

Abstract
We and others previously showed that signaling through cSrc or atypical protein kinase C (aPKC) pathway regulates the proliferation of prostate cancer cells and is associated with their progression to castrate-resistance in vivo. However, the interrelation of these two kinases has been largely unexplored. In the present study, we show that androgen-induced activation of cSrc regulates the activity of aPKC through the small molecular weight G protein Rac1 in androgen-dependent LNCaP cells. Knockdown of cSrc in those cells reduces the phosphorylation of aPKC and the abundance of activated form of Rac1. Additionally, the treatment of those cells with Rac1 inhibitor repressed cell cycle progression at G1/S transition. In fact, forced expression of a constitutively active Rac1 mutant in LNCaP cells promoted cell proliferation under androgen-depleted conditions both in vitro and in vivo. Moreover, LNCaP C4-2 and AILNCaP cells, the syngeneic androgen-independent sublines from LNCaP cells, harbored abundant Rac1-GTP. Importantly, the inhibition of Rac1 suppressed cell proliferation and induced apoptotic cell death in all prostate cancer cell lines tested irrespective of their androgen-dependence. In immunohistochemical evaluation of tumor specimens from prostate cancer patients, Rac1 pathway appeared to be activated in the majority of castrate-resistant diseases. Collectively, our present results both in vitro and in vivo highly implicate that Rac1 can be a potential therapeutic target for patients with advanced prostate cancer, especially those with castrate-resistant status.
Androgen dependent or -independent Rac1 activation is required and sufficient for cell cycle progression at G1/S phase transition in prostate cancer cells.
Prostate cancer is the most commonly diagnosed cancer among men in many industrialized countries and is the second leading cause of cancer death in the United States (1, 2). Most untreated prostate cancer cells, as well as normal prostatic epithelial cells, require androgen stimulation to proliferate and maintain cellular viability, as first described by Huggins and Hodges (3) and later studied by Heinlein and Chang (4). Therefore, androgen deprivation or antiandrogen therapy has been the standard treatment modality for disseminated or relapsed prostate cancer. However, most prostate cancers eventually escape and proliferate under androgen depleted conditions, at which point they are called castrate-resistant prostate cancer (CRPC). To date, no curative treatment for CRPC exists and therefore the prognosis of patients in this status is extremely poor. Thus, there is an urgent need to understand the mechanisms of progression to CRPC in the hope of developing novel treatments for this disease.
The best characterized function of androgen and the androgen receptor (AR) is genotropic action, which involves their translocation from the cytoplasm to the nucleus upon ligand binding, binding to specific DNA sequences, and transcriptional regulation of specific genes such as prostate-specific antigen. Besides this type of action, several cellular signal transduction pathways are also induced by the androgen stimulation (5, 6, 7). Typically, activation of these pathways is provoked rapidly within 5 min after the androgen stimulation (8). For example, it was reported that androgen induces cell cycle progression of LNCaP cells through the ligand-dependent physical interaction between proline-rich region of AR and Src homology 3 domain of cSrc (8). It was also shown that R1881 or dihydrotestosterone activates extracellularly regulated kinase (Erk) in LNCaP cells, leading to enhanced cellular proliferation. Importantly, this effect was reversed by the treatment with the Src inhibitor PP1 (5, 8). As for another important role of cSrc, it has been reported that activated cSrc phosphorylates the tyrosine residues of AR and enhances its transcriptional activity (9, 10). cSrc-mediated up-regulation of the transcriptional activity of AR leads to androgen-independent cell proliferation (9, 10). Indeed, signals through cSrc are known to regulate apoptosis, migration, and adhesion of prostate cancer cells (11), and early data with Src inhibitors for the treatment of prostate cancer appear promising (12, 13). However, how cSrc regulates the proliferation and viability of prostate cancer cells remains controversial. For example, although the activation of Erk has been shown to be a main downstream target of androgen-induced activation of cSrc in LNCaP cells (5, 6, 8), the inhibition of Src signals by Dasatinib caused the cell cycle arrest and the apoptotic cell death in the same cells without affecting the activity of Erk (13).
Previously, we reported that atypical protein kinase C (aPKC) and S6 kinase (S6K) were activated in an androgen-dependent manner in LNCaP cells and the effect of androgen stimulation became most distinctive after 72 h, suggesting that genotropic action also involved in the activation of cellular signal transduction pathways. In addition, this pathway was also constitutively activated in androgen-independent prostate cancer cell lines such as PC3 and DU145 (14). Importantly, inhibition of aPKC-S6K pathways induced cell cycle arrest and apoptotic cell death in prostate cancer cell lines, including LNCaP, PC3, and DU145 cells, indicating that signals through aPKC-S6K were required for cell proliferation and survival of cultured prostate cancer cells. However, the signals regulating this pathway remain unclear.
Here, we report for the first time that androgen-induced signal transduction through cSrc activates an aPKC-S6K pathway in LNCaP cells. Importantly, we also clarified that Rac1, a member of Rho family small guanosine triphosphatases (GTPases), mediates signal transduction pathways between cSrc and aPKC. It was also revealed that constitutive activation of the signal transduction by the forced expression of dominant-positive cSrc (cSrcY530F) or Rac1 (Rac1V12) enabled LNCaP cells to proliferate under androgen-depleted conditions. In fact, inhibition of Rac1 induced both apoptosis and repression of cell cycle in prostate cancer cells irrespective of their androgen dependence.
Immunohistochemical study suggested that the signal through Rac1 was also activated in human prostate cancer specimens in castrate-resistant status. Collectively, our results suggest that activation of Rac1 is a common phenomenon in androgen-independent progression of prostate cancer cells in vivo and indicate that Rac1 is a potent target for the treatment of patients with CRPC.
Results
Androgen-dependent activation of aPKC is regulated by signals through cSrc via Rac1 in LNCaP cells
We first asked whether the activation of cSrc is regulated by the androgen stimulation in LNCaP cells. LNCaP cells were grown in charcoal-stripped fetal bovine serum (CSFBS), which was significantly reduced in steroid hormones, including androgen, and then stimulated with the synthetic androgen R1881. The levels of phosphorylation at the activation site of cSrc (Y419) were examined by Western blot with the use of phospho-specific antibodies. As previously reported (5), the activation of cSrc was observed by the stimulation with R1881. The activation of cSrc was first seen 5 min after treatment and continued to be activated for 72 h. Importantly, the androgen-induced increase of activated form of aPKC (T410) began to increase after 5 min and became more pronounced after longer periods of treatment with a maximum effect reached at 72 h. These results suggested that activity of aPKC was dependent on the genotropic effects of the AR (Fig. 1A).
Fig. 1.

Androgen stimulation activates aPKC signals through cSrc via Rac1. The number below each band represents the mean value from densitometry reading. Densitometry readings normalized to total proteins are reported as averages from three independent experiments. A, LNCaP cells were left untreated or treated with CSFBS for 72 h followed by stimulation with 1 nm R1881 for indicated durations, and 60 μg of total cellular protein was separated by SDS-PAGE to examine indicated proteins by immunoblotting. B, LNCaP cells were treated with dimethylsulfoxide (DMSO) or SU6656 at indicated concentrations for 48 h, and total cell lysate containing 60 μg of protein was separated by SDS-PAGE to examine indicated proteins by immunoblotting. C, LNCaP cells were treated as indicated and cultured for 48 h. Then total cell protein was subjected to Rac1 pull-down assay or SDS-PAGE to examine indicated proteins by immunoblotting. D, LNCaP cells were treated with 100 μm Rac1 inhibitor for indicated durations (hr). Total cell lysate (60 μg) were separated by SDS-PAGE, and indicated proteins were examined by immunoblotting. E, LNCaP cells were cultured in phenol red-free RPMI 1640 supplemented with 10% CSFBS for 72 h and then treated ethanol or, R1881 (1 nm) with or without Rac1 inhibitor (100 μm) for 24 h. Cells were subjected to Rac1 pull-down assay and SDS-PAGE or flow cytometry analysis. Histograms represent triplicate independent experiments, and the column chart indicates the mean and sd values (*, P < 0.01). F, LNCaP cells were transfected with siRNA for indicated genes and harvested 48 h later for SDS-PAGE. The number below the band represents the mean value from densitometry reading.
To clarify whether cSrc is an upstream regulator of aPKC in LNCaP cells, we first examined the effect of the Src family inhibitor SU6656. In fact, the treatment with this inhibitor successfully reduced the abundance of activated form of cSrc and the phosphorylated form of aPKC was also decreased in a dose-dependent manner (Fig. 1B). In contrast, treatment of LNCaP cells with an aPKC inhibitor, which effectively reduced the signals of phosphorylated form of aPKC, did not grossly affect the level of activated form of cSrc (see Supplemental Fig. 1, published on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org). Taken together, these results suggested that aPKC is a downstream target of cSrc in LNCaP cells. Then, we examined the mediator regulating the activity of aPKC downstream of cSrc. Several groups reported that cSrc regulates the activity of Rac1 guanine-exchanging factors such as Tiam1 and Vav (15, 16). It was also reported that Rho-family small G protein Rac1 regulates the activity of aPKC in cell lines such as Hela and hematopoietic progenitor cells (17, 18). Motivated by these findings, we asked whether cSrc regulates the activity of aPKC through Rac1 in LNCaP cells. To assess the activity of Rac1, pull-down assays were performed to estimate the abundance of the GTP form of Rac1 (Rac1-GTP). As shown in Fig. 1C, the amount of Rac1-GTP was diminished by androgen withdrawal (growth in CSFBS) and increased by the stimulation with R1881 similar to that of activated form of cSrc (Fig. 1C, lanes 1–3). Notably, treatment with SU6656 inhibited androgen induced activation of Rac1 almost comparable level with that of bicalutamide (Fig. 1C, compare lanes 4 and 5). Additionally, a specific Rac1 inhibitor scarcely affected the abundance of activated form of cSrc (Fig. 1C, lane 6). Therefore, these results highly suggested that the activation of Rac1 was regulated by the androgen stimulation through cSrc. In fact, the abundance of the activated forms of cSrc and Rac1 was reduced by the treatment with the AR antagonist (Fig. 1C, compare lanes 3 and 4).
Next, we examined whether Rac1 regulates the activity of aPKC. Treatment of LNCaP cells with a Rac1 inhibitor (19) caused a time-dependent reduction in the abundance of the activated form of aPKC and its downstream target S6K (T389), whereas it scarcely affected the level of the activated form of cSrc (Fig. 1D). Collectively, these results suggested that cSrc regulated the activity of aPKC through Rac1 in LNCaP cells.
Previously, we reported that the activation of aPKC is required for androgen-dependent cell proliferation of LNCaP cells (14). To extend these findings, we next examined the roles of Rac1 activation on androgen-dependent cell cycle progression of those cells. LNCaP cells were cultured under androgen-depleted conditions for 48 h and stimulated with R1881 in the presence or absence of the Rac1 inhibitor. Treatment with the Rac1 inhibitor significantly inhibited the effect of R1881 on a cell cycle progression at G1/S phase and reduced S phase population consequently, as determined by fluorescence-activated cell sorting (Fig. 1E).
To confirm that androgen stimulation resulted in the activation of aPKC through cSrc via Rac1, we have examined the effect of siRNA knockdown of AR, cSrc, and Rac1. As expected, knockdown of AR significantly decreased the abundance of activated form of cSrc, Rac1, and aPKC (Fig. 1F, lane 2). The knockdown of cSrc resulted in the down-regulation of both Rac1 and aPKC, whereas that of Rac1 reduced the signal of activated form of aPKC but not cSrc (Fig. 1F, lanes 3 and 4). Collectively, these results indicate that androgen stimulation activate serial cSrc-Rac1-aPKC pathway in LNCaP cells and the activation of Rac1 is required for the G1/S transition in androgen-dependent cell cycle progression of these cells.
Activation of Rac1 promoted the cell proliferation of LNCaP cells under androgen-depleted conditions without affecting the AR transactivity
Next we examined whether the activation of Rac1 could promote cell proliferation of LNCaP cells under androgen-depleted conditions. Toward this end, we established two different LNCaP monoclones that were engineered to express constitutively active Rac1 mutant (Rac1V12). Both the subclones expressed 2.8–5.1 times more abundant phosphorylated form of aPKC compared with that of parental mock-transfected cells under androgen-depleted condition (Fig. 2A). Cell cycle analysis confirmed that a significant greater proportion of LNCaP Rac1V12 cells successfully entered into S phase compared with control cells under 10% CSFBS, whereas no significant differences were observed in the cell cycle distribution among them when they were cultured under 10% FBS (Fig. 2B). Consistent with the results of cell cycle analysis, LNCaP Rac1V12 cells acquired the ability to proliferate under androgen-depleted conditions (Fig. 2C). Importantly, mouse xenograft of these cells expressing constitutively active Rac1 successfully grew in castrated mice, whereas that of control cells did not (Fig. 2D). These results indicate that the activation of Rac1 successfully promote androgen-independent cell proliferation of LNCaP cells in vitro and in vivo.
Fig. 2.

LNCaP cells with forced expression of a constitutively active form of Rac1 (Rac1V12) acquire an ability of androgen-independent cell growth. LNCaP Mock and LNCaP Rac1V12 cells were incubated in RPMI 1640 supplemented with 10% FBS. Then, the medium was exchanged to RPMI 1640 supplemented with 10% CSFBS and were incubated for 72 h. A, Cells were harvested and 60 μg of total cellular protein was separated by SDS-PAGE. The indicated proteins were examined by immunoblotting. The number below each band represents the mean value from densitometry reading. Densitometry readings normalized to total proteins are reported as averages from two independent experiments. *, Exogenous HA-tagged Rac1; **, endogenous Rac1. B, Cell cycle was examined by flow cytometry analysis. Column chart indicates mean and sd (error bars) of them (*, P < 0.01). C, Cells were seeded into 6-cm dishes at 5.0 × 105 cells/plate in RPMI 1640 supplemented with 10% FBS. After 24-h incubation for adhesion, cells on one dish were harvested, and the number of cells was counted as baseline (d 0). Then, the medium on other dish was exchanged to RPMI 1640 supplemented with 10% CSFBS, and the number of cells were counted on d 2, 4, 6, and 8. Results are shown as mean and sd of triplicate independent experiments (*, P < 0.01). D, Indicated cells were injected sc on a flank of male mice (n = 5 for each subline), and mice were surgically castrated after tumor formation was confirmed (see Materials and Methods). Then, the tumor volume was measured weekly. Results are shown as the mean and sd values (*, P < 0.01; bottom). E and F, Transactivity of endogenous AR in LNCaP (E) and exogeneous wt-AR in DU145 cells (F) was examined by dual-luciferase reporter assay (Promega). Cells were untreated (control) or treated with 10−6 m bicalutamide, 1 μm SU6656, or 100 μm Rac1 inhibitor, and the reporter activity was examined in the absence or presence of 10−11 m R1881 (left). Cells were transfected with pcDNA3.1(+) Mock, cSrcY530F, or HA-Rac1V12, and their effect on the reporter activity was examined in the absence or presence of R1881 (10−11 m) (right). In each column, relative luciferase activities to controls are shown with the sd values. Protein expressions under the same conditions for reporter assays were examined by Western blotting.
Previously, it was reported that activation of cSrc was responsible for the development of hormone-refractory prostate cancer (10). The authors showed that cSrc played critical roles in the androgen-independent cell proliferation of prostate cancer cells by augmenting AR transcriptional activity through the phosphorylation of AR at Y534 site. We therefore next asked whether the activation of Rac1 affects the AR transcriptional activity as a downstream effecter of cSrc. LNCaP cells were transiently transfected with a luciferase reporter plasmid containing androgen response elements. As shown in Fig. 2E, the reporter activity provoked by R1881 was reduced by AR antagonist, bicalutamide, or cSrc inhibitor SU6656, whereas the Rac1 inhibitor had no significant effect on it (Fig. 2E, left). Similarly, forced expression of constitutive active mutant of cSrc (cSrcY530F) in those cells successfully enhanced the reporter activity, whereas that of Rac1V12 showed no significant effect on it (Fig. 2E, right). Because LNCaP cells harbor T877A mutation in AR (20), AR-negative DU145 cells were transfected with a plasmid expressing wild-type AR (wt-AR), and the effect of Rac1 activation or inhibition on the reporter activity was examined. As shown in Fig. 2F, SU6656, but not the Rac1 inhibitor, reduced the enhanced reporter activity of wt-AR induced by R1881 and the activation of cSrc signals by the expression of cSrcY530F induced the transactivation of wt-AR, whereas that of Rac1 by Rac1V12 did not. Collectively, these results indicated that the activity of Rac1 had no significant effect on the transcriptional activity of either the wt- or mutated (T877A)-AR.
Activation of Rac1-aPKC pathway is required for cSrc-mediated androgen-independent cell proliferation of cultured LNCaP cells
Previously, it was reported that LNCaP cells could acquire androgen-independence in vivo through the activation of cSrc (10). To further examine roles of cSrc-Rac1-aPKC pathway in cSrc-mediated androgen-independent cell proliferation of LNCaP cells, we established two different monoclones that expressed constitutively active mutant of cSrc (cSrcY530F). As expected, both subclones could successfully proliferate under androgen-depleted conditions in culture (Fig. 3A). Importantly, LNCaP cSrcY530F cells cultured under these conditions retained over 2.2–2.8 times more abundant Rac1-GTP and phosphorylated aPKC, respectively, compared with those of control cells (Fig. 3B). In fact, treatment with Rac1 inhibitor reduced the abundance of activated form of aPKC in those cells (Fig. 3C). In cell cycle assays, a significantly greater proportion of LNCaP cSrcY530F cells could enter proliferative cycle and progress through G1 and into S phase in comparison with control cells. Importantly, inhibition of the activity of Rac1 or aPKC caused cell cycle arrest at G1 phase in LNCaP cSrcY530F cells as well (Fig. 3D). Collectively, these results suggest that the activation of Rac1-aPKC pathway is required for G1/S cell cycle transition in cSrc-mediated androgen-independent cell proliferation of LNCaP cells.
Fig. 3.

Activation of Rac1-aPKC pathway is required for androgen-independent proliferation of cultured LNCaP cSrcY530F cells. A, LNCaP Mock and LNCaP cSrcY530F cells were seeded into 6-cm dishes at 5.0 × 105 cells/plate in RPMI 1640 supplemented with 10% FBS. After 24 h incubation for adhesion, cells on one dish were harvested, and the number of cells was counted as baseline (d 0). Then medium of the other dishes were replaced by phenol red-free RPMI 1640 supplemented with 10% CSFBS, and the number of cells was counted on the d 2, 4, 6, and 8. Results are shown as the mean and sd values of triplicate independent experiments (*, P < 0.001). B, Cells were treated with CSFBS for 72 h, and total protein was extracted. Rac1-GTP was separated by pull-down assay using 2 mg of total cellular protein and examined immunoblotting after SDS-PAGE. Other indicated proteins were separated by SDS-PAGE and examined by immunoblotting using 60 μg of total cellular protein. The number below each band represents the mean value from densitometry reading. Densitometry readings are normalized to total protein and reported as averages from two independent experiments. C, LNCaP Mock and cSrcY530F clones were cultured in CSFBS for 48 h and followed by the treatment with DDW or 100 μm Rac1 inhibitor for 48 h. Total cellular protein (60 μg) was separated by SDS-PAGE, and indicated proteins were examined by immunoblotting. The number below each band represents the mean value from densitometry reading. Densitometry readings are normalized to total protein and reported as averages from two independent experiments. D, Cell cycle was examined by flow cytometry analysis. Cells were seeded into six dishes at 5.0 × 105 cells/plate in RPMI 1640 supplemented with 10% FBS. After 24 h incubation, medium was replaced by phenol red-free RPMI 1640 supplemented with 10% CSFBS. They were cultured for 24 h in the existence of 100 μm Rac1 inhibitor or 20 μm PKCζ pseudosubstrate inhibitor and harvested by trypsinization for flow cytometry. Column chart indicates the mean and sd values.
Activation of Rac1 is related to androgen-independent progression and survival of prostate cancer cells in culture
Next we assessed the roles of cSrc-Rac1-aPKC pathway in androgen-independent progression of LNCaP cells by using their syngeneic androgen-independent sublines, LNCaP C4-2 and AILNCaP (14, 20). AILNCaP cells were established by the long-term cultures of parental LNCaP cells in the absence of androgen stimulation (14). Under androgen depleted condition, C4-2 and AILNCaP cells expressed 3.4 and 3.9 times more abundant Rac1-GTP compared with LNCaP cells cultured in the same condition, respectively. Accordingly, both cells expressed more than 2.8 times abundant activated form of aPKC compared with the parental cells (Fig. 4A, lanes 1–3). It should be noted that AILNCaP cells expressed less abundant phosphorylated cSrc compared with parental LNCaP cells, whereas C4-2 cells harbored 1.3 times more abundant phosphorylated cSrc.
Fig. 4.

Activation of Rac1 is related to androgen-independent progression and survival of prostate cancer cells in culture. A, Total cellular protein (60 μg) of LNCaP, LNCaP C4-2, and AILNCaP cells were separated by SDS-PAGE, and indicated proteins were examined by immunoblotting. Rac1-GTP was separated by pull-down assay using 2 mg of total cellular protein and examined by immunoblotting after SDS-PAGE. The number below each band represents the mean value from densitometry reading. Densitometry readings are normalized to total protein and reported as averages from two independent experiments. B, C4-2 and AILNCaP cells were cultured in CSFBS and treated with dimethylsulfoxide (DMSO; ctl), SU6656 (1 μm) or Rac1 inhibitor (100 μm) 48 h before the extraction of total protein. Rac1-GTP was separated by pull-down assay with 2 mg of total cellular protein and examined immunoblotting after SDS-PAGE. Other indicated proteins were separated by SDS-PAGE and examined by immunoblotting with 60 μg of total cellular protein. The number below each band represents the mean value from densitometry reading. Densitometry readings are normalized to total protein and reported as averages from three independent experiments. C, C4-2 and AILNCaP cells were cultured in CSFBS and treated with DMSO, SU6656 (1 μm), or Rac1 inhibitor (100 μm) for 48 h before the harvest for flow cytometry. Column charts indicate the mean and sd values (*, P < 0.01). D, Cells were seeded into 6-cm dishes and transfected by 1 μg of pEGFP-N1 expressing H2B-enhanced GFP fusion protein alone or along with 12.5 ng of control (scramble) or Rac1 siRNA. Nuclear fragmentation (arrowheads) and chromatin condensation (arrows) were scored 48 h later using fluorescent microscopy. Column chart represents results of three independent experiments, in which at least 100 fluorescent cells were scored for chromatin condensation and nuclear fragmentation. Scale bar, 50 μm. E, LNCaP, C4-2 and AILNCaP cells were grown on 6-cm dishes, transfected with 8 μg pcDNA3.1(+) Mock (left dishes) or pcDNA3.1(+) HA-Rac1N17 (right dishes) with Lipofectamine 2000 reagent (Invitrogen) according to supplier’s protocol and cultured in RPMI 1640 medium supplemented with 10% FBS (LNCaP) or CSFBS (C4-2 and AILNCaP) containing 1 mg/ml of G418 for 2 wk. Colonies were visualized with 0.1% crystal violet staining.
To examine the significance of cSrc and Rac1 activities in androgen-independent proliferation in these cells, we treated the cells with Src family kinase inhibitor SU6656 and Rac1 inhibitor since sufficient transfection efficiency was not achieved by the use of specific siRNAs especially in AILNCaP cells. In both cells, the treatment with Rac1 inhibitor (100 μm) resulted in the reduction in the abundance of activated aPKC, whereas that with SU6656 (1 μm) decreased the signals of Rac1-GTP in C4-2 cells but not in AILNCaP cells (Fig. 4B). In C4-2 cells, attenuation of cSrc by siRNA resulted in substantial decrease in the abundance of activated forms of Rac1 and aPKC (data not shown). Thus, these results indicate that the activation of Rac1-aPKC pathway is regulated by the signals other than cSrc in the latter cells. In fact, inhibition of Rac1 successfully repressed cell cycle progression at G1/S transition in both cells, whereas that of cSrc failed to inhibit it in AILNCaP cells (Fig. 4C).
Previously, we reported that inhibition of the activity of aPKC in LNCaP, PC3, and DU145 cells caused apoptotic cell death (14). So, we next asked whether the knockdown of Rac1 induces apoptosis in prostate cancer cells. We confirmed that another Rac1 siRNA (no. 2) successfully reduced total amount of Rac1 (Supplemental Fig. 2). The cells were transfected with two independent Rac1 or control siRNAs along with the plasmid encoding histone-green fluorescent protein (GFP) to detect the chromatin condensation or nuclear fragmentation. Both Rac1 siRNAs induced a significant increase in the number of cells exhibiting the hallmarks of apoptotic cell death in LNCaP cells and their syngeneic androgen-independent sublines (Fig. 4D).
There have been reports indicating that short-term assays do not correlate with the total cell kill and therefore long-term viability assays often more accurately reflect the in vivo chemosensitivity profiles of tumors (21). Thus, we performed clonogenic colony-forming assays. Cells were transfected with empty vector or a plasmid encoding dominant negative Rac1 [hemagglutinin (HA)-Rac1N17]. Cells transfected with Rac1N17 formed fewer colonies than those of mock transfected cells (Fig. 4E). Collectively, these results indicate that the activation of Rac1 is required for the cell cycle progression and survival of LNCaP cells and their syngeneic androgen-independent sublines.
Then, we further examined the significance of Rac1 activation in other androgen-independent prostate cancer cell lines PC3 and DU145. These lines do not produce detectable levels of AR and proliferate in an androgen-independent manner (20). Similar to C4-2 and AILNCaP cells, PC3 and DU145 cells expressed 5.7 and 7.2 times more abundant Rac1-GTP compared with LNCaP cells, respectively. Importantly, the degree of Rac1 activation vastly exceeded that of cSrc activation in the latter cells (Supplemental Fig. 3A, compare lanes 1 with 2 and 3). When they were treated with SU6656 (1 μm), the abundance of Rac1-GTP and an activated form of aPKC decreased in PC3 cells, whereas neither of them was affected by the same procedure in DU145 cells (Supplemental Fig. 3B). In contrast, Rac1 inhibitor successfully repressed the activation of Rac1 and aPKC similarly in both cells (Supplemental Fig. 3C). Accordingly, the cell cycle analysis demonstrated that the treatment with Rac1 inhibitor repressed cell cycle progression at G1/S transition in both cells, whereas the treatment with SU6656 affected that only in PC3 but not in DU145 cells (Supplemental Fig. 3D). In fact, the IC50 of SU6656 for cell proliferation was approximately 3-fold lower in PC3 cells than in DU145 cells (IC50 = 0.74 and 2.4 μm for PC3 and DU145 cells, respectively; P < 0.01) (Supplemental Fig. 3E, upper panel). Consistent with higher Rac1 activity, DU145 cells were the most sensitive to Rac1 inhibitor in their cell proliferation (IC50 = 20.0, 18.7, and 14.9 μm for LNCaP, PC3, and DU145 cells, respectively) (Supplemental Fig. 3E, lower panel). Intriguingly, Rac1 siRNAs induced apoptotic cell death to those cells including LNCaP cSrcY530F cells (Supplemental Fig. 3F).
Although the exact mechanism underlying the different sensitivity to the inhibition of cSrc remains to be clarified, these results indicate that the activation of Rac1 is required for cell cycle progression and survival of cultured prostate cancer cells irrespective of their AR expression or androgen dependence. Thus, Rac1 can be a potent therapeutic target of prostate cancer.
Signaling through Rac1 is activated in castrate-resistant human prostate cancer specimens
Then, we next examined whether signaling through Rac1 was activated in human prostate cancer tissue. For this purpose, we performed immunohistochemical evaluation of surgical specimens from patients with no prior hormone therapy and CRPC. The expression and activation of molecules involved in this pathway was evaluated with antibodies specific for AR, phospho-cSrc Y419, phospho-aPKC T410, and the cell proliferation-associated antigen, Ki-67. In addition, we examined the abundance of phosphorylated Pak1 (T212), the most distinguishing effecter molecule of Rac1 (22, 23), to assess the activity of Rac1. We confirmed that the antiphosphorylated Pak1 (T212) antibody signal in western blot assays reflected the activity of Rac1 in HEK293 cells (Supplemental Fig. 4).
Cases 1 and 2 show the representative immunostaining of specimens from hormone-naive and CRPC, respectively (Fig. 5). As for the former population, immunohistochemical evaluation was performed by using a tissue microarray (TMA) consisting of samples from 67 Japanese patients (184 spots) (14). Our TMA series showed that positive immunostaining was observed in 105 (57%), 97 (52%), 147 (80%), 85 (46%), and 101 (54%) spots for anti-AR, phospho-cSrc Y419, Rac1, phospho-Pak1 T212, and phospho-aPKC T410 antibodies, respectively. Immunostaining intensity of these molecules was significantly stronger in prostate cancer cells than in adjacent normal epithelial cells (P < 0.001) (Fig. 5 and Table 1). To clarify the significance of the activation of cSrc-Rac1-aPKC pathway in the cell proliferation of this population, we examined the correlation of the results of immunostaining with Ki-67 labeling index (LI). When the spots were stratified by Ki-67 LI with a cutoff of 5% according to previous reports (24, 25), 52 out of 55 (93%) samples that demonstrated positive staining for all of four antibodies exhibited high Ki-67 LI, whereas 37 out of 38 spots (97%) with negative staining for all antibodies showed low Ki-67 LI. Cochran-Armitage test for trend demonstrated a significant association between an increasing number of positive stainings of the antibodies evaluated and high Ki-67 LI (P < 0.0001; Supplemental Table 1). Therefore, these results suggested that the activation of cSrc-Rac1-aPKC pathway might be associated with higher ratio of proliferating cells in hormone-naive cancer.
Fig. 5.
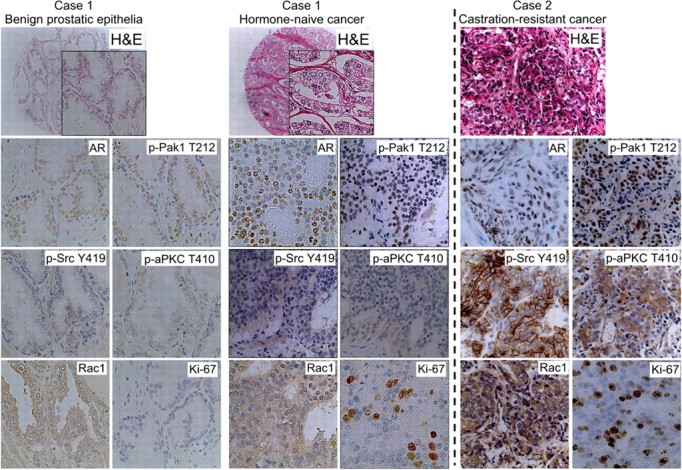
Expression of AR, phospho-Src (Y419), phospho-Pak1 (T212), and phospho-aPKC (T410) in benign prostatic epithelial, hormone-naïve, and castrate-resistant human prostate cancer tissues. Representative immunohistochemical stainings of prostate cancer specimens from two individual patients are shown. Case 1, Gleason 3+3 hormone-naive prostate cancer, and adjacent benign prostatic epithelia obtained by radical prostatectomy undergone for prostate cancer as the primary treatment. Case 2, CRPC specimen obtained by transurethral resection for local relapse after androgen-deprivation therapy. Original magnification, ×200.
Table 1.
Immunostainability of the molecules in AR-cSrc-Rac1-aPKC axis in hormone-naive (n = 184) and castration-resistant (n = 30) human prostate cancer specimen
| Antibody | Benign epithelium (n = 74) | Hormone-naive (n = 184) | Castration-resistant (n = 30) | |||
|---|---|---|---|---|---|---|
| AR1 | ||||||
| (−) | 48 (0.65) | 79 (0.43) | 4 (0.13) | |||
| (+) | 26 (0.35) | 91 (0.49) | 11 (0.37) | |||
| (2+) or (3+) | 0 (0) | 14 (0.08) | 15 (0.50) | |||
| Phospho-cSrc Y4191 | ||||||
| (−) | 62 (0.84) | 87 (0.47) | 0 (0) | |||
| (+) | 12 (0.16) | 76 (0.41) | 5 (0.17) | |||
| (2+) or (3+) | 0 (0) | 21 (0.11) | 25 (0.83) | |||
| Rac11 | ||||||
| (−) | 54 (0.73) | 37 (0.20) | 0 (0) | |||
| (+) | 20 (0.27) | 136 (0.74) | 2 (0.07) | |||
| (2+) or (3+) | 0 (0) | 11 (0.06) | 28 (0.93) | |||
| Phospho-Pak1 T2121 | ||||||
| (−) | 63 (0.85) | 99 (0.54) | 0 (0) | |||
| (+) | 11 (0.15) | 77 (0.42) | 8 (0.27) | |||
| (2+) or (3+) | 0 (0) | 8 (0.04) | 22 (0.73) | |||
| Phospho-aPKC T4101 | ||||||
| (−) | 57 (0.77) | 83 (0.45) | 0 (0) | |||
| (+) | 17 (0.23) | 76 (0.41) | 7 (0.23) | |||
| (2+) or (3+) | 0 (0) | 25 (0.14) | 23 (0.77) | |||
| Ki-67 labeling index1 | ||||||
| <5.0% | 56 (0.76) | 96 (0.52) | 0 (0) | |||
| 5.0% or more | 18 (0.24) | 88 (0.48) | 30 (1.00) | |||
Figures in parentheses represent the proportion.
P < 0.001, Fisher’s exact test.
As for CRPC samples, we observed moderately to strongly positive immunostaining by antiphospho-cSrcY419 antibody in 83% of cases in accordance with a previous report (10). Importantly, more than 90% of samples were positively stained by Rac1 in the same degree. In fact, moderate to strong immunostaining was observed in more than 70% of cases by antibodies to phospho-Pak1 T212 and phospho-aPKC T410 with high Ki-67 LI. Additionally, the intensity of immunostaining was significantly higher in CRPC samples than those from hormone-naive cancer (Table 1). Collectively, molecules involved in the activation of Rac1 appeared to be activated in the majority of specimens in our series of CRPC. Although the numbers of specimens are too small to draw firm conclusions, these results suggested that this pathway could be a potent therapeutic target in the treatment of CRPC. Further analyses of additional samples from different populations will be necessary to confirm the clinical relevance of this pathway in CRPC.
Discussion
Previously, we reported that activation of aPKC-S6K pathway is required for the proliferation of prostate cancer cells, LNCaP, PC3, and DU145 cells (14). Our present study showed for the first time that Rac1 is an upstream regulator of aPKC in prostate cancer cells. It was also revealed that cSrc-Rac1-aPKC pathway constituted a serial signal transduction cascade in androgen-dependent proliferation of LNCaP cells. Importantly, androgen- dependent activation of cSrc was reported to induce activation of Rac1 and to cause changes in the actin assembly in NIH3T3 cells, expressing endogenous AR (26). As for the relationship between AR and Rac1 in LNCaP cells, it was revealed that the activation of this small G protein was observed in 1 h after androgen stimulation (Fig. 6), and this result highly suggested that AR activate Rac1 via nongenotropic action, possibly through cSrc. Importantly, inactive cSrc is localized at perinuclear sites and activated cSrc is translocated to the cell periphery (27, 28). Notably, G proteins such as Rac1 and CDC42 regulate the intracellular localization of cSrc and Src’s biological effects (29). Thus, cSrc and Rac1 build a feed forward loop.
Fig. 6.

Effect of androgen stimulation and Rac1 inhibition on the phosphorylation levels of aPKC in LNCaP cells. LNCaP cells were grown in FBS or CSFBS flowed by stimulation with R1881 and/or Rac1 inhibitor. Total cellular protein was separated by SDS-PAGE, and then indicated proteins were examined by immunoblotting. Densitometry readings are normalized to total protein and reported as averages from two independent experiments.
At present, it is still unclear how signals through AR activate aPKC. Because a maximum effect of AR reached at 72 h after androgen stimulation, these results suggested that activity of aPKC was dependent on the genotropic effects of the AR. For example, it is reported that aPKC is activated by signals through phosphatidylinositol kinase, Ras, IL-1, or TNF-α (30). Additionally, it is suggested that the binding of partitioning defective 6 homolog α (PAR-6) to aPKC causes the suppression of its kinase activity and the binding of activated Rac1 to PAR-6 releases this suppression (30). Therefore, one possible mechanism is that nongenotropic action of AR releases the PAR-6-mediated repression of aPKC via Rac1, and then, the genotropic mechanism of AR activates upstream signals of this kinase.
Rac1 is a member of Rho family small GTPases and had been reported to regulate actin reorganization, membrane ruffling, and the formation of lamellipodia (31). In fact, it has been reported that cSrc regulates the activation of Rac1 in NIH3T3 and HEK293 cells (15, 16). Rac1 is also known to regulate cellular signal transduction pathways that control gene expression, cell cycle progression, cell growth, apoptosis, and differentiation (19, 32, 33, 34, 35, 36, 37).
Previously, it was reported that the activation of cSrc could promote prostate cancer cell growth under androgen-depleted conditions by the up-regulation of AR transcriptional activity through the phosphorylation of AR Y534 (10). According to more recent reports, Src family kinases are stimulated by a kind of neuropeptides and activate various downstream effectors promoting oncogenic cell growth and migration of prostate cancer cells in the absence of androgen (38, 39). Because cSrc activates Rac1 as well, we examined the roles of Rac1 in cSrc-mediated androgen-independent cell proliferation in LNCaP cells. We found that Rac1 activation had little effect on AR transactivation function. In contrast, the treatment of LNCaP cSrcY530F cells with Rac1 inhibitor decreased the proportion of cells in S phase in cell cycle analysis and reduced the cell proliferation. Thus, these results indicate that Rac1 activation is required for cSrc-mediated androgen-dependent cell proliferation of LNCaP cells.
Currently, there is limited knowledge on the role of Rac1 in prostate cancer carcinogenesis and progression. Engers et al. (40, 41) reported that expression of both Rac1 and Tiam1, a guanine-exchanging factor of Rac1, were elevated in prostate cancer and intraepithelial neoplasia compared with benign epithelium and that the level of their expression was positively associated with poor postoperative disease-free survival. In addition, it was reported that lower gene expression of Rac GTPase-activating protein in epithelial cultures freshly isolated from the prostatectomy tissues was one of the significant predictors for aggressive clinical behavior of the disease (42). With regard to androgen-independent progression in cultured prostate cancer cells, Knight-Krajewski et al. (43) reported that androgen-independent prostate cancer cells showed higher activity of Rac1 compared with those retaining an androgen dependency. It was also demonstrated that Rac1 regulated the activity of nuclear factor-κB (NF-κB) (44). Interestingly, the activity of NF-κB was up-regulated in androgen-independent prostate cancer xenografts compared with their androgen-dependent counterparts (45). In accordance with these reports, LNCaP C4-2 and AILNCaP cells, the syngeneic androgen-independent sublines from LNCaP cells harbored abundant Rac1-GTP. Furthermore, we showed that forced expression of a constitutively active form of Rac1 promoted G1/S transition and caused androgen-independent cell proliferation in LNCaP cells. Thus, these results suggest that the activation of Rac1 might be closely related to the androgen-independent progression of prostate cancer cells. Importantly, Wang et al. (46) reported that AR selectively up-regulates M phase cell-cycle genes in androgen-independent cells, including UBEC2, a gene that inactivates the M phase checkpoint. Thus, androgen-independent cell proliferation might be up-regulated by both the activation Rac1 for promotion of G1/S transition and expression of UBEC2 for escaping from M phase checkpoint.
Notably, more than 70% of specimens from CRPC patients were stained strongly with Rac1 and activated form of aPKC. As for aPKC, we previously presented that aPKC regulated the activity of p70 S6K (S6K1) in LNCaP cells (14). Intriguingly, Cinar et al. (47) reported that siRNA knocking down of S6K1 reduced the abundance of AR in the same cells. Therefore, these results imply that AR-aPKC also build a feed forward loop. Collectively, cSrc-Rac1-aPKC pathway might reinforce the roles of AR in CPRC. Thus, the most important implication derived from the present study is that Rac1 can be a potent therapeutic target in the management of advanced prostate cancers, especially those in castrate-resistant status. In fact, the inhibition of Rac1 repressed cell cycle progression at G1/S transition in androgen-independent, C4-2, AILNCaP, PC3, and DU145 prostate cancer cells.
Although LNCaP C4-2 and AILNCaP cells harbored almost comparable levels of Rac1-GTP and the sensitivity to the inhibition of Rac1, a significant difference was observed in their sensitivity to Src family kinase inhibitor to the cell proliferation. These results suggest that molecules other than cSrc might also regulate the activity of Rac1 in AILNCaP cells. In this regard, it is reported that Rac1 is activated by various upstream signals through other than cSrc such as focal adhesion kinase, Ras, and phosphatidylinositol kinase (48, 49, 50). To date, unlike Ras, direct mutational activation of Rac1 and other Rho GTPases in human cancers has never been identified (23, 51, 52). Consistently, no mutations or transcript variants of Rac1 were identified in prostate cancer cell lines used in the present study (data not shown).
Collectively, our present results in both in vitro and in vivo highly suggested that Rac1 and signaling pathways through this molecule could be a potent therapeutic target of prostate cancer especially for that in castration resistant status. For this purpose, it is further required to clarify the mechanisms of activation of Rac1 other than cSrc and the roles of its downstream target (i.e. aPKC, S6K, Pak1, or NF-κB) in prostate cancer cells (13, 22, 23, 44, 53, 54).
Materials and Methods
Antibody and reagents
Antiphospho-p70S6K (T389) and antiphospho-Src (Y419) antibodies were obtained from Cell Signaling Technology (Beverly, MA). Anti-AR, antiphospho-PKCζ (T410) for phospho-aPKC, anti-PKCζ for aPKC, anti-cSrc (C-18), anti-β-tubulin and anti-S6K were obtained from Santa Cruz Biotechnology, (Santa Cruz, CA). Anti-Rac1 was obtained from Upstate Biotechnology (Lake Placid, NY). Anti-HA was purchased from Covance (Berkeley, CA), and antiphospho-Pak1 (T212) from Sigma (St. Louis, MO). Myristoylated PKCζ-pseudosubstrate inhibitor, SU6656, and Rac1 inhibitor were purchased from Calbiochem (San Diego, CA). Bicalutamide was obtained from Toronto Research Chemicals (Toronto, Ontario, Canada) and R1881 (methyltrienolone) from DuPont Merck Pharmaceutical (Boston, MA).
Cell culture
LNCaP, PC3, DU145, and HEK293 cells were obtained from the American Type Culture Collection (Rockville, MD). AILNCaP cells were described previously (14). The cells were cultured routinely in RPMI 1640 (LNCaP, PC3, DU145, and LNCaP C4-2; Invitrogen, Carlsbad, CA) or DMEM (HEK293; Invitrogen) supplemented with 10% (vol/vol) FBS at 37 C in incubators with humidified air and 5% CO2. For androgen depletion, phenol red-free RPMI 1640 (GIBCO, Freiburg, Germany) supplemented with 10% (vol/vol) CSFBS (Hyclone, Logan, UT) was used. AILNCaP was cultured in androgen- depleted condition. For cell counting, cells were collected by trypsinization, and viable cells were counted by trypan blue dye exclusion using hemocytometer.
Expression construct and transfection
Retroviral plasmids pBABE-puro HA-Rac1V12 or N17 were generated by subcloning Rac1V12/N17 from pMxRac1V12/N17 (kind gift from M. Hattori and N. Minato, Kyoto, Japan) into the BamHI and EcoRI sites of pBABE-puro HA (kind gift from Dr. Kondo, Yokohama, Japan) (55). pcDNA3.1(+) HA-Rac1V12/N17 was generated by subcloning HA-Rac1V12/N17 into the HindIII and EcoRI sites of the mammalian expression vector pcDNA3.1(+)/myc-His C (Invitrogen) as it does not express myc and polyhistidine epitopes. The coding sequence of human cSrcY530F was PCR amplified using primers with Y530F mutation and BglII/EcoRI overhangs and cloned into the BamHI and EcoRI sites of the mammalian expression vector pcDNA3.1(+)/myc-His C (Invitrogen) because it does not express myc and polyhistidine epitopes. pEGFP-N1 H2B was generated as described previously (56). Nuclear expression of histone-GFP fusion protein was confirmed by transient transfection to HEK293 cells (data not shown). All constructs were amplified by PCR, and DNA sequences were verified using ABI PRISM 310 genetic analyzer. Transfection of pcDNA3.1(+) and pEGFP was performed in DMEM or RPMI 1640 with or without 10% FBS using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
Colony-forming assay
Cells (1 × 105) were grown on 6-cm dishes, transfected with 8 μg pcDNA3.1 as described above, and cultured in appropriate medium containing 1 mg/ml of G418 for 2–4 wk. The cells were washed with PBS, fixed with neutral-buffered formaldehyde (10%), and stained with 0.1% of crystal violet-H2O.
Immunoblotting and Rac1 pull-down assay
Cell lysis for SDS-PAGE followed by immunoblotting was done as previously reported (14). Results of quantitative densitometric analysis for the abundances of indicated proteins are also determined. All densitometry readings were normalized to total protein or b-actin and reported as averages from three independent experiments. The abundance of Rac1-GTP was examined by pull-down assay using Rac1 activation assay Biochem kit (no. BK035; Cytoskeleton, Inc., Denver, CO) following manufacturer’s instructions.
Flow cytometry and apoptosis assay
Flow cytometry for cell cycle analysis was done as previously reported (14). In apoptosis assays, cells (1 × 105) were grown on 6-cm dishes, transfected by 1 μg pEGFP-N1 H2B with or without 12.5 ng of control or Rac1 (Ambion ID120600 and 120602) siRNA, using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. We confirmed that expression of Rac1 protein satisfactorily knocked down by the siRNA transfection in HEK293 cells (Supplemental Fig. 4). Apoptosis was assessed 48 h later in more than 200 cells with the use of a fluorescent microscope with a ×40 objective. Apoptotic cells were determined by the nuclear morphology with chromatin condensation and/or nuclear fragmentation, compared with normal cell nuclei stained homogeneously.
AR reporter assays
AR reporter assays to determine AR transactivity were performed as previously described (57). Briefly, LNCaP, PC3, or DU145 human prostate cancer cells were inoculated into 24-well plates at 1.5 × 105 per well in phenol red-free RPMI 1640 supplemented with charcoal/dextran-treated fetal calf serum and were transiently cotransfected with 500 ng of p5.3PSAp-Luc and 5 ng of pTK-RL with (DU145) or without (LNCaP) 50 ng of human wt-AR expression vector (pCMV AR-wt), by the use of Lipofectamine 2000 reagent (Invitrogen). Expression vector (50 ng) for mock, cSrcY530F or Rac1V12 were also cotransfected in some experiments as indicated. Cells were stimulated with synthesized androgen R1881 24 h after transfection along with indicated inhibitors. An additional 24 h later, luciferase activity of the cell lysate at the indicated concentration of R1881, bicalutamide, SU6656, or Rac1 inhibitor was determined using Dual-Luciferase Reporter Assay System (Promega, Madison, WI) with a luminometer (MicroLumat Plus LB96V; Berthold Technologies, Bad Wildbad, Germany).
In vivo xenograft model for tumorigenesis
Cells were collected by trypsinization and 1 × 107 cells were suspended in 100 μl RPMI 1640/BD Matrigel (1:1) and injected sc to the right flank of male BALB/c AnNCrj nude mice. Tumor volumes were measured weekly with a caliper using the formula, a × b2 × 0.52, where a is the largest diameter and b is the largest diameter perpendicular to a. When the tumor volume reached 150 mm3 (∼6–7 mm in diameter), the tumor-bearing mouse was surgically castrated (d 0). All experiments involving laboratory animals were done in accordance with the Guideline for Animal Experiments of Kyoto University and approved by Animal Research Committee at Kyoto University Graduate School of Medicine.
Prostate cancers TMA and immunohistochemistry
Hormone-naive prostate cancer tissues were derived from radical prostatectomy specimens of 67 localized prostate cancer patients at Kyoto University Hospital. TMAs were constructed from these specimens as previously described (14, 24). Hormone-refractory prostate cancer tissues were derived from specimen of palliative transurethral resection (n = 29) or lymph node dissection (n = 1) of the prostate gland of 30 patients who had developed hormone-refractory prostate cancer at least against androgen deprivation therapy. Standard indirect immunoperoxidase procedures with monoclonal and polyclonal antibodies were applied to detect phospho-Src Y419 (1:75), anti-Rac1 (1:500), phospho-Pak1 T212 (1:200), and phospho-aPKC T410 (1:100).
Immunopositivity of phospho-cSrc was graded into negative, weak, moderate, and strong as shown in Supplemental Fig. 5, and the strongest staining for more than 10% cells was scored for each case. AR immunopositivity was graded semiquantitatively as negative (no staining), weak (light immunostaining involving <10% of the cells), moderate (light-to-moderate immunostaining involving 10–50%), and strong (moderate-to-strong immunostaining involving >50%). Immunostainability of Ki-67 was evaluated as previously described and reported as Ki-67 LI (24, 25). With regard to Rac1 (58), phospho-Pak1 (59), and phospho-aPKC (14), immunopositivity was graded as described previously. All pathological evaluations were performed by two of the authors (T.Ko. and Y.M.) independently.
Statistical analysis
Data are presented as mean ± sd. Statistical differences between two groups of data were analyzed using the Student’s t test. Statistical significances of immunopositivity were examined by Fisher’s exact test and Cochran-Armitage test for trend as indicated. In the experiments of pharmacological inhibition of cell proliferation, 50% inhibitory concentrations (IC50) were calculated with XLfit 4 (IDBS, Emeryville, CA). Statistical significance was applied to P values of less than 0.05.
Acknowledgments
We thank Dr. Masakazu Hattori and Nagahiro Minato (Kyoto, Japan) for supplying pMx Rac1V12; Dr. Keiichi Kondo (Yokohama, Japan) for pBABE-puro HA; Dr. Isao Kanatani, Dr. Masayuki Imamura, and Dr. Hiromitsu Negoro for the technical assistance on the luminometer MicroLumat Plus LB96V for the AR transactivation assay; Dr. Asumi Yokota for cell cycle analysis using fluorescence-activated cell sorting; Dr. William G. Kaelin, Jr. (Boston, MA) for critical perusal of the manuscript; Drs. Makoto Mark Taketo and Michiyuki Matsuda for helpful advice and discussion; members of Cancer Research Course in Graduated Courses for Integrated Research Training, including Dr. Hisato Jingami; all members of the Ogawa’s lab (Kyoto University Graduate School of Medicine, Kyoto, Japan); and the skilful technical assistance of Tomoko Matsushita, Chie Hagihara, and Megumi Kuraguchi.
Footnotes
This work was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Yamaguchi Endocrine Research Association, the Japanese Foundation for Prostate Research, the Organon Urology Academia, the First Young Research Grant from Japanese Urological Association, the Kobayashi Institute for Innovative Cancer Chemotherapy, the Takeda Science Foundation, and the Formation for Genomic Analysis of Disease Model Animals with Multiple Genetic (Center of Excellence Program), Ministry of Education, Culture, Sports, Science, and Technology, Japan. T.Ko. is a Japan Society for the Promotion of Science Research Fellow.
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 4, 2010
Abbreviations: aPKC, Atypical protein kinase C; AR, androgen receptor; CRPC, castrate-resistant prostate cancer; CSFBS, charcoal-stripped fetal bovine serum; Erk, extracellularly regulated kinase; GFP, green fluorescent protein; GTPases, guanosine triphosphatases; HA, hemagglutinin; LI, labeling index; NF-κB, nuclear factor-κB; PAR-6, partitioning defective 6 homolog α; S6K, S6 kinase; TMA, tissue microarray; wt-AR, wild-type AR.
References
- 1.Ferlay J, Bray F, Pisani P, Parkin D2001. Globocan 2000: cancer incidence, mortality and prevalence worldwide. Version 1.0. Lyon, France: IARC Press
- 2.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ2006. Cancer statistics, 2006. CA-Cancer J Clin 56:106–130 [DOI] [PubMed] [Google Scholar]
- 3.Huggins C, Hodges CV1941. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res 1:293–297 [DOI] [PubMed] [Google Scholar]
- 4.Heinlein CA, Chang C2004. Androgen receptor in prostate cancer. Endocr Rev 25:276–308 [DOI] [PubMed] [Google Scholar]
- 5.Unni E, Sun S, Nan B, McPhaul MJ, Cheskis B, Mancini MA, Marcelli M2004. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res 64:7156–7168 [DOI] [PubMed] [Google Scholar]
- 6.Zhoul J, Hernandez G, Tu SW, Huang CL, Tseng CP, Hsieh JT2005. The role of DOC-2/DAB2 in modulating androgen receptor-mediated cell growth via the nongenomic c-Src-mediated pathway in normal prostatic epithelium and cancer. Cancer Res 65:9906–9913 [DOI] [PubMed] [Google Scholar]
- 7.Inoue T, Kobayashi T, Terada N, Shimizu Y, Kamoto T, Ogawa O, Nakamura E2007. Roles of androgen-dependent and -independent activation of signal transduction pathways for cell proliferation of prostate cancer cells. Expert Rev Endocrinol Metab 2:689–704 [DOI] [PubMed] [Google Scholar]
- 8.Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F2000. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J 19:5406–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai SJ, Ma AH, Tepper CG, Chen HW, Kung HJ2006. Inappropriate activation of the androgen receptor by nonsteroids: involvement of the Src kinase pathway and its therapeutic implications. Cancer Res 66:10449–10459 [DOI] [PubMed] [Google Scholar]
- 10.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, Veenstra TD, Chen H, Qiu Y2006. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10:309–319 [DOI] [PubMed] [Google Scholar]
- 11.Chang YM, Kung HJ, Evans CP2007. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia 9:90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans CP, Lara PN, Kung H, Yang JC, Activity of the Src-kinase inhibitor AZD0530 in androgen-independent prostate cancer (AIPC): pre-clinical rationale for a phase II trial. Proc ASCO Annual Meeting, Atlanta, GA, 2006
- 13.Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, Mirosevich J, Lee FY, Jove R2005. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res 65:9185–9189 [DOI] [PubMed] [Google Scholar]
- 14.Inoue T, Yoshida T, Shimizu Y, Kobayashi T, Yamasaki T, Toda Y, Segawa T, Kamoto T, Nakamura E, Ogawa O2006. Requirement of androgen-dependent activation of protein kinase Cζ for androgen-dependent cell proliferation in LNCaP Cells and its roles in transition to androgen-independent cells. Mol Endocrinol 20:3053–3069 [DOI] [PubMed] [Google Scholar]
- 15.Marignani PA, Carpenter CL2001. Vav2 is required for cell spreading. J Cell Biol 154:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Servitja JM, Marinissen MJ, Sodhi A, Bustelo XR, Gutkind JS2003. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem 278:34339–34346 [DOI] [PubMed] [Google Scholar]
- 17.Goichberg P, Kalinkovich A, Borodovsky N, Tesio M, Petit I, Nagler A, Hardan I, Lapidot T2006. cAMP-induced PKCζ activation increases functional CXCR4 expression on human CD34+ hematopoietic progenitors. Blood 107:870–879 [DOI] [PubMed] [Google Scholar]
- 18.Noda Y, Takeya R, Ohno S, Naito S, Ito T, Sumimoto H2001. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells 6:107–119 [DOI] [PubMed] [Google Scholar]
- 19.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y2004. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA 101:7618–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobel RE, Sadar MD2005. Cell lines used in prostate cancer research: a compendium of old and new lines—part 1. J Urol 173:342–359 [DOI] [PubMed] [Google Scholar]
- 21.Brown JM, Wouters BG1999. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 59:1391–1399 [PubMed] [Google Scholar]
- 22.Kumar R, Gururaj AE, Barnes CJ2006. p21-activated kinases in cancer. Nat Rev Cancer 6:459–471 [DOI] [PubMed] [Google Scholar]
- 23.Sahai E, Marshall CJ2002. RHO-GTPases and cancer. Nat Rev Cancer 2:133–142 [DOI] [PubMed] [Google Scholar]
- 24.Inoue T, Segawa T, Shiraishi T, Yoshida T, Toda Y, Yamada T, Kinukawa N, Kinoshita H, Kamoto T, Ogawa O2005. Androgen receptor, Ki67, and p53 expression in radical prostatectomy specimens predict treatment failure in Japanese population. Urology 66:332–337 [DOI] [PubMed] [Google Scholar]
- 25.Bubendorf L, Sauter G, Moch H, Schmid HP, Gasser TC, Jordan P, Mihatsch MJ1996. Ki67 labelling index: an independent predictor of progression in prostate cancer treated by radical prostatectomy. J Pathol 178:437–441 [DOI] [PubMed] [Google Scholar]
- 26.Castoria G, Lombardi M, Barone MV, Bilancio A, Di Domenico M, De Falco A, Varricchio L, Bottero D, Nanayakkara M, Migliaccio A, Auricchio F2004. Rapid signalling pathway activation by androgens in epithelial and stromal cells. Steroids 69:517–522 [DOI] [PubMed] [Google Scholar]
- 27.Sefton BM, Trowbridge IS, Cooper JA, Scolnick EM1982. The transforming proteins of Rous sarcoma virus, Harvey sarcoma virus and Abelson virus contain tightly bound lipid. Cell 31:465–474 [DOI] [PubMed] [Google Scholar]
- 28.Yeatman TJ2004. A renaissance for SRC. Nat Rev Cancer 4:470–480 [DOI] [PubMed] [Google Scholar]
- 29.Timpson P, Jones GE, Frame MC, Brunton VG2001. Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Curr Biol 11:1836–1846 [DOI] [PubMed] [Google Scholar]
- 30.Suzuki A, Akimoto K, Ohno S2003. Protein kinase C λ/ι (PKCλ/ι): a PKC isotype essential for the development of multicellular organisms. J Biochem 133:9–16 [DOI] [PubMed] [Google Scholar]
- 31.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70:401–410 [DOI] [PubMed] [Google Scholar]
- 32.Bar-Sagi D, Hall A2000. Ras and Rho GTPases: a family reunion. Cell 103:227–238 [DOI] [PubMed] [Google Scholar]
- 33.Benitah SA, Frye M, Glogauer M, Watt FM2005. Stem cell depletion through epidermal deletion of Rac1. Science 309:933–935 [DOI] [PubMed] [Google Scholar]
- 34.Buscemi N, Murray C, Doherty-Kirby A, Lajoie G, Sussman MA, Van Eyk JE2005. Myocardial subproteomic analysis of a constitutively active Rac1-expressing transgenic mouse with lethal myocardial hypertrophy. Am J Physiol Heart Circ Physiol 289:H2325–H2333 [DOI] [PubMed]
- 35.Etienne-Manneville S, Hall A2002. Rho GTPases in cell biology. Nature 420:629–635 [DOI] [PubMed] [Google Scholar]
- 36.Stappenbeck TS, Gordon JI2000. Rac1 mutations produce aberrant epithelial differentiation in the developing and adult mouse small intestine. Development 127:2629–2642 [DOI] [PubMed] [Google Scholar]
- 37.Van Aelst L, D'Souza-Schorey C1997. Rho GTPases and signaling networks. Genes Dev 11:2295–2322 [DOI] [PubMed] [Google Scholar]
- 38.Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP2008. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene 27:6365–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang JC, Ok JH, Busby JE, Borowsky AD, Kung HJ, Evans CP2009. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res 69:151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engers R, Mueller M, Walter A, Collard JG, Willers R, Gabbert HE2006. Prognostic relevance of Tiam1 protein expression in prostate carcinomas. Br J Cancer 95:1081–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engers R, Ziegler S, Mueller M, Walter A, Willers R, Gabbert HE2007. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr Relat Cancer 14:245–256 [DOI] [PubMed] [Google Scholar]
- 42.Nanni S, Priolo C, Grasselli A, D'Eletto M, Merola R, Moretti F, Gallucci M, De Carli P, Sentinelli S, Cianciulli AM, Mottolese M, Carlini P, Arcelli D, Helmer-Citterich M, Gaetano C, Loda M, Pontecorvi A, Bacchetti S, Sacchi A, Farsetti A2006. Epithelial-restricted gene profile of primary cultures from human prostate tumors: a molecular approach to predict clinical behavior of prostate cancer. Mol Cancer Res 4:79–92 [DOI] [PubMed] [Google Scholar]
- 43.Knight-Krajewski S, Welsh CF, Liu Y, Lyons LS, Faysal JM, Yang ES, Burnstein KL2004. Deregulation of the Rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21(CIP1) levels in androgen-independent human prostate cancer cells. Oncogene 23:5513–5522 [DOI] [PubMed] [Google Scholar]
- 44.Cammarano MS, Minden A2001. Dbl and the Rho GTPases activate NFκB by IκB kinase (IKK)-dependent and IKK-independent pathways. J Biol Chem 276:25876–25882 [DOI] [PubMed] [Google Scholar]
- 45.Chen CD, Sawyers CL2002. NF-κB activates prostate-specific antigen expression and is up-regulated in androgen-independent prostate cancer. Mol Cell Biol 22:2862–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, JänneOA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M2009. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cinar B, De Benedetti A, Freeman MR2005. Post-transcriptional regulation of the androgen receptor by mammalian target of rapamycin. Cancer Res 65:2547–2553 [DOI] [PubMed] [Google Scholar]
- 48.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC2005. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer 5:505–515 [DOI] [PubMed] [Google Scholar]
- 49.Auer KL, Contessa J, Brenz-Verca S, Pirola L, Rusconi S, Cooper G, Abo A, Wymann MP, Davis RJ, Birrer M, Dent P1998. The Ras/Rac1/Cdc42/SEK/JNK/c-Jun cascade is a key pathway by which agonists stimulate DNA synthesis in primary cultures of rat hepatocytes. Mol Biol Cell 9:561–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aoki K, Nakamura T, Fujikawa K, Matsuda M2005. Local phosphatidylinositol 3,4,5-trisphosphate accumulation recruits Vav2 and Vav3 to activate Rac1/Cdc42 and initiate neurite outgrowth in nerve growth factor-stimulated PC12 cells. Mol Biol Cell 16:2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boettner B, Van Aelst L2002. The role of Rho GTPases in disease development. Gene 286:155–174 [DOI] [PubMed] [Google Scholar]
- 52.Jaffe AB, Hall A2002. Rho GTPases in transformation and metastasis. Adv Cancer Res 84:57–80 [DOI] [PubMed] [Google Scholar]
- 53.Chou MM, Blenis J1996. The 70 kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell 85:573–583 [DOI] [PubMed] [Google Scholar]
- 54.Lambert JM, Karnoub AE, Graves LM, Campbell SL, Der CJ2002. Role of MLK3-mediated activation of p70 S6 kinase in Rac1 transformation. J Biol Chem 277:4770–4777 [DOI] [PubMed] [Google Scholar]
- 55.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin Jr WG2002. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 1:237–246 [DOI] [PubMed] [Google Scholar]
- 56.Kanda T, Sullivan KF, Wahl GM1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8:377–385 [DOI] [PubMed] [Google Scholar]
- 57.Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, Kamoto T, Ogawa O2005. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res 65:9611–9616 [DOI] [PubMed] [Google Scholar]
- 58.Liu SY, Yen CY, Yang SC, Chiang WF, Chang KW2004. Overexpression of Rac-1 small GTPase binding protein in oral squamous cell carcinoma. J Oral Maxillofac Surg 62:702–707 [DOI] [PubMed] [Google Scholar]
- 59.Wang RA, Zhang H, Balasenthil S, Medina D, Kumar R2006. PAK1 hyperactivation is sufficient for mammary gland tumor formation. Oncogene 25:2931–2936 [DOI] [PubMed] [Google Scholar]