

Abstract
The active estrogen estradiol (E2) stimulates breast cancer cell (BCC) growth, whereas the androgen dihydrotestosterone (DHT) has shown an antiproliferative effect. The principal product synthesized by the 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) is E2, although we have demonstrated that the purified enzyme also inactivates DHT. However, the direct roles of 17β-HSD1 in sex-hormone regulation and BCC proliferation have not been completely established. Here, we show that 17β-HSD1 inhibition suppresses DHT catabolism by 19%, whereas knockdown of the gene expression increases the concentration of DHT by 41% in the T47D BCC line. The 17β-HSD1/DHT complex crystal structure reveals that DHT binds in both normal and reverse modes, but the latter mode leading to O3 reduction is preferred with stronger interactions. Using RNA interference and an inhibitor of 17β-HSD1, we demonstrate that 17β-HSD1 expression is negatively correlated to DHT levels in BCC but positively correlated to estrone reduction, E2 levels, and cell proliferation. 17β-HSD1 inhibition reduces DHT inactivation, increasing the antiproliferative effect by DHT in T47D cells after 8 d treatment. Thus, 17β-HSD1 up-regulates BCC growth by a dual action on estradiol synthesis and DHT inactivation. We have further demonstrated that 17β-HSD1 can enhance the E2-induced expression of the endogenous estrogen-responsive gene pS2, providing an important information regarding the modulation of the estrogen responsiveness by 17β-HSD1 that may also contribute to BCC growth. These results strongly support the rationale for inhibiting 17β-HSD1 in breast cancer therapy to eliminate estrogen activation via the sulfatase pathway while avoiding the deprivation of DHT.
17β-HSD Type 1 activates estrogen, inactivates androgen and modulates the transcription of the endogenous estrogen-responsive gene pS2.
Breast cancer is the most frequent cancer in women and one of the most prevalent cancers in humans (1, 2). In the Western world, breast cancer affects approximately one in 10 women and is the leading cause of death among those in their 40s (1). Most of breast cancer is initially hormone dependent (3, 4). The potent estrogenic hormone estradiol (E2) stimulates the growth of cancer cells and is obligatory for the induction and progression of hormone-dependent breast cancers (5, 6). Indeed, a high concentration of E2 in breast cancer cells (BCC) increases their proliferation rate (7). For therapeutic applications, several strategies including the inhibition of the synthesis of E2 have been deployed to reduce its mitogenic effect.
Two principal pathways are implicated in the final steps of E2 activation in breast cancer tissue. The aromatase pathway transforms androgens into estrogens (8), and the sulfatase pathway converts estrone sulfate (E1S) into estrone (E1) by estrone sulfatase (9, 10), followed by E1 conversion into the potent E2 by the action of reductive 17β-hydroxysteroid dehydrogenases (17β-HSD) (11, 12, 13). Quantitative evaluation indicates that in human breast tumors, E1S via sulfatase is a much more likely precursor for E2 than androgens via aromatase (14). 17β-HSD type 1 (17β-HSD1) remains an important enzyme for E2 production because it can use E1 as substrate from both aromatase and sulfatase pathways, and it principally synthesizes E2 using reduced nicotinamide adenine dinucleotide (NADPH) as cofactor (11, 15). Moreover, the expression and activity of 17β-HSD1 are significantly higher in breast cancer than in normal breast tissue (12), and it has been suggested that this higher expression could explain the elevated E2 concentration in breast tumors (16). Previously, we reported the first observation of the inactivation of the potent androgen dihydrotestosterone (DHT) into 5α-androstane-3β,17β-diol (3β-diol) and androstanedione (A-dione) by homogeneous 17β-HSD1 (17). An antiproliferative effect of DHT has been observed in MCF7 and ZR-75-1 estrogen-dependent BCC lines (18, 19, 20). In fact, in the presence of estrogens, androgens inhibit the estrogenic-stimulated growth of estrogen-dependent tumors via androgen receptor (AR) mediation (21, 22). Until now, despite all of these observations, the direct contribution of 17β-HSD1 to the proliferation of BCC and the regulation of the two potent steroid hormones E2 and DHT has not been completely defined. RNA interference has already been successfully used to knock down genes and allow the study of their biological role in target cells (23). Using small interfering RNA (siRNA), in conjunction with a steroidal inhibitor of 17β-HSD1 previously reported (24), we established here the direct role of 17β-HSD1 in 1) DHT inactivation in BCC, 2) the resulting DHT level, 3) the E2 level in cells, and 4) the proliferation of BCC.
Because the structure of 17β-HSD1 was determined as the first example of any human steroid-converting enzyme (25, 26, 27), the structure-function relationship for this enzyme has been studied (17, 28). Recent progress in crystallization techniques has provided us with better tools to directly demonstrate the structural basis for the substrate binding site and the multispecificity of 17β-HSD1. It was of particular importance to study the binding of DHT with this enzyme, because this androgen plays an important role in decreasing breast cancer proliferation. Thus, in this study, a high-quality structure of the 17β-HSD1/DHT complex was solved to determine how 17β-HSD1 can bind the most potent androgen DHT as a substrate in addition of its cognate estrogens and catalyze DHT conversion.
Results
17β-HSD1 expression and inhibition in different human BCC lines
The mRNA transcription of endogenous 17β-HSD1 in two hormone-dependent BCC lines (MCF7 and T47D), in which both estrogen receptor (ER) and AR are expressed, was evaluated by semiquantitative RT-PCR. Analysis showed higher endogenous 17β-HSD1 mRNA expression in T47D cells than in MCF7 cells (Fig. 1A).
Fig. 1.

17β-HSD1 expression and knockdown by siRNA. A, Semiquantitative RT-PCR was performed using 17β-HSD1 and β-actin primers and total RNA extracted from T47D and MCF7 cells. T47D expressed significantly higher 17β-HSD1 mRNA than MCF7 cells. B, Western blot was carried out with protein extracted from MCF7 cells simultaneously cotransfected with 17β-HSD1 plasmid and different concentrations of three 17β-HSD1 siRNA or control siRNA (SNC1 siRNA) using 17β-HSD1 antibody. The band represents 17β-HSD1 protein.
To determine whether siRNA can effectively knock down the expression of 17β-HSD1 in cells, three siRNA against 17β-HSD1 and negative control siRNA were selected and used. MCF7 cells were simultaneously cotransfected with both 17β-HSD1 plasmid and specific siRNA or 17β-HSD1 plasmid and control siRNA. Total protein was extracted from cells 48 h after transfection and was analyzed by Western blot using a 17β-HSD1 antibody. The siRNA were used at several concentrations (1–25 nm) to evaluate their specificity in detail. The specificity and knockdown effect of siRNA were confirmed, because no 17β-HSD1 band or a very weak band was obtained when 17β-HSD1 siRNA were used. In contrast, a 17β-HSD1 band of high intensity was obtained in the presence of control siRNA (Fig. 1B).
DHT inactivation in BCC by endogenous 17β-HSD1
Using homogeneous 17β-HSD1 protein and HEK-293 cells stably transfected with 17β-HSD1, we previously showed that 17β-HSD1 inactivated DHT (17). Here, we performed a complete analysis including the use of 17β-HSD1 inhibitor and transfected and nontransfected cells to test whether 17β-HSD1 inactivates DHT in intact cells, particularly in BCC. First, DHT was used as the substrate for an enzyme activity assay, without the addition of any cofactor, in wild-type (wt) HEK-293 cells (used as control cells) and in HEK-293 cells stably transfected with 17β-HSD1 recombinant plasmid. Western blot showed a significant increase in 17β-HSD1 protein level in transfected HEK-293 cells (Fig. 2A). The latter cells were cultured in the absence and presence of the 17β-HSD1 inhibitor. The reduced products [as a mixture of 3β-diol and 5α-androstane-3α,17β-diol (3α-diol)] formed by the stably transfected HEK-293 cells were five times higher than wt HEK-293 cells (Fig. 2B, left). The mixture of 3β-diol and 3α-diol (3β/3α-diol) formed in stably transfected HEK-293 cells dropped by 81 and 85% in the presence of 1 and 10 μm inhibitor, respectively (Fig. 2B, left), confirming that DHT reduction is predominantly catalyzed by 17β-HSD1 in these cells. The 3β-reduction (inactivation) of DHT by 17β-HSD1 is supported by the observation that 240 min after incubation of the cells with DHT, the amount of DHT remaining in the medium of the stably transfected HEK-293 cells cultured in the absence of inhibitor decreased by 20% (94 to 74%) compared with less than 2% when the cells were cultured in the presence of the 17β-HSD1 inhibitor (Fig. 2B, right).
Fig. 2.

DHT inactivation by 17β-HSD1. A, Western blot was carried out using a 17β-HSD1 antibody and total protein extracted from wt HEK-293 and HEK-293 cells stably transfected with 17β-HSD1. B, Activity assays were carried out in wt and stably transfected HEK-293 cells using [14C]DHT as substrate. The 17β-HSD1 inhibitor was used to inhibit its activity in the stably transfected cells. The assay was carried out for four incubation times. The percentage of [14C]DHT substrate remaining in the cell supernatant after 240 min incubation is shown on the left. C, Total protein extracts from MCF7 cells were used in activity assays (c1 and c2) and Western blot (c3) to evaluate the correlation between 17β-HSD1 expression, DHT reduction, and E1 conversion to E2: c1, conversion of [14C]E1 into [14C]E2 after 30 min incubation; c2, reduction of [14C]DHT after 60 min incubation. Numbers (1–3), represent, respectively, protein from untransfected MCF7 cells, protein from MCF7 cells cotransfected with both 17β-HSD1 plasmid and control siRNA (SNC1 siRNA), and protein from MCF7 cells cotransfected with both 17β-HSD1 plasmid and specific siRNA (siRNA2). D, Activity assay carried out in wt T47D cells using [14C]DHT as substrate. CTL represents the control cells incubated without inhibitor. The spots representing the oxidized and reduced products on TLC plate after activity assay in the control cells were shown in E. Numbers (0–3), Periods (in hours) of cell incubation. Error bars, sd. *, P < 0.05 by Student’s t test. Note that 3β/3α-diol represents a mixture of 3β-diol and 3α-diol.
To evaluate the correlation between 17β-HSD1 expression and DHT inactivation as well as E2 synthesis, MCF7 cells were cotransfected with both 17β-HSD1 plasmid and siRNA, and activity assays (conversion of E1 into E2 after 30 min incubation and reduction of DHT after 60 min incubation) were performed along with Western blot. Analysis revealed that MCF7 cells in which 17β-HSD1 expression was knocked down by its specific siRNA did not exhibit a 17β-HSD1 band, whereas MCF7 cotransfected with 17β-HSD1 and control siRNA exhibited a strong 17β-HSD1 band. This consequently led to significantly less DHT inactivation as well as less E2 production in the former cells than in the latter (Fig. 2C). Thus, the expression of 17β-HSD1 is positively correlated with DHT inactivation as well as with E2 production, but the effect of 17β-HSD1 on this latter activity is, at least, 8.3 (based on the activities per milligram protein, i.e. the specific activity) times higher than its effect on DHT inactivation under these experimental conditions. In fact, the specific activities of the enzyme from MCF7 cells cotransfected with both 17β-HSD1 plasmid and control siRNA (data obtained from Fig. 2C) were 49.9 and 6.0 nmol/min · mg total protein for E1 conversion into E2 and DHT reduction, respectively. Although the estrogenic activity is much higher, it is important to study this androgen inactivation, because nanomolar level DHT plays an important role in BCC proliferation (19, 20).
We then investigated whether endogenous 17β-HSD1 is able to inactivate DHT in wt BCC. T47D cells were chosen because the expression of endogenous 17β-HSD1 is higher in this cell line than in MCF7 cells. Using DHT as the substrate, activity assays were performed in the presence and absence of the 17β-HSD1 inhibitor. DHT reduction into 3β/3α-diol was inhibited by 19% in the presence of 10 μm inhibitor (Fig. 2D), indicating that endogenous 17β-HSD1 inactivates DHT in BCC. DHT was also oxidized into A-dione, but the analysis was focused on the reduction of DHT because the oxidized product was negligible compared with the reduced product (Fig. 2E).
Because 3β-diol and 3α-diol migrated to the same level on thin-layer chromatography (TLC) plates, they were indistinguishable by this method. To examine whether endogenous 17β-HSD1 in BCC reduces DHT into 3β-diol or 3α-diol, gas chromatography/mass spectrometry (GC/MS) analysis was performed using the reduced product collected from the TLC plates. In wt MCF7 cells expressing a low quantity of 17β-HSD1, the reduced products of DHT were comprised of 6% 3β-diol and 94% 3α-diol. In cells that expressed high levels of 17β-HSD1 (HEK-293 stably transfected with 17β-HSD1 and MCF7 stably transfected with 17β-HSD1 and T47D), the quantity of 3β-diol formed increased to 30%, whereas the quantity of 3α-diol formed decreased to 70% (Table 1). These new results were intriguing because the present study showed that 17β-HSD1 contributes to more than 85% DHT inactivation in stably transfected HEK-293, whereas liquid chromatography-mass spectrometry analysis indicated that the homogeneous 17β-HSD1 reduces DHT into 3β-diol in the presence of NADPH (17). We thus used the crystallizable 17β-HSD1 protein for an in vitro activity assay, and the reduced product was analyzed by GC/MS after 35 and 85% DHT conversion. For these two DHT conversions by homogeneous 17β-HSD1, the reduced products were comprised of 91% 3β-diol and only 9% 3α-diol (Table 1).
Table 1.
Percentage of 3β-diol vs. 3α-diol formed after DHT reduction: quantification by GC/MS analysis
| 3β-Diol concentration (%) | 3α-Diol concentration (%) | |
|---|---|---|
| Activity assays in intact cells | ||
| wt MCF7 | 6 | 94 |
| MCF7 stably transfected with 17β-HSD1 | 17 | 83 |
| wt T47D | 30 | 70 |
| HEK-293 stably transfected with 17β-HSD1 | 30 | 70 |
| In vitro activity assay with purified 17β-HSD1 protein (NADPH used as cofactor) | 91 | 9 |
Effects of 17β-HSD1 on the concentrations of DHT and E2 in BCC
To establish the direct impact of 17β-HSD1 expression on DHT and E2 levels in BCC, enzyme immunoassay and ELISA were carried out using cell culture supernatants (cell medium). The E2 concentrations in the medium of wt MCF7 cells and MCF7 stably transfected with 17β-HSD1 recombinant plasmid were first compared. Western blot showed an increased level of 17β-HSD1 protein in stably transfected MCF7 cells (Fig. 3A). The level of E2 in the medium of stably transfected MCF7 cells was significantly higher than that of wt MCF7 cells. The E2 increase in stably transfected MCF7 was significant (2-fold higher) at 48 h incubation (Fig. 3B). From 24–48 h incubation, the E2 level increased by 20% in wt MCF7 cells but by 120% in stably transfected MCF7 cells (Fig. 3B), demonstrating a rapid increase in E2 catalyzed by the overexpressed 17β-HSD1 in the latter cells.
Fig. 3.

Hormone level modulations by 17β-HSD1. A, Western blot showing 17β-HSD1 expression in wt MCF7 and MCF7 cells stably transfected with 17β-HSD1. B, E2 level in MCF7 cells after 17β-HSD1 overexpression. The wt MCF7 cells and stably transfected MCF7 cells were cultured under the same conditions, and 24 and 48 h after cell plating, E2 level in cell supernatants was measured by enzyme immunoassay. C, Western blot (top) and semiquantitative RT-PCR (bottom) showing 17β-HSD1 expression after transfection of T47D cells with 17β-HSD1 siRNA (mixed siRNA2 and siRNA3) or control siRNA (mixed SNC1 siRNA and scramble siRNA). D and E, E2 (D) and DHT (E) levels in T47D cells after the knockdown of 17β-HSD1 gene expression. T47D cells were transfected with 17β-HSD1 siRNA and control siRNA, and 24 and 48 h after transfection, E2 and DHT levels in cell supernatants were measured by enzyme immunoassay and ELISA analysis, respectively. All experiments were repeated three times so that each data point, expressed in picograms per milliliter, is a mean of six replicates. Error bars represent sd. *, P < 0.05 by Student’s t test.
The effects of the knockdown of endogenous 17β-HSD1 expression (by siRNA) on E2 and DHT levels were then tested in T47D cells. To demonstrate the knockdown of endogenous 17β-HSD1 expression by siRNA, T47D cells were transfected with 50–100 nm mixed 17β-HSD1 siRNA or mixed control siRNA, and total protein and RNA were extracted from cells 48 h after transfection and analyzed by Western blot or semiquantitative RT-PCR, respectively. The use of mixed 17β-HSD1-specific siRNAs was to achieve higher inhibition of endogenous expression. 17β-HSD1 protein was decreased by about 70% (Fig. 3C, top), whereas its mRNA was decreased by more than 90% (Fig. 3C, bottom) when 17β-HSD1-specific siRNA were used, indicating that siRNA efficiently knocked down the expression of endogenous 17β-HSD1 in T47D cells. When 17β-HSD1-specific siRNA was used to knock down 17β-HSD1 expression for 48 h, the level of E2 in the T47D cell medium was 66% lower than when control siRNA was used (Fig. 3D). However, the DHT concentration in these 17β-HSD1-siRNA-transfected T47D cells was 41% higher than in T47D transfected with control siRNA (Fig. 3E). These results revealed that 17β-HSD1 positively modulates E2 concentration and negatively modulates DHT concentration in BCC. This positive and negative modulation was observed from 24–48 h after transfection, the time course over which E2 concentration increased and DHT concentration decreased in the control siRNA-transfected T47D cell medium. Meanwhile, the E2 concentration remained almost unchanged in the 17β-HSD1 siRNA-transfected T47D cell medium (Fig. 3, D and E). These observations suggest a time-course effect of 17β-HSD1 in the modulation of these two hormones.
Effects of E2 and DHT on BCC growth
To evaluate the direct impact of E2 and DHT on BCC, T47D cells were cultured for 6 d in steroid-deprived medium (dextran-coated charcoal-treated medium) and in the same medium supplemented with 0.1 nm E2. The addition of E2 to the medium was based on previous reports (21, 22, 29) stating that androgens inhibit the estrogenic-stimulated growth of estrogen-dependent tumors in the presence of estrogens. Results showed that addition of E2 (0.01, 0.1, and 1 nm) significantly increased (92–328%) T47D cell proliferation, whereas addition of 0.01 and 0.1 nm DHT had no significant effect on the cell proliferation in the steroid-deprived medium. However, both DHT concentrations (0.01 and 0.1 nm) significantly decreased by 35 and 26% the growth of T47D cells induced by E2 in the E2-supplemented medium, respectively (Fig. 4A).
Fig. 4.

Effect of E2 and DHT on BCC cell growth. A, T47D cells (40,000) were cultivated 6 d in dextran-coated charcoal-treated medium or in this medium containing 0.1 nm E2 and supplemented with 0.01–1 nm E2 or DHT. Media were changed every 2 d. MTT analyses were carried out after incubation time to evaluate cell growth. B, T47D cells (40,000) were transfected with 200 nm 17β-HSD1 siRNA or control siRNA for 48 h, and then cells were incubated with the indicated concentrations of E2 and DHT or with both steroids for 8 d before evaluation of cell growth by MTT analysis. C, One day after plating 40,000 cells per well in 24-well plates, wt MCF7 and stably transfected MCF7 cells were incubated with the indicated concentrations of E2 and DHT or with DHT plus 0.1 nm E2 for 4 d. Cell growths were then evaluated by MTT analysis. CTL represents the control cells incubated without any steroid. The dotted line indicates the growth level of cells when cultivated without any steroid. All experiments were repeated two to three times with three replicates at each time and were carried out in dextran-coated charcoal-treated medium. Error bars represent sd. *, P < 0.05 vs. 0 or 0.1 nm E2 by Student’s t test.
We further evaluated the correlation between 17β-HSD1 expression and the decrease effect of DHT on E2-stimulated growth. T47D cells were transfected with 200 nm 17β-HSD1-specific siRNA (mixed siRNA1-3, Table 2) or with control siRNA for 48 h. Then, cells were incubated with E2 or DHT or with the mixture of both steroids in charcoal-treated medium for 8 d before the evaluation of cell growth. The concentration of 0.01 nm DHT was used for this analysis, because it demonstrates a significant effect in this cell line after 6 d treatment (Fig. 4A). Under these new experimental conditions, results revealed that in 17β-HSD1 siRNA- and control siRNA-transfected cells, DHT alone (0.01 nm) did not affect cell growth, whereas E2 alone (0.01 or 0.1 nm) significantly increased cell growth. DHT addition in the medium with 0.1 nm E2 decreases the E2-stimulated growth by a more significant effect (17.4%) in the 17β-HSD1 siRNA-transfected cells compared with the control siRNA-transfected cells (Fig. 4B), showing that diminution of 17β-HSD1 expression can enhance the DHT effect in inhibiting E2-stimulated growth. We then evaluated the effect of DHT on the E2-stimulated growth in wt MCF7 cells and in stably transfected MCF7 cells overexpressing 17β-HSD1. Cells were cultured during 4 d in charcoal-treated medium supplemented with DHT or DHT plus 0.1 nm E2. Analyses showed that both 0.1 and 1 nm DHT significantly decrease the E2-stimulated growth in MCF7 cells (about 23–24% decrease), whereas no significant effect of DHT was observed at 0.1 nm in the stably transfected cells expressing high a level of 17β-HSD1 (Fig. 4C). Thus, in the presence of low DHT concentration (0.1 nm), 17β-HSD1 can modulate the DHT effect in inhibiting E2-stimulated growth in MCF7 cells.
Table 2.
siRNA sequences for 17β-HSD1
| siRNA name | Sense sequence (5′–3′) | Antisense sequence (5′–3′) |
|---|---|---|
| siRNA1 | GCUGGACGUGAAUGUAGUA[dT][dT] | UACUACAUUCACGUCCAGC[dT][dT] |
| siRNA2 | GCCUUUCAAUGACGUUUAU[dT][dT] | AUAAACGUCAUUGAAAGGC[dT][dT] |
| siRNA3 | CCACAGCAAGCAAGUCUUU[dT][dT] | AAAGACUUGCUUGCUGUGG[dT][dT] |
Effects of 17β-HSD1 expression and activities on BCC proliferation
To investigate the impact of 17β-HSD1 activities on BCC proliferation, T47D cells were treated for 12 d with varying concentrations of 17β-HSD1 inhibitor in complete growth medium. The growth of T47D cells was reduced by 16 and 53% with 1 and 5 μm 17β-HSD1 inhibitor, respectively (Fig. 5A, left). Decreased cell proliferation in the presence of the inhibitor was not due to cell death (no dead cells were found in the medium when cells were observed under an optic microscope, before each change of the medium) or to an antiestrogenic effect [a previous study (24) showed that the inhibitor was not antiestrogenic]. To verify whether the 17β-HSD1 inhibitor itself stimulates apoptosis or induce cytotoxicity at the used concentrations, fetal bovine serum (FBS) was treated with dextran-coated charcoal to remove any steroid present in the serum and medium, and cells were cultivated in the presence of the inhibitor in the resulting charcoal-treated medium. Both 1 and 5 μm inhibitor stimulates cell growth in the absence of any steroid, with a significant and higher stimulation observed at 5 μm (Fig. 5A, right). These observations revealed that the inhibitor does not stimulate apoptosis or induce cytotoxicity at 1 and 5 μm. Thus, the decreased cell proliferation in the complete growth medium in the presence of these two concentrations of the inhibitor should be attributed to the inhibition of 17β-HSD1 activities, rather than cell toxicity.
Fig. 5.
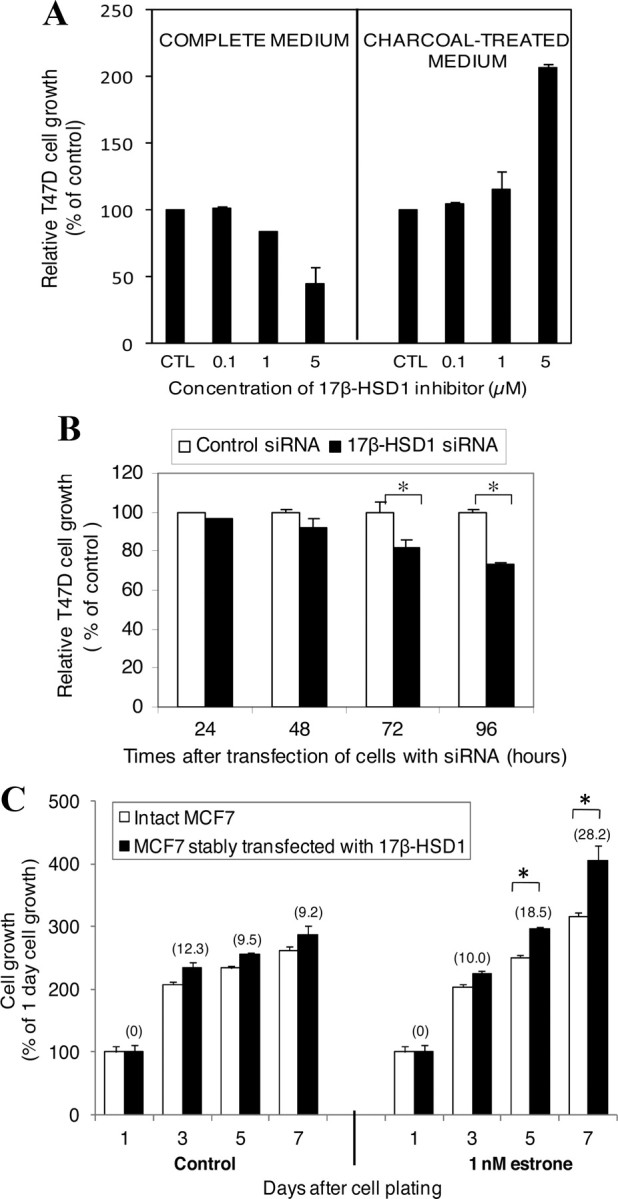
BCC proliferation after 17β-HSD1 inhibition and overexpression. A, T47D cells (7,500–15,000) were cultured for 12 d in complete medium or in dextran-coated charcoal-treated medium containing the 17β-HSD1 inhibitor at different concentrations (0.1–5 μm), and cell growth was compared by MTT analysis. CTL represents the control cells incubated without inhibitor. Control is fixed as 100%. B, T47D cells (50,000) were transfected with 100 nm 17β-HSD1 siRNA and control siRNA, and cell growth was compared by MTT analysis 24, 48, 72, and 96 h after transfection. C, Wild-type MCF7 cells (25,000) and stably transfected MCF7 cells were cultivated under the same conditions in dextran-coated charcoal-treated medium and plated in several wells of 24-well plates for incubation. At 24 h after cell plating, triplicate wells were used to evaluate the cell growth 1 d after plating (1 d cell growth) by MTT analysis. In parallel, old medium in the remaining wells was replaced by charcoal-treated medium (control) or this medium supplemented with 1 nm E1, and incubation was continued with a medium change every 2 d. MTT analyses were performed to evaluate cell growth several times (3–7 d) after cell plating. The numbers in parentheses represent the percentage of increase of transfected MCF7 cell growth compared with wt MCF7 cells for each time interval. Each treatment was carried out in triplicate, and the tests were repeated at least two times. Error bars represent sd. *, P < 0.05 by Student’s t test.
The contribution of 17β-HSD1 expression to BCC proliferation was investigated by knockdown of 17β-HSD1 expression in T47D cells and also studied by comparing the proliferation rates of wt MCF7 cells, and MCF7 cells in which the enzyme was overexpressed. T47D cells transfected with 17β-HSD1-specific siRNA exhibited significantly reduced growth rates (18 and 27% reduction at 72 and 96 h after transfection, respectively) compared with T47D cells transfected with control siRNA (Fig. 5B). When wt MCF7 cells and MCF7 cells stably transfected with 17β-HSD1 were cultivated under the same conditions in dextran-coated charcoal-treated medium supplemented with 1 nm E1, the stably transfected cells were observed to have a significant 18.5 and 28.2% higher proliferation rate than the wt cells 5 and 7 d after cell plating, respectively. In a steroid-deprived medium (dextran-coated charcoal-treated medium), the stably transfected cells showed a higher but nonsignificant proliferation than the wt cells (Fig. 5C). These observations show the direct enhancement of BCC proliferation by 17β-HSD1 expression, which activates E1.
Effect of 17β-HSD1 on the estrogen responsiveness of the pS2 gene
When cells were treated with E2, T47D cells transfected with 17β-HSD1 siRNA grew slower than cells transfected with control siRNA while increasing 17β-HSD1 expression by stable transfection in MCF7 cells led to a more pronounced cell growth increase compared with the nontransfected wt cells (Fig. 6A, left and right). To further confirm the effect of 17β-HSD1 expression on BCC proliferation, the changes in the expression of an estrogen-responsive gene (ERG) were studied. A well known ERG, pS2, was chosen for this experiment. T47D cells were transfected with 17β-HSD1-specific siRNA or control siRNA for 48 h. Then, cells were incubated with 1 nm E2 in charcoal-treated medium for an additional 48 h. Total RNA was extracted from cells and used for the evaluation of pS2 mRNA expression by quantitative real-time RT-PCR analysis. Treatment of control siRNA-transfected cells with E2 led to an 11-fold higher pS2 mRNA than in the absence of E2, confirming the estrogen responsiveness of this gene. In the absence of E2, knockdown of 17β-HSD1 by its specific siRNA led to a 20-fold decrease of pS2 mRNA compared with control siRNA. In the presence of E2, the mRNA level of pS2 gene was significantly decreased (by 14 times) when 17β-HSD1 was knocked down (Fig. 6B). These results showed that 17β-HSD1 enhances the endogen expression and the estrogen responsiveness of the pS2 gene.
Fig. 6.

Estrogen responsiveness modulation by 17β-HSD1. A, T47D cells were transfected with 17β-HSD1 siRNA or control siRNA for 48 h, and then cells were incubated with 0.01 and 0.1 nm E2 for 8 d (left); MCF7 cells and stably transfected MCF7 cells were cultivated in the presence of 1 nm E2 for 1 and 7 d (right). Experiments were carried out in charcoal-treated medium, and cell growth was evaluated by MTT analysis. CTL represents the control cells incubated without any steroid. B, T47D cells were transfected with 17β-HSD1-specific siRNA or with control siRNA for 48 h, and then cells were incubated with 1 nm E2 or ethanol vehicle (EtOH) for 48 h more. Total RNA was extracted from cells and used for the evaluation of pS2 mRNA expression by quantitative real-time RT-PCR analysis. Error bars represent sd. *, P < 0.05 by Student’s t test.
Crystal complex structure of 17β-HSD1/DHT
The 17β-HSD1/DHT complex crystals were obtained by soaking the apoenzyme crystals, and the complex formation was confirmed after structure determination. The complex structure, solved at 1.7 Å resolution, included one molecule of 17β-HSD1 in the asymmetric unit with one molecule of DHT, one molecule of glycerol, and 85 molecules of water and a final R-factor (Rfactor) of 21.6% and free R-factor (Rfree) of 24%. The electron density of the loop, consisting of residues 190–201 was missing, similar to other 17β-HSD1 complexes (26, 27) and so was not included in the final refined structure. DHT was introduced in the second round of refinement. It was first introduced in the normal binding mode as was reported in the previous structure (PDB id 1DHT). The refinement of the complex structure with DHT in the normal mode resulted in a B-factor for the ligand as high as 90 Å2, whereas the occupancy for the ligand was 1.0, and the B-factor for the surrounding residues was 47 Å2. This hinted at the possibility of the presence of an alternative binding mode of DHT and prompted us to refine the reverse-oriented DHT with occupancy 1.0. The refinement of the B-factor still resulted in values as high as 80 Å2. Such a high B-factor in either conformation meant a high perturbation for the ligand. In the present binary complex, the occupancy for DHT in the normal mode was reduced to 0.5, which resulted in lowering the B-factor to 55 Å2, and for the reverse orientation at occupancy of 0.5 the B-factor was 45 Å2. The refinement for both DHT orientations was carried out separately, and the electron density showed a perfect fit for either binding mode except for the B-ring. This suggested a higher occupancy for the reverse binding mode (Fig. 7, A and B). The H-bonding as seen in Fig. 7C shows the possibility of an H-bond only with His221 in the reverse binding orientation with no H-bonds present in the normal binding mode of DHT with the enzyme. The cofactor NADP was introduced to both structures by modeling and the distances between the O3/O17 (reverse/normal mode) with the NC4 of the nicotinamide of NADP was 4.35/3.75 Å and between Tyr155 to NC4 of NADP was 5.4 Å (Fig. 7D).
Fig. 7.

Crystal complex structure of 17β-HSD1/DHT. A and B, Electronic density of DHT for 2Fo-Fc map seen at 0.8ς cutoff in reverse binding mode (A) and normal binding mode (B). C, Stereo representation showing the H-bond of DHT with the residues His221 in the reverse binding mode (DHT represented in blue), whereas in the normal binding mode, there is no H-bond interaction present (DHT in green). D, Distances between DHT, Tyr155, and the cofactor NADP in 1) reverse mode (the distance between the O3 of DHT with NC4 of NADP as 4.35 Å and between Tyr155 to NC4 of NADP as 5.4 Å) and in 2) normal mode (the distance between the O17 of DHT with NC4 of NADP as 3.75 Å and between Tyr155 to NC4 of NADP as 5.4 Å). Note that the final model of the complex has been submitted with the protein data bank with PDB code 3KLM.
Discussion
Contribution of 17β-HSD1 to E2 accumulation in BCC
Measuring the level of E2 in BCC is important because it is the most potent estrogen playing a major role in the development and progression of hormone-dependent breast cancer. In breast cancers, 17β-HSD1 is involved in the final step of E2 synthesis (11). In contrast to 17β-HSD1, 17β-HSD type 2 (17β-HSD2) inactivates E2 by catalyzing its oxidation into E1. Our results show that knockdown of 17β-HSD1 expression in T47D cells significantly reduces the E2 level (by 66%), whereas overexpression of 17β-HSD1 in MCF7 cells increases this steroid concentration 2-fold. Under our experimental conditions, the cell number may not significantly change to affect the E2 and DHT measurements after 24 and 48 h cell incubation when there were not significant differences in the growths of cells from different treatments (see Fig. 4, B and C). Thus, these results demonstrate a positive correlation between 17β-HSD1 expression and the level of E2 in BCC that could be important. To evaluate the contribution of 17β-HSD2 in the reduction of E2 level, this enzyme was overexpressed in T47D cells to achieve a higher level of expression than 17β-HSD1 (30). The E2 level remained unchanged in such 17β-HSD2-overexpressed T47D cells when compared with the wt T47D cells in which the 17β-HSD1 expression was higher than 17β-HSD2. Taken together, these results indicate that the reductive activity of 17β-HSD1 predominates over the effect of the oxidative activity of 17β-HSD2 for the regulation of the E2 level in BCC because we found that knockdown or overexpression of 17β-HSD1 alone was sufficient to significantly change the E2 level in BCC medium.
Physiological significance of DHT inactivation by 17β-HSD1
Our results showed that endogenous 17β-HSD1 enzyme inhibition by one of its inhibitors results in a 19% decrease of DHT reduction into a mixture of 3β-diol and 3α-diol in wt T47D cells. This demonstrates that endogenous 17β-HSD1 contributes to at least 19% of DHT inactivation in BCC. Quantitative analysis showed that the reduced product of DHT contains 30% 3β-diol and 70% 3α-diol for T47D and transfected HEK-293 cells, whereas purified 17β-HSD1 reduces DHT mostly (91%) to 3β-diol. Our RT-PCR result showed that endogenous 17β-HSD1 is more highly expressed in T47D cells than in MCF7 cells, and Western blot revealed the presence of the enzyme in MCF7 but not in HEK-293 cells. Taking into account previously published works (30, 31), it is reasonable to assume that 17β-HSD1 is not endogenously expressed in HEK-293 cells. Thus, in the light of these facts, our results suggested that 1) 17β-HSD1 reduces DHT into both 3β-diol and 3α-diol, but 2) 3β-diol may be preferentially synthesized, and 3) the 3β-diol formed may be further transformed in intact cells due to the activity of other endogenous enzymes. Indeed, HEK-293 cells express a high level of cytochrome P540-7B1 (CYP7B1), which can rapidly hydroxylate 3β-diol into 6α- or 7α-triol (32, 33). The 85% inhibition of reduction of DHT in transfected HEK-293 cells compared with 19% inhibition in wt T47D cells and the high percentage of 3α-diol formed in wt MCF7 (94%) cells and in T47D cells (70%) may be due to the presence of other enzymes that reduce DHT, principally into 3α-diol, in these latter two cell types. However, the physiological significance of the reduction of DHT into 3α-diol may be less than the preferential reduction of DHT into 3β-diol by 17β-HSD1 because, in vivo, the formation of 3α-diol is reversible. It can be converted back to DHT via 3α-hydroxysteroid oxidase activity, whereas the formation of the metabolite 3β-diol is irreversible (33, 34). Thus, in contrast to 3α-diol, transformation of DHT into 3β-diol leads to a definite loss of DHT. Moreover, it has been shown that 3β-diol itself possesses some low estrogenicity and induces BCC proliferation and does not bind to the AR but rather to ERα and ERβ (35, 36). The physiological concentration of DHT in BCC is maintained at the nanomolar level (37, 38). The maintenance of this concentration appears to be important for limiting hormone-dependent BCC growth because the addition of physiological levels of DHT to BCC culture decreases the proliferation of these cells (18, 20, 39). In our study, it has been demonstrated that a DHT concentration from 0.01–0.1 nm is extremely important to limit the BCC growth induced by E2, a DHT concentration in the working capacity of 17β-HSD1. This constitutes an important argument to inhibit this estrogenic enzyme.
Structural evidence of the use of DHT as a substrate by 17β-HSD1
The DHT binary complexes solved in the present study revealed the presence of both normal and reverse (alternative) binding modes. The A-dione ternary structure was previously reported during an attempt to obtain ternary complex of DHT/NADP with the 17β-HSD1 enzyme (28), which suggested oxidation of DHT at the O17 position. However, the two orientations observed in the current structure revealed that both the reduction at position O3 as well as oxidation at position O17 were possible. Kinetic studies reporting that DHT could be oxidized to A-dione (Km = 26 ± 6 μm) or reduced to 3β-diol (Km = 32 ± 9 μm) by purified 17β-HSD1 (17) hinted that DHT was able to bind to the enzyme in either mode. This may result in lower occupancy for either mode in the crystal structure. Moreover, activity assays carried out in intact cells (in the present study) showed that the quantity of reduced product (3β/3α-diol) formed is higher than that of the oxidized product (A-dione) in stably transfected HEK-293 cells (data not shown). The current structure supports these observations, because the H-bond interaction is present only in the reverse orientation. The more prominent fitting of the electron density suggests that the reverse orientation (O3) is the preferred mode of binding with a stronger interaction compared with the normal orientation (O17). This is the first time that both binding modes of DHT, as well as the preference for the reverse mode in 17β-HSD1, have been confirmed by crystallography. The dual binding modes were not identified in the earlier 17β-HSD1/DHT structure at 2.24 Å with a B-factor of 70 Å2 for the ligand (PDB id 1DHT). The modeling studies corroborate all the observations as the distance between the cofactor (nicotinamide of NADP), Tyr155, and the O3/O17 (reverse/normal mode) of DHT are within the distance that catalysis can take place.
17β-HSD1 stimulates BCC proliferation via a dual function
Other studies relating to 17β-HSD1 evaluated the estrogenicity of inhibitors (40, 41), effect of E1 administration (7, 42), or effect of 17β-HSD2 (30) on cell proliferation. Here, we have demonstrated the direct impact of 17β-HSD1 itself on the proliferation of BCC: 17β-HSD1 down-regulation results in the decrease of BCC proliferation by at least 27%, whereas increasing the expression of 17β-HSD1 leads to the stimulation of their growth. Moreover, this is the first study that demonstrates the influence of 17β-HSD1 on the level of DHT in BCC: knockdown of the 17β-HSD1 gene expression by siRNA leads to a 41% increase of the DHT level in T47D cells. In fact, 17β-HSD1 increases the level of E2 but diminishes the level of DHT by catalyzing its conversion into 3β/3α-diol. The 19% contribution of 17β-HSD1 to the inactivation of DHT into 3β/3α-diol compared with the 41% change in the level of DHT may be due to the increased expression of the enzyme after its blockade by the inhibitor. Such an effect has been described (43). The dual roles of 17β-HSD1 reveal its important physiological implication in breast cancer, because both contribute to the amplification of the proliferative effect of 17β-HSD1 on BCC growth. In fact, it is well known that E2 increases BCC growth mediated by ER, and DHT was proposed to decrease such estrogen-dependent BCC growth by an AR-mediated mechanisms (18, 20, 21, 22, 44, 45). The present study is in concordance with this statement, because DHT decreases T47D cell growth (by 35 and 26% at 0.01 and 0.1 nm DHT, respectively) stimulated by E2 after 6 d culture in charcoal-treated medium. In addition, inhibition of 17β-HSD1 leads to better demonstration of the 0.01 nm DHT efficacy in decreasing E2-stimulated growth after 8 d treatment in T47D cells, whereas overexpression of the enzyme leads to a loss of the 0.1 nm DHT efficacy in stably transfected MCF7 cells. Taken together, these results indicate that 17β-HSD1 can negatively modulate the DHT effect, which is to decrease BCC growth. A decrease in AR-mediated effects might further stimulate tumor development because a positive AR status is generally considered a good prognosis marker for breast cancer progression (22). Amplification of the proliferative effect of 17β-HSD1 on BCC is further supported by the fact that endogenous 17β-HSD1 inactivates DHT preferentially into 3β-diol, favoring an immediate and irreversible diminution of DHT level. Based on all these observations, we propose that the stimulatory effect of 17β-HSD1 on BCC growth is associated with dual effects on E2 accumulation and DHT inactivation: the interrelated 17β-HSD1 expression and E2 and DHT levels may lead to the control of BCC growth. Thus, in addition to increased estrogen-dependent proliferation [as suggested in the literature (46)], our study indicates that the predominance of 17β-HSD1 in malignant breast tissue may lead to a decreased androgen-dependent antiproliferative effect in hormone-dependent BCC with physiological concentrations of DHT, which is moderate and around 0.01–0.1 nm. Consequently, the rate of breast cancer progression may be accelerated.
Estrogen activity is mediated by its cognate receptor (ER), and the mechanism involves the induction of the receptor activation by E2: occupancy of ER by E2 induces ER conformational changes that allow its interaction with specific enhancers known as estrogen-responsive elements (ERE) present in ERG and with general transcription factors. This may ultimately lead to the expression of ERG as well as stimulation of cell growth and proliferation by altered expression of genes responsible for controlling cell cycle and proliferation (47, 48). The breast cancer estrogen-inducible protein (pS2), which is strongly expressed in breast cancer but barely expressed in normal breast, was chosen in the present study to examine whether 17β-HSD1 can modulate the expression of an ERG. The endogenous pS2 gene expression was down-regulated and its estrogen responsiveness was decreased by 14 times after the knockdown of endogenous 17β-HSD1 expression by siRNA in T47D cells. The two ER isoforms are implicated in cell proliferation in T47D cells, and high levels of ERβ stimulation may lead to decreased cell proliferation, whereas high levels of ERα lead to increased cell proliferation (49). Curtis et al. (50) showed that in contrast with the progesterone receptor (PR) gene, the estrogen responsiveness of pS2 gene was not affected by the knockdown of the endogenous tumor metastasis suppressor protein (nm23-H1). The authors further observed that nm23-H1 interacts with ERα and increases the ERα-ERE complex formation in MCF7 cells and associates with the promoter region of the PR but not with the pS2 ERE region. After our present work, a further study needs to be carried out to elucidate how 17β-HSD1 affects the expression and the estrogen responsiveness of pS2 gene. Therefore, in addition to E2 synthesis, the modulation of the estrogen responsiveness by 17β-HSD1 shown in the present study, may also contribute to BCC growth. This may explain the results obtained in Fig. 6A.
Our decision to use T47D and MCF7 cells as models to study the impact of 17β-HSD1 on BCC was justified not only by the fact that they are the most commonly used cell lines but also because T47D and MCF7 may be considered as reasonable models for breast tumor cells in luminal epithelial-like/ER+/ERBB2− tumors (51). Thus, by comparing the cell lines to breast tumors, we demonstrate that 17β-HSD1 may play a critical role in the cell growth of hormone-dependent breast cancers in patients with a proteomic profile similar to T47D. In such breast cancers, 17β-HSD1 is the major enzyme that produces E2.
In summary, the present study shows that the proliferation of some BCC is significantly ascribable to their high content of 17β-HSD1. The result from this study may be applicable to hormone-dependent BCC expressing a high level of 17β-HSD1 and in which both ER and AR are expressed. In view of these results, it is reasonable to assume that designing and using a highly potent inhibitor of 17β-HSD1 can open new possibilities in clinical applications for breast cancer by blocking E2 formation via the sulfatase pathway and avoiding the deprivation of DHT by the same enzyme. Until now, only selective estrogen receptor modulators and aromatase inhibitors are used in the treatment of hormone-dependent breast cancer. Integrating a potent selective 17β-HSD1 inhibitor in adjuvant therapy or in combination with an existing treatment may help in the regression of related breast cancers.
Materials and Methods
Cell culture
T47D, MCF7, and HEK-293 cells were from the American Type Culture Collection (ATCC, Manassas,VA). MCF7 cells were maintained in DME low-glucose medium supplemented with 1 nm β-estradiol. T47D cells were propagated in DME high-glucose medium containing 7.5 mg/liter bovine insulin (Sigma, Oakville, Ontario, Canada). HEK-293 cells were cultured in MEM. All cells were cultured in the presence of 10% FBS at 37 C in a humidified atmosphere of 95% air and 5% CO2. When indicated, FBS was treated overnight at 4 C with 2% dextran-coated charcoal to remove the remaining steroids present in the serum. For all experiments, phenol red-free media were used with MCF7 and T47D cells, two hormone-dependent BCC lines.
Plasmid construction and stable transfection
Specific primers for the 17β-HSD1 gene (gene HSD17B1; GenBank accession no. NM_000413) were synthesized and used in PCR to amplify human 17β-HSD1 cDNA that had been previously subcloned (52, 53). The fragment thus obtained was inserted in the HindIII/EcoRI site of the pcDNA3.1(+) vector and cloned in bacterial strain TOP10 (Invitrogen, Burlington, Ontario, Canada). The recombinant plasmid was purified from positive colonies by MaxiPrep, followed by phenol-chloroform extraction and ethanol precipitation. A total of 1 × 106 MCF7 and HEK-293 cells were then transfected in six-well plates with the recombinant plasmid (1 and 4 μg, respectively) by using Lipofectamine 2000 (Invitrogen). Stable transfectants were selected for 4 wk with 500 μg/ml G-418 (Invitrogen).
siRNA synthesis and transfections
The sense and antisense sequences of three 17β-HSD1 siRNA (Table 2, siRNA1, siRNA2, and siRNA3) were selected by using the program BLOCK-iT RNAi Designer (Invitrogen). The specificity of siRNA sequences was checked by a BLAST search of the GenBank database. Duplex siRNA of 17β-HSD1 were synthesized and purified by HPLC by GenePharma (Shanghai, China). Transfection of T47D cells with siRNA was carried out using Lipofectamine siRNAMax (Invitrogen), 50–200 nm mixed siRNA duplexes (Table 2), and unless otherwise stated, 40,000–50,000 cells were seeded in 24-well plates with 0.5 ml medium. Simultaneous cotransfection of MCF7 cells with both recombinant plasmid (17β-HSD1) and siRNA was performed using Lipofectamine 2000 and 1 × 106 cells in six-well plates. Silencer Negative Control 1 (SNC1) siRNA (Ambion, Austin, TX) and scramble siRNA (GenePharma) were used as control siRNA in the transfection experiments.
17β-HSD1 steroidal inhibitor
The 2-methoxy-E2 C-16 derivative inhibitor of 17β-HSD1 (24) (compound 18) was used in the present study. This steroidal inhibitor shows a potent inhibition of 17β-HSD1 activity (transformation of [14C]E1 into [14C]E2) in wt T47D cells (81, 69, and 37% inhibition at 10, 1, and 0.1 μm inhibitor, respectively) (24).
Western blot
Total proteins were extracted from cells with CelLytic-M cell lysis reagent (Sigma) supplemented with 1 mm phenylmethylsulfonyl fluoride and 1% protease inhibitors cocktail (EMD Chemicals, Gibbs-town, NJ). Proteins were quantified using the Bradford method, and equal amounts of proteins from each sample were separated on a 12% SDS-PAGE and then electroblotted onto nitrocellulose membranes. The membranes were blocked with 5% nonfat milk in PBS-Tween 20 for 1 h at room temperature. Membranes were then incubated 2 h at room temperature in blocking buffer containing 1:100,000 dilution of the primary anti-17β-HSD1 rabbit monoclonal antibody (clone EP1682Y) (Abcam, Cambridge, MA). For loading control, a 1:7500 dilution of monoclonal anti-β-actin antibody produced in mouse (Sigma) was used. Afterward, membranes were washed and incubated for 1 h with the respective horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 10,000 times. Protein signals were visualized with chemiluminescence reagent (PerkinElmer, Waltham, MA), and bands were quantified by using a Storm imaging system (Molecular Dynamics, Sunnyvale, CA).
Semiquantitative RT-PCR
Total RNA was isolated from cells using Trizol Reagent (Invitogen) and treated by recombinant DNase I (Ambion). Two hundred nanograms of total RNA was subjected to a one-step semiquantitative RT-PCR (54) using Titanium Taq RT Enzyme Mix (Clontech, Mountain View, CA), two primers (55) for human β-actin (used as internal control), and the following 17β-HSD1 primers: 5′-498ATG CGA GAG TCT GGC GGT TCT G519-3′ and 5′-870AGC CTC GGC CCC AGC CTC GGC CTT T894-3′. The RT-PCR program was carried out in the Eppendorf Mastercycler Gradient (Eppendorf, Mississauga, Ontario, Canada). The reverse transcription (RT) was carried out at 50 C for 60 min followed by initial denaturation at 94 C for 5 min. The PCR program was 30 sec at 94 C for denaturation, 30 sec at 65 C for annealing, and 1 min at 68 C for elongation, followed by 10 min final elongation. The number of cycles used for PCR was 40 when comparing 17β-HSD1 RNA levels in T47D and MCF7 cells and 33 for RNA from T47D cells transfected with siRNA. This ensured that the reaction was stopped when amplification of 17β-HSD1 RNA was still in the exponential phase and not in the plateau phase. The PCR products were run on 3% agarose gel in Tris/Borate/EDTA buffer and stained with ethidium bromide. Bands were viewed and photographed on UV light and were quantified by using a Storm device. The ratio between 17β-HSD1 signal and β-actin signal was calculated to determine RT-PCR value.
Quantitative real-time RT-PCR
T47D cells were seeded in six-well plates at 3 × 105 cells per well in charcoal-treated medium and transfected with 200 nm 17β-HSD1-specific siRNA (mixed siRNA1, siRNA2, and siRNA3) or control siRNA using Lipofectamine siRNAMAx. At 48 h after transfection, transfection medium was replaced by fresh charcoal-treated medium containing ethanol vehicle or 1 nm E2. Cell incubation was continued for another 48 h, after which total RNA was extracted from cells using Trizol reagent and sent to the Q_RTPCR Platform service (research center of the Laval University Hospital Center, Québec, Canada) for the mRNA quantification of pS2 by real-time RT-PCR. Total RNA was treated with deoxyribonuclease I before performing the mRNA quantifications as previously described (56) using primers 5′-acg ccc tcc cag tgt gca aat-3′ and 5′-aat ctg tgt tgt gag ccg agg-3′.
Activity assay and GC/MS analysis
Homogeneous 17β-HSD1 protein (obtained as described below) and total protein extracted from MCF7 cells, 72 h after cotransfection with 17β-HSD1 plasmid and siRNA, were used for in vitro activity assays using previous conditions (16). Activity assays with intact HEK-293 and T47D cells were performed at 37 C in a 5% CO2 incubator using 0.5–1 × 106 cells in six-well plates with medium containing charcoal-treated fetal calf serum and 1 μm [14C]E1 or [14C]DHT (American Radiolabeled Chemicals, St. Louis, MO). Medium was supplemented with the 17β-HSD1 inhibitor when testing inhibition of 17β-HSD1 activity. Reaction mixture aliquots were removed at various time intervals, and steroids were extracted using 3 vol diethyl ether and transferred on a TLC plate using 80 μl dichloromethane. Separation of steroids on the TLC plate was done in toluene/acetone (4:1) mixture. TLC plates were allowed to dry and then exposed, and spots were quantified by using a Storm imaging system.
Spots representing the reduced products of DHT were removed from the TLC plate by scraping, steroids present in the spots were extracted and dissolved with a solution of 1% Trisil TBT in isooctane, and a derivatization reaction was performed at 80 C for 15 min. An aliquot (3 μl) was analyzed by GC/MS using an Agilent chromatographic column (DB-17ht, 30 m × 0.250 mm internal diameter; film thickness = 0.15 μm, Agilent Technologies, Mississauga, Ontario, Canada). Gas chromatograph and mass spectrometer were from Agilent, model HP6890 and HP5973, respectively. All the GC/MS analysis was performed by the bioanalytical platform (research center of the Laval University Hospital Center, Québec, Canada).
Determination of E2 and DHT levels
A total of 50,000 wt MCF7 and stably transfected MCF7 cells with the same number of passages were cultured in complete growth media. Also, 50,000 T47D cells were transfected with 100 nm control and 17β-HSD1 siRNA as described above. Cell supernatants were collected from wells at two incubation times (24 and 48 h), split into two equal 200-μl volumes, and immediately frozen at −80 C until time of analyses. The levels of E2 and DHT in the BCC lines supernatant were determined by using a commercial E2 enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI) and DHT ELISA kit (Alpha Diagnostic International, San Antonio, TX) according to the supplier’s protocol. The media aliquots were defrosted and mixed with 500 μl EIA buffer (Cayman Chemical) before the E2 analysis, whereas the aliquots were used without dilution for DHT analysis. Fresh media were used as controls to determine the basal levels of E2 and DHT in the medium before incubation with the cells. Duplicate wells were prepared for each medium to be tested, and plates were read at 412 nm (E2 analysis) and 450 nm (DHT analysis) in a plate reader (Spectra Max 340PC; Molecular Devices, Sunnyvale, CA). The values of fresh medium were removed from values obtained after incubation to evaluate the effect of 17β-HSD1.
Cell proliferation assay
Cell proliferation was evaluated by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (57). Briefly, after the culture period of T47D and MCF7 cells in 24-well plates, the existing medium was replaced by 200 μl fresh medium and 20 μl MTT reagent (ATCC) for 4 h at 37 C. Then, 200 μl detergent reagent (ATCC) were added for overnight incubation at room temperature. Absorbency of each well was measured at 570 nm.
Protein purification, crystallization, and structure determination
The 17β-HSD1 was purified from human placenta as previously described (52, 58), and 16 mg/ml of the obtained homogeneous 17β-HSD1 protein with a specific activity of 7.1 μmol/min · mg was used for crystallization. Apoenzyme crystals were obtained using the vapor diffusion method in a hanging drop setup with 1000 μl well solution consisting of 27% polyethylene glycol (PEG) 4K, 100 mm HEPES buffer (pH 7.5), 0.16 mm MgCl2, and 20% glycerol at 27 C. Crystals appeared after 4 d and were then soaked by gradual addition of DHT over 1 wk until a final concentration of 3 mm was reached.
Data collection was carried out at the X25 beam line at the Brookhaven National Synchrotron facility to 1.7 Å resolution. Data were reduced using HKL and SCALA, and the structures were solved by the difference Fourier method using the protein atomic coordinates for 17β-HSD1 binary complex with E2 (PDB code 1IOL). The structural parameters for DHT were obtained from the PDB. The complex structure was subjected to multiple rounds of refinement using Refmac, alternating with manual inspection in program Coot.
Acknowledgments
We thank Dr. C. Doillon for his advice in the cell culture, from the research center of the Laval University Hospital Center, Québec, Canada. We acknowledge Dr. P. Rehse and Dr. M. Kelly for their critical reading and editing of the manuscript.
NURSA Molecule Pages:
Ligands: 17β-estradiol | Dihydrotestosterone.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) grant MOP57892 (PI: S.-X.L.) and a consortium for operation of a protein crystallographic synchrotron beam-line (by CIHR and Natural Science and Engineering Research Council of Canada).
C.-Q.C was from the Institute of Biochemistry and Cell Biology, Shanghai Institutes of Biological Sciences, the People’s Republic of China, where S.-X.L. is an adjunct professor.
Disclosure Summary: The authors have nothing to disclose.
First Published Online February 19, 2010
Abbreviations: A-dione, Androstanedione; AR, androgen receptor; BCC, breast cancer cell; DHT, dihydrotestosterone; 3α-diol, 5α-androstane-3α,17β-diol; 3β-diol, 5α-androstane-3β,17β-diol; E1, estrone; E2, estradiol; ER, estrogen receptor; ERE, estrogen-responsive element; ERG, estrogen-responsive gene; E1S, E1 sulfate; FBS, fetal bovine serum; GC/MS, gas chromatography/mass spectrometry; 17β-HSD, 17β-hydroxysteroid dehydrogenase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NADPH, reduced nicotinamide adenine dinucleotide; siRNA, small interfering RNA; TLC, thin-layer chromatography; wt, wild type.
References
- 1.Sasco AJ2003. Breast cancer and the environment. Horm Res 60(Suppl 3):50 [DOI] [PubMed]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P2005. Global cancer statistics, 2002. CA Cancer J Clin 55:74–108 [DOI] [PubMed] [Google Scholar]
- 3.Segaloff A1978. Hormones and mammary carcinogenesis. In: McGuire WL, ed. Breast cancer 2: advances in research and treatment. New York: Plenum Press; 1–22
- 4.Kirschner MA1979. The role of hormones in the development of human breast cancer. In: McGuire WL, ed. Breast cancer 3: advances in research and treatment current topics. New York: Plenum Press; 199–226
- 5.Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA2001. Mechanisms of estrogen action. Physiol Rev 81:1535–1565 [DOI] [PubMed] [Google Scholar]
- 6.Pasqualini JR, Chetrite GS2005. Recent insight on the control of enzymes involved in estrogen formation and transformation in human breast cancer. J Steroid Biochem Mol Biol 93:221–236 [DOI] [PubMed] [Google Scholar]
- 7.Miettinen MM, Poutanen MH, Vihko RK1996. Characterization of estrogen-dependent growth of cultured MCF-7 human breast-cancer cells expressing 17β-hydroxysteroid dehydrogenase type 1. Int J Cancer 68:600–604 [DOI] [PubMed] [Google Scholar]
- 8.Bhatnagar AS, Batzl C, Häusler A, Schieweck K, Lang M, Trunet PF1996. Pharmacology of non-steroidal aromatase inhibitors. In: Pasqualini JR, Katzenellenbogen BS, eds. Hormone-dependent cancer. New York: Marcel Dekker; 155–168
- 9.MacIndoe JH1988. The hydrolysis of estrone sulfate and dehydroepiandrosterone sulfate by MCF-7 human breast cancer cells. Endocrinology 123:1281–1287 [DOI] [PubMed] [Google Scholar]
- 10.Pasqualini JR, Gelly C, Nguyen BL, Vella C1989. Importance of estrogen sulfates in breast cancer. J Steroid Biochem 34:155–163 [DOI] [PubMed] [Google Scholar]
- 11.Nguyen BL, Chetrite G, Pasqualini JR1995. Transformation of estrone and estradiol in hormone-dependent and hormone-independent human breast cancer cells. Effects of the antiestrogen ICI 164,384, danazol, and promegestone (R-5020). Breast Cancer Res Treat 34:139–146 [DOI] [PubMed] [Google Scholar]
- 12.Pasqualini JR2004. The selective estrogen enzyme modulators in breast cancer: a review. Biochim Biophys Acta 1654:123–143 [DOI] [PubMed] [Google Scholar]
- 13.Aka JA, Mausumi M, Lin SX2009. Reductive 17β-hydroxysteroid dehydrogenases in the sulfatase pathway: critical in the cell proliferation of breast cancer. Mol Cell Endocrinol 301:183–190 [DOI] [PubMed] [Google Scholar]
- 14.Santner SJ, Feil PD, Santen RJ1984In situ estrogen production via the estrone sulfatase pathway in breast tumors: relative importance versus the aromatase pathway. J Clin Endocrinol Metab 59:29–33 [DOI] [PubMed] [Google Scholar]
- 15.Poutanen M, Isomaa V, Peltoketo H, Vihko R1995. Role of 17β-hydroxysteroid dehydrogenase type 1 in endocrine and intracrine estradiol biosynthesis. J Steroid Biochem Mol Biol 55:525–532 [DOI] [PubMed] [Google Scholar]
- 16.Vermeulen, A, Deslypere, JP, Paridaens R1986. Steroid dynamics in the normal and carcinomatous mammary gland. J Steroid Biochem 25:799–802 [DOI] [PubMed] [Google Scholar]
- 17.Gangloff A, Shi R, Nahoum V, Lin SX2003. Pseudo-symmetry of C19 steroids, alternative binding orientations, and multispecificity in human estrogenic 17β-hydroxysteroid dehydrogenase. FASEB J 17:274–276 [DOI] [PubMed] [Google Scholar]
- 18.Couture P, Thériault C, Simard J, Labrie F1993. Androgen receptor-mediated stimulation of 17β-hydroxysteroid dehydrogenase activity by dihydrotestosterone and medroxyprogesterone acetate in ZR-75-1 human breast cancer cells. Endocrinology 132:179–185 [DOI] [PubMed] [Google Scholar]
- 19.Birrell SN, Hall RE, Tilley WD1998. Role of the androgen receptor in human breast cancer. J Mammary Gland Biol Neoplasia 3:95–103 [DOI] [PubMed] [Google Scholar]
- 20.Greeve MA, Allan RK, Harvey JM, Bentel JM2004. Inhibition of MCF-7 breast cancer cell proliferation by 5α-dihydrotestosterone; a role for p21(Cip1/Waf1). J Mol Endocrinol 32:793–810 [DOI] [PubMed] [Google Scholar]
- 21.von Schoultz B2007. Androgens and the breast. Maturitas 57:47–49 [DOI] [PubMed] [Google Scholar]
- 22.Nicolás Díaz-Chico B, Germán Rodríguez F, González A, Ramírez R, Bilbao C, Cabrera de León A, Aguirre Jaime A, Chirino R, Navarro D, Díaz-Chico JC2007. Androgens and androgen receptors in breast cancer. J Steroid Biochem Mol Biol 105:1–15 [DOI] [PubMed] [Google Scholar]
- 23.Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK2003. RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev 67:657–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laplante Y, Cadot C, Fournier MA, Poirier D2008. Estradiol and estrone C-16 derivatives as inhibitors of type 1 17β-hydroxysteroid dehydrogenase: blocking of ER+ breast cancer cell proliferation induced by estrone. Bioorg Med Chem 16:1849–1860 [DOI] [PubMed] [Google Scholar]
- 25.Ghosh D, Pletnev VZ, Zhu DW, Wawrzak Z, Duax WL, Pangborn W, Labrie F, Lin SX1995. Structure of human estrogenic 17β-hydroxysteroid dehydrogenase at 220 Å resolution. Structure 3:503–513 [DOI] [PubMed] [Google Scholar]
- 26.Azzi A, Rehse PH, Zhu DW, Campbell RL, Labrie F, Lin SX1996. Crystal structure of human estrogenic 17β-hydroxysteroid dehydrogenase complexed with 17β-estradiol. Nat Struct Biol 3:665–668 [DOI] [PubMed] [Google Scholar]
- 27.Breton R, Housset D, Mazza C, Fontecilla-Camps JC1996. The structure of a complex of human 17β-hydroxysteroid dehydrogenase with estradiol and NADP+ identifies two principal targets for the design of inhibitors. Structure 4:905–915 [DOI] [PubMed] [Google Scholar]
- 28.Shi R, Lin SX2004. Cofactor hydrogen bonding onto the protein main chain is conserved in the short chain dehydrogenase/reductase family and contributes to nicotinamide orientation. J Biol Chem 279:16778–16785 [DOI] [PubMed] [Google Scholar]
- 29.Jiang C, Guo J, Wang Z, Xiao B, Lee HJ, Lee EO, Kim SH, Lu J2007. Decursin and decursinol angelate inhibit estrogen-stimulated and estrogen-independent growth and survival of breast cancer cells. Breast Cancer Res 9:R77 [DOI] [PMC free article] [PubMed]
- 30.Jansson A, Gunnarsson C, Stål O2006. Proliferative responses to altered 17β-hydroxysteroid dehydrogenase (17HSD) type 2 expression in human breast cancer cells are dependent on endogenous expression of 17HSD type 1 and the oestradiol receptors. Endocr Relat Cancer 13:875–884 [DOI] [PubMed] [Google Scholar]
- 31.Day JM, Tutill HJ, Newman SP, Purohit A, Lawrence HR, Vicker N, Potter BV, Reed MJ2006. 17β-Hydroxysteroid dehydrogenase type 1 and type 2: association between mRNA expression and activity in cell lines. Mol Cell Endocrinol 248:246–249 [DOI] [PubMed] [Google Scholar]
- 32.Tang W, Norlin M2006. Regulation of steroid hydroxylase CYP7B1 by androgens and estrogens in prostate cancer LNCaP cells. Biochem Biophys Res Commun 344:540–546 [DOI] [PubMed] [Google Scholar]
- 33.Oliveira AG, Coelho PH, Guedes FD, Mahecha GA, Hess RA, Oliveira CA2007. 5α-Androstane-3β,17β-diol (3β-diol), an estrogenic metabolite of 5α-dihydrotestosterone, is a potent modulator of estrogen receptor ERβ expression in the ventral prostrate of adult rats. Steroids 72914–72922 [DOI] [PubMed]
- 34.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM2004. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem 279:10784–10795 [DOI] [PubMed] [Google Scholar]
- 35.Picciarelli-Lima P, Oliveira AG, Reis AM, Kalapothakis E, Mahecha GA, Hess RA, Oliveira CA2006. Effects of 3-β-diol, an androgen metabolite with intrinsic estrogen-like effects, in modulating the aquaporin-9 expression in the rat efferent ductules. Reprod Biol Endocrinol 4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sikora MJ, Cordero KE, Larios JM, Johnson MD, Lippman ME, Rae JM2009. The androgen metabolite 5α-androstane-3β,17β-diol (3βAdiol) induces breast cancer growth via estrogen receptor: implications for aromatase inhibitor resistance. Breast Cancer Res Treat 115:289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zava DT, McGuire WL1978. Human breast cancer: androgen action mediated by estrogen receptor. Science 199:787–788 [DOI] [PubMed] [Google Scholar]
- 38.Hackenberg R, Hawighorst T, Filmer A, Slater EP, Bock K, Beato M, Schulz KD1992. Regulation of androgen receptor mRNA and protein level by steroid hormones in human mammary cancer cells. J Steroid Biochem Mol Biol 43:599–607 [DOI] [PubMed] [Google Scholar]
- 39.Dauvois S, Geng CS, Lévesque C, Mérand Y, Labrie F1991. Additive inhibitory effects of an androgen and the antiestrogen EM-170 on estradiol-stimulated growth of human ZR-75-1 breast tumors in athymic mice. Cancer Res 51:3131–3135 [PubMed] [Google Scholar]
- 40.Pelletier JD, Poirier D1996. Synthesis and evaluation of estradiol derivatives with 16α-(bromoalkylamide), 16α-(bromoalkyl) or 16α-(bromoalkynyl) side chain as inhibitors of 17β-hydroxysteroid dehydrogenase type 1 without estrogenic activity. Bioorg Med Chem 4:1617–1628 [DOI] [PubMed] [Google Scholar]
- 41.Tremblay MR, Boivin RP, Luu-The V, Poirier D2005. Inhibitors of type 1 17β-hydroxysteroid dehydrogenase with reduced estrogenic activity: modifications of the positions 3 and 6 of estradiol. J Enzyme Inhib Med Chem 20:153–163 [DOI] [PubMed] [Google Scholar]
- 42.Day JM, Foster PA, Tutill HJ, Parsons MF, Newman SP, Chander SK, Allan GM, Lawrence HR, Vicker N, Potter BV, Reed MJ, Purohit A2008. 17β-Hydroxysteroid dehydrogenase type 1, and not type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int J Cancer 122:1931–1940 [DOI] [PubMed] [Google Scholar]
- 43.Lin JT, Connelly MB, Amolo C, Otani S, Yaver DS2005. Global transcriptional response of Bacillus subtilis to treatment with subinhibitory concentrations of antibiotics that inhibit protein synthesis. Antimicrob Agents Chemother 49:1915–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aspinall SR, Stamp S, Davison A, Shenton BK, Lennard TW2004. The proliferative effects of 5-androstene-3β,17β-diol and 5α-dihydrotestosterone on cell cycle analysis and cell proliferation in MCF7, T47D and MDAMB231 breast cancer cell lines. J Steroid Biochem Mol Biol 88:37–51 [DOI] [PubMed] [Google Scholar]
- 45.Suzuki T, Miki Y, Nakamura Y, Moriya T, Ito K, Ohuchi N, Sasano H2005. Sex steroid-producing enzymes in human breast cancer. Endocr Relat Cancer 12:701–720 [DOI] [PubMed] [Google Scholar]
- 46.Vihko P, Isomaa V, Ghosh D2001. Structure and function of 17β-hydroxysteroid dehydrogenase type 1 and type 2. Mol Cell Endocrinol 171:71–76 [DOI] [PubMed] [Google Scholar]
- 47.Perillo B, Sasso A, Abbondanza C, Palumbo G2000. 17β-Estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol 20:2890–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mense SM, Remotti F, Bhan A, Singh B, El-Tamer M, Hei TK, Bhat HK2008. Estrogen-induced breast cancer: alterations in breast morphology and oxidative stress as a function of estrogen exposure. Toxicol Appl Pharmacol 232:78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sotoca AM, van den Berg H, Vervoort J, van der Saag P, Ström A, Gustafsson JA, Rietjens I, Murk AJ2008. Influence of cellular ERα/ERβ ratio on the ERα-agonist induced proliferation of human T47D breast cancer cells. Toxicol Sci 105:303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curtis CD, Likhite VS, McLeod IX, Yates JR, Nardulli AM2007. Interaction of the tumor metastasis suppressor nonmetastatic protein 23 homologue H1 and estrogen receptor α alters estrogen-responsive gene expression. Cancer Res 67:10600–10607 [DOI] [PubMed] [Google Scholar]
- 51.Lacroix M, Leclercq G2004. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat 83:249–289 [DOI] [PubMed] [Google Scholar]
- 52.Lin SX, Yang F, Jin JZ, Breton R, Zhu DW, Luu-The V, Labrie F1992. Subunit identity of the dimeric 17β-hydroxysteroid dehydrogenase from human placenta. J Biol Chem 267:16182–16187 [PubMed] [Google Scholar]
- 53.Huang YW, Pineau I, Chang HJ, Azzi A, Bellemare V, Laberge S, Lin SX2001. Critical residues for the specificity of cofactors and substrates in human estrogenic 17β-hydroxysteroid dehydrogenase 1: variants designed from the three-dimensional structure of the enzyme. Mol Endocrinol 15:2010–2020 [DOI] [PubMed] [Google Scholar]
- 54.Aatsinki JT, Lakkakorpi JT, Pietilä EM, Rajaniemi HJ1994. A coupled one-step reverse transcription PCR procedure for generation of full-length open reading frames. Biotechniques 16:282–284, 286–288 [PubMed] [Google Scholar]
- 55.Wen CY, Ito M, Wang H, Chen LD, Xu ZM, Matsuu M, Shichijo K, Nakayama T, Nakashima M, Sekine I2003. IL-11 up-regulates Tie-2 expression during the healing of gastric ulcers in rats. World J Gastroenterol 9:788–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luu-The V, Paquet N, Calvo E, Cumps J2005. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques 38:287–293 [DOI] [PubMed] [Google Scholar]
- 57.van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM1994. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods 174:311–320 [DOI] [PubMed] [Google Scholar]
- 58.Zhu DW, Lee X, Breton R, Ghosh D, Pangborn W, Daux WL, Lin SX1993. Crystallization and preliminary X-ray diffraction analysis of the complex of human placental 17β-hydroxysteroid dehydrogenase with NADP+ J Mol Biol 234:242–244 [DOI] [PubMed] [Google Scholar]