

Abstract
Genome-wide analysis of gene expression has changed the RNA world. Recent techniques leading to this revolution have been the use of cross-linking and immunoprecipitation (CLIP) combined with high-throughput sequencing (HITS-CLIP) to determine sites on nascent mRNAs to which RNA-binding proteins bind. Several researchers (including us) have been examining the role of RNA-binding proteins in polyadenylation, including the role of the 64,000 Mr component of the cleavage stimulation factor, CstF- 64. In this chapter, we present our optimizations of the CLIP procedure for examination of CstF-64 binding to nascent pre-mRNAs expressed in testis. For CstF-64 CLIP, we use a well-characterized monoclonal antibody (3A7) that recognizes CstF-64. Rather than optimizing tricky but essential RNA fragment cloning schemes, we illustrate the use of the proprietary Illumina TruSeq Small RNA Sample Preparation kit for this step. Other techniques such as SDS-PAGE and the transfer to the nitrocellulose membrane techniques follow the original Illumina protocol (though we point out potential pitfalls). Finally, we discuss the options for high-throughput sequencing and some general suggestions for bioinformatic analysis of the data.
Keywords: Polyadenylation, CstF-64, RNA-binding protein, UV cross-linking, Immunoprecipitation, CLIP, RNA-seq
1 Introduction
Polyadenylation site choice is governed by the coordinated action of a swarm of RNA-binding proteins [1 – 3] that bind to signals in the nascent mRNA [4]. Molecular techniques have been used to map some of these interactions to individual nascent mRNAs [5 – 7], but newer techniques are required to examine protein–RNA interactions that encompass genes expressed from the entire genome. The approach that combines ultraviolet cross-linking and immunoprecipitation (CLIP, [8]) with high-throughput sequencing of cDNAs generated from the bound RNA fragments (HITS-CLIP, [9]) is ideal for global analysis of these protein–RNA interactions to reveal functional insights [10].
Immunoprecipitation can have greater or lesser specificity, depending on the antibody and conditions. Therefore, optimization steps should precede HITS-CLIP. In this chapter, we present our optimizations for the 3A7 monoclonal antibody that recognizes the 64,000 Mr subunit of the cleavage stimulation factor (CstF-64, [11]) in HITS-CLIP from mouse testis. Because 3A7 is highly specific for the somatic form of CstF-64 in mice [12] and shows low nonspecific background immunoprecipitation [5], it has proven to be ideal for HITS-CLIP [13]. Other antibodies may work, as well [10, 14]. We further give tips on high-throughput sequencing of the resulting cDNA libraries.
2 Materials
2.1 Reagents
All reagents, buffers, and other solutions were prepared using double- deionized water and stored as indicated. General laboratory reagents (e.g., Tris base, NaCl, SDS, MgCl2), if not specifically indicated, were purchased from one of the major bioscientific vendors located in the United States: Fisher Scientific or Sigma-Aldrich. Other items purchased included the following:
NuPAGE® Novex 10 % Bis–Tris Gel 1.0 mm, 10 Well SDS-PAGE gels (NP0301Box, Life Technologies).
NuPAGE® Sample Reducing Agent (10×, NP0009, Life Technologies).
NuPage Antioxidant (NP0005, Life Technologies).
Protran BA-85, 0.45 μm (10 402 594, Whatman) nitrocellulose membrane (Sigma-Aldrich).
Water-saturated phenol, pH 6.6 (AM9710, Life Technologies).
Agencourt AMPure XP beads (A63880, Beckman Coulter).
Corning® Costar® Spin-X® centrifuge tube filters (CLS8161, Sigma-Aldrich).
SequaGel–UreaGel Concentrate (EC-830, National Diagnostic).
30 % acrylamide and bis-acrylamide solution, 37.5:1 ratio (161-0158, Bio-Rad).
20 mg/mL UltraPure™ Glycogen (10814-010, Life Technologies).
Dynabeads® Protein G for Immunoprecipitation (100-04D, Life Technologies).
EasyTide [γ-32 P]-ATP, 3,000 Ci/mmol, 10 mCi/mL (BLU502A100UC, PerkinElmer).
Precision Plus Protein™ Dual Color Standards (161-0374, Bio-Rad).
Kaleidoscope Prestained Standards (161-0324, Bio-Rad).
Low Molecular Weight DNA Ladder (N3233S, New England Biolabs).
Adenosine 5′-triphosphate (ATP, R0441, Fermentas) was prepared as a 10 mM solution in aliquots, which were stored at −80 °C and discarded after each use.
2.2 Buffers
The buffers were made from stock solutions with double- deionized water prior to the beginning of the experiment. The following buffers and solutions were obtained from commercial vendors:
10× Dulbecco’s phosphate-buffered saline (DPBS), no calcium, no magnesium (14200-075, Life Technologies).
Buffers in a Box, Premixed TBE Buffer, 10× (1666703001, Roche Applied Science).
NuPAGE® MOPS SDS Running Buffer (for Bis–Tris Gels only, 20×, NP0001, Life Technologies).
NuPAGE® Transfer Buffer (20×, NP0006-1, Life Technologies).
NuPAGE® LDS Sample Buffer (4×, NP0007, Life Technologies).
3 M Sodium Acetate, pH 5.5 (AM9740, Life Technologies).
Blue/Orange Loading Dye, 6× (G1881, Promega).
SequaGel–UreaGel Diluent (EC-830 National Diagnostic).
Gel Loading Buffer II (Denaturing PAGE, AM8547G, Life Technologies).
Whenever possible we used the buffers supplied with the corresponding enzymes or systems. Some of the general laboratory stock supply buffers are not listed. However, the following buffers were always prepared “in-house”:
Low-salt immunoprecipitation buffer—1× DPBS, 0.1 % SDS (w/v), 0.5 % NP-40 (v/v), 0.5 % deoxycholic acid sodium salt (w/v).
High-salt immunoprecipitation buffer—5× DPBS, 0.1 % SDS, 0.5 % NP-40, 0.5 % deoxycholic acid, sodium salt.
Dephosphorylation buffer—50 mM Tris–HCl pH 7.9, 100 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol (added fresh before use).
Phosphatase wash buffer—50 mM Tris–HCl pH 7.5, 20 mM EGTA, 0.5 % NP-40 (v/v).
Polynucleotide kinase buffer—50 mM Tris–HCl pH 7.9, 50 mM NaCl, 10 mM MgCl2.
Proteinase K reconstitution buffer—100 mM Tris–HCl pH 7.9, 50 mM NaCl.
Proteinase K dilution buffer—100 mM Tris–HCl pH 7.9, 50 mM NaCl, 10 mM EDTA.
Proteinase K urea buffer—6 M Urea, 100 mM Tris–HCl pH 7.9, 50 mM NaCl, 5 mM EDTA.
RNA PAGE elution buffer—1.66 mg/mL proteinase K, 50 mM Tris–HCl pH 7.5, 25 mM NaCl, 0.3 M sodium acetate pH 5.5, 0.5 % SDS.
DNA PAGE elution buffer—50 mM Tris–HCl pH 7.5, 25 mM NaCl/0.3 M sodium acetate pH 5.5, 0.5 % SDS.
2.3 DNA and RNA Adapters
Below are listed the RNA and DNA adapters and primers used in the CLIP protocol. For the RNA PCR primer (RPI1) containing index 1, the index sequence is underlined. Illumina’s oligonucleotides are modified and purified in a proprietary manner (see Note1):
RNA 5′ adapter (RA5): 5′-GUUCAGAGUUCUACAGUCCGACGAUC-3′.
RNA 3′ adapter (RA3): 5′-TGGAATTCTCGGGTGCCAAGG-3′.
Stop oligo (STP): 5′-GAAUUCCACCACGUUCCCGUGG-3′.
RNA RT primer (RTP): 5′-GCCTTGGCACCCGAGAATTCCA-3′.
RNA PCR primer (RP1): 5′-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3′.
RNA PCR primer, index 1 (RPI1): 5′-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3′.
2.4 Enzymes
The following enzymes and kits were used as recommended by the suppliers:
SUPERase ⋅ In (AM2696, Life Technologies).
RQ1 RNase-Free DNase (M610A, Promega).
RNase I (Cloned), 100 U/μL (AM2294, Life Technologies).
Calf intestinal alkaline phosphatase (CIP, M0290S, New England Biolabs).
T4 polynucleotide kinase (M0201L, New England Biolabs).
T4 RNA Ligase 2, truncated (M0242L, New England Biolabs).
SuperScript® III Reverse Transcriptase (18080-044, Life Technologies).
Proteinase K was prepared as follows: 25 mg of Proteinase K, recombinant, PCR Grade Lyophilizate from Pichia pastoris (03115836001, Roche Applied Science) was dissolved in 6.25 mL of Proteinase K reconstitution buffer to produce a 4 mg/mL solution, and 0.25 mL aliquots were frozen at −80 °C. Prior to use, a working solution was prepared by mixing 0.25 mL of the frozen aliquot with 0.25 mL Proteinase K dilution buffer and incubating for 10 min at 37 °C.
2.5 Equipment and Kits
UV Crosslinker CL-1000 (UVP LLC.) or equivalent.
Thermomixer® R (Eppendorf, North America).
Sonic Dismembrator Model 60 (Fisher Scientific).
DynaMag™-2 Magnetic Particle Concentrator (Invitrogen) or equivalent.
XCell SureLock™ Electrophoresis Cell/Novex Mini-Cell (Invitrogen).
TruSeq small RNA prep kit (Indexes 1–12, RS-200-0012, Illumina).
PicoFuge® microcentrifuge (Stratagene) or equivalent.
2.6 Animals
25-day-old male C57BL/6 mice were obtained from litters bred in-house. All animal protocols were performed as approved by the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee.
3 Methods
3.1 Tissue Disruption and UV Cross-Linking
The amount of material required for CLIP varies depending on the tissue source, protein abundance, and efficacy of the antibody. For this protocol (Fig. 1), we describe CLIP using the 3A7 monoclonal antibody to isolate CstF-64–RNA complexes from mouse testes. Guidelines to optimize the procedure for other tissues and conditions are discussed in Note2.
Sacrifice two male mice in the manner approved by your Institutional Animal Care and Use Committee. Collect the seminiferous tubules by making incision into the tunica albuginea of the testes and expressing the content in 3–5 mL ice-cold 1× DPBS in a 15 mL conical tube. Disrupt the seminiferous tubules using vigorous shaking. Let the material settle down for ~3 min on ice. Remove the supernatant. Add 3–5 mL fresh ice-cold 1× DPBS. Repeat the procedure for a total of three times.
Resuspend the seminiferous tubules in 1 mL of 1× DPBS and transfer to a single well of 12-well plate, pre-chilled on ice.
Place the 12-well plate in the UVP UV Crosslinker CL-1000, and irradiate the tissues with 200 mJ/cm2 at 254 nm three times (for a total of 600 mJ/cm2). Mix between irradiations by swirling the plate. Transfer the samples to 2 mL microcentrifuge tubes, and rinse the well with an additional 1 mL of 1× DBPS.
Collect the samples by low-speed centrifugation (1,000 × g) for 5 min at 4 °C.
At this stage, you can store the tissue at −80 °C or continue directly with tissue lysis. For freezing, snap-freeze the samples in liquid nitrogen or dry ice; slower freezing (say, by transferring to a −80 °C freezer) is less desirable, since it might promote proteolysis.
Fig. 1.
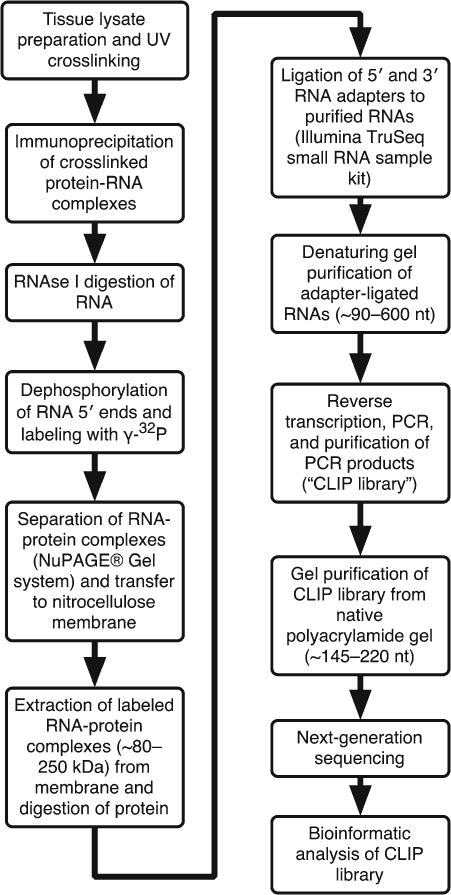
Summary of steps in CLIP protocol. Shown is a flow chart of the steps described in this chapter
3.2 Antibody Coupling to the Paramagnetic Dynabeads Protein G
Transfer 100 μL of Dynabeads Protein G in each of the four 1.5 mL microcentrifuge tubes and resuspend by vortexing. The 3A7 mouse monoclonal antibody we use here is of the IgG1 isotype, which binds best to Protein G. If your antibody is of a different isotype, choose the appropriate Dynabeads.
- Wash Dynabeads twice with 1 mL low-salt immunoprecipitation buffer:
- Add 1 mL of low-salt immunoprecipitation buffer.
- Vortex briefly.
- Clear liquid quickly by microcentrifugation.
- Place tube in the magnetic particle concentrator for 30–60 s.
- Remove supernatant by vacuum aspiration.
- Repeat.
Resuspend Dynabeads in 500 μL low-salt immunoprecipitation buffer.
Add 20 μg of IgG 1 (~500 μL of tissue culture supernatant) of mouse monoclonal anti-CstF-64 clone 3A7 antibody.
Incubate the tubes on a Thermomixer R (1,100 rpm, shaking for 20 s and resting for 30 s) for 2 h at 23 °C.
Wash three times with 1 mL low-salt immunoprecipitation buffer. See Note3 for antibody-coupled Dynabeads storage conditions.
3.3 Total Cell Lysate Preparation, RNAse I and DNase I Digestions, and Immunoprecipitation of the CstF-64 Protein
Prepare and chill (4 °C) the low-salt immunoprecipitation buffer, at least 1 mL/testis. Usually, we have the testes from one animal collected in one 2 mL tube (see Subheading 3.1). Immediately prior to use, add 2 μL SUPERase ⋅ In per testis and ~5 mg of phenylmethylsulfonyl fluoride (PMSF) powder to 10 mL of buffer (see Note4).
Prepare 1:500 (low RNase) and 1:50 (high RNase) dilutions of RNase I in low-salt immunoprecipitation buffer and store on ice (see Note5).
If tissue/cell pellets have been frozen at −80 °C, thaw on ice.
As soon as the tissue is thawed, add 0.7 mL ice-cold low-salt immunoprecipitation buffer. Pipette up and down several times until most of the tissue cells are lysed. Add an additional 0.8 mL buffer, and mix by pipetting. The lysate should become extremely viscous.
Incubate on ice for 5 min.
Sonicate using the Sonic Dismembrator small probe three times on ice with five pulses each time. Avoid foaming or warming the lysate.
Clear the lysates by centrifugation at maximum speed (15,800 × g) at tabletop microcentrifuge for 3 min at 4 °C.
Transfer the supernatant (~1.5 mL) to a fresh 2 mL tube. Avoid disturbing the pellet.
Optional: Incubate the lysate for 5 min at 75 °C and then cool on ice (see Note6).
Add 40 μL RQ1 RNase-free DNase to each of the samples and vortex.
Split the volume of each tissue lysate into two tubes, ~700 μL each. Save a small amount of lysate for examination by Western blot (see Fig. 2a). For mice, we split the lysate from both testes of a single animal into two tubes, one of which will be digested with the high-RNase dilution (“tube 1”) and the other digested with the low-RNase dilution (“tube 2”).
Add 7 μL of high-RNase dilution to tube 1 and of the low-RNase dilution to tube 2.
Mix by vortexing, microcentrifuge briefly to clear droplets, and incubate for 5 min at 37 °C.
Centrifuge the lysates at 15,800 × g for 3 min at 4 °C.
Transfer the supernatant (~700 μL) to the antibody-coupled Dynabeads Protein G. Vortex and spin down briefly to collect the liquid from the sides and the lid of the tubes.
Incubate on a Thermomixer R (1,100 rpm shaking for 20 s and resting for 30 s) for 1 h at 4 °C (see Note7).
Collect the antibody-coupled Dynabeads on the magnetic particle concentrator.
Save the supernatant as flow-through for a Western blot (see Fig. 2a).
Wash the beads once with 1 mL low-salt immunoprecipitation buffer as for step 2 in Subheading 3.2. Take care not to remove Dynabeads from the tube.
Wash the antibody-coupled Dynabeads Protein G four times with 1 mL high-salt immunoprecipitation buffer.
Wash the beads once with 1 mL low-salt immunoprecipitation buffer, and leave them in the buffer until all necessary reagents are ready to be used in the next step.
Fig. 2.
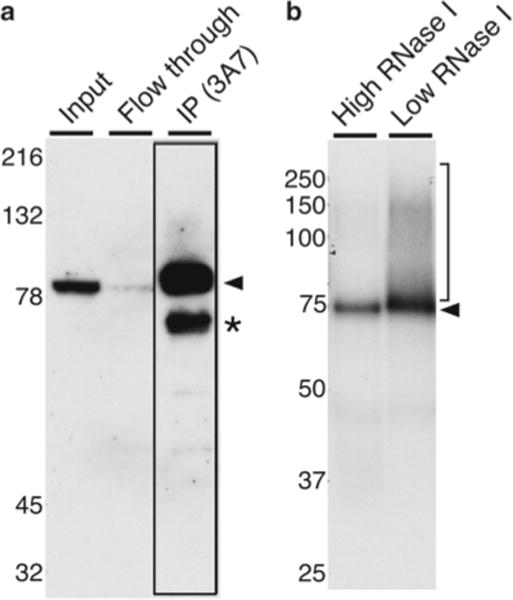
The 3A7 monoclonal antibody recognizes CstF-64 cross-linked to RNA. (a) A Western blot of CstF-64 from testis lysate showing total extract (input), unbound (flow-through), and immunoprecipitate-bound material [IP (3A7)], corresponding to Subheading 3.3. The arrowhead indicates the mobility of the CstF- 64 protein; the asterisk indicates a breakdown product we have observed previously (cf., [23]); the boxed lane highlights the immunoprecipitated material. Approximate molecular weights in kilodaltons are indicated to the left. (b) Representative autoradiograph after the CLIP procedure using 3A7 antibody against CstF-64 protein (Subheading 3.7). Shown are radioactively labeled RNA oligomers cross-linked to the immunoprecipitated CstF-64 protein. The arrowhead indicates the mobility of CstF-64 after digestion with either high or low RNase I (see text). The region of the membrane that was excised from the low-RNase I lane is indicated by the bracket on the right. Approximate molecular weights in kilodaltons are indicated to the left. Note the slight difference in the mobility of the CstF-64 protein resolved on 10 % SDS-PAGE (a) and NuPAGE® Novex 10 % Bis–Tris gels (b)
3.4 Dephosphorylation of the 5′ RNA Ends with Calf Intestinal Alkaline Phosphatase
Wash Dynabeads twice with 1 mL of dephosphorylation buffer as in step 2 in Subheading 3.2. After the second wash, remove the supernatant with a pipette and discard it.
Prepare the dephosphorylation reaction mix by adding 2 μL/tube CIP (10 U/μL) to 50 μL dephosphorylation buffer (supplied by the manufacturer, 45 μL of water and 5 μL of 10× buffer 3).
Resuspend beads in dephosphorylation reaction mix.
Incubate the suspension for 10 min at 37 °C in the Thermomixer R (1,100 rpm shaking for 20 s and resting for 30 s).
Wash the antibody-coupled Dynabeads Protein G twice with 1 mL phosphatase wash buffer as in step 2 in Subheading 3.2.
3.5 γ-32 P ATP Labeling of the RNA 5′ Ends Using T4 Polynucleotide Kinase
Wash the antibody-coupled Dynabeads Protein G twice in 1 mL polynucleotide kinase buffer, and discard the supernatant.
Working behind a Plexiglas shield and taking precautions for radioactive use, prepare the T4 polynucleotide kinase mix by adding 3 μL of T4 polynucleotide kinase (10 U/μL) to 47 μL of 5 μL 10× T4 polynucleotide kinase buffer, 0.5 μL SUPERase ⋅ In, 0.5 μL γ-32 P-ATP, and 41 μL of water.
Add 50 μL of the mix to the tube and mix by vortexing.
Incubate the tubes for 30 min at 37 °C in the Thermomixer R (1,100 rpm shaking for 20 s and resting for 30 s).
Add 0.75 μL of 10 mM ATP to each tube and incubate for an additional 5 min at 37 °C.
Collect the antibody-coupled Dynabeads Protein G on a magnetic collector.
Remove the T4 polynucleotide kinase reaction according to the safety regulations for disposal of radioactive materials.
Wash the antibody-coupled Dynabeads Protein G five times with 0.8 mL polynucleotide kinase buffer, and discard the supernatant as radioactive waste.
The beads can be stored at −20 °C overnight in a Plexiglas-shielded container.
3.6 Separation of Protein–RNA Complexes by SDS-PAGE Using the NuPAGE® Novex Bis–Tris Gel System
In order to prevent alkaline hydrolysis of RNA oligonucleotides, it is necessary to use an electrophoresis system that buffers to pH 7.0. Therefore, Ule et al. [15] recommend the Novex NuPAGE 10 % Bis–Tris system (for homemade alternatives, see Note8).
Prepare 1× NuPAGE® MOPS SDS Running Buffer by diluting the 20× buffer. Add 0.5 mL NuPAGE® antioxidant to 200 mL pre-chilled 1× MOPS SDS running buffer before use. Remove the sealing sticker from the bottom of the NuPAGE® Novex 10 % Bis–Tris Gel 1.0 mm, 10-well polyacrylamide gel. Remove the comb, and wash the wells two to three times with water and three to four times with the 1× NuPAGE® MOPS SDS running buffer supplemented with the NuPAGE Antioxidant. Assemble the electrophoretic unit as recommended. If you run only a single gel, use the “dam” to block one of the sides. Pour the 1× NuPAGE® MOPS SDS running buffer supplemented with the NuPAGE® Antioxidant in the “top” inner chamber of the assembled unit.
Resuspend the Dynabeads Protein G from the T4 polynucleotide kinase labeling step in 25 μL of 2× NuPAGE® LDS Sample Buffer supplemented with 1× NuPAGE® Sample Reducing Agent.
Incubate for 10 min at 75 °C to elute the 32P-labeled CstF-64– oligoribonucleotide complexes from the antibody. Mix twice during the heating by gently flicking the tubes (do not invert the tube, as that can spread the radioactive material to the tube lid). Allow the samples to cool briefly on ice.
Spin the tubes briefly, and collect the Dynabeads on the magnetic particle collector.
Load 10 μL of Precision Plus Protein™ Dual Color Standard and 15 μL of LDS sample buffer to the first gel well. Load subsequent wells with supernatants from each sample (~25 μL). Take care to prevent sample spillover between the wells. Fill all “empty” lanes with 25 μL of 2× NuPAGE® LDS Sample Buffer.
Run the gel at 180 V for 1–2 h with cooling at 4 °C (possibly in a cold room).
3.7 Transfer the Separated 32P-Labeled Protein–RNA Complexes to a Nitrocellulose Membrane
Pre-chill 1 L of 1× transfer buffer (50 mL 20× NuPAGE® Transfer buffer, 100 mL methanol, 850 mL water). Add 1 mL NuPAGE® Antioxidant for efficient transfer to the 1× transfer buffer just before use.
Cut a nitrocellulose membrane (BA-85) to the size of the gel (7.5 × 9 cm) and pre-wet in 1× transfer buffer in a separate container. Wet the transfer sponges and assemble on a wet transfer electrophoresis cassette. While submerged, squeeze the sponges to remove any trapped air.
Make a “transfer sandwich” by stacking three sheets of pre-wetted thick chromatographic paper (7.9 × 9.4 cm) and the nitrocellulose membrane on top of the transfer sponge. Be sure to eliminate bubbles at every stage. If necessary, assemble the “transfer sandwich” while submerged to eliminate the possibility of trapped air bubbles between the layers.
Disassemble the electrophoresis unit, and release the gel from the glass plates. Be aware that the gel is radioactive, so take appropriate precautions.
Wet your gloves in the 1× transfer buffer to prevent sticking to the gel. Using the gel releaser wedge as carrying and support tool, place the gel on top of the nitrocellulose membrane in the transfer cassette.
Cover the gel with three sheets of pre-wetted thick chromatographic paper, and place a wet transfer sponge on top of the paper. To eliminate residual bubbles, roll a small pipette or 15 mL tube over the “sandwich” and close the transfer cassette.
Place the “sandwich” in the electrophoretic transfer unit. Make note of the cassette orientation to ensure correct direction of transfer.
Fill the electrophoresis tank with ~1 L of cold 1× transfer buffer.
Transfer at 60 V for 2.5–3 h at 4 °C in a cold room on ice.
At the end of the transfer, disassemble the electrophoretic cassette and discard the buffer and gel as radioactive waste.
Rinse the membrane once with 1× DPBS.
Wrap the wet membrane with clear plastic wrap. Expose it to X-ray film at −80 °C. We include luminescent stickers (such as Glogos, Agilent) on the plastic backing to correctly align the developed film to the membrane.
Expose to film for 6–24 h (see Note9).
3.8 Extract the Labeled RNAs, Which Are Bound to the CstF-64 Protein from the Membrane Using Proteinase K
Operating behind a Plexiglas shield, align the developed film over the membrane using the Glogos as a guide (Fig. 2b). Identify the band that corresponds to the size of RNA-binding protein in the sample digested with the high concentration of RNase I (high RNase, arrow, Fig. 2b), and mark the plastic wrap. The position of the peak of radioactive signal in the high-RNase lane will serve to indicate the approximate size of the CstF-64–oligoribonucleotide complex.
Compare the low-RNase lane to the high-RNase lane, and determine where the CstF-64–oligoribonucleotide complex will migrate (right bracket, Fig. 2b). Mark the plastic wrap. Using a clean razor or scalpel, cut out the bracketed region of the membrane corresponding to the low RNase (right bracket, Fig. 2b). Transfer excised membrane to a sterile microcentrifuge tube.
Dilute a 250 μL aliquot of Proteinase K (PK, 4 mg/mL) with a 250 μL of Proteinase K dilution buffer to yield a 2 mg/mL PK solution in 100 mM Tris–HCl pH 7.5, 50 mM NaCl, and 5 mM EDTA.
Incubate the PK solution at 37 °C for 10 min.
Add 200 μL of PK solution to each tube containing the excised membranes. Make sure that the membrane is soaked in the PK solution.
Incubate at 37 °C in the Thermomixer R (1,100 rpm shaking for 20 s and resting for 30 s) for 20 min.
Add 200 μL Proteinase K urea buffer; incubate at 55 °C in the Thermomixer R (1,100 rpm shaking for 20 s and resting for 30 s) for 20 min.
Reset the Thermomixer R to 37 °C and allow to cool (~6–7 min). Add 400 μL of water-saturated phenol (pH 6.6) and 130 μL chloroform and incubate at 37 °C in the Thermomixer R (1,100 rpm shaking for 20 s and resting for 30 s) for 5 min.
Separate the aqueous and organic phases by brief centrifugation at maximum speed in a tabletop centrifuge.
Transfer 400–450 μL of the aqueous phase to a fresh microcentrifuge tube, add 50 μL of 3 M sodium acetate pH 5.2, 0.5 μL of glycogen (20 mg/mL), and 1 mL of ice-cold 100 % ethanol. Allow the precipitate to form at −80 °C for at least 20 min. Alternatively, you could split the ethanol precipitation in two tubes.
Centrifuge the samples at maximum speed at 4 °C for 15 min.
Wash the pellet twice with 0.5 mL of cold 75 % ethanol.
Let the pellet dry at room temperature for 5–10 min or until no ethanol can be detected. Do not over-dry the pellet.
Add 5 μL of RNase-free water to the pellet.
Incubate at 37 °C for 5 min with some occasional vortexing and pipetting to resuspend the pellet.
Spin the tube briefly to settle the droplets.
3.9 Adapter Ligation to the Isolated RNAs Using Illumina’s TruSeq Small RNA Sample Prep Kit
In this section, we present an abbreviated version of a procedure, which is outlined in detail in the Illumina TruSeq small RNA prep kit manual (available through the Illumina, Inc. website [16]).
Dilute the original adapters 5′ (RA5) and 3′ (RA3) 1:5 (4 μL RNAse-free water and 1 μL of each adapter).
- Ligation of the 3′ adapter:
- Transfer 5 μL of the dissolved RNA from the extracting step into a 0.2 mL PCR tube.
- Add 1 μL diluted RA3 to the isolated RNA.
- Incubate the tube for 2 min at 70 °C and then cool on ice.
- Prepare the following reaction mix in a fresh tube (scale up as needed for multiple reactions):
- 2 μL ligation buffer (HML, Illumina).
- 1 μL RNase inhibitor.
- 1 μL T4 RNA ligase 2, truncated.
- Transfer the above mixture to the tube containing the RNA and RA3 adapter.
- Mix by pipetting up and down eight to ten times.
- Briefly spin down the tube to collect all the droplets.
- Incubate for 1 h at 28 °C.
- Add 1 μL of the Stop Oligo (STP, Illumina); continue to incubate for an additional 15 min at 28 °C.
- Place the tube on ice.
-
Ligation of the 5′ adapter (RA5).
Add 1 μL of diluted 5′ adapter in a new tube.
Incubate the tube at 70 °C for 2 min.
Immediately place the tube on ice.
- Add to the RA5:
- 1 μL 10 mM ATP.
- 1 μL T4 RNA ligase.
Mix by pipetting up and down.
Transfer 3 μL of the RA5 adapter mix to the RNA/RA3 reaction mix from step 2j; mix by pipetting up and down eight to ten times.
Incubate at 28 °C for 1 h.
Place on ice.
Add 15 μL of 2× gel loading buffer II.
Incubate at 75 °C for 10 min.
Place on ice.
3.10 Denaturing 9.3 % Polyacrylamide Gel Electrophoresis
- Prepare a 9.3 % denaturing polyacrylamide gel (20 mL) using 16 × 17 cm glass plates with 0.75 mm thick spacers and the corresponding 15-well comb:
- 2 mL 10× TBE.
- 7.5 mL SequaGel–UreaGel Concentrate.
- 10.5 mL SequaGel–UreaGel Diluent.
- 0.12 mL 10 % ammonium persulfate.
- 0.02 mL N, N, N′, N′-tetramethylethane-1,2-diamine (TEMED).
Assemble the gel in a vertical gel apparatus; fill cathode and anode reservoirs with 1× TBE running buffer. Remove the comb, and pre-run the gel at 10 W for ~1 h until glass plates are warm to touch.
Immediately before loading your sample, clean accumulated urea from the wells of the gel using a syringe and a fine-gauge needle.
Incubate the samples at 75 °C for 10 min, spin down briefly, and place on ice. Load samples into each lane. Include the radioactively pre-labeled low-molecular-weight DNA ladder.
Run the gel with the loaded samples until the bromophenol blue dye nears the bottom of the gel.
Wrap the gel in plastic wrap and expose to X-ray film overnight at −80 °C (see Subheading 3.7, step 12).
3.11 Extract the RNA with Ligated Adapters from Denaturing Polyacrylamide Gel
Develop the film, and mark the sizes of the radioactively pre-labeled low-molecular-weight DNA ladder on the film using a permanent marker pen. Align the film to the gel using the Glogo dots. Mark on the plastic wrap the position of the DNA ladder and migration of the loading dyes. Cut out the radioactive material above 90 nt (usually above the xylene cyanol FF dye) all the way to the beginning of the well (~600 nt, see Fig. 3a). You may also verify the size of your RNA fragments using as a guide the sizes of the radioactively pre-labeled low-molecular-weight DNA ladder (see Note10).
Prepare a “gel crushing tube,” by making two holes on the bottom of 0.5 mL microfuge tube with an 18-gauge needle.
Place the gel crushing tube inside a fresh 2 mL microfuge tube.
Place the cutout gel fragment inside the gel crushing tube and centrifuge at 6,000 × g. At the end of this process, the gel fragment will be crushed and extruded into the 2 mL tube.
Discard the 0.5 mL crushing tube as radioactive waste. Add 600 μL of RNA PAGE elution buffer to the 2 mL tube containing the crushed gel fragments. Incubate with end-over-end tumbling for 3–4 h at room temperature to elute the adapter-ligated RNA fragments.
Transfer the slurry to a Spin-X tube and centrifuge at 16,000 × g for 5 min or until the majority of the solution enters the collection tube.
Phenol/chloroform extract the flow-through material: Add 400 μL of water-saturated phenol, pH 6.6 and 130 μL of chloroform. Vigorously shake the samples for 15 s, collect the aqueous phase, add 1 μL glycogen (20 mg/mL), and split the volume equally into two tubes (~300 μL/tube). Discard phenol/chloroform as radioactive organic waste.
Add 900 μL of ice-cold 100 % ethanol. Precipitate the samples overnight at −20 °C.
Collect the precipitated ligated RNA mixture by centrifugation at maximum speed at 4 °C for 15 min.
Discard the supernatant as radioactive waste. Wash the pellet twice with 0.5 mL of ice-cold 75 % ethanol.
Let the pellet dry for 5–10 min at room temperature or until no more ethanol can be detected. Do not over-dry the pellet. Proceed immediately to Subheading 3.12.
Fig. 3.
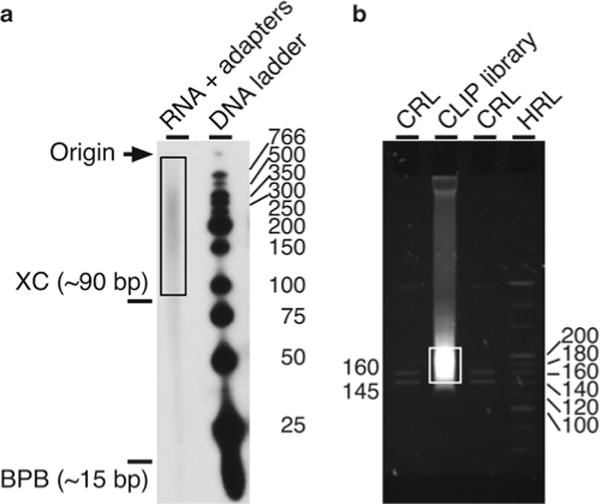
Purification of RNA oligomers and DNA amplimers after the CLIP procedure. (a) Autoradiograph of the 32P-labeled RNA oligonucleotides with various sizes isolated as described in the text and subsequently ligated to the RA3 and RA5 adapters using the Illumina’s TruSeq small RNA prep kit (Subheading 3.11). The box indicates radioactive RNA with the ligated adapters (RNA + adapters) that is extracted to produce the CLIP DNA library. Shown are sizes in nucleotides of the radioactively labeled low-molecular-weight DNA ladder (DNA ladder, right) and approximate mobilities of the xylene cyanol (XC) and bromophenol blue (BPB, left). (b) Representative fluorescent image of ethidium bromide-stained native PAGE of the CLIP library (Subheading 3.14). The box indicates the region of the final CLIP library that was extracted from the gel. Lanes are the custom RNA ladder (CRL), CLIP library, and high-resolution ladder (HRL). Ladder fragment sizes are indicated in base pairs. Please note that the theoretical size of a PCR product without an insert is 117 bp
3.12 Reverse Transcriptase (RT) Reaction and PCR Using Illumina’s TruSeq Small RNA Sample Prep Kit
Resuspend the RNA pellets in 6 μL water. Transfer the solution in a 0.2 mL tube.
- Perform the RT reaction as follows:
- Add 1 μL of the RTP to 6 μL of the RNA, mix, and incubate for 2 min at 70 °C. Place immediately on ice.
- Prepare 12.5 mM dNTPs by mixing 0.5 μL of water with 0.5 μL 25 mM dNTPs provided with the Illumina kit.
- In a separate tube mix the following (“enzyme mix”):
- 2 μL 5× Buffer (Illumina).
- 1 μL 0.1 M DTT.
- 0.5 μL of 12.5 mM dNTPs.
- 1 μL SUPERase ⋅ In.
- 1 μL SuperScript III Reverse Transcriptase.
- Add 5.5 μL of the enzyme mix to the RNA/RTP sample. The total volume will be now 12.5 μL.
- Incubate the samples as follows using a thermocycler: 25 °C for 5 min; 42 °C for 20 min; 50 °C for 1 h; and 4 °C unlimited.
-
Prepare PCR reactions using the indexed RPI1 and RP1 primers. Be sure to record the RPI1 indices for each corresponding sample. A “master mix” can be prepared that includes the PCR master mix from the Illumina kit and RP1 primer (prepare at least 10 % extra “master mix” to account for pipetting errors):
(a) TruSeq small RNA kit PCR mix 25 μL
(b) 10 μM RP1 primer 2 μL
(c) 10 μM RPI1 primer 2 μL
(d) cDNA total reaction 12.5 μL
(e) Water 8.5 μL
PCR cycle conditions:
1× 98 °C, 1 min
25–28× 98 °C, 15 s 60 °C, 30 s 72 °C, 30 s
1× 72 °C, 10 min 1× Hold at 10 °C
3.13 Purify the PCR Product Using Agencourt AMPure XP Beads
The purification step is important for concentrating the sample and reducing the salt concentration, which otherwise would interfere with the electrophoretic mobility of the DNA. Alternatively, this step may be omitted and the samples can be treated as recommended in the Illumina manual for the TruSeq small RNA prep kit. However, we recommend performing this concentration and purification step.
Bring the AMPure XP magnetic beads to room temperature (about 30 min).
Resuspend the beads by vortexing briefly.
Transfer the PCR reaction mix (Subheading 3.12) to a 1.7 mL tube, add 90 μL of AMPure beads, and mix by pipetting gently ten times.
Incubate for 10 min at room temperature.
Place the tube on the magnetic concentrator stand for 2 min.
Gently remove the clear solution using a pipette; dispose of as radioactive waste.
Add 200 μL of 80 % ethanol, being careful not to disturb the beads. Remove the wash solution using a pipette.
Repeat the 80 % ethanol wash for a total of two times.
Allow the pellet to dry with an open lid on the magnetic stand (about 5–10 min or until the pellet looks dry).
Resuspend the dried beads in 10 μL of 10 mM Tris–HCl, pH 8.0. Ensure that the beads are in suspension by vortexing or pipetting up and down.
Incubate at room temperature for 2 min. Spin briefly to settle the liquid.
Place the tube on a magnetic stand for 2 min.
Remove 8.5–10 μL of the clear solution and place in a new tube.
Add 3 μL of 6× Blue/Orange Loading Dye Buffer (Promega).
3.14 Resolve the PCR Product Using Native PAGE
- Prepare a 6 % native polyacrylamide gel (see the recipe for 10 mL below). We prepare these gels using the same glass plates as for the protein mini gels (8.6 × 6.8 cm) with 1.00 mm thick spacers and the corresponding 10-well comb:
1 mL 10× TBE
2 mL 30 % acrylamide and bis-acrylamide solution, 37.5:1 ratio
120 μL 10 % ammonium persulfate
20 μL TEMED
6.88 mL Water
Prepare the custom RNA and high-resolution DNA ladders (supplied with the Illumina TruSeq small RNA prep kit) as recommended in the Illumina manual (Fig. 3b).
Load the ladders on the side and your sample in the middle (Fig. 3b). Leave empty lanes between samples to minimize cross-contamination. Perform electrophoresis at 110 V for ~1 h or until the bromophenol blue dye reaches the bottom of the gel. The majority of the PCR product will be between 145 and 220 bp, approximately 1.5–2.5 cm slower than the bromophenol blue dye when it reaches the bottom of the gel (Fig. 3b).
3.15 Extract the PCR Product with Molecular Weight ~145–220 nt from the Native Polyacrylamide Gel
Stain the native polyacrylamide gel with 0.5 μg/mL ethidium bromide in water.
Use a UV transilluminator to visualize the DNA (see Fig. 3b).
Excise the gel slices using a clean scalpel or razor blade for each sample. For CLIP products in the 35–45 nt range, excise a broad region between 145 and 220 bp. Try to make the gel piece as small as possible without sacrificing yield.
Prepare a “gel crushing tube” as for Subheading 3.11.
Place the crushing tube inside a fresh 2 mL microfuge tube.
Place the cutout gel fragment inside the gel crushing tube and centrifuge at 6,000 × g to crush the gel and elute the fragments into the larger tube.
Discard the 0.5 mL crushing tube. Add 400 μL of DNA PAGE elution buffer to the 2 mL tube containing the crushed gel and incubate with end-over-end tumbling for 3–4 h at room temperature.
Transfer the slurry to a Spin-X tube and centrifuge at maximum speed (~16,000 × g) until majority of the solution enters the collection tube.
Add 1 μL of glycogen (20 mg/mL) and 900 μL 100 % ethanol. Precipitate overnight at −20 °C.
Collect the precipitated PCR products by centrifugation at maximum speed at 4 °C for 15 min.
Wash the pellet twice with 0.5 mL cold 75 % ethanol.
Let the pellet dry for 5–10 min at room temperature or until no more ethanol can be detected. Do not over-dry the pellet.
Dissolve the pellet in 20 μL of 10 mM Tris–HCl, pH 7.9. This is the final CLIP library.
Assess the size and quantity of the CLIP library using an Agilent 2100 Bioanalyzer with a DNA 1000 kit. To obtain good-quality data from the sequencing on Illumina platform, a concentration of at least 1.5 nM and total of 35 ng is needed.
3.16 Illumina Sequencing of the CLIP Library
We recommend single-end (SE) sequencing with the Illumina platform for the CLIP library, performing 50 cycles of sequencing of the inserts (“50SE”). In the case of multiple samples, a multiplexing (use of several short sequence tags in the adapters to uniquely identify different samples) can be performed. 1 Gb (~20 million reads) of clean data (de-multiplexed and adapter sequences removed) is usually sufficient for analysis of a single CLIP experiment. Currently, there are many commercial vendors who will perform the sequencing for a reasonable fee. Before choosing a specific vendor, consider the following: the platform that they are using for sequencing, the physical location of their facility (which will affect shipping costs), the turnaround time, and, last but not least, the price.
3.17 Bioinformatic Analysis of the CLIP Library
A detailed workflow of bioinformatic analysis of CLIP data is beyond the scope of this chapter and requires specialized skills [13, 17]. As a general outline, the sequencing CLIP data is first aligned to a reference genome (i.e., of the organism from which you obtained your samples, the Mus musculus genome in our example) and any ambiguous localizations of reads removed. Next, filter all identical RNA sequences, thus eliminating duplicates that may have arisen during the PCR steps. Thus filtered, sequence reads should be analyzed with regard to their annotated locations in the genome—e.g., in exons, introns, intergenic sequences, and 5′ or 3′ UTRs. Any further analysis probably needs to be tailored to the characteristic of your specific RNA-binding protein and to your hypotheses. Also (and way beyond the scope of this chapter), you should consider comparing your HITS-CLIP data to RNA-seq [17, 18] or high-throughput technologies to identify polyadenylation site [13, 19] data from the same samples. Those correlations can be used to address functional aspects and suggest new hypotheses to test.
Footnotes
For more information please refer to the Illumina custom sequence letter as of September 7, 2012, available through their website for free after registration (https://www.illumina.com/). Oligonucleotide sequences © 2007–2012 Illumina, Inc. All rights reserved [20].
Generally, the amount of tissue to consider sufficient for a CLIP procedure needs to be in the range that the radioactivity from the 32P-labeled RNAs can be visualized in an overnight exposure on X-ray film as indicated in Subheading 3.7. In our experience, 1–2 testes (about 80 mg each) obtained from a single 25-day-old male animal were sufficient for the CLIP procedure with 3A7 antibody. However, pilot experiments should be performed to optimize the amount of the starting material.
(a) In our experience the physical disruption of the tissue by vigorous shaking, triturating, and pipetting techniques is not problematic and lead to efficient cross-linking between the CstF-64 protein and RNA. Since the UV can penetrate several layers of cells obtaining single-cell suspension may not be necessary. Some tissues (e.g., liver, heart, muscle) may be more resistant to physical disruption. Therefore, an enzymatic digestion (e.g., by collagenase) of the tissue may be considered. If you work with a tissue requiring enzymatic digestion, use appropriate techniques to prevent uncontrolled digestion of the RNA.
(b) We determined that irradiating thrice with the 200 mJ/cm2 254 nm UV source was optimal by performing pilot experiments to examine patterns of cross-linked 32P-labeled proteins (see Fig. 2b). RNA-binding proteins may have different RNA-interacting surfaces and therefore different cross-linking efficiencies. Therefore, we recommend determining the optimal cross-linking conditions for each RNA-binding protein empirically.
We also recommend optimizing the initial immunoprecipitation conditions by trying different detergents (RIPA, NP-40, etc.), salts, and times. Determine the yield by Western blot. Once the immunoprecipitation conditions are determined, we suggest proceeding and performing a pilot CLIP experiment to verify that the conditions are indeed optimal for CLIP. If you have any doubts about the conditions, you can further optimize them by performing CLIP up to Subheading 3.8, which visualizes the protein–RNA complexes (Fig. 2b). In this protocol, we exclusively describe immunoprecipitation conditions that work best with the 3A7 antibody, which recognizes the mammalian CstF-64 protein.
You can prepare the antibody-coupled Dynabeads Protein G a few days ahead by washing once with 1 mL low-salt immunoprecipitation buffer and storing the coupled antibody in low-salt immunoprecipitation buffer supplemented with 0.05 % sodium azide at 4 °C. Prior to the immunoprecipitation wash the Dynabeads Protein G twice with 1 mL low-salt immunoprecipitation buffer.
PMSF has a short half-life in aqueous solution. Therefore, rather than preparing a stock PMSF solution, we find that adding a “pinch” of powder (enough to cover the tip of a metal spatula, ~5–10 mg) is sufficient to saturate a 10 mL aqueous solution. In our experience PMSF was sufficient to prevent degradation of CstF-64 in our testis samples. In tissues with greater endogenous proteases, consider using a cocktail of proteinase inhibitors.
RNase dilutions were prepared as follows: to 499 and 49 μL of low-salt immunoprecipitation buffer 1 μL of RNase I (see Subheading 2.4) was added to create the low- and high-RNase solutions, respectively. Tubes with the corresponding dilutions are vortexed and briefly spun down to clear the droplets.
Heating at 75 °C is an antibody-specific step and many antibody–protein interactions will not benefit from it. Because the 3A7 monoclonal antibody recognizes an epitope [21] that is revealed upon denaturation [11], we find that heating the lysate prior to immunoprecipitation increases yield while decreasing background.
We recommend the use of the Thermomixer. In lieu of using the Thermomixer, one could agitate the samples on a rocker platform at 37 °C.
We recommend using Novex® NuPAGE® SDS-PAGE Gel System from Life Technologies. The use of the NuPAGE system will greatly reduce variability during the SDS-PAGE electrophoresis. However, if you cannot obtain the Novex® NuPAGE® SDS-PAGE Gel System, homemade gels and buffers can be prepared and used according to the protocols published by the laboratory of Dr. Robert T. Sauer at the Department of Biology at the Massachusetts Institute of Technology [22].
Exposure times will vary greatly depending on the amount of the starting material, specific activity of the radioactivity, and immunoprecipitation efficiency of the antibody. However, if it takes longer than overnight at −80 °C to visualize the signal on an X-ray film with a fresh radioactivity, you should consider increasing the amount of the starting tissue/cells (see Note2).
The size of the RA3 and RA5 adapters ligated to each other is 47 nt. If one wants to obtain mainly insert sizes of 13 nt one should cut a band between 58 and 62 nt. However, in practice it is much better to collect as much material as possible above 90–100 nt. In our experience, if we do not perform this step, majority of our products contain adapters ligated to each other. In addition, nucleic acid sequences smaller than 20–30 nt are difficult to align unequivocally to the reference genome.
References
- 1.Änko ML, Neugebauer KM. RNA-protein interactions in vivo: global gets specific. Trends Biochem Sci. 2012;37:255–262. doi: 10.1016/j.tibs.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Darmon SK, Lutz CS. mRNA 3′ end processing factors: a phylogenetic comparison. Comp Funct Genomics. 2012;2012:876893. doi: 10.1155/2012/876893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010;38:2757–2774. doi: 10.1093/nar/gkp1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian B, Graber JH. Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip Rev RNA. 2012;3:385–396. doi: 10.1002/wrna.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacDonald CC, Wilusz J, Shenk T. The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNAs downstream of the cleavage site and influences cleavage site location. Mol Cell Biol. 1994;14:6647–6654. doi: 10.1128/mcb.14.10.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murthy KGK, Manley JL. The 160-kD subunit of human cleavage-polyadenylation specificity factor coordinates pre-mRNA 3′-end formation. Genes Dev. 1995;9:2672–2683. doi: 10.1101/gad.9.21.2672. [DOI] [PubMed] [Google Scholar]
- 7.Brown KM, Gilmartin GM. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor I m. Mol Cell. 2003;12:1467–1476. doi: 10.1016/s1097-2765(03)00453-2. [DOI] [PubMed] [Google Scholar]
- 8.Ule J, Jensen KB, Ruggiu M, et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 9.Licatalosi DD, Mele A, Fak JJ, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin G, Gruber AR, Keller W, et al. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012;1:753–763. doi: 10.1016/j.celrep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Takagaki Y, Manley JL, MacDonald CC, et al. A multisubunit factor CstF is required for polyadenylation of mammalian pre-mRNAs. Genes Dev. 1990;4:2112–2120. doi: 10.1101/gad.4.12a.2112. [DOI] [PubMed] [Google Scholar]
- 12.Wallace AM, Dass B, Ravnik SE, et al. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proc Natl Acad Sci U S A. 1999;96:6763–6768. doi: 10.1073/pnas.96.12.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoque M, Ji Z, Zheng D, et al. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods. 2012;10(2):133–139. doi: 10.1038/nmeth.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao C, Biesinger J, Wan J, et al. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci U S A. 2012;109:18773–18778. doi: 10.1073/pnas.1211101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ule J, Jensen K, Mele A, et al. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 16.Illumina Inc. Illumina website. 2012 Available from http://www.illumina.com/. Accessed 15 Jan 2013.
- 17.Li W, Yeh HJ, Shankarling GS, et al. The τCstF-64 polyadenylation protein controls genome expression in testis. PLoS One. 2012;7:e48373. doi: 10.1371/journal.pone.0048373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derti A, Garrett-Engele P, Macisaac KD, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Illumina Inc. Customer letter. 2012 Available from https://www.illumina.com/. Accessed 15 Jan 2013.
- 21.Dass B, McDaniel L, Schultz RA, et al. The gene CSTF2T encoding the human variant CstF-64 polyadenylation protein τCstF-64 is intronless and may be associated with male sterility. Genomics. 2002;80:509–514. [PubMed] [Google Scholar]
- 22.OpenWetWare contributors. Sauer: bis-Tris SDS-PAGE, the very best. 2009 Available from http://openwetware.org/index.php?title=Sauer:bis-Tris_SDS-PAGE%2C_the_very_best&oldid=300293. Accessed 15 Jan 2013.
- 23.Dass B, Tardif S, Park JY, et al. Loss of polyadenylation protein τCstF-64 causes spermatogenic defects and male infertility. Proc Natl Acad Sci U S A. 2007;104:20374–20379. doi: 10.1073/pnas.0707589104. [DOI] [PMC free article] [PubMed] [Google Scholar]