

Abstract
Polyadenylation is an essential cellular process in eukaryotic cells (Edmonds M and Abrams R, J Biol Chem 235, 1142–1149, 1960; Zhao J et al., Microbiol Mol Biol Rev 63, 405–445, 1999; Edmonds M, Progr Nucleic Acid Res Mol Biol 71, 285–389, 2002). For this reason, it has been difficult to examine the functions of specific polyadenylation proteins in vivo. Here, we describe a cell culture assay that allows structure-function experiments on CstF-64, a protein that binds to pre-mRNAs downstream of the cleavage site for accurate and efficient polyadenylation. We also demonstrate that the stem-loop luciferase assay for polyadenylation (SLAP) accurately reflects CstF-64-dependent polyadenylation. This assay could be easily adapted to the study of other important RNA-binding proteins in polyadenylation.
Keywords: Polyadenylation, CstF-64, Luciferase reporter gene, Dual-Luciferase Assay, RNA-binding protein, Epitope tagging
1 Introduction
Many stages in gene expression require the action of RNA-binding proteins. One such stage, polyadenylation, requires at least five separate protein complexes [4 – 6] requiring the action of many RNA-binding proteins [7, 8]. One of the best studied of these proteins is the 64,000 Mr subunit of the cleavage stimulation factor, CstF-64 [9, 10]. CstF-64 binds to the downstream G/U-rich sequence element in pre-mRNAs [11, 12] and is essential for polyadenylation of the pre-mRNA [6, 13, 14].
Because it is essential for normal cellular growth and development, examination of the structure-function relationships of the CstF-64 protein by disrupting its activities in polyadenylation in vivo has been difficult [13, 15, 16]. However, in vitro studies have allowed descriptions of the linear domains of CstF-64: the N-terminal RNA-binding domain (RBD), the hinge region, the MEAR(A/G) region, and the highly conserved C-terminal domain [12, 17 – 23]. But these proposed functions of putative domains had not been demonstrated in vivo due to the absence of a straightforward assay system.
Here, we present the stem-loop luciferase assay for polyadenylation (SLAP) that allows structure-function studies of CstF-64 in vivo. The SLAP procedure involves the downstream G/U-rich sequence element of a luciferase reporter gene to be replaced with two copies of the MS2 bacteriophage stem-loop element [24]. This modification makes luciferase expression dependent on the co-expression of a CstF-64-MS2 coat protein RNA-binding domain [25] fusion protein. These constructs are expressed in HeLa cells and the varying levels of luciferase expression correlate with changes in mRNA polyadenylation. The SLAP system has allowed us to verify not only that SLAP is a valid method of measuring polyadenylation in vivo but also implicated that the RBD, hinge, and CTD domains of CstF-64 were critical for polyadenylation [26].
2 Materials
All DNA, buffers, and other solutions were prepared using double deionized water. All reagents were stored as indicated.
2.1 Transfection Components (Store at −20 °C)
SL-Luc luciferase reporter: Generated by removing the SV40 polyadenylation cassette from pRL-SV40 (Promega, Madison, Wisconsin) using NotI and ClaI restriction enzymes. A SV40 polyadenylation cassette with MS2 stem loops replacing the G/U-rich downstream polyadenylation element was cloned in by PCR from p3Z SVL-MS2 (Maciolek and McNally (Medical College of Wisconsin)) also using NotI and ClaI primers (primer sequences available upon request). Additionally, two other SL-Luc constructs were made: one that contains neither the G/U-rich downstream element nor the MS2 stem loops (SL-Luc ΔSL) and one in which the AAUAAA polyadenylation signal was mutated by site-directed mutagenesis to AGGAGA (SL-LucAGGAGA). All SL-Luc constructs are shown in Fig. 1b.
MS2-CstF-64 fusion plasmids were obtained from Maciolek and McNally (Medical College of Wisconsin, ref. 24) and then cloned into p3XFLAG 7.1 (Sigma-Aldrich, St Louis, Missouri) using NotI, BglII, and KpnI restriction enzymes. Various primer combinations were used to generate different CstF-64 deletion constructs (primers available upon request). CstF-64 constructs that were made are as follows and diagramed in Fig. 1c: MS2-CstF-64, MS2-CstF-64 ΔRBD, MS2-CstF-64 ΔHinge, MS2-CstF-64 ΔP/G, MS2-CstF-64 ΔMEARA, MS2-CstF-64 ΔCTD, and MS2-CstF-64Y85A-L86S (point mutations demonstrated to inactivate the MS2 coat protein RNA-binding domain [27]).
pGL3-Control (firefly luciferase plasmid, Promega, Madison, Wisconsin).
p3XFLAG 7.1 empty vector (Sigma-Aldrich, St. Louis, Missouri).
Fig. 1.
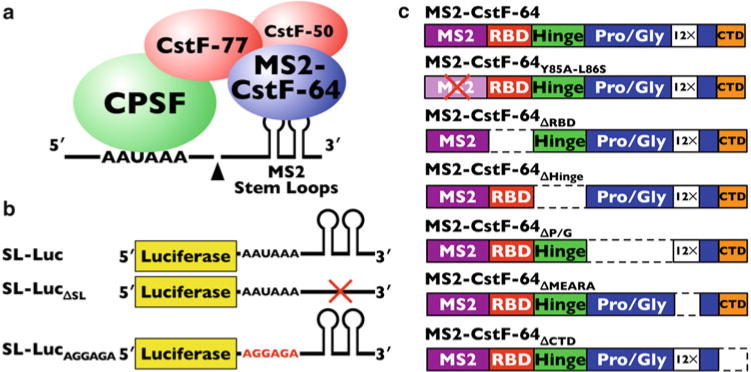
The SLAP method and components. (a) Simplified cartoon of components in polyadenylation. Shown are the pre-mRNA (line) with the MS2 stem-loop RNA elements, the cleavage and polyadenylation specificity factor (CPSF, green ball), the subunits of the cleavage stimulation factor (CstF-77, CstF-50, red balls), and the MS2-CstF-64 (blue ball). (b) Stem-loop luciferase (SL-Luc) constructs used in SLAP. (c) MS2-CstF-64 and deletion constructs
2.2 HeLa Cell Culture (Store at 4 °C Except Where Indicated)
Cell culture media: DMEM with 4.5 g/L-glucose, L -glutamine, and sodium pyruvate. DMEM was supplemented with 10 % Cosmic Calf Serum (Hyclone, Logan, Utah), 100 U/mL penicillin, and 100 μg streptomycin/mL.
Cell culture media without penicillin and streptomycin.
Serum-free cell culture media: DMEM with 4.5 g/L-glucose, L-glutamine, and sodium pyruvate.
Cell culture media for post-transfection: Media from #2 with 20 % Cosmic Calf Serum.
Opti-MEM I Reduced Serum Medium (Invitrogen).
Lipofectamine (Invitrogen).
HeLa cells (ATCC, Manassas, Virginia)—grow at 37 °C in 5 % CO2 (see below) and store in liquid nitrogen.
2.3 Luciferase Assay Components (Store at −80 °C Except Where Indicated)
Passive Lysis Buffer from Dual-Luciferase® Reporter Assay System (Promega, Madison, Wisconsin). Prepare a 1× working concentration by adding 1 volume of 5× PLB to 4 volumes of distilled water (see Note1).
Phosphate buffered saline (PBS; 1×): 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4 pH 7.4 in 1 L H2O. Store at room temperature.
Luciferase Assay Reagent II (LAR II) (Promega, Madison, Wisconsin). Resuspend the provided lyophilized Luciferase Assay Substrate in 10 mL of Luciferase Assay Buffer II (Promega, Madison, Wisconsin). Store for 1 month at −20 °C or 1 year at −80 °C (see Notes2 – 4).
Stop & Glo Reagent (Promega, Madison, Wisconsin). Stop & Glo is supplied at a 50× concentration. Add 1 volume of 50× substrate to 50 volumes of Stop & Glo buffer in a polypropylene tube (see Notes5 and 6).
Turner Designs (Sunnyvale, California) TD-20/20 Luminometer or equivalent. Store at room temperature.
3 Methods
Carry out all procedures at room temperature unless otherwise specified.
3.1 HeLa Cell Transfection (All Procedures to Be Carried Out Under a Sterile Hood)
Approximately 24 h before transfection, plate 2.75×104 HeLa cells per well on 24-well plates. Cells are to be plated in 500 μL of media without antibiotics. 15 wells will need to be plated for each experimental data point (one triplicate for each experiment and five replicates of each experiment, see Note7).
On the day of transfection, set up 1.5 mL tubes for each experiment (each triplicate) as follows: 100 μL of Opti-MEM with 48 ng of SL-Luc reporter, 16 ng of pGL3 control, 400 ng of MS2-CstF-64 fusion plasmid, and 536 ng of p3XFLAG 7.1 for a total of 1,000 ng of DNA (see Notes8 and 9).
In a separate 1.5 mL tube for each experiment, add 100 μL of Opti-MEM with 8 μL of Lipofectamine Transfection Reagent (see Note10).
Pipette the Opti-MEM/Lipofectamine mixture into the tube containing the DNA, mix gently with a pipetter, and incubate for 15 min at room temperature.
Add 600 μL of Opti-MEM to the combined DNA/Lipofectamine mix and mix gently (see Note11).
Under a tissue culture hood, remove growth medium from the HeLa cells and replace it with 200 μL of serum-free DMEM in each well. Add 250 μL of DNA/Lipofectamine/Opti-MEM (from step 5) to each well (three times for each triplicate). Mix by rocking the plate gently.
Incubate the plate for 5 h at 37 °C.
Add 400 μL of DMEM with 20 % Cosmic Calf Serum to each well without removing the transfection media. Incubate at 37 °C for approximately 19 h.
Remove transfection media and replace with 500 μL per well of regular cell culture media with 10 % Cosmic Calf Serum and antibiotics (penicillin and streptomycin). Incubate plates at 37 °C for 24 h.
3.2 Cell Lysis
Label enough 1.5 mL tubes for each well of the transfection (see Note12).
Prepare enough Passive Lysis Buffer by diluting 5×PLB with water (see Note1).
Remove media from the cells and gently add approximately 500 μL of 1×PBS to each well to wash media from the cells.
Wash cells a second time with 1×PBS.
Remove PBS from cells and add 100 μL of Passive Lysis Buffer per well.
Place the plates onto an orbital shaker platform and agitate gently for 15 min at room temperature.
Transfer the lysate into the appropriate labeled 1.5 mL tube (from step 1).
3.3 Luciferase Assay
Prepare Luciferase Assay Reagent II as described above (see Note2).
Prepare Stop & Glo Reagent as described above (see Note3).
Set up enough 1.5 mL tubes for each sample in the experiment (15 for each data point, see Note13).
To each tube (from step 3), add 100 μL of LAR II.
Program the luminometer to perform a 2-s premeasurement delay, followed by a 10-s measurement period for each assay.
Transfer 20 μL of cell lysate (from Subheading 3.2, step 7) into the luminometer tube containing LAR II. Mix by pipet-ting four or five times.
Place tube in the luminometer and initiate reading immediately after mixing.
If the luminometer is not connected to a printer or computer, record the firefly luciferase activity measurement (see Note14).
Remove the tube and dispense 100 μL of Stop & Glo Reagent. Mix by pipetting, and replace the tube in the luminometer and initiate reading.
Record the Renilla luciferase activity measurement (see Note15).
Discard the reaction tube and proceed to the next tube.
- When all samples have been measured, analyze as follows (Table 1):
- Divide each Renilla activity value by its corresponding firefly luciferase activity value. This produces the luciferase activity for each transfected well.
- Then average the triplicate luciferase values (three transfected well values).
- Then average the five triplicate groups that are the replicates for each MS2 or MS2-CstF-64 construct.
- Use the values for the replicates to determine the standard deviation for each construct.
- Graphs for these results can be produced using a program such as Microsoft Excel (Fig. 2).
Use the values for the replicates along with a statistics program such as InStat (Graphpad Software, Inc, San Diego, California) to determine the validity of the results using an ANOVA and a Tukey Post Test.
Table 1.
Typical Luciferase Values
| Renilla a | Firefly | Renilla/firefly | Average Renilla/firefly | Standard deviation | |
|---|---|---|---|---|---|
| SL-Luc | 390.3 | 138.9 | 2.810 | 2.889 | 0.993 |
| 994.9 | 253.8 | 3.920 | |||
| 556.2 | 287.0 | 1.938 | |||
|
| |||||
| SL-Luc + MS2-CstF-64 | 8,966.0 | 211.5 | 42.392 | 41.066 | 1.152 |
| 9,238.0 | 229.2 | 40.305 | |||
| 11,466.0 | 283.1 | 40.502 | |||
|
| |||||
| SL-LucAGGAGA | 131.2 | 318.6 | 0.412 | 0.427 | 0.016 |
| 124.8 | 292.9 | 0.426 | |||
| 123.2 | 278.0 | 0.443 | |||
|
| |||||
| SL-LucAGGAGA + MS2-CstF-64 | 508.7 | 158.5 | 3.209 | 3.021 | 0.187 |
| 646.1 | 227.9 | 2.835 | |||
| 589.4 | 195.2 | 3.019 | |||
Shown are typical luciferase values from three technical replicates using HeLa cells transfected as indicated using the Dual-Luciferase ® Reporter Assay System and the TD-20/20 Luminometer. For simplicity, only technical replicates are shown. However, we routinely perform five independent measurements (independently transfected wells) with three technical replicates each
Fig. 2.
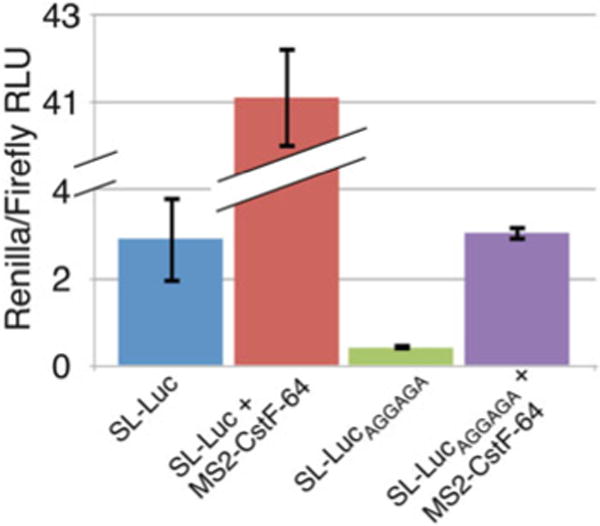
Renilla/firefly ratios as calculated from the data in Table 1. Shown are relative luciferase units calculated from the Renilla/firefly ratios for SL-Luc, SL-Luc + MS2-CstF-64, SL-LucAGGAGA, and SL-LucAGGAGA + MS2-CstF-64. Note that the Y-axis is broken to accommodate the higher values for SL-Luc + MS2-CstF-64
Footnotes
Prepare 1×PLB on the day of the cell lysis and make sure that enough is made for all samples plus 10 % for overage.
When planning SLAP experiments, it is best to plan a number of wells so that all of the LAR II is used. With each data point representing 15 transfected wells and each well using 100 μL of LAR II, only about 6 or 7 data points (constructs) are needed to use this amount of LAR II.
Prepare the LAR II reagent on the day of the luciferase assay (it should not be refrozen).
Prepare a new LAR II solution for each new experiment. However, because the LAR II reagent is expensive, if you need to use LAR II that has been frozen (left over from a previous assay), make sure to mix it with the new LAR II so that each sample gets the same LAR II for consistency.
Make Stop & Glo on the day of the luciferase assay. Fresh reagent always works best.
Make enough Stop & Glo for all samples plus 10 % to ensure that each sample gets enough of the reagent.
For organizational purposes it is easiest to plate cells by experiment (each one of the five replicates for each data point being tested). For example, if transfecting MS2 alone, MS2-CstF-64, ΔRBD, ΔHinge, ΔP/G, ΔMEARA, and ΔCTD wells can be arranged for each triplicate (three wells of MS2 alone, etc.) until all triplicates are done for each replicate, then do the same for the next replicate. This breaks the samples into different replicates for ease of organization.
It is easiest to make one large master mix tube for each data point (experimental construct) that you will be testing for. So prepare five times the amount of Opti-MEM and DNA plus 10 %. This provides each well with the same amount of Opti-MEM and DNA.
When making the master mix, it is most convenient to use a 5 mL (or 15 mL) tube to ensure that all components can be mixed.
When using a master mix, add 550 μL of Opti-MEM with 44 μL of Lipofectamine.
If making a master mix (see Note8), add 3,300 μL of Opti-MEM to the master mix tubes.
Keep the same organization for the sample tubes as for the wells on the plates. Not all of the sample will be used in each luciferase assay, so samples can be retained and stored at −70 °C and used for additional luciferase assays.
Prepare enough tubes to do all samples. All luciferase samples from one transfection should be performed on the same day. This eliminates day-to-day variation between the samples and the LAR II and Stop & Glo Reagents that can occur.
Record the firefly luciferase activity as quickly as possible if not using a printer or make sure to take the same amount of time between samples. For greatest reproducibility, we find it most effective to use a stopwatch to time the addition of the reagents and luciferase readings.
Record the Renilla luciferase activity quickly to ensure timely transfer from one sample to the next. For greatest reproducibility, we find it most effective to use a stopwatch to time the addition of the reagents and luciferase readings.
References
- 1.Edmonds M, Abrams R. Polynucleotide biosynthesis: formation of a sequence of ade-nylate units from adenosine triphosphate by an enzyme from thymus nuclei. J Biol Chem. 1960;235:1142–1149. [PubMed] [Google Scholar]
- 2.Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edmonds M. A history of poly A sequences: from formation to factors to function. Prog Nucleic Acid Res Mol Biol. 2002;71:285–389. doi: 10.1016/s0079-6603(02)71046-5. [DOI] [PubMed] [Google Scholar]
- 4.Christofori G, Keller W. 3′ cleavage and polyadenylation of mRNA precursors in vitro requires a poly(A) polymerase, a cleavage factor, and a snRNP. Cell. 1998;54:875–889. doi: 10.1016/s0092-8674(88)91263-9. [DOI] [PubMed] [Google Scholar]
- 5.Takagaki Y, Ryner LC, Manley JL. Four factors are required for 3′-end cleavage of pre-mRNAs. Genes Dev. 1989;3:1711–1724. doi: 10.1101/gad.3.11.1711. [DOI] [PubMed] [Google Scholar]
- 6.Gilmartin GM, Nevins JR. An ordered pathway of assembly of components required for polyadenylation site recognition and processing. Genes Dev. 1989;3:2180–2190. doi: 10.1101/gad.3.12b.2180. [DOI] [PubMed] [Google Scholar]
- 7.Mandel CR, Bai Y, Tong L. Protein factors in pre-mRNA 3′-end processing. Cell Mol Life Sci. 2008;65:1099–1122. doi: 10.1007/s00018-007-7474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, Di Giammartino DC, Taylor D, et al. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 2009;33:365–376. doi: 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore CL, Chen J, Whoriskey J. Two proteins crosslinked to RNA containing the adenovirus L3 poly(A) site requires the AAUAAA sequence for binding. EMBO J. 1988;7:3159–3169. doi: 10.1002/j.1460-2075.1988.tb03183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilusz J, Shenk T. A 64 kd nuclear protein binds to RNA segments that include the AAUAAA polyadenylation motif. Cell. 1988;52:221–228. doi: 10.1016/0092-8674(88)90510-7. [DOI] [PubMed] [Google Scholar]
- 11.Chou ZF, Chen F, Wilusz J. Sequence and position requirements for uridylate-rich downstream elements of polyadenylation signals. Nucleic Acids Res. 1994;22:2525–2531. doi: 10.1093/nar/22.13.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacDonald CC, Wilusz J, Shenk T. The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNA downstream of the cleavage site and influences cleavage site location. Mol Cell Biol. 1994;14:6647–6654. doi: 10.1128/mcb.14.10.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takagaki Y, Manley JL. Levels of poly-adenylation factor CstF-64 control IgM heavy chain mRNA accumulation and other events associated with B cell differentiation. Mol Cell. 1998;2:761–771. doi: 10.1016/s1097-2765(00)80291-9. [DOI] [PubMed] [Google Scholar]
- 14.Proudfoot NJ, O’Sullivan J. Polyadenylation: a tail of two complexes. Curr Biol. 2002;12:855–857. doi: 10.1016/s0960-9822(02)01353-2. [DOI] [PubMed] [Google Scholar]
- 15.Minvielle-Sebastia L, Winsor B, Bonneaud N, et al. Mutations in the yeast RNA14 and RNA15 genes result in an abnormal mRNA decay rate; sequence analysis reveals an RNA-binding domain in the RNA15 protein. Mol Cell Biol. 1991;11:3075–3087. doi: 10.1128/mcb.11.6.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dass B, Tardif S, Park JY, et al. Loss of polyadenylation protein τCstF-64 causes spermatogenic defects and male infertility. Proc Natl Acad Sci U S A. 2007;104:20374–20379. doi: 10.1073/pnas.0707589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pérez Cañadillas JM, Varani G. Recognition of GU-rich polyadenylation regulatory elements by human CstF-64 protein. EMBO J. 2003;22:2821–2830. doi: 10.1093/emboj/cdg259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takagaki Y, Manley JL. RNA recognition by the human polyadenylation factor CstF. Mol Cell Biol. 1997;17:3907–3914. doi: 10.1128/mcb.17.7.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takagaki Y, Manley JL. Complex protein interactions within the human polyadenylation machinery identify a novel component. Mol Cell Biol. 2000;20:1515–1525. doi: 10.1128/mcb.20.5.1515-1525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai Y, Auperin TC, Chou CY, et al. Crystal structure of murine CstF-77: dimeric association and implications for polyadenylation of mRNA precursors. Mol Cell. 2007;25:863–875. doi: 10.1016/j.molcel.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 21.Richardson JM, McMahon KW, MacDonald CC, et al. MEARA sequence repeat of human CstF-64 polyadenylation factor is helical in solution. A spectroscopic and calorimet-ric study. Biochemistry. 1999;38:1403–1407. doi: 10.1021/bi990724r. [DOI] [PubMed] [Google Scholar]
- 22.Qu X, Pérez Cañadillas JM, Agarawal S, et al. The C-terminal domains of vertebrate CstF-64 and its yeast orthologue Rna15 form a new structure critical for mRNA 3′-end processing. J Biol Chem. 2007;282:2101–2115. doi: 10.1074/jbc.M609981200. [DOI] [PubMed] [Google Scholar]
- 23.Ruepp MD, Schweingruber C, Kleinschmidt N, et al. Interactions of CstF-64, CstF-77, and symplekin: implications on localisation and function. Mol Biol Cell. 2011;22:91–104. doi: 10.1091/mbc.E10-06-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maciolek NL, McNally MT. Characterization of Rous sarcoma virus polyadenylation site use in vitro. Virology. 2008;374:468–476. doi: 10.1016/j.virol.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lago H, Fonseca SA, Murrau JB, et al. Dissecting the key recognition features of the MS2 bacteriophage translational repression complex. Nucleic Acids Res. 1998;26:1337–1344. doi: 10.1093/nar/26.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hockert JA, Hsiang-Jui Y, MacDonald CC. The hinge domain of the cleavage stimulation factor protein CstF-64 is essential for CstF-77 interaction, nuclear localization, and polyadenylation. J Biol Chem. 2010;285:695–704. doi: 10.1074/jbc.M109.061705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valegard K, Murrary JB, Stonehouse NJ, et al. The three-dimensional structures of two complexes between recombinant MS2 capsids and RNA operator fragments reveal sequence-specific protein-RNA interactions. J Mol Biol. 1997;270:724–738. doi: 10.1006/jmbi.1997.1144. [DOI] [PubMed] [Google Scholar]