

Abstract
The mammalian target of rapamycin complex 1 (mTORC)1 pathway has emerged as a critical signaling component in the modulation of insulin’s metabolic action. This effect is triggered by a nutrient- and insulin-mediated negative feedback loop in which mTOR and S6 kinase (S6K)1 phosphorylate insulin receptor substrate (IRS)-1 on serine residues, which blunts phosphatidylinositol 3-kinase (PI3K) activation. Acute inhibition of mTORC1/S6K1 by rapamycin increases insulin signaling and glucose uptake in myocytes and adipocytes, but whether these effects can be maintained under chronic inhibition of mTORC1 or S6K1 remains unclear. Here, we analyzed the effect of chronic rapamycin inhibition or small interfering RNA-based down-regulation of specific elements of the mTORC1/S6K1 pathway on insulin signaling and glucose transport in adipocytes. Both chronic inhibition of mTORC1 by rapamycin or knockdown of either mTOR, raptor, or S6K1 reduced inhibitory serine phosphorylation of IRS-1, while increasing its insulin-stimulated tyrosine phosphorylation and associated PI3K activity. However, knockdown of either mTOR or raptor selectively blunted IRS-1 phosphorylation on Ser636/639, whereas only S6K1 knockdown was found to reduce phosphorylation of IRS-1 on Ser1101. Unexpectedly, insulin-induced activation of Akt2 and glucose transporter 4 expression were reduced after chronic disruption of the mTORC1/S6K1 pathway, impairing insulin-mediated glucose uptake despite increased PI3K activation. In conclusion, these data indicate that both mTORC1 and S6K1 are key elements of the negative feedback loop but inhibit insulin-induced PI3K activity through phosphorylation of specific serine residues in IRS-1. However, this study also shows that chronic inhibition of the mTORC1/S6K1 pathway uncouples IRS-1/PI3K signaling from insulin-induced glucose transport due to impaired activation of Akt2 and blunted glucose transporter 4 expression.
mTORC1 and S6K1 inhibit PI3K activity in adipocytes through IRS-1 phosphorylation on specific serine residues. However, chronically inhibiting mTORC1/S6K1 impairs Akt2 activation and GLUT4 expression.
The evolutionary conserved serine/threonine kinase mammalian target of rapamycin (mTOR) pathway is largely known for its central role in translational initiation, and therefore is a major determinant of cell growth and cell proliferation (1). This pathway acts as a nutrient sensor, which integrates a number of environmental signals such as insulin, glucose, and amino acids (1, 2). Insulin-induced activation of mTOR is relayed through the insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3-kinase (PI3K)/Akt pathway and is synergistically increased in the presence of amino acids (1, 2, 3).
mTOR is part of two discrete multiprotein complexes named mTORC1 and mTORC2 characterized by the presence of the scaffold proteins raptor and rictor, respectively (4, 5). Although mTORC2 is largely insensitive to the immunosuppressive drug rapamycin, the hormonal and nutrient-induced activation of the mTORC1 complex and its downstream effector ribosomal S6 kinase 1 (S6K1) are completely inhibited by this inhibitor (4, 5). In addition to its well-known translational role, recent studies have proposed a new function of the mTORC1/S6K1 pathway in the control of insulin’s metabolic actions. Indeed, we and others have shown that mTORC1 operates an inhibitory feedback loop on insulin-induced glucose uptake through promoting inhibitory phosphorylation of multiple serine residues in IRS-1 (reviewed in Refs. 6 and 7). Many studies have identified Ser1101 (8, 9), Ser636/639 (10, 11, 12, 13), Ser307 (14, 15), and Ser312 (14) as potential targets of the Ser/Thr kinase activity of mTOR and/or S6K1 (Ser1101, Ser636/639, Ser307, and Ser312 in human IRS-1 are respectively the equivalent of Ser1097, Ser632/635, Ser302, and Ser307 in mouse IRS-1; the human notation is used throughout the paper). Phosphorylation of IRS-1 on serine residues interferes with activation of the PI3K/Akt pathway, causing inhibition of insulin action on glucose uptake in both skeletal muscle, liver, and adipose cells (3, 8, 12). It was therefore postulated that the mTORC1/S6K1 pathway plays an essential role in glucose homeostasis by fine tuning of insulin signaling to metabolic pathways (6, 7). We have also shown that this pathway modulates insulin signaling to the PI3K/Akt pathway as well as glucose uptake in human skeletal muscle and adipocytes (12, 16).
mTOR and S6K1 are overactivated in animal models of obesity and insulin resistance (13, 17). Moreover, lack of S6K1 protects against diet-induced obesity and insulin resistance in mice (13). This suggests that chronic inhibition of the mTORC1/S6K1 pathway may protect against obesity-linked insulin resistance. In the present study, we have sought to determine the effect of chronic inhibition of the mTORC1/S6K1 pathway with rapamycin on insulin signaling to IRS-1/PI3K/Akt and on glucose uptake in 3T3-L1 adipocytes. We have also used RNA interference to further clarify the role of mTOR, raptor, and S6K1 in the operation of this feedback loop mechanism.
Results
Disruption of the mTORC1/S6K1 pathway in 3T3-L1 adipocytes by small interfering RNA (siRNA) and chronic rapamycin treatment
Transfection of 3T3-L1 adipocytes with siRNA designed against mTOR, S6K1, and raptor reduced target protein expression by 67 ± 7, 71 ± 8, and 77 ± 5%, respectively (P < 0.01; Fig. 1, A and B). Down-regulation of these proteins by siRNA was highly specific except for raptor siRNA, which also decreased mTOR protein levels by 68 ± 6% (P < 0.01; Fig. 1, A and B), which has been previously reported in vitro (18) and in vivo (19). No effects were observed on the expression of rictor, which is part of the mTORC2 complex (Fig. 1A) and of S6K2 (data not shown). Long-term rapamycin treatment did not alter the expression of analyzed proteins compared with vehicle-treated controls (Fig. 1, A and B).
Fig. 1.
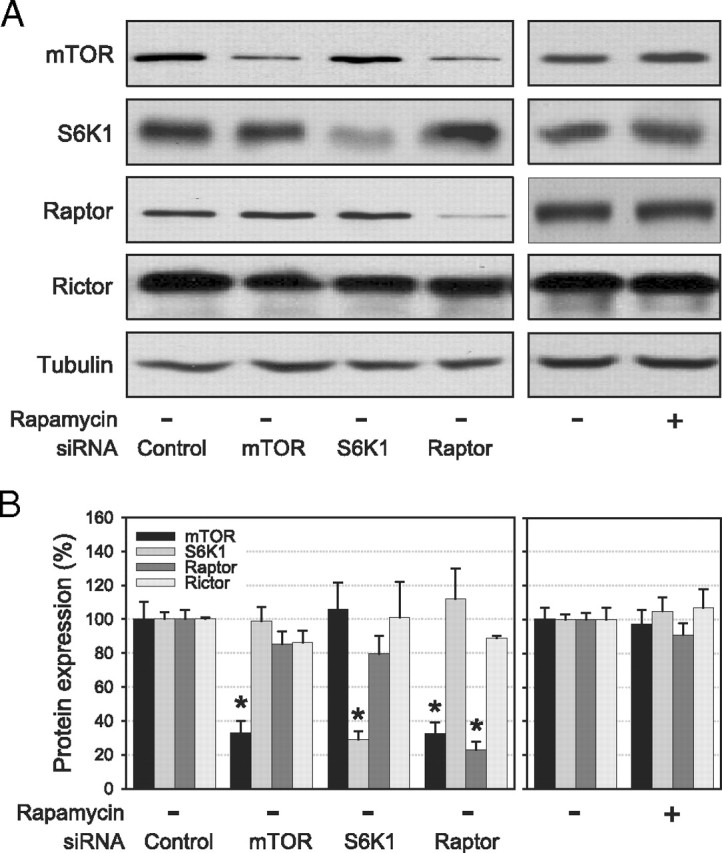
mTOR, S6K1, raptor, and rictor protein levels after electroporation of siRNAs or chronic rapamycin treatment of 3T3-L1 adipocytes. Cells were electroporated with the indicated control or target siRNA or treated 24 h with vehicle (DMSO, 0.01%) or rapamycin (25 nm). A, Cell lysates were analyzed by immunoblotting for the expression of mTOR (n = 6), S6K1 (n = 6), raptor (n = 3), rictor (n = 3), and tubulin. B, Densitometric analysis of protein expression normalized for the expression of tubulin. Results in panel B are represented as means ± se (*, P < 0.05 vs. control siRNA).
S6K1 Thr389 phosphorylation was measured to determine the activation of the mTORC1/S6K1 pathway after insulin stimulation for 45 min (1). As expected, rapamycin abolished insulin-induced activation of the pathway as shown by insulin failure to increase S6K1 phosphorylation (P < 0.01; Fig. 2A) and S6K1 activity (P < 0.01; Fig. 2B). Similarly, mTOR, S6K1, and raptor protein knockdown by RNA interference significantly reduced insulin-mediated activation of the mTORC1/S6K1 pathway by at least 50% (P < 0.05; Fig. 2, A and B). The residual S6K1 activation in cells treated with siRNA was probably due to the incomplete disruption of the target protein expression (Fig. 1B).
Fig. 2.

S6K1 activation after RNA interference or chronic rapamycin treatment of 3T3-L1 adipocytes. Cells were electroporated with control or target siRNAs or treated (24 h) with rapamycin (25 nm) or DMSO and serum deprived before 45 min stimulation with 10 nm insulin. A, Western blotting of cell lysates was performed with phosphospecific antibody against S6K1 Thr389 (n = 5). B, S6K1 kinase activity was measured using S6 peptide (RRRLSSLRA) as substrate (n = 3). Densitometry measures of phosphorylation and kinase activity were normalized for the expression of S6K1 and for protein content, respectively. Results in panel A show representative gels and densitometric analysis. Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Chronic inhibition of the mTORC1/S6K1 pathway impedes the negative feedback loop toward the IRS-1/PI3K pathway
Studies in cultured cells and animal models have shown that the mTORC1 pathway increases inhibitory phosphorylation of IRS-1 on multiple serine residues (including Ser636/639 and Ser1101), uncoupling insulin receptor signaling to IRS-1 tyrosine phosphorylation, and associated PI3K activity (3, 8, 12, 13, 17, 20, 21). We therefore next assessed whether chronic inhibition of this pathway impedes the negative feedback loop against insulin signaling to IRS-1/PI3K and used Ser636/639 and Ser1101 as surrogate markers of mTORC1- and S6K1-mediated inhibition of IRS-1 signaling. Chronic rapamycin treatment completely blocked phosphorylation on IRS-1 Ser636/639 (Fig. 3A) and significantly reduced insulin-induced Ser1101 phosphorylation (Fig. 3B). This was associated with increased insulin stimulation of both IRS-1 tyrosine phosphorylation (Fig. 3C) and IRS-1-associated PI3K activity (Fig. 3D). We next sought to identify which component of the mTORC1/S6K1 pathway is responsible for this effect. siRNA-mediated knockdown of mTOR, S6K1, or raptor all reduced inhibitory Ser636/639 IRS-1 phosphorylation. However, raptor knockdown fully abrogated Ser636/639 phosphorylation, whereas S6K1 silencing only partially reduced phosphorylation of this site (P < 0.05; Fig. 3A). Conversely, insulin-induced Ser1101 phosphorylation of IRS-1 was only significantly reduced by S6K1 knockdown (P < 0.05; Fig. 3B) because neither mTOR nor raptor siRNAs were found to significantly decrease Ser1101 phosphorylation. siRNA-mediated disruption of the mTORC1/S6K1 pathway increased insulin-induced IRS-1 tyrosine phosphorylation (+53, +35, and +63% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 3C) as well as insulin-induced IRS-1-associated PI3K activity (+50, +33, and +45% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 3D).
Fig. 3.

Effect of chronic down-regulation of the mTORC1/S6K1 pathway on the activation of the IRS-1/PI3K pathway in 3T3-L1 adipocytes. After siRNA or rapamycin treatment, cells were serum deprived and stimulated for 45 min with 10 nm insulin before lysis and subsequent analysis of IRS-1 phosphorylation and PI3K activity. Western blotting of cell lysates (A and B) or immunoprecipitated IRS-1 (C) with antibodies against (A) IRS-1 Ser636/639 (n = 5), (B) IRS-1 Ser1101 (n = 3), and (C) phosphotyrosine (n = 3). D, IRS-1 associated PI3K activity (n = 8). Densitometry measures of phosphorylation and kinase activity were normalized for the expression of IRS-1 and for protein content, respectively. Results show representative gels and densitometric analysis. Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Chronic inhibition of the mTORC1/S6K1 pathway in 3T3-L1 adipocytes impairs insulin-stimulated Akt activation
We next examined whether chronic down-regulation of the mTORC1/S6K1 pathway increases insulin activation of Akt, a downstream target of PI3K and a key effector of insulin’s metabolic actions. Surprisingly, we found that Akt phosphorylation was markedly suppressed with the chronic loss of a functional mTORC1/S6K1 pathway (Fig. 4, A and B). Immunoblot quantification revealed a decrease of at least 50% in the insulin-stimulated phosphorylation of both Ser473 (P < 0.05; Fig. 4A) and Thr308 (P < 0.05; Fig. 4B) compared with untreated cells. Similar results were obtained using both siRNA and rapamycin to disrupt the mTORC1/S6K1 pathway. The reduced phosphorylation of Ser473 was not related to increased expression or activity of protein phosphatase 2A (PP2A) in rapamycin-treated cells (see Supplemental Fig. 1, A and B, published on The Endocrine Society’s Journals Online web site at http://endo.endojournals. org). In agreement with regulatory sites phosphorylation, insulin-stimulated Akt activity, measured with an antibody that cross-reacts with both Akt1 and Akt2 isoforms, was significantly decreased in insulin-stimulated adipocytes treated with all siRNAs (−41, −52, and −51% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 5A). Because the Akt2 isoform is predominantly responsible for insulin-mediated glucose uptake in adipocytes (22, 23), we next determined whether chronic disruption of the mTORC1/S6K1 pathway affects Akt1 and Akt2 isoforms differently. Using isoform-specific antibodies, we found that Akt1 activity was only marginally affected, with only the raptor siRNA causing a small but significant decrease in Akt1 activity (−22%, P < 0.05; Fig. 5B). In sharp contrast, Akt2 activity was markedly blunted in the same cells (−57, −94, and −75% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 5C).
Fig. 4.
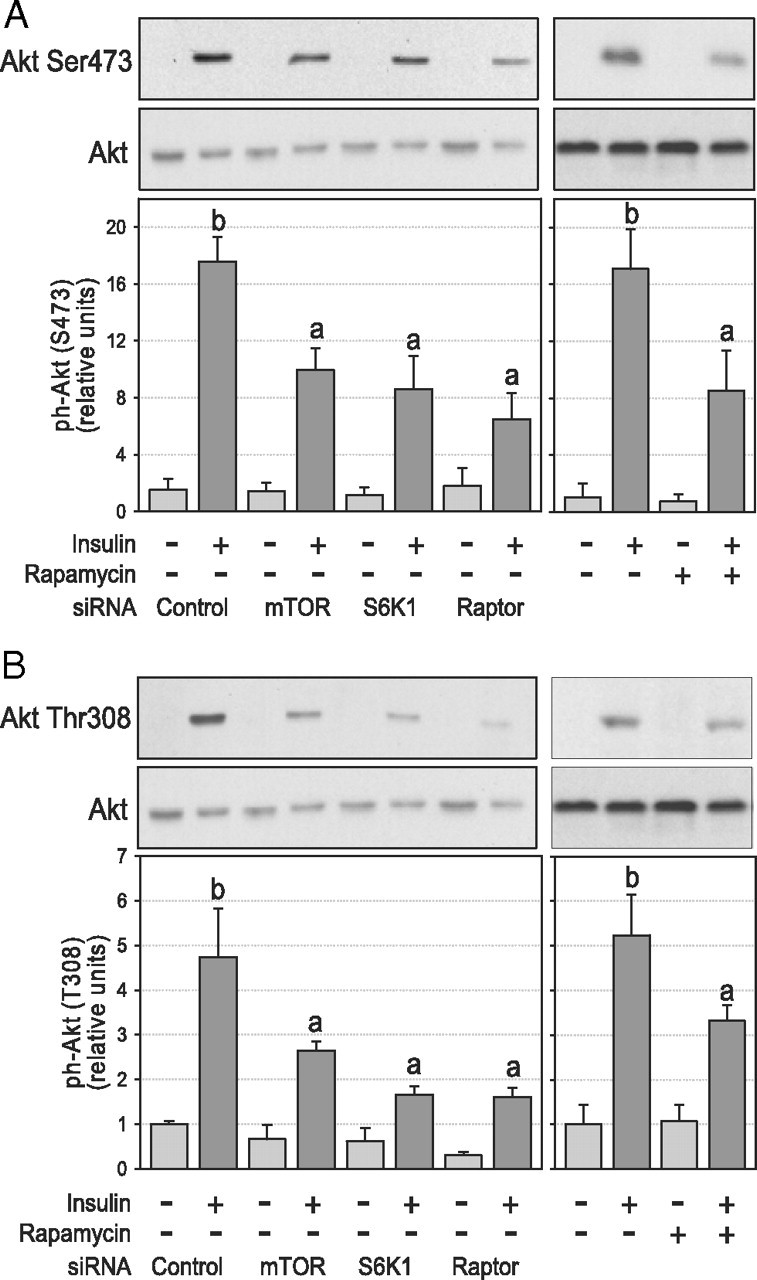
Effect of chronic inhibition of the mTORC1/S6K1 pathway on insulin-stimulated Akt phosphorylation in 3T3-L1 adipocytes. After siRNA or rapamycin treatment, cells were serum deprived and stimulated for 45 min with 10 nm insulin before lysis and subsequent analysis of Akt phosphorylation. Western blotting of cell lysates with phosphospecific antibodies against (A) Akt Ser473 (n = 6) and (B) Akt Thr308 (n = 5). Densitometry measures of phosphorylation were normalized for the expression of Akt. Results show representative gels and densitometric analysis. Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Fig. 5.
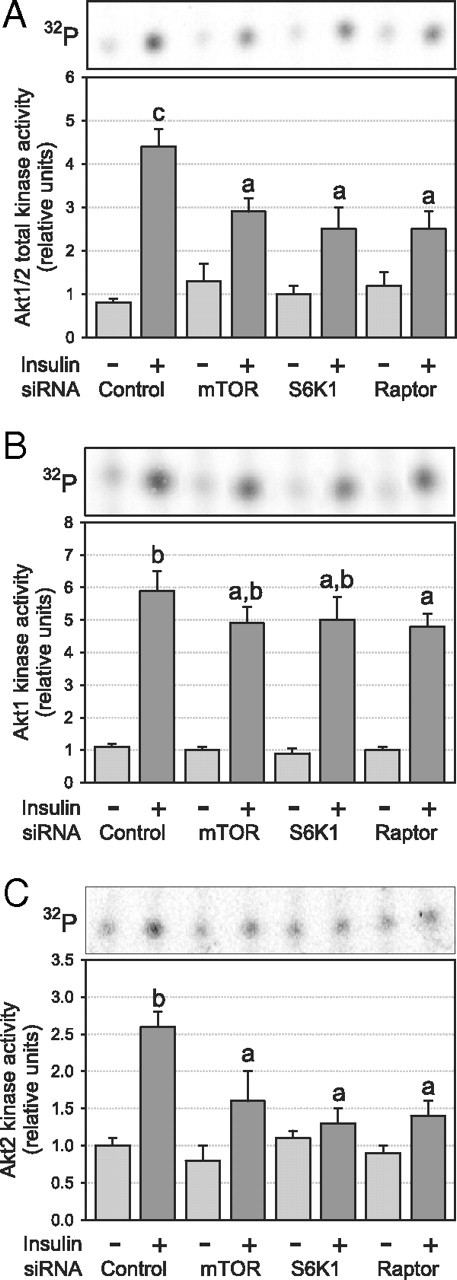
Effect of chronic inhibition of the mTORC1/S6K1 pathway on insulin-stimulated Akt activation in 3T3-L1 adipocytes. After siRNA or rapamycin treatment, cells were serum deprived and stimulated for 45 min with 10 nm insulin before lysis and measurement of Akt activity after immunoprecipitation of (A) Akt1/2 (n = 5), (B) Akt1 (n = 3), or (C) Akt2 (n = 3). Kinase activity was normalized for protein content. Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Chronic inhibition of the mTORC1/S6K1 pathway did not impede Akt2 recruitment to the plasma membrane in 3T3-L1 adipocytes
To further explore the mechanism involved in the uncoupling of PI3K signaling to Akt2, we first examined the effect of chronic rapamycin treatment on phosphatidylinositol (3, 4, 5)-triphosphate (PIP3) cellular levels. As expected from the augmented PI3K activation, we found that chronic rapamycin treatment increased PIP3 production in 3T3-L1 adipocytes (P < 0.05; Fig. 6A). This is in accordance with the lack of effect of chronic rapamycin treatment on the expression of the PIP3 phosphatases, namely phosphatase and tensin homolog (PTEN) and SH2-domain-containing inositol 5-phosphatase (SHIP)-2 in these cells (Supplemental Fig. 1, C and D). We next assessed insulin-induced PIP3 accumulation and its ability to recruit downstream effectors to the plasma membrane by monitoring the cellular redistribution of green fluorescent protein (GFP)-tagged Bruton’s tyrosine kinase (Btk)-pleckstrin homology (PH) by confocal microscopy (24). As expected, insulin treatment increased the membrane recruitment of Btk-PH (Fig. 6B). After chronic rapamycin treatment, we observed a slight increase in the number of cells displaying strong insulin-induced plasma membrane translocation of Btk-PH (Fig. 6B). Furthermore, insulin-induced plasma membrane translocation of Akt2 was not altered by chronic rapamycin treatment (Fig. 6C). Thus neither PIP3 production nor the PIP3-dependent activation and plasma membrane recruitment of Akt2 were impeded by chronic rapamycin treatment, suggesting that chronic inhibition of the mTORC1/S6K1 pathway impedes insulin signaling downstream of Akt2 recruitment to the plasma membrane.
Fig. 6.
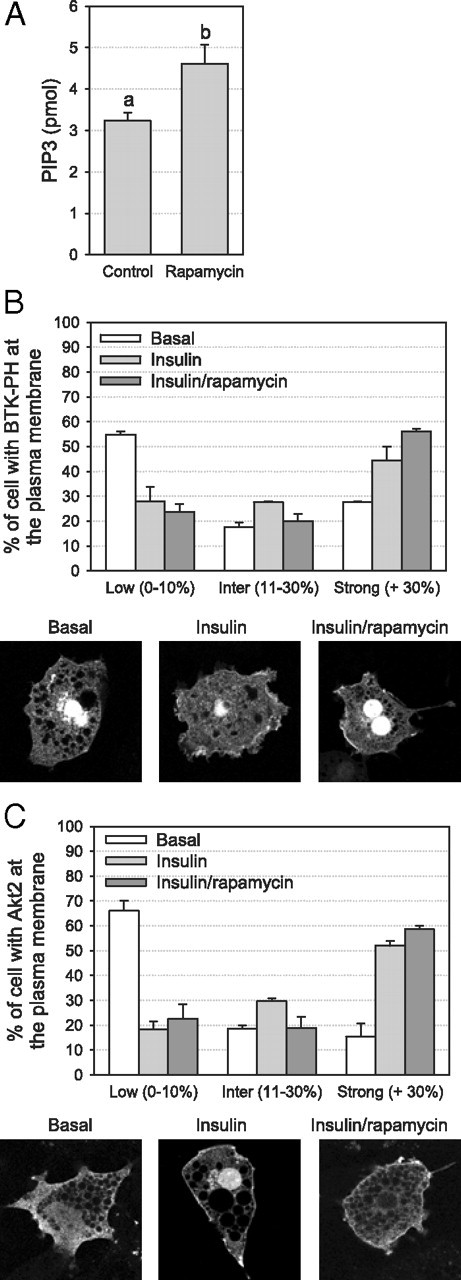
Effect of chronic inhibition of the mTORC1/S6K1 pathway on insulin-stimulated PIP3 accumulation and Akt2 recruitment to the plasma membrane in 3T3-L1 adipocytes. After 24 h rapamycin treatment, cells were serum deprived and stimulated for 45 min with 10 nm insulin. A, PIP3 levels were measured by competitive ELISA (n = 2). B, Cellular localization of Btk-PH-GFP was assessed by confocal microscopy. C, Cellular localization of Akt2-HA was assessed by confocal microscopy after staining with an HA antibody. The percentage of cells with low (0–10%), intermediate (11–30%), or strong (>30%) localization of Btk-PH-GFP and Akt2-HA at the plasma membrane was evaluated in a total of at least 100 cells from two independent experiments (panels B and C). Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Chronic inhibition of the mTORC1/S6K1 pathway inhibits insulin-induced glucose uptake in 3T3-L1 adipocytes
Because molecular or pharmacological inactivation of the mTORC1/S6K1 pathway inversely modulated PI3K and Akt, two key effectors of insulin’s metabolic actions, we next assessed the effect of chronic inhibition of this pathway on the ability of insulin to augment glucose uptake. Insulin-induced 2-deoxyglucose uptake was inhibited by siRNAs targeting the mTORC1/S6K1 pathway (−28, −59, and −38% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 7A), and a similar effect was observed with rapamycin (−28%, P < 0.05; Fig. 7A). Because 2-deoxyglucose uptake results from two independent steps (i.e. facilitated 2-deoxyglucose transport and its phosphorylation by hexokinase), we next specifically assessed the transport step using 3-O-methylglucose, a nonmetabolizable analog of glucose. As for 2-deoxyglucose, we observed a significant decrease in insulin-mediated 3-O-methylglucose uptake after chronic disruption of the mTORC1/S6K1 pathway (P < 0.05; Fig. 7B). These results indicate that the reduction of insulin-induced glucose uptake was mainly caused by a reduced capacity to transport glucose. Because 3T3-L1 adipocytes express glucose transporter (GLUT)1 and GLUT4 and both are involved in the stimulatory action of insulin on glucose transport, we next determined 2- deoxyglucose uptake in the presence of indinavir, a potent and selective GLUT4 inhibitor (25). The GLUT4-mediated glucose uptake, corresponding to the uptake inhibited by indinavir (i.e. about 65% of total uptake in nontreated cells), was reduced to the same extent as total glucose uptake by all siRNAs (−29, −51, and −42% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 7C) and chronic rapamycin treatment (−24%; P < 0.05; Fig. 7C), suggesting that chronic inhibition of the mTORC1/S6K1 pathway specifically reduces GLUT4-mediated hexose uptake.
Fig. 7.
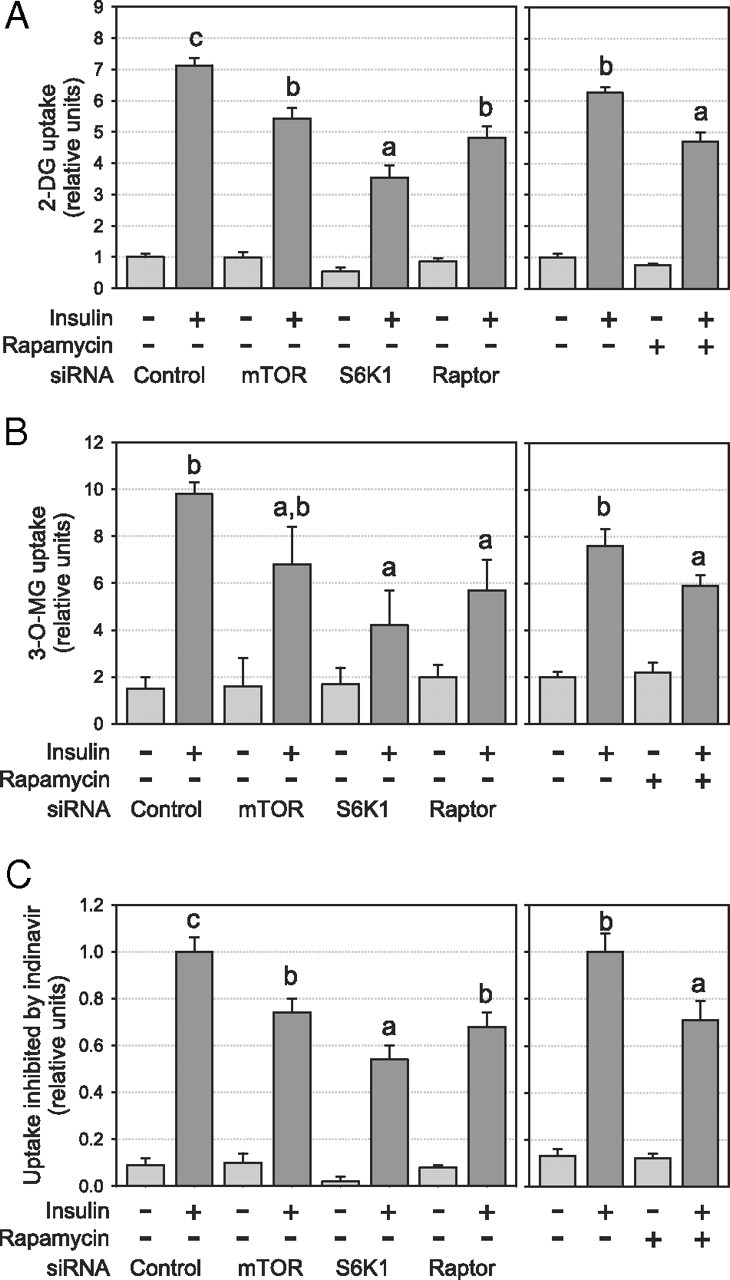
Chronic inhibition of the mTORC1/S6K1 pathway reduces insulin-induced glucose uptake in 3T3-L1 adipocytes. After siRNA or chronic rapamycin treatments, cells were serum deprived and stimulated for 45 min with 10 nm insulin before glucose uptake was measured using (A) 2-D-deoxyglucose uptake (n = 10), (B) 3-O-methylglucose uptake (n = 3), and (C) 2-D-deoxyglucose in the presence of the GLUT4 inhibitor indinavir (n = 4). Glucose uptake inhibited by indinavir therefore represents GLUT4-mediated glucose uptake. Glucose uptake was normalized for protein content. Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
GLUT4 but not GLUT1 expression is reduced by chronic inhibition of the mTORC1/S6K1 pathway
We next evaluated whether disruption of the mTORC1/ S6K1 pathway alters the expression of GLUT4 in 3T3-L1 adipocytes. In accordance with indinavir-inhibited glucose uptake data, we found a significant decrease in cellular GLUT4 protein expression after siRNA treatment (−24, −51, and −43% for mTOR, S6K1, and raptor siRNA, respectively; P < 0.05; Fig. 8A) and rapamycin treatment (−19%, P < 0.05; Fig. 8A). GLUT4 mRNA expression was similarly affected (P < 0.05; Fig. 8B), suggesting that chronic inhibition of the mTORC1/S6K1 pathway alters GLUT4 gene expression. As predicted from the indinavir experiments, GLUT1 protein expression was not affected under the same experimental conditions (Fig. 8C).
Fig. 8.
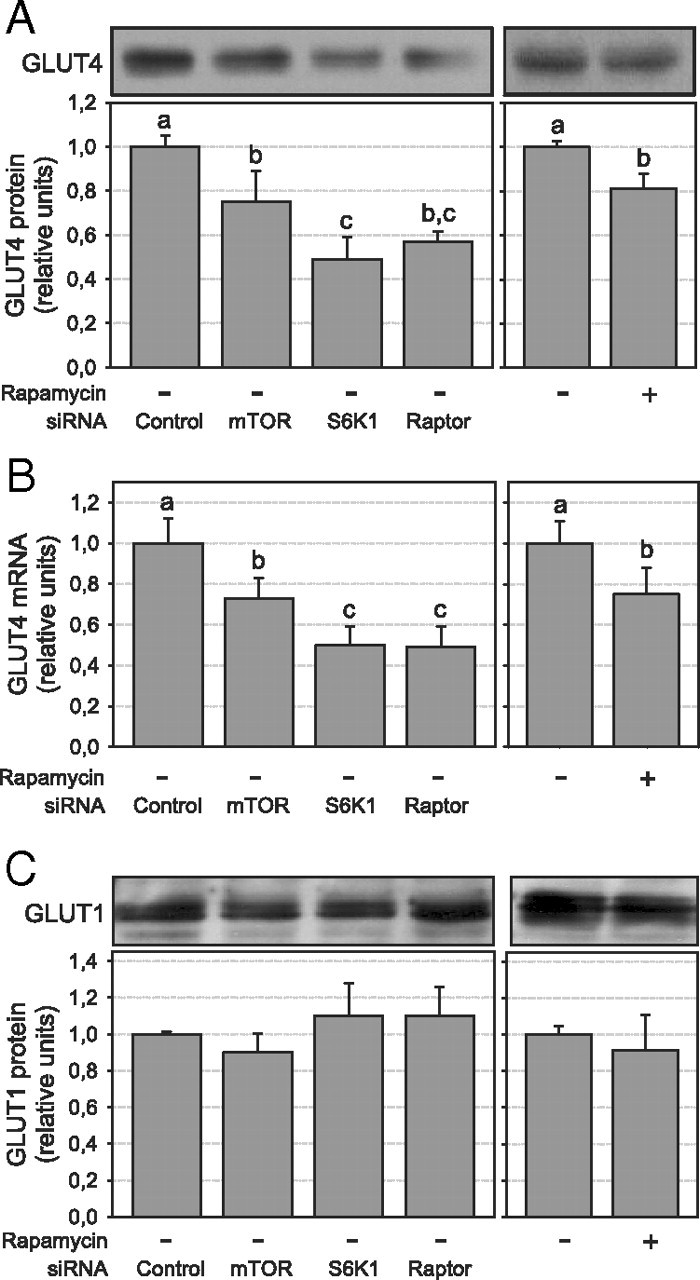
Chronic disruption of the mTORC1/S6K1 pathway blunts GLUT4 expression in 3T3-L1 adipocytes. GLUT4 expression after siRNA or rapamycin treatment was assessed (A) by Western blotting using a specific antibody against GLUT4 (n = 4) or (B) by quantitative real-time PCR (n = 3). C, GLUT1 protein expression was determined by Western blotting using a GLUT1-specific antibody (n = 3). Densitometry measures were normalized for the expression of tubulin. Results in A and C show representative gels and densitometric analysis. Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Chronic inhibition of the mTORC1/S6K1 pathway has little effect on protein and lipid contents in 3T3-L1 adipocytes
Treatment with either siRNAs or rapamycin did not affect cell viability and morphology. However, because the mTORC1 pathway has been implicated in the control of protein synthesis, cell growth and adipocyte differentiation (26) and that S6K1−/− mice have a reduced adipose tissue mass (13), we assessed the potential impact of the siRNA and pharmacological treatments on both protein and lipid contents of the adipocytes. We observed a slight decrease in the protein content (10–17%, P < 0.05; Fig. 9A) when the mTORC1 pathway was inhibited with rapamycin and after S6K1 or raptor siRNA treatment, but this effect was not observed with the mTOR siRNA. Moreover, determination of cellular triacylglycerol levels using Oil Red O staining revealed a modest reduction in adipocyte lipid content after treatments with rapamycin or with S6K1 and raptor siRNA (12–20%, P < 0.05; Fig. 9B); but again, this was not observed with the mTOR siRNA. Finally, we quantified the mRNA content of the adipogenic transcription factor peroxisome proliferator-activated receptor γ (PPARγ). Although the mTOR siRNA had no effect on PPARγ mRNA content, siRNA targeting of S6K1 and raptor slightly reduce its mRNA expression (∼30%, P < 0.05; Fig. 9C). Conversely, chronic rapamycin treatment increased PPARγ mRNA expression by 31% (P < 0.05; Fig. 9C). Also, we did not observe any effect of mTORC1 inhibition on the phosphorylation of AMPK or of ACC, a key downstream mediator of AMPK in the modulation of lipogenesis (Supplemental Fig. 2) (27). Thus, chronic inhibition of the mTORC1/S6K1 pathway has a modest influence on the adipocyte phenotype as compared with the marked reduction of both Akt2 kinase activity and GLUT4 expression in these cells.
Fig. 9.
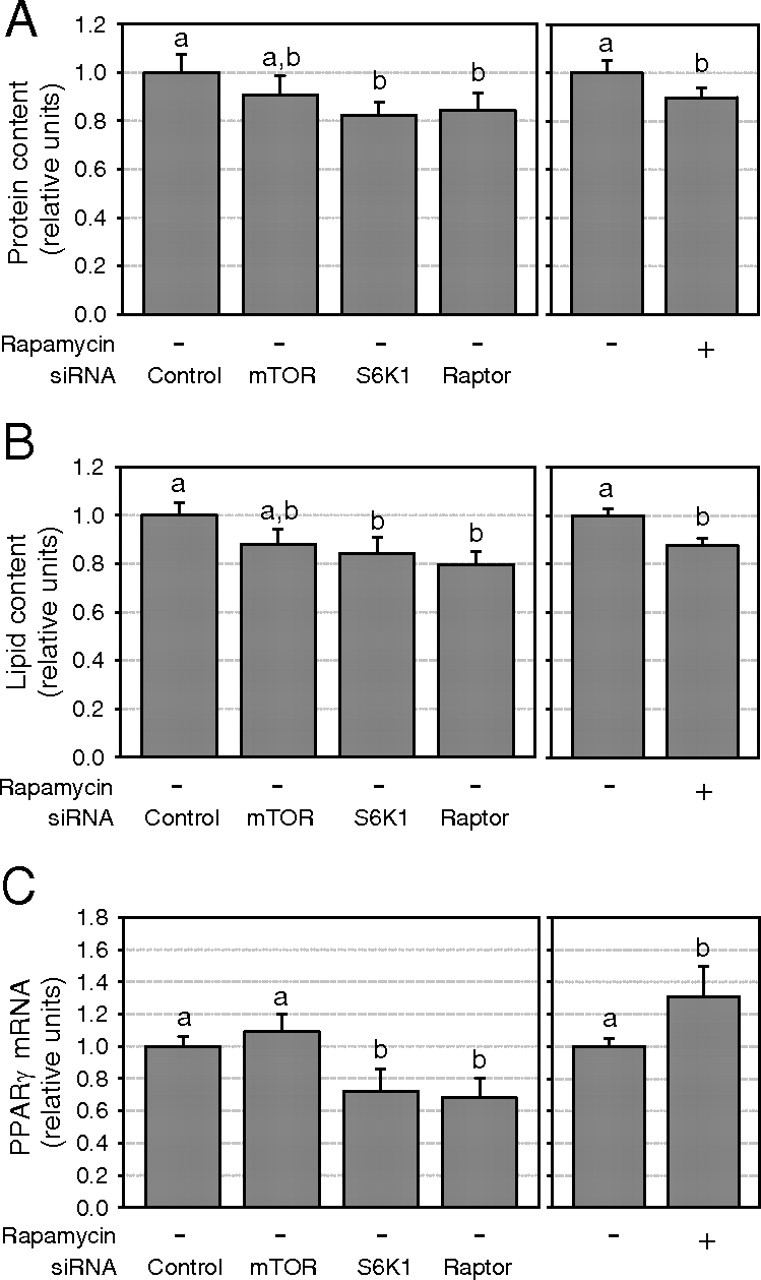
Chronic mTORC1/S6K1 pathway disruption slightly affects protein and lipid contents in 3T3-L1 adipocytes. A, Protein content was measured by colorimetry (n = 7). B, Lipid accumulation in 3T3-L1 adipocytes was measured by Oil-Red-O staining (n = 4). C, PPARγ mRNA expression was measured by quantitative real-time PCR (n = 3). Results are shown as means ± se. Groups with different letters are significantly different (P < 0.05).
Discussion
We and others have characterized a negative feedback loop in which components of the mTORC1/S6K1 pathway uncouple IRS-1 from activation of the PI3K/Akt pathway (3, 8, 12, 13, 15, 17, 28). Rapid activation of this feedback loop by increased nutrient availability and insulin acutely modulates insulin action on glucose metabolism in muscle, adipose, and liver cells (3, 8, 12, 17, 29). Moreover, chronic nutrient excess and hyperinsulinemia, two features of obesity, both induce this nutrient/hormonal sensing feedback mechanism, promoting insulin resistance for glucose metabolism via inhibitory serine phosphorylation of IRS-1 and impairment of PI3K activation (7). In agreement with previous studies using acute rapamycin treatment (12), we show here that chronic incubation with the inhibitor also suppresses this negative feedback loop in 3T3-L1 adipocytes, by preventing IRS-1 Ser636/639 and Ser1101 phosphorylation, thus allowing optimal activation of IRS-1-associated PI3K activity by insulin. The importance of mTORC1 complex for the function of the feedback loop was further demonstrated by specifically silencing mTOR, or both mTOR and raptor expression, using RNA interference. Silencing mTOR and/or raptor increased insulin-stimulated IRS-1-dependent PI3K as much as chronic rapamycin treatment, confirming that the drug improves insulin signaling by inhibition of mTORC1. These results agree with previous studies showing the role of mTORC1 in mediating inhibition of insulin signaling (30) and with the recent report that raptor interacts with the SAIN domain of IRS-1 allowing mTOR to phosphorylate Ser636/639 in IRS-1 (21).
S6K1 is a key downstream mTORC1 effector, but its potential role in the operation of the mTOR-dependent negative feedback loop toward IRS-1/PI3K signaling remains unclear. S6K1 disruption protects against diet-induced obesity and insulin resistance in mice (13). However, these mice are also leaner and this could indirectly contribute to the protection from insulin resistance. Moreover, RNA interference or loss-of-function studies have yielded contradictory findings as to the role of S6K1 in mediating IRS-1 serine phosphorylation and inhibition of insulin signaling (8, 16, 17, 30). However, none of those studies specifically compared the role of individual components of the mTORC1 complex vs. that of S6K1 in 3T3-L1 adipocytes or other classical metabolic targets of insulin action. We show here that specifically interfering with S6K1 expression reduced IRS-1 Ser1101 and Ser636/639 phosphorylation and increased IRS-1/PI3K signaling, thus revealing a key role for S6K1 as a downstream effector of mTORC1-linked inhibition of insulin signaling. Our results are consistent with the recent observation that S6K1 can promote insulin resistance by directly phosphorylating IRS-1 at Ser1101 (8). Our data also suggest that this site is not targeted by mTORC1 because neither mTOR nor raptor silencing was found to blunt Ser1101 phosphorylation. However, raptor knockdown (equivalent to mTORC1 inhibition in our studies) abrogated Ser636/639 phosphorylation by insulin, which is in agreement with previous studies (21, 30). Our data showing partial reduction of Ser636/639 phosphorylation after S6K1 knockdown are also in accordance with previous observations in HeLa cells (7) and L6 myocytes (8) and with the finding of reduced IRS-1 Ser636/639 phosphorylation in adipose tissue of S6K1 knockout mice (13). Ser636/639 phosphorylation, although not directly mediated by S6K1, is dependent on S6K1 phosphorylation of IRS-1 (14).
Chronic inhibition of the mTORC1/S6K1 pathway by rapamycin or RNA interference increased insulin action on IRS-1 associated PI3K activity, a key effector of insulin’s metabolic actions. Surprisingly, however, insulin-induced glucose uptake was significantly impeded by both chronic rapamycin treatment and after targeting individual components of the mTORC1/S6K1 pathway by RNA interference. This unexpected finding appears to be explained by the fact that chronic inhibition of the mTORC1 pathway impairs both the activation of Akt (in particular Akt2) by insulin as well as the expression of GLUT4, the main GLUT isoform responsible for insulin action on glucose uptake in 3T3-L1 adipocytes. The role of impaired GLUT4 expression in the inhibition of insulin action by chronic mTORC1 inhibition was confirmed by assessing GLUT4-specific glucose uptake in the presence of the GLUT4-specific inhibitor indinavir. These effects cannot be attributed to a possible dedifferentiation of the adipocytes after chronic disruption of the mTORC1 pathway because of the following reasons: 1) insulin-induced PI3K activation was inversely modulated in the same cells, 2) the defects in both Akt2 activation and GLUT4 expression are greater than the small effects of chronic mTORC1 inhibition on lipid and protein contents, and 3) selective mTOR knockdown had a similar effect on Akt2 activation and GLUT4 expression while not affecting protein and lipid contents or PPARγ expression.
The present study shows that insulin-induced activation of Akt2, but not Akt1, is severely affected by the disruption of the mTORC1/S6K1 pathway. This may explain why glucose metabolism is severely affected in adipocytes as compared with other mTORC1/S6K1-regulated functions such as protein synthesis. Indeed, both cellular and animal studies have shown that Akt1 mainly mediates the effect of insulin on cell proliferation, protein synthesis, and apoptosis, whereas Akt2 appears to relay insulin’s metabolic signaling and is critical for glucose homeostasis (22, 23, 31). This is consistent with our observation that a defect in Akt2 activation is associated with reduced insulin-induced glucose uptake in adipocytes lacking mTOR, raptor, or S6K1 after RNA interference.
Among potential mechanisms involved in the uncoupling of PI3K activation and Akt2 activation, we first explored the potential involvement of PP2A because this enzyme can specifically dephosphorylate Ser473 on Akt (32) and was previously reported to be inhibited by mTOR (33, 34). However, neither the expression nor the phosphatase activity of PP2A are increased by rapamycin. We also explored whether the chronic inhibition of the mTORC1/S6K1 pathway causes activation of the lipid phosphatases PTEN and SHIP-2 leading to degradation of PIP3 despite elevated PI3K activity. Both PTEN and SHIP-2 have been implicated in the development of insulin resistance in 3T3-L1 adipocytes (35, 36, 37). However, we confirmed that cellular PIP3 levels were increased after rapamycin treatment and the mTORC1 inhibitor failed to modulate either PTEN or SHIP-2 protein expression, suggesting that modulation of these lipid phosphatases is not the mechanism underlying the defective Akt2 activation after chronic blockade of the mTORC1/S6K1 pathway. Using confocal microscopy, we further confirmed that insulin-induced accumulation of PIP3 in the plasma membrane, as revealed by GFP-tagged BTK-PH recruitment, as well as Akt2 translocation to the plasma membrane, are not impeded after chronic exposure of 3T3-L1 adipocytes to rapamycin. Although we found no evidence for reduced phosphoinositide dependent protein kinase 1 plasma membrane recruitment (data not shown), we cannot exclude the possibility that phosphoinositide dependent protein kinase 1 kinase activity is affected by rapamycin. However, this is unlikely to be the main mechanism behind reduced Akt2 activation because the phosphorylation of both Thr308 and Ser473 are affected by the mTORC1 inhibitor and full activation of Akt requires phosphorylation on both residues (38).
Because mTORC2 was recently proposed to phosphorylate Akt on Ser473 (39), we also considered the possibility that chronic rapamycin treatment as well as mTOR or raptor siRNAs may impede Akt2 activation through alterations of mTORC2. Indeed, long-term rapamycin treatment has been shown to inhibit the mTOR/rictor complex assembly and Akt signaling (40). However, we ruled out this possibility by selectively targeting S6K1 downstream of mTORC1. S6K1 knockdown similarly affected Akt2 and insulin-induced glucose uptake as compared with siRNAs targeting mTOR or raptor, suggesting that secondary inhibition of the mTORC2 complex is likely not involved in the disruption of Akt2 activation upon chronic inhibition of mTORC1.
The present study also reveals a novel role for the mTORC1/S6K1 pathway in the chronic regulation of GLUT4 expression in adipocytes. A previous study in 3T3-L1 adipocytes has shown that insulin chronically increased GLUT1 but not GLUT4 protein expression and that this effect was linked to a rapamycin-sensitive increase in GLUT1 mRNA translation (41). However, as shown here, basal GLUT1 protein expression was not reduced in adipocytes chronically exposed to rapamycin. In contrast to GLUT1, the expression of GLUT4 appears to be dependent on an intact mTORC1/S6K1 pathway because both rapamycin and siRNA-mediated inhibition of specific elements in this pathway reduced GLUT4 protein levels. The fact that GLUT4 mRNA was similarly affected suggests that this nutrient-sensing pathway may be essential for the expression of transcriptional factors involved in GLUT4 gene promoter activity. A recent study has shown that PPARγ suppression in adipocytes reduces GLUT4 expression (42). Thus, the reduction of PPARγ levels after silencing S6K1 or raptor may contribute to GLUT4 repression in the present study. In fact, both the S6K1 and raptor siRNAs lead to a comparable loss of PPARγ and GLUT4 expression, whereas silencing mTOR by chronic incubation with rapamycin failed to decrease PPARγ expression and only modestly reduced GLUT4 expression (compare Fig. 7, A and B, and Fig. 8C). GLUT4 gene expression is regulated in a complex and tissue-specific manner. In adipose tissue, GLUT4 transcription is driven by multiple transcription factors (e.g. by sterol regulatory element binding protein-1c, CCAAT-enhancer-binding protein-α, liver X receptor α, PPARγ, olfactory neuron-specific transcription factor-1/early B cell factor-1, and neurofibromatosis type-1) (43, 44, 45), and the focus of future investigations will be to test whether mTOR and S6K1 are key modulators of their transcriptional activities.
Our results are also of clinical relevance because the chronic use of rapamycin as an immunosuppressive agent was found to be associated with an increased incidence of impaired glucose tolerance and of type 2 diabetes in patients (46). Moreover, long-term exposure to rapamycin caused a marked decrease of insulin-induced Akt phosphorylation in peripheral blood mononuclear cells and increased the prevalence of insulin resistance in human subjects (47). Thus, the adverse effects of chronic mTORC1 inhibition on insulin sensitivity and glucose tolerance may be linked to defective activation of Akt in multiple tissues and reduced adipose expression of GLUT4, as seen in 3T3-L1 adipocytes.
In summary, this study shows the crucial role of both mTORC1 and its downstream effector S6K1 in the operation of the negative feedback loop toward insulin-induced glucose uptake in adipocytes. Our results suggest that mTORC1 negatively modulates insulin action on PI3K activation via inhibitory phosphorylation of IRS-1 Ser636/639, whereas S6K1 is key to phosphorylation of both Ser636/639 and Ser1101 residues. However, chronic inhibition of mTORC1 or S6K1 also blunts insulin-stimulated glucose uptake by uncoupling PI3K activity from Akt2 activation as well as by decreasing GLUT4 expression. These data thus help explain previous observations of impaired glucose homeostasis in rapamycin-treated subjects and indicate that chronic inhibition of the mTORC1/S6K1 pathway may not be a useful strategy for combating insulin resistance in obesity.
Materials and Methods
Materials
Cell culture media were purchased from Life Technologies, Inc. (Burlington, Ontario, Canada). Fetal bovine serum (FBS) and bovine calf serum were respectively purchased from Hyclone (Logan, UT) and Wisent (St. Bruno, CA). The 2-D-[3H]deoxyglucose was from NEN Life Science Products (Boston, MA). [32P]ATP, protein A- and G-Sepharose were from GE Healthcare (Little Chalfont, UK). Polyclonal antibodies against IRS-1 (H-165), AKT 1/2 (H-136), Akt 1 (C-20), S6K1 (C-18), and S6 peptide (RRRLSSLRA) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antiphospho-specific antibodies against Akt (Ser473 and Thr308), IRS-1 (Ser1101), and p70S6K1 (Thr389) were from Cell Signaling Technology (Boston, MA), and IRS-1 (Ser636/639) was from Abcam (Cambridge, MA). Akt/SGK peptides (RPRAATF) and antibodies against phosphotyrosine (4G10), p85 subunits, and Akt2 were obtained from Upstate Biotechnology (Lake Placid, NY). Anti-GLUT4 polyclonal antibody was purchased from Chemicon (Temecula, CA), and anti-GLUT1 polyclonal antibody was a generous gift from Dr. Amira Klip (Hospital for Sick Children, Toronto, Canada). Recombinant human insulin was obtained from Ely Lilly & Co. (Toronto, Canada). Rapamycin was purchased from Biomol (Plymouth Meeting, PA). PIP3 measurement was performed using a competitive ELISA kit (K-2500; Echelon, Salt Lake City, UT) following the manufacturer’s instructions.
Cell culture
The 3T3-L1 cells, kindly provided by Dr. A. Klip (Hospital for Sick Children, Toronto, Canada), were grown and differentiated as previously described (12).
RNAi and electroporation
HPLC purified siRNAs were synthesized by Integrated DNA Technologies (IDT, Coralville, IA). The sequences of the 21-mer oligos were: mTOR, 5′-AGUCCGUUCCUUCUCCUUCdTdG-3′; S6K1, 5′-AAGGCGAAUAGGAGGGCAGdTdT-3′; raptor, 5′-UUAAUCGCAGCUACCUCUAdTdG-3′. The control was an irrelevant siRNA against luciferase designed with a similar GC ratio compared with other siRNAs, as previously described (48, 49), siRNAs were electroporated in fully differentiated 3T3-L1 adipocytes as described by Nguyen et al. (50). Completely differentiated 3T3-L1 (d 7) were trypsinized, centrifuged at 1200 rpm for 5 min, and resuspended in 650 μl of serum-free DMEM. The cells were immediately electroporated with 2.5 nmol of siRNA with the Bio-Rad Gene Pulser Xcell (250 V, 950 μF, and ∞ Ω; Bio-Rad, Mississauga, Ontario, Canada). Rapidly, electroporated cells were resuspended in prewarmed DMEM 10% FBS and replated in tissue culture plate. Three days after electroporation, cells were serum deprived for 4 h and stimulated as indicated in the figure legends. Using Cy3-labeled siRNA, we have demonstrated that at least 90% of the cells were transfected (data not shown). In agreement with a previous study (50), cell morphology and function (e.g. glucose uptake and insulin signaling) were not altered by the electroporation procedure.
Rapamycin treatment
Fully differentiated 3T3-L1 adipocytes were treated with rapamycin (25 nm) or vehicle [dimethylsulfoxide (DMSO), 0.01%] in DMEM 10% FBS for 24 h. Cells were then serum deprived for the remaining 4 h in presence of rapamycin (25 nm) or vehicle (DMSO, 0.01%) and stimulated with insulin (10 nm) for 45 min.
Lipid quantification
Cells were rinsed twice in ice-cold PBS and fixed with 10% formalin for 1 h at room temperature. Cells were washed three times with H2O and incubated with filtered Oil Red O solution [0.2% (wt/vol) Oil Red O, 60% (vol/vol) isopropanol, 40% (vol/vol) dH2O]. Cells were washed three times with H2O and dried at 37 C before elution in isopropanol solution containing 1% Igepal. Coloration was quantified by absorbance at λ = 500 nm.
Protein extraction and Western blotting
After treatments, cells were lysed in solubilisation buffer (50 mm HEPES, pH 7.5; 150 mm NaCl; 1 mm EGTA; 20 mm β-glycerophosphate; 1% Nonidet P-40; 10 mm NaF; 2 mm Na3VO4; and protease inhibitors). Western blotting was performed as previously described (10). Protein phosphorylation and protein levels detected by immunoblotting were normalized for the expression of the protein and tubulin, respectively.
Enzyme activity assay
PI3K, Akt, and S6K1 activity was measured by incorporation of 32P to L-α-phosphatidylinositol, Akt/SGK substrate, and S6 peptide, respectively, as previously described (10). Ak1 and Akt2 were specifically immunoprecipitated using isoform-specific antibodies. Kinase activities were normalized for the protein content.
PIP3 measurement
Fully differentiated 3T3-L1 were treated with rapamycin for 24 h, serum deprived for 4 h, and stimulated with 10 nm insulin for 45 min. The cells were washed twice with ice-cold PBS, and PIP3 was extracted and measured accordingly to the manufacturer’s instructions (PIP3 Mass ELISA kit; Echelon).
Plasmids electroporation and immunofluorescence
Fully differentiated 3T3-L1 were transfected by electroporation using 106 cells in 50 μl of hypoosmolar buffer (Brinkmann, Mississauga, Ontario, Canada) (400 V, 100 μsec, eight pulses) in a Bio-Rad Gene Pulser Xcell with 3 μg of Btk-PH-GFP (24) or Akt2-HA plasmid (51). After electroporation, the cells were seeded on collagen-coated glass slides and were kept for 6 h in CO2-independent medium (Invitrogen, Carlsbad, CA) followed by 24-h incubation in normal cell culture medium. Electroporated 3T3-L1 were treated with rapamycin (25 nm). After 24 h of treatment, cells were serum deprived for 4 h and stimulated with 10 nm insulin for 45 min. The cells were washed twice with PBS and fixed in 3.7% formaldehyde in PBS for 15 min. For the Akt2-HA plasmid, the cells were incubated 1 h at room temperature in blocking buffer (0.1% saponin and 3% BSA in PBS) and 1 h at 37 C in blocking buffer containing the primary antibody (mouse anti-HA; Covance, Princeton, NJ). The cells were washed three times in PBS and incubated 1 h at 37 C with the secondary antibody (goat antimouse Alexa 594; Invitrogen). Observations were made and confocal images were taken with an MRC-1024 confocal system (Bio-Rad) on a Diaphot inverted microscope (Nikon, Tokyo, Japan) and captured with Laser Sharp software (Bio-Rad). The recruitment of Btk-PH and Akt2-HA to the plasma membrane was determined semiquantitatively by estimating by eye in at least 100 cells the percentage of the cell perimeter staining positively for Btk-PH-GFP and Akt2-HA within the arbitrarily defined ranges of 0–10% (low), 11–30% (intermediate), and more than 30% (strong).
Measurement of glucose uptake
The 2-D-[3H]deoxyglucose uptake was measured with or without indinavir as previously described (10). The 3-O-methylglucose uptake was measured as follows: 50 μm 3-O-[3H]methylglucose (4 μCi/ml) was added to HEPES-buffered saline and uptake allowed to occur for 30 sec and then washed three times with ice-cold saline containing 1 mm HgCl2. Glucose uptake was normalized for the protein content.
Quantitative real-time PCR
Total RNA was obtained using trizol (Invitrogen) and purified with RNeasy Plus (QIAGEN, Hilden, Germany). RNA was reverse transcribed with poly-dT primers, and the resultant cDNA was analyzed by real-time PCR (Rotor-Gene RG3000A; Corbett Research, Montréal, Quebec, Canada) with FastStart SyBR Green Master mix (Roche, Mississauga, Ontario, Canada) followed by comparative quantification. Glyceraldehyde-3-phosphate dehydrogenase was used to normalize mRNA expression of GLUT4 and PPARγ. The primers used were (forward/reverse): GLUT4, 5′-GATGATGAACCTGTTGGCCT-3′/5′-AGCGGAACAGCTCCAAGATG-3′; PPARγ, 5′-CGCTGATGCACTGCCTATGA-3′/5′-AGAGGTCCACAGAGCTGATTCC-3′; and glyceraldehyde-3-phosphate dehydrogenase, 5′-CCACCACCCTGTTGCTGTAG-3′/5′-CTTGGGCTACACTGAGGACC-3′.
Statistical analyses
Densitometric analysis was performed with ImageQuant TL software (GE Healthcare). siRNA and rapamycin treatments were compared with vehicle using ANOVA followed by Fisher’s post hoc test. For clarity reasons, only the effects of siRNA and rapamycin were statistically represented in the figures. Differences were considered to be statistically significant at P < 0.05.
Acknowledgments
We thank Audrey Nguyen (University of California, San Diego, CA) for her help with the design of siRNAs and with the electroporation protocol; Dr. J. Lavoie for providing plasmids; and Dr. Rita Kohen for critical reading of the manuscript.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR) Grant 144336. A.M. is the recipient of a CIHR/Pfizer Research Chair on the pathogenesis of insulin resistance and cardiovascular diseases. A.V. and V.P.H were supported by studentships from the Fonds de Recherche en Santé du Québec. A.V. was also supported by studentships from the Association Diabète Québec.
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 4, 2010
A.V. and V.P.H contributed equally to this work.
Abbreviations: DMSO, Dimethylsulfoxide; FBS, fetal bovine serum; GFP, green fluorescent protein; GLUT, glucose transporter; IRS-1, insulin receptor substrate-1; mTOR, mammalian target of rapamycin; PH, pleckstrin homology; PI3K, phosphatidylinositol 3-kinase; PIP3, phosphatidylinositol (3 4 5 )-triphosphate; PP2A.protein phosphatase 2A; PPARγ, peroxisome proliferator-activated receptor γ; PTEN, phosphatase and tensin homolog; S6K1, S6 kinase 1; SHIP, SH2-domain-containing inositol 5-phosphatase; siRNA, small interfering RNA.
References
- 1.Hay N, Sonenberg N2004. Upstream and downstream of mTOR. Genes Dev 18:1926–1945 [DOI] [PubMed] [Google Scholar]
- 2.Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, Yonezawa K2006. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 25:6361– 6372 [DOI] [PubMed] [Google Scholar]
- 3.Tremblay F, Marette A2001. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276:38052–38060 [DOI] [PubMed] [Google Scholar]
- 4.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14:1296–1302 [DOI] [PubMed] [Google Scholar]
- 5.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K2002. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189 [DOI] [PubMed] [Google Scholar]
- 6.Tremblay F, Jacques H, Marette A2005. Modulation of insulin action by dietary proteins and amino acids: role of the mammalian target of rapamycin nutrient sensing pathway. Curr Opin Clin Nutr Metab Care 8:457–462 [DOI] [PubMed] [Google Scholar]
- 7.Um SH, D'Alessio D, Thomas G2006. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 3:393–402 [DOI] [PubMed] [Google Scholar]
- 8.Tremblay F, BrûléS, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A2007. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci USA 104:14056–14061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Gao Z, Yin J, Quon MJ, Ye J2008. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(α) signaling through IKK2. J Biol Chem 283: 35375–35382 [DOI] [PMC free article] [PubMed]
- 10.Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, Donner DB2001. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci USA 98:4640–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson CJ, White MF, Rondinone CM2004. Mammalian target of rapamycin regulates IRS-1 serine 307 phosphorylation. Biochem Biophys Res Commun 316:533–539 [DOI] [PubMed] [Google Scholar]
- 12.Tremblay F, Gagnon A, Veilleux A, Sorisky A, Marette A2005. Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3-L1 and human adipocytes. Endocrinology 146:1328–1337 [DOI] [PubMed] [Google Scholar]
- 13.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G2004. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205 [DOI] [PubMed] [Google Scholar]
- 14.Shah OJ, Hunter T2006. Turnover of the active fraction of IRS1 involves raptor-mTOR- and S6K1-dependent serine phosphorylation in cell culture models of tuberous sclerosis. Mol Cell Biol 26:6425–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF2004. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 166:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, Waldhäusl W, Marette A, Roden M2005. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes 54:2674–2684 [DOI] [PubMed] [Google Scholar]
- 17.Khamzina L, Veilleux A, Bergeron S, Marette A2005. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146:1473–1481 [DOI] [PubMed] [Google Scholar]
- 18.Tzatsos A2009. Raptor binds the SAIN (Shc and IRS-1 NPXY binding) domain of insulin receptor substrate-1 (IRS-1) and regulates the phosphorylation of IRS-1 at Ser-636/639 by mTOR. J Biol Chem 284:22525–22534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN2008. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8:399–410 [DOI] [PubMed] [Google Scholar]
- 20.Kimball SR2006. Interaction between the AMP-activated protein kinase and mTOR signaling pathways. Med Sci Sports Exerc 38:1958–1964 [DOI] [PubMed] [Google Scholar]
- 21.Tzatsos A2009. Raptor binds the SAIN (Shc and IRS-1 NPXY-binding) domain of IRS-1 and regulates the phosphorylation of IRS-1 at Ser636/639 by mTOR. J Biol Chem 284:22525–22534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ2001. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276:38349–38352 [DOI] [PubMed] [Google Scholar]
- 23.Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP2003. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci USA 100:7569–7574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Várnai P, Rother KI, Balla T1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem 274:10983–10989 [DOI] [PubMed] [Google Scholar]
- 25.Rudich A, Konrad D, Török D, Ben-Romano R, Huang C, Niu W, Garg RR, Wijesekara N, Germinario RJ, Bilan PJ, Klip A2003. Indinavir uncovers different contributions of GLUT4 and GLUT1 towards glucose uptake in muscle and fat cells and tissues. Diabetologia 46:649–658 [DOI] [PubMed] [Google Scholar]
- 26.Gagnon A, Lau S, Sorisky A2001. Rapamycin-sensitive phase of 3T3-L1 preadipocyte differentiation after clonal expansion. J Cell Physiol 189:14–22 [DOI] [PubMed] [Google Scholar]
- 27.Berggreen C, Gormand A, Omar B, Degerman E, Göransson O2009. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J Physiol Endocrinol Metab 296:E635–E646 [DOI] [PubMed]
- 28.Shah OJ, Wang Z, Hunter T2004. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 14:1650–1656 [DOI] [PubMed] [Google Scholar]
- 29.Patti ME, Brambilla E, Luzi L, Landaker EJ, Kahn CR1998. Bidirectional modulation of insulin action by amino acids. J Clin Invest 101:1519–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzatsos A, Kandror KV2006. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol 26:63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw 3rd EB, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ2001. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β). Science 292: 1728–1731 [DOI] [PubMed]
- 32.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM2004. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol 24:8778–8789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson RT, Desai BN, Hardwick JS, Schreiber SL1999. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci USA 96:4438–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartley D, Cooper GM2002. Role of mTOR in the degradation of IRS-1: regulation of PP2A activity. J Cell Biochem 85:304–314 [DOI] [PubMed] [Google Scholar]
- 35.Wada T, Sasaoka T, Funaki M, Hori H, Murakami S, Ishiki M, Haruta T, Asano T, Ogawa W, Ishihara H, Kobayashi M2001. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5′-phosphatase catalytic activity. Mol Cell Biol 21:1633–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson KA, Buse MG2008. Mechanisms of high-glucose/insulin-mediated desensitization of acute insulin-stimulated glucose transport and Akt activation. Am J Physiol Endocrinol Metab 294:E870–E881 [DOI] [PMC free article] [PubMed]
- 37.Sasaoka T, Wada T, Fukui K, Murakami S, Ishihara H, Suzuki R, Tobe K, Kadowaki T, Kobayashi M2004. SH2-containing inositol phosphatase 2 predominantly regulates Akt2, and not Akt1, phosphorylation at the plasma membrane in response to insulin in 3T3-L1 adipocytes. J Biol Chem 279:14835–14843 [DOI] [PubMed] [Google Scholar]
- 38.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA1996. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 39.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 40.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168 [DOI] [PubMed] [Google Scholar]
- 41.Taha C, Liu Z, Jin J, Al-Hasani H, Sonenberg N, Klip A1999. Opposite translational control of GLUT1 and GLUT4 glucose transporter mRNAs in response to insulin. Role of mammalian target of rapamycin, protein kinase B, and phosphatidylinositol 3-kinase in GLUT1 mRNA translation. J Biol Chem 274:33085–33091 [DOI] [PubMed] [Google Scholar]
- 42.Liao W, Nguyen MT, Yoshizaki T, Favelyukis S, Patsouris D, Imamura T, Verma IM, Olefsky JM2007. Suppression of PPAR{γ} attenuates insulin-stimulated glucose uptake by affecting both GLUT1 and GLUT4 in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 293:E219–E227 [DOI] [PubMed]
- 43.Olson AL, Knight JB2003. Regulation of GLUT4 expression in vivo and in vitro. Front Biosci 8:s401–s409 [DOI] [PubMed]
- 44.Miura S, Tsunoda N, Ikeda S, Kai Y, Cooke DW, Lane MD, Ezaki O2004. Nuclear factor 1 regulates adipose tissue-specific expression in the mouse GLUT4 gene. Biochem Biophys Res Commun 325:812–818 [DOI] [PubMed] [Google Scholar]
- 45.Im SS, Kwon SK, Kang SY, Kim TH, Kim HI, Hur MW, Kim KS, Ahn YH2006. Regulation of GLUT4 gene expression by SREBP-1c in adipocytes. Biochem J 399:131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teutonico A, Schena PF, Di Paolo S2005. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol 16:3128–3135 [DOI] [PubMed] [Google Scholar]
- 47.Di Paolo S, Teutonico A, Leogrande D, Capobianco C, Schena PF2006. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: a crossroad between cancer and diabetes? J Am Soc Nephrol 17:2236–2244 [DOI] [PubMed] [Google Scholar]
- 48.Liao W, Nguyen MT, Yoshizaki T, Favelyukis S, Patsouris D, Imamura T, Verma IM, Olefsky JM2007. Suppression of PPAR-γ attenuates insulin-stimulated glucose uptake by affecting both GLUT1 and GLUT4 in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 293:E219–E227 [DOI] [PubMed]
- 49.Liao W, Nguyen MT, Imamura T, Singer O, Verma IM, Olefsky JM2006. Lentiviral short hairpin ribonucleic acid-mediated knockdown of GLUT4 in 3T3-L1 adipocytes. Endocrinology 147:2245–2252 [DOI] [PubMed] [Google Scholar]
- 50.Nguyen MT, Satoh H, Favelyukis S, Babendure JL, Imamura T, Sbodio JI, Zalevsky J, Dahiyat BI, Chi NW, Olefsky JM2005. JNK and tumor necrosis factor-α mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol Chem 280: 35361–35371 [DOI] [PubMed]
- 51.Mitsuuchi Y, Johnson SW, Moonblatt S, Testa JR1998. Translocation and activation of AKT2 in response to stimulation by insulin. J Cell Biochem 70:433–441 [PubMed] [Google Scholar]