

Abstract
Two sibling DNA polymerases synthesize most of the eukaryotic nuclear genome. A new study provides insights into the distinct protein interactions that deliver these replicases for asymmetric leading- and lagging-strand replication and reveals possible cross-talk between DNA replication and other cellular processes.
Among the many DNA polymerases used to replicate and maintain eukaryotic genomes, two have the major responsibility for replicating nuclear DNA1. DNA polymerase (Pol) ε synthesizes most of the nascent leading strand in a largely continuous manner, and Pol δ synthesizes most of the nascent lagging strand as a series of ~200-bp Okazaki fragments. Pol ε and Pol δ are members of the same family of B polymerases and catalyze the same 5′-polymerization and 3′-exonucleolytic-proofreading reactions, yet they differ in structure, subunit composition, biochemical properties and protein partnerships (Fig. 1). Although these differences undoubtedly reflect their strand-specific roles in replication, uncertainty remains regarding how they are differentially delivered to the two strands. An exciting study by O’Donnell and colleagues2 now offers new insights into the mechanisms responsible for this asymmetric polymerase targeting.
Figure 1.
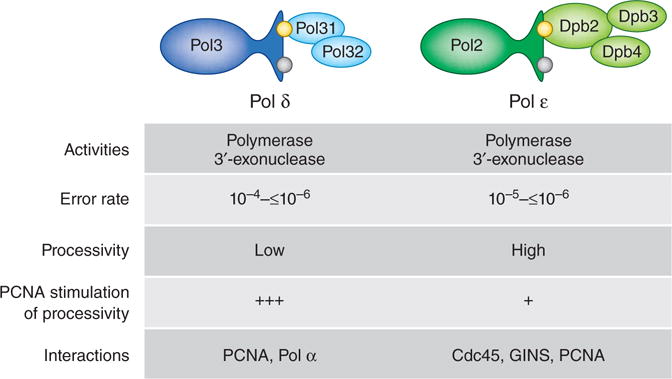
Comparison of DNA polymerases δ and ε. The four subunits of Pol ε (green) and the three subunits of yeast Pol δ (blue) are shown; a fourth subunit (p12) is present in the human enzyme. The catalytic (Pol3 and Pol2) subunits of both polymerases contain a 3′ exonuclease that can proofread replication errors to achieve the in vitro error rates shown above. The highest error rates are for single base deletions in long homonucleotide runs, which are proofread with low efficiency23. Consistent with a role in largely continuous leading-strand replication, Pol ε synthesizes DNA processively, i.e., without dissociating after nucleotide incorporation. Pol δ is highly processive only when assisted by PCNA. Proteins that interact with the individual polymerases are also listed.
Replication requires the CMG complex, which is composed of Cdc45, Mcm2–7 and GINS. Assembly of this 11-protein complex at replication origins activates the helicase activity of Mcm2–7, a six-member ring that encircles leading-strand DNA3. A replisome then forms, which contains Pols α, δ and ε as well as many other proteins, including the RFC clamp loader, the DNA-encircling PCNA clamp and the single-strand DNA–binding protein RPA. In a tour de force display of protein biochemistry, O’Donnell and colleagues2 purified more than two dozen Saccharomyces cerevisiae replication proteins and reconstituted two distinct replication reactions. In CMG-dependent reactions mimicking leading-strand replication, Pol ε synthesized DNA more processively and ten times faster than Pol δ, the latter of which is distributive with CMG. When the two polymerases were placed in competition, Pol ε was selectively recruited over Pol δ, and when Pol δ was loaded in a complex with CMG, it was displaced by Pol ε. In complementary studies of PCNA- and RPA-dependent synthesis of primed, single-stranded DNA mimicking lagging-strand replication, Pol δ synthesized DNA more quickly and processively than Pol ε (also ref. 4), and when Pol δ and Pol ε were placed in competition for PCNA binding, Pol δ was by far the favored partner. These results strongly imply that CMG selectively recruits Pol ε over Pol δ for leading-strand replication, whereas PCNA selectively recruits Pol δ over Pol ε for lagging-strand replication2 (Fig. 2). These observations provide key insights into how eukaryotic cells deliver two nonidentical polymerase siblings to synthesize the nascent leading and lagging strands.
Figure 2.
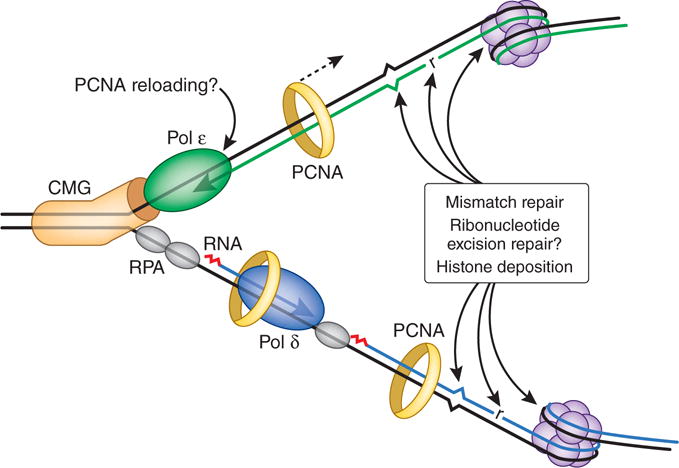
The eukaryotic replication fork. Modeled after ref. 2, but with postreplicative DNA transactions included. The CMG complex (orange) unwinds parental DNA to permit continuous leading-strand synthesis by Pol ε (green). The lagging strand is synthesized as a series of Okazaki fragments that are initiated by an RNA primer (red) made by a primase. This RNA primer is then extended by Pol α to generate a DNA primer that is extended by Pol δ (blue), which in turn synthesizes most of the nascent lagging strand with the assistance of PCNA (yellow ring). Given the weak PCNA–Pol ε interactions, we indicate the possibility of occasional dissociation of PCNA in order to mark the leading strand4 for postreplicative events that require PCNA. The most frequent of these events is histone deposition to initiate assembly of nucleosomes (purple), which are present every few hundred base pairs. Ribonucleotides (r) are incorporated during replication once every 1,000 to 10,000 bp and are removed by the ribonucleotide excision repair pathway, which could theoretically be coordinated with replication. Least abundant yet highly important are those mismatches generated by the replicases that escape proofreading and that must be removed by mismatch repair to suppress genome instability and tumorigenesis. RPA, replication protein A.
As impressive as it already is in size and complexity, the reconstituted leading-strand apparatus carries out replication about ten-fold more slowly than it does within a cell, perhaps because it lacks other proteins that are known to act at the replication fork in vivo. The new study thus provides a unique opportunity to determine the role of these missing proteins in mediating the increased speed and efficiency of fork movement. For example, the replication-progression complex contains several proteins, such as Ctf4 and Mcm10, which associate with the CMG complex and may promote more efficient coupling between lagging and leading strands5.
What are the protein-protein interactions that underlie the selective delivery of the two polymerases? The authors suggest two candidates: (i) interaction of the noncatalytic Dpb2 subunit of Pol ε with the Psf1 subunit of GINS for leading-strand replication and (ii) preferential interactions of the PCNA clamp with Pol δ for lagging-strand replication. But then what about the PCNA ring on the leading strand? Should it not recruit Pol δ in favor of Pol ε? Apparently, interactions with CMG override those with PCNA. However, a functional fork doesn’t always need to be built up as described. This follows from prior studies showing that the catalytic domain of Pol ε is dispensable for yeast cell growth6,7, indicating that Pol δ and/or Pol α can synthesize both DNA strands. Nonetheless, Pol ε catalytic-domain mutants grow very poorly, and this reinforces the idea that Pol ε is normally the favored enzyme for leading-strand replication.
Although coordinated replication of duplex DNA is intrinsically asymmetric, evolution does not necessarily require the use of different replicases for leading- and lagging-strand synthesis. For example, the same catalytic subunit of Pol III replicates both strands of the Escherichia coli genome8. This observation, and the evolutionary conservation of Pol ε, Pol δ and other components of the eukaryotic replisome, imply that there are advantages to using two different replicases for nuclear DNA replication. Given that nuclear DNA is wrapped around nucleosomes, one possibility is that continuous leading-strand replication by CMG–Pol ε has been optimized to promote nucleosome disruption ahead of the fork. The use of two different polymerases with different accessory-protein requirements may allow refined coordination of replication with repair of DNA mismatches9 (which are generated at a rate of ~10−8), with repair of ribonucleotides (which are incorporated once per 1,000–10,000 bp (ref. 10)) and with histone reloading behind replication forks (which occurs about every 200 bp (ref. 11)). All three processes use PCNA, thus rationalizing its presence on the nascent leading-strand duplex although it binds only weakly to Pol ε (ref. 4) and has only a minimal effect on the efficiency of leading-strand replication by Pol ε (ref. 2). The authors suggest that while Pol ε holds onto CMG during leading-strand synthesis, it can cycle on and off PCNA–DNA to periodically provide RFC access for repeated loading of new PCNA clamps on the leading strand. This idea is attractive for several reasons. Pol ε cycling off the primer terminus after making a mismatch could provide an opportunity for ‘extrinsic proofreading’ by a different exonuclease, such as the exonuclease domain of Pol δ (ref. 12) or another exonuclease13. Pol ε cycling off the primer terminus after making a mismatch could also provide an opportunity to asymmetrically load the essential mismatch-repair protein MutLα onto DNA, to enable its endonuclease activity to nick the nascent strand for eventual mismatch excision9,14. The new data showing that lagging-strand replication benefits more from PCNA function are also particularly intriguing given that PCNA interacts with chromatin-assembly factor 1 (CAF1) to load histones H3 and H4 onto newly replicated DNA15. Indeed, a defect in CAF1 results in longer-than-normal Okazaki fragments16. Is it possible that parental and/or newly synthesized histones are asymmetrically reloaded after replication in a manner somehow related to the use of different eukaryotic replicases for leading- and lagging-strand replication? If so, this could have interesting epigenetic implications in mammals. This speculation raises the additional issue of whether the observations reported here for yeast proteins are conserved in mammals (discussed in ref. 2).
The use of different strand-specific replicases also has implications for genome stability. Pol ε is somewhat more accurate than Pol δ (Fig. 1) and is much more accurate than proofreading-deficient Pol α. Evidence in yeast suggests that replication errors are generated at different rates and are repaired with different efficiencies during replication of lagging strands as compared to leading strands17. It follows that perturbing replicase activity or replicase-specific error correction should reduce genome stability in a strand-specific manner. On an evolutionary time scale, strand-specific effects on genome instability could influence genome composition. Polymerase-specific, and therefore strand-specific, replication error rates could differentially affect cancer susceptibility, as already indicated by differences in tissue-specific tumors in mice deficient in proofreading by Pol ε as compared to Pol δ (ref. 18).
Studies of how Pol ε and Pol δ are delivered to the two DNA strands during normal replication also raise questions about what happens when replication-fork progression is impeded19,20 to activate cellular checkpoints and about whether it matters if the impediment is located in the leading- or lagging-strand template. Given the complexity of delivering Pol ε and Pol δ for replication that usually begins at origins, important questions are: what enzymology operates on the leading and lagging strands after stalled forks resume replication, how long does this enzymology persist, and what are the consequences for downstream events in order to preserve the integrity of the nuclear genome?
For many years, detailed biochemical analysis of eukaryotic replication initiation and replisome function largely used the Xenopus extract system21. Now, with initiation of replication recapitulated in yeast extracts just a few years ago22, and the new study of leading- and lagging-strand replication with purified proteins2, yeast will become an even more powerful system to integrate biochemistry with elegant genetic analysis.
Acknowledgments
Research in the authors’ laboratories is supported by Project Z01 ES065070 from the Division of Intramural Research of the US National Institutes of Health (National Institute of Environmental Health Sciences) to T.A.K. and by grant GM032431 from the US National Institutes of Health to P.M.B.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Thomas A Kunkel, Laboratory of Molecular Genetics and Laboratory of Structural Biology, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, North Carolina, USA.
Peter M Burgers, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, Missouri, USA.
References
- 1.Johansson E, Dixon N. Cold Spring Harb Perspect Biol. 2013;5:a012799. doi: 10.1101/cshperspect.a012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgescu RE, et al. Nat Struct Mol Biol. 2014;21:664–670. doi: 10.1038/nsmb.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell SD, Botchan MR. Cold Spring Harb Perspect Biol. 2013;5:a012807. doi: 10.1101/cshperspect.a012807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chilkova O, et al. Nucleic Acids Res. 2007;35:6588–6597. doi: 10.1093/nar/gkm741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gambus A, et al. EMBO J. 2009;28:2992–3004. doi: 10.1038/emboj.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kesti T, Flick K, Keranen S, Syvaoja JE, Wittenberg C. Mol Cell. 1999;3:679–685. doi: 10.1016/s1097-2765(00)80361-5. [DOI] [PubMed] [Google Scholar]
- 7.Dua R, Levy DL, Campbell JL. J Biol Chem. 1999;274:22283–22288. doi: 10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell M, Langston L, Stillman B. Cold Spring Harb Perspect Biol. 2013;5:a010108. doi: 10.1101/cshperspect.a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiricny J. Cold Spring Harb Perspect Biol. 2013;5:a012633. doi: 10.1101/cshperspect.a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams JS, Kunkel TA. DNA Repair (Amst) 2014;19:27–37. doi: 10.1016/j.dnarep.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacAlpine DM, Almouzni G. Cold Spring Harb Perspect Biol. 2013;5:a010207. doi: 10.1101/cshperspect.a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin YH, et al. Mol Cell Biol. 2005;25:461–471. doi: 10.1128/MCB.25.1.461-471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nick McElhinny SA, Pavlov YI, Kunkel TA. Cell Cycle. 2006;5:958–962. doi: 10.4161/cc.5.9.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pluciennik A, et al. Proc Natl Acad Sci USA. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibahara K, Stillman B. Cell. 1999;96:575–585. doi: 10.1016/s0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- 16.Smith DJ, Whitehouse I. Nature. 2012;483:434–438. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lujan SA, et al. PLoS Genet. 2012;8:e1003016. doi: 10.1371/journal.pgen.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albertson TM, et al. Proc Natl Acad Sci USA. 2009;106:17101–17104. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeeles JT, Poli J, Marians KJ, Pasero P. Cold Spring Harb Perspect Biol. 2013;5:a012815. doi: 10.1101/cshperspect.a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosal G, Chen J. Transl Cancer Res. 2013;2:107–129. doi: 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pacek M, Walter JC. EMBO J. 2004;23:3667–3676. doi: 10.1038/sj.emboj.7600369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heller RC, et al. Cell. 2011;146:80–91. doi: 10.1016/j.cell.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Diaz M, Kunkel TA. Trends Biochem Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]