primers was verified by examining the melting curve as well as the sequences of the PCR products. The primer sequences for amplifying human cPLA2α and β-actin genes are illustrated in Table 1.
Table 1.
Primer sequences used for qRT-PCR
| Genes | Primer sequences (5′–3′) | GenBank accession no. | Size of PCR products (bp) |
|---|---|---|---|
| cPLA2α | |||
| Forward | ATGGCCTTGGTGAGTGATTC | NM_024420 | 179 |
| Reverse | TCAGGATCTGCTACAGCTGC | ||
| COX-2 | |||
| Forward | TGTGCAACACTTGAGTGGCT | NM_000963 | 297 |
| Reverse | ACTTTCTGTACTGCGGGTGG | ||
| β-Actin | |||
| Forward | GGGAAATCGTGCGTGACATTAAG | NM_001101 | 275 |
| Reverse | TGTGTTGGCGTACAGGTCTTTG |
Determination of cPLA2α, COX-2, Gαs, and phosphorylated CREB-1 protein levels with Western blotting
After removal of the culture medium, the cells were washed and collected in PBS containing phosphatase inhibitor. After spinning down, total cellular protein was extracted using a kit from Active Motif (Carlsbad, CA). The expression levels of cPLA2α, COX-2, Gαs, and phosphorylated CREB-1 proteins were examined using a standard Western blotting protocol. Briefly, 50 μg protein of each sample was electrophoresed in 7.5% SDS-polyacrylamide gel and transferred to the nitrocellulose blot. The blot was blocked with nonfat milk solution and incubated with 1:500 dilutions of antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) against cPLA2α (sc-454), COX-2 (sc-19999), Gαs (sc-823), and phosphorylated CREB-1 (ser133) (sc-101663) overnight. After washing, the blot was incubated with the corresponding secondary antibodies conjugated with horseradish peroxidase (Santa Cruz) for 1 h. The enhanced chemiluminescence detection system (Millipore, Billerica, CA) was used to detect the bands with peroxidase activity. The same blot was reprobed with β-actin antibody (sc-47778; Santa Cruz) as a loading control.
5′ Progressive deletion and site-directed mutagenesis of the cPLA2α gene promoter and construction into the pGL3-enhancer plasmid
The pGL2 basic plasmid containing the −595-bp cPLA2α promoter was kindly provided by Dr. Shelhamer (National Institutes of Health, Bethesda, MD). Progressive deletion of the cloned cPLA2α gene promoter was carried out with PCR using the combination of a common antisense primer and a series of 5′ sense primers (progressing toward the transcription start site) as paired primer sets. The annealing positions of these primers are shown in Fig. 3. Restriction sites for KpnI and BglII were designed into the sense and antisense primers, respectively. In addition to −595 bp, a series of 5′-deleted cPLA2α gene promoters was produced (−505 and −458 bp). Based upon the 5′ deletion study, site-directed triple-nucleotide mutations were introduced into the predicted GREs at −485 to −490 bp by designing the mutation sites into the sense primer for cloning the −505-bp promoter fragment (Fig. 5A). The PCR products were cut with KpnI and BglII, and ligated into the polycloning sites of pGL3-enhancer plasmid. All of the above cloned promoter fragments were verified by sequencing.
Transient transfection of human amnion fibroblasts and HFL-1 cells with pGL3-enhancer plasmid carrying cPLA2α promoter-driven luciferase reporter gene
Both human amnion fibroblasts and HFL-1 cells were grown to approximately 70% confluence in complete medium containing 10% NCS and antibiotic-antimycotic before the start of transfection. On the day of transfection, the cells were washed with PBS, and the media were changed to antibiotic-free media containing 10% NCS. The cells were cotransfected with 0.5 μg/well pGL3-enhancer plasmid carrying cPLA2α promoter and 0.05 μg/well pSV-β-galactosidase plasmid using Lipofectamine LTX in Opti-MEM (Invitrogen, Carlsbad, CA). The above plasmids were extracted from the transformed bacteria using endotoxin-free Nucleobond PC500 EF kit (Macherey-Nagel, Duren, Germany). The pSV-β-galactosidase plasmid was used for transfection efficiency control. Transfection was allowed for 12 h. The media were changed to complete media, and the cells were left to recover for 24 h. The cells were then treated with cortisol in the presence and absence of RU486 (1 μm) in serum-free medium for another 24 h. The cells were then lysed for subsequent measurement of luciferase and β-galactosidase activity using luciferase assay and β-galactosidase enzyme assay systems (Promega), respectively. The ratio of cPLA2α promoter-driven luciferase activity against β-galactosidase activity was obtained to correct differential transfection efficiency in each well and to express the promoter activity. The transfection method for RSV-SG plasmid was the same as the reporter gene plasmid.
Coimmunoprecipitation assay
Coimmunoprecipitation assays were carried out to study the interaction of GR and CREB-1 in human amnion fibroblasts. Briefly, upon cortisol (1 μm) stimulation for 24 h, total cellular protein was extracted and immunoprecipitated by GR (sc-1003X; Santa Cruz) or CREB-1 antibodies (sc-186X; Santa Cruz) and protein A (Upstate Biotechnology, Temecula, CA). The protein complex was washed adequately and collected for Western blotting with GR antibodies to detect GR in the complex.
ChIP assay demonstrating the binding of GR or CREB-1 to cPLA2α promoter in human amnion fibroblasts and HFL-1 cells
Primary human amnion fibroblasts and HFL-1 cells were prepared as described above. The cells were treated with cortisol (1 μm) for 24 h. Upon termination of treatment, ChIP assay was conducted using a kit from Upstate Biotechnology and a method modified from the manufacturer’s protocol. Briefly, the cells were fixed with 1% formaldehyde to cross-link the transcription factors to chromatin DNA. After washing with PBS, the cells were incubated with glycine and then scraped off the dish in PBS containing protease inhibitor cocktail. After spinning down, the cells were resuspended with lysis buffer supplemented with protease inhibitor cocktail and broken up using a Dounce homogenizer to aid nuclei release. After spinning down, the nuclei were resuspended in digestion buffer supplemented with protease inhibitor cocktail. The shearing of chromatin DNA was carried out by sonication to produce an optimized size of input DNA around 500 bp. The sheared DNA was collected for subsequent immunoprecipitation with GR antibody (Santa Cruz) or CREB-1 antibody (Santa Cruz) or normal serum as negative control. The immunoprecipitate was then incubated with protein A agarose/salmon sperm DNA and the antibody/protein/DNA/agarose complex was washed and collected for subsequent reverse cross-linking. The sheared DNA recovered from reverse cross-linking was extracted with a DNA extraction kit for further PCR analysis. The positions and sequences of the primers spanning the predicted GRE promoter region used for PCR are illustrated in Figs. 5 and 8B). Real-time PCR was performed on the sheared DNA fragments and was stopped before saturation according to the amplification curve. PCR products were analyzed with 2% agarose gel electrophoresis.
Statistical analysis
All data are reported as mean ± sem. Paired Student’s t test or one-way ANOVA test followed by the Student-Newman-Keuls test was used to assess significant differences where appropriate. Significance was set at P < 0.05.
NURSA Molecule Pages:
Ligands: Hydrocortisone.
Footnotes
This work was supported by the Natural Science Foundation of China (30570680, 30870935, and 30911120485).
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 4, 2010
Abbreviations: ChIP, Chromatin immunoprecipitation; CHX, cycloheximide; COX-2, cyclooxygenase-2; cPLA2α, cytosolic phospholipase A2α; CRE, cAMP response element; CREB-1, CRE-binding protein-1; dn, dominant-negative; DRB, 5,6-dichlorobenzimidazole riboside; GR, glucocorticoid receptor; GRE, glucocorticoid response element; NCS, newborn calf serum; PG, prostaglandin; PKA, protein kinase A; qRT-PCR, quantitative real-time PCR.
References
- 1.Saigal S, Doyle LW2008. Preterm birth 3: an overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet 371:261–269 [DOI] [PubMed] [Google Scholar]
- 2.Creasy RK1991. Preventing preterm birth. N Engl J Med 325:727–729 [DOI] [PubMed] [Google Scholar]
- 3.Gibb W1998. The role of prostaglandins in human parturition. Ann Med 30:235–241 [DOI] [PubMed] [Google Scholar]
- 4.Challis JR, Lye SJ, Gibb W1997. Prostaglandins and parturition. Ann NY Acad Sci 828:254–267 [DOI] [PubMed] [Google Scholar]
- 5.Duchesne MJ, Thaler-Dao H, de Paulet AC1978. Prostaglandin synthesis in human placenta and fetal membranes. Prostaglandins 15:19–42 [DOI] [PubMed] [Google Scholar]
- 6.Economopoulos P, Sun M, Purgina B, Gibb W1996. Glucocorticoids stimulate prostaglandin H synthase type-2 (PGHS-2) in the fibroblast cells in human amnion cultures. Mol Cell Endocrinol 117:141–147 [DOI] [PubMed] [Google Scholar]
- 7.Sun K, Ma R, Cui X, Campos B, Webster R, Brockman D, Myatt L2003. Glucocorticoids induce cytosolic phospholipase A2 and prostaglandin H synthase type 2 but not microsomal prostaglandin E synthase (PGES) and cytosolic PGES expression in cultured primary human amnion cells. J Clin Endocrinol Metab 88:5564–5571 [DOI] [PubMed] [Google Scholar]
- 8.Challis J, Sloboda D, Matthews S, Holloway A, Alfaidy N, Howe D, Fraser M, Newnham J2000. Fetal hypothalamic-pituitary adrenal (HPA) development and activation as a determinant of the timing of birth, and of postnatal disease. Endocr Res 26:489–504 [DOI] [PubMed] [Google Scholar]
- 9.Sippell WG, Müller-Holve W, Dörr HG, Bidlingmaier F, Knorr D1981. Concentrations of aldosterone, corticosterone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogesterone, 11-deoxycortisol, cortisol, and cortisone determined simultaneously in human amniotic fluid throughout gestation. J Clin Endocrinol Metab 52:385–392 [DOI] [PubMed] [Google Scholar]
- 10.Irvine RF1982. How is the level of free arachidonic acid controlled in mammalian cells. Biochem J 204:3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhen T, Cidlowski JA2005. Antiinflammatory action of glucocorticoids: new mechanisms for old drugs. N Engl J Med 353:1711–1723 [DOI] [PubMed] [Google Scholar]
- 12.Newton R, Kuitert LM, Slater DM, Adcock IM, Barnes PJ1997. Cytokine induction of cytosolic phospholipase A2 and cyclooxygenase-2 mRNA is suppressed by glucocorticoids in human epithelial cells. Life Sci 60:67–78 [DOI] [PubMed] [Google Scholar]
- 13.Xue S, Slater DM, Bennett PR, Myatt L1996. Induction of both cytosolic phospholipase A2 and prostaglandin H synthase-2 by interleukin-1β in WISH cells is inhibited by dexamethasone. Prostaglandins 51:107–124 [DOI] [PubMed] [Google Scholar]
- 14.Casey ML, MacDonald PC, Mitchell MD1985. Despite a massive increase in cortisol secretion in women during parturition, there is an equally massive increase in prostaglandin synthesis: a paradox. J Clin Invest 75:1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potestio FA, Zakar T, Olson DM1988. Glucocorticoids stimulate prostaglandin synthesis in human amnion cells by a receptor-mediated mechanism. J Clin Endocrinol Metab 67:1205–1210 [DOI] [PubMed] [Google Scholar]
- 16.Zhu XO, Yang Z, Guo CM, Ni XT, Li JN, Ge YC, Myatt L, Sun K2009. Paradoxical stimulation of cyclooxygenase-2 expression by glucocorticoids via a cyclic-AMP response element in human amnion fibroblasts. Mol Endocrinol 23:1839–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freed KA, Moses EK, Brennecke SP, Rice GE1997. Differential expression of type II, IV and cytosolic PLA2 messenger RNA in human intrauterine tissues at term. Mol Hum Reprod 3:493–499 [DOI] [PubMed] [Google Scholar]
- 18.Skannal DG, Brockman DE, Eis AL, Xue S, Siddiqi TA, Myatt L1997. Changes in activity of cytosolic phospholipase A2 in human amnion at parturition. Am J Obstet Gynecol 177:179–184 [DOI] [PubMed] [Google Scholar]
- 19.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T1997. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390:618–622 [DOI] [PubMed] [Google Scholar]
- 20.Strähle U, Klock G, Schütz G1987. A DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. Proc Natl Acad Sci USA 84:7871–7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohenegger M, Waldhoer M, Beindl W, Böing B, Kreimeyer A, Nickel P, Nanoff C, Freissmuth M1998. Gsα-selective G protein antagonists. Proc Natl Acad Sci USA 95:346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lappas M, Rice GE2004. Phospholipase A2 isozymes in pregnancy and parturition. Prostaglandins Leukot Essent Fatty Acids 70:87–100 [DOI] [PubMed] [Google Scholar]
- 23.Lappas M, Permezel M, Georgiou HM, Rice GE2001. Type II phospholipase A2 in preterm human gestational tissues. Placenta 22:64–69 [DOI] [PubMed] [Google Scholar]
- 24.Lin LL, Lin AY, DeWitt DL1992. Interleukin-1-α induces the accumulation of cytosolic phospholipase-A2 and the release of prostaglandin-E2 in human fibroblasts. J Biol Chem 267:23451–23454 [PubMed] [Google Scholar]
- 25.Hoeck WG, Ramesha CS, Chang DJ, Fan N, Heller RA1993. Cytoplasmic phospholipase-A2 activity and gene expression are stimulated by tumor necrosis factor: dexamethasone blocks the induced synthesis. Proc Natl Acad Sci USA 90:4475–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolan-O'Keefe M, Chow V, Monnier J, Visner GA, Nick HS2000. Transcriptional regulation and structural organization of the human cytosolic phospholipase A2 gene. Am J Physiol 278:L649–L657 [DOI] [PubMed]
- 27.Goppelt-Struebe M1997. Molecular mechanisms involved in the regulation of prostaglandin biosynthesis by glucocorticoids. Biochem Pharmacol 53:1389–1395 [DOI] [PubMed] [Google Scholar]
- 28.Kol S, Ben-Shlomo I, Payne DW, Ando M, Rohan RM, Adashi EY1998. Glucocorticoids suppress basal (but not interleukin-1-supported) ovarian phospholipase A2 activity: evidence for glucocorticoid receptor-mediated regulation. Mol Cell Endocrinol 137:117–125 [DOI] [PubMed] [Google Scholar]
- 29.Sun HP, Xu B, Inoue H, Chen QM2008. p38 MAPK mediates COX-2 gene expression by corticosterone in cardiomyocytes. Cell Signal 20:1952–1959 [DOI] [PubMed] [Google Scholar]
- 30.Tokudome S, Sano M, Shinmura K, Matsuhashi T, Morizane S, Moriyama H, Tamaki K, Hayashida K, Nakanishi H, Yoshikawa N, Shimizu N, Endo J, Katayama T, Murata M, Yuasa S, Kaneda R, Tomita K, Eguchi N, Urade Y, Asano K, Utsunomiya Y, Suzuki T, Taguchi R, Tanaka H, Fukuda K2009. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J Clin Invest 119:1477–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo C, Yang Z, Li W, Zhu P, Myatt L, Sun K2008. Paradox of glucocorticoid-induced cytosolic phospholipase A2 group IVA messenger RNA expression involves glucocorticoid receptor binding to the promoter in human amnion fibroblasts. Biol Reprod 78:193–197 [DOI] [PubMed] [Google Scholar]
- 32.Wu T, Ikezono T, Angus CW, Shelhamer JH1994. Characterization of the promoter for the human 85-kDa cytosolic phospholipase A2 gene. Nucleic Acids Res 22:5093–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kassel O, Herrlich P2007. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol 275:13–29 [DOI] [PubMed] [Google Scholar]
- 34.Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK1993. Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem 268:5353–5356 [PubMed] [Google Scholar]
- 35.Liu JL, Papachristou DN, Patel YC1994. Glucocorticoids activate somatostatin gene transcription through cooperative interaction with the cyclic-AMP signaling pathway. Biochem J 301:863–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wettschureck N, Offermanns S2005. Mammalian G proteins and their cell type specific functions. Physiol Rev 85:1159–1204 [DOI] [PubMed] [Google Scholar]
- 37.Kalavantavanich K, Schramm CM2000. Dexamethasone potentiates high-affinity β-agonist binding and Gsα protein expression in airway smooth muscle. Am J Physiol 278:L1101–L1106 [DOI] [PubMed]
- 38.Saito N, Guitart X, Hayward M, Tallman JF, Duman RS, Nestler EJ1989. Corticosterone differentially regulates the expression of Gs α and Gi α messenger RNA and protein in rat cerebral cortex. Proc Natl Acad Sci USA 86:3906–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grigsby PL, Sooranna SR, Adu-Amankwa B, Pitzer B, Brockman DE, Johnson MR, Myatt L2006. Regional expression of prostaglandin E2 and F2α receptors in human myometrium, amnion, and choriodecidua with advancing gestation and labor. Biol Reprod 75:297–305 [DOI] [PubMed] [Google Scholar]
- 40.Grigsby PL, Sooranna SR, Brockman DE, Johnson MR, Myatt L2006. Localization and expression of prostaglandin E2 receptors in human placenta and corresponding fetal membranes with labor. Am J Obstet Gynecol 195:260–269 [DOI] [PubMed] [Google Scholar]