

Abstract
Class I phosphoinositide 3-kinases (PI3Ks) catalyze production of the lipid messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3), which plays a central role in a complex signaling network regulating cell growth, survival, and movement. This network is overactivated in cancer and inflammation, and there is interest in determining the PI3K catalytic subunit (p110α, p110β, p110γ, or p110δ) that should be targeted in different therapeutic contexts. Previous studies have defined unique regulatory inputs for p110β, including direct interaction with Gβγ subunits, Rac, and Rab5. We generated mice with knock-in mutations of p110β that selectively blocked the interaction with Gβγ and investigated its contribution to the PI3K isoform dependency of receptor tyrosine kinase (RTK) and G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor (GPCR) responses in primary macrophages and neutrophils. We discovered a unique role for p110β in supporting synergistic PIP3 formation in response to the coactivation of macrophages by macrophage colony-stimulating factor (M-CSF) and the complement protein C5a. In contrast, we found partially redundant roles for p110α, p110β, and p110δ downstream of M-CSF alone and a nonredundant role for p110γ downstream of C5a alone. This role for p110β completely depended on direct interaction with Gβγ, suggesting that p110β transduces GPCR signals in the context of coincident activation by an RTK. The p110β-Gβγ interaction was also required for neutrophils to generate reactive oxygen species in response to the Fcγ receptor–dependent recognition of immune complexes and for their β2 integrin–mediated adhesion to fibrinogen or poly-RGD+, directly implicating heterotrimeric G proteins in these two responses.
INTRODUCTION
Class I phosphoinositide 3-kinases (PI3Ks) are a family of signaling enzymes that synthesize the second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3) and are involved in regulating an array of critical cellular functions, such as growth, proliferation, and cell survival (1–3). PIP3 exerts its effects through the recruitment and activation of various effector proteins by specific PIP3-binding domains, the best studied being the pleckstrin homology domain, which is present in PDK1 (phosphoinositide-dependent protein kinase 1), Akt1 and Akt2, P-Rex1 (PIP3-dependent rac exchanger 1), Grp1 (general receptor for phosphoinositides 1), and BTK (Bruton’s tyrosine kinase) (4, 5). The class I family of PI3K enzymes consists of three class Ia members, which are heterodimers consisting of a p110 catalytic subunit (p110α, p110β, or p110δ) combined with one of a shared group of “p85” regulatory subunits (p85α, its splice variants p50-and p55α, p85β, or p55γ), whereas the single class Ib member, PI3Kγ, consists of a p110γ catalytic subunit coupled to either a p101 or p84 regulatory protein (3, 6). The p110α and p110β subunits are ubiquitously present, whereas p110δ and p110γ are most abundant in hematopoietic cells (7, 8). Class Ia PI3Ks are classically engaged downstream of the activation of receptor tyrosine kinases (RTKs) through the direct binding of two Src homology 2 (SH2) domains in the p85 regulatory subunits with sequence-specific phosphorylated tyrosine residues (phosphotyrosines) in the receptors or their adaptor proteins. PI3Kγ activation occurs downstream of G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor (GPCR) signaling through the binding of Gβγ subunits to both p110γ and either of the p101 or p84 regulatory subunits (1, 3, 6). Class I PI3Ks can be further regulated through binding to the small guanosine triphosphatases of the Ras, Rho, and Rab families (1, 3, 9).
Whereas all class I PI3K family members produce PIP3, and class Ia members share high sequence homology and similar domain structures, as well as a common pool of p55 and p85 regulatory subunits, it is becoming increasingly clear that individual PI3K isoforms can play distinct roles in both physiology and pathology (1, 3, 10–12). To some extent, the selective involvement of p110γ and p110δ in cellular responses can be explained by their possession of a unique regulatory subunit or differential tissue distribution, respectively, but the ubiquitous p110α and p110β subunits can also play remarkably selective roles in cell signaling. For example, p110α positively regulates angiogenesis, growth, and anabolic metabolism, and it is a prevalent oncoprotein (13–15), whereas p110β is required for efficient platelet activation (16, 17), osteoclast-mediated bone resorption (18), and spermatogenesis (19, 20), and it drives tumorigenesis in cells lacking the phosphatase PTEN (phosphatase and tensin homolog) (21, 22).
Detailed investigations of the molecular properties of p110α and p110β have revealed a plausible mechanism by which mutation in the gene encoding p110α more commonly creates hyperactive proteins, but how p110α is selectively recruited by several RTKs, particularly receptors for insulin-like growth factor 1 (IGF1), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF), is still unclear (23). In contrast, there has been substantial progress in understanding how p110β might be selectively regulated. Gβγ subunits bind to and activate p110β, and this interaction supports substantial, synergistic activation of p110β-p85 dimers by Gβγ and phosphotyrosine peptides in vitro (24, 25). This interaction has been suggested to drive the p110β-dependent activation of Akt in response to GPCRs invarious model cell systems, including mouse embryonic fibroblasts (MEFs) and primary macrophages (26–28). The Gβγ-binding site in p110β is localized to the C2-helical domain linker, and point mutations (KK532/533DD) and a cell-permeable peptide inhibitor have been defined that selectively disrupt this interaction (24). This work has confirmed that direct binding between Gβγ and p110β is essential for the activation of p110β by GPCRs in model cells and PTEN−/− tumor cell lines (24).
Furthermore, the Ras-binding domain (RBD) of p110β preferentially binds to guanosine triphosphate (GTP)–bound (active) Rac and Cdc42 proteins, whereas the RBD of p110α selectively binds to GTP-bound Ras proteins (9). Knock-in mutations in the RBD of p110β that disrupt its binding to Rac and Cdc42 (S205D/K224A) abolish the p110β-dependent activation of Akt in response to GPCR stimulation in MEFs and ablate the contribution of p110β to a mouse model of lung fibrosis (9). In MEFs, GPCRs stimulate guanine nucleotide exchange on Rac through the Gβγ-dependent regulation of ELMO1-DOCK180 (engulfment and cell motility 1/dedicator of cytokinesis 180), thus providing an alternative input by Gβγ into the regulation of p110β (9). The helical domain of p110β selectively binds to Rab5, and this interaction appears to be important for supporting both kinase-dependent and kinase-independent roles for p110β in the control of endocytosis and the induction of autophagy (29, 30).
However, the extent to which the individual properties of p110β described earlier define unique roles for PI3Kβ heterodimers in vivo is still unclear, particularly with respect to its role as a direct transducer of GPCR signaling. We have attempted to better understand the importance of Gβγ binding to p110β by generating a knock-in mouse strain in which this interaction is selectively ablated (p110βKK526/527DD). We characterized the activation of PI3K isoforms by GPCRs and RTKs in primary macrophages and neutrophils isolated from these mice and compared them to cells isolated from mice with a defective p110β RBD (p110βS205D/K224A) or lacking active p110δ (p110δD910A) or p110γ (p110γKO). We assessed PI3K activity by measuring PIP3 directly, thus avoiding the potentially nonlinear readouts from measuring effector responses further downstream in the signaling pathway (for example, the phosphorylation of Akt). We found that stimulation of GPCRs alone resulted in the predominant use of p110γ, but coincident stimulation of a GPCR and an RTK resulted in the direct binding of Gβγ to p110β to drive a synergistic PIP3 response. These conclusions were supported by measuring additional PI3K-dependent functions of neutrophils, including the production of reactive oxygen species (ROS) in response to immune complexes, a response that depends on the coincident activation of FcγRs and BLT1 (31).
RESULTS
Mice expressing a mutant p110β isoform that cannot bind to Gβγ were generated
To investigate the importance of the direct binding of Gβγ to p110β in vivo, we generated knock-in mice (termed β-Gβγ mice) with two point mutations (KK526/527DD) in the gene encoding p110β that block this interaction (fig. S1) (24). These mutations affect neither the activation of p85-p110β heterodimers by phosphotyrosine peptides in vitro nor the binding of Rac, Cdc42, or Rab5 to p110β (24, 32). The β-Gβγ mice were generated with a self-deleting tAceCre-PGK-EM7-Neo resistance cassette (for the detailed targeting strategy, see fig. S1B and the Supplementary Materials), and correct targeting was confirmed by Southern blotting analysis (fig. S2A). The presence of the correct mutations was confirmed by polymerase chain reaction analysis and the sequencing of genomic DNA from mouse tissue (fig. S2, B and C). The β-Gβγ mice were healthy and fertile and displayed no overt phenotype. The abundance of p110β in bone marrow–derived macrophages (BMDMs) or bone marrow neutrophils (BMNs) from these mice was unaffected by the introduction of these point mutations (fig. S3). The β-Gβγ mice were used in experiments together with β-RBD (RBD-deficient p110β; p110βS205D/K224A) (9), β-KO (p110β-deficient) (31), γ-KO (p110γ-deficient) (33), and δ-KI (kinase-inactive p110δ; p110δD910A) (34) mice, as well as with p110β-selective (TGX221) (17), p110δ-selective (IC87114) (35), and p110α-selective (BYL719) (36) inhibitors to evaluate the contributions of individual PI3K isoforms to the generation of PIP3 in response to receptor stimulation. We did not perform experiments with α-KO (p110α-deficient) mice because they exhibit a block in early embryonic development (13).
The C5a-stimulated generation of PIP3 in macrophages is driven by p110γ, whereas the M-CSF–stimulated generation of PIP3 is driven by a combination of p110α, p110β, and p110δ
Previous work showed that primary BMDMs exhibit substantial class I PI3K–dependent signaling in response to stimulation by macrophage colony-stimulating factor (M-CSF), which acts through the RTK CSF1R (c-fms), or by C5a, which acts through the GPCR C5AR1 (also known as CD88) (28, 33, 37). We initially established the kinetics and scale of PIP3 generation in response to these agonists (the “PIP3 response”) in BMDMs isolated from wild-type mice. Stimulation of serum-starved primary BMDMs with a submaximal dose of C5a (30 nM; fig. S4A) produced rapid and substantial accumulation of PIP3, which was maximal between 15 and 30 s after stimulation (Fig. 1, A and B). Stimulation with an analogous concentration of M-CSF (30 ng/ml; fig. S4B) generated slightly greater amounts of PIP3 but with very similar kinetics (Fig. 1, C and D). The PIP3 responses to C5a and M-CSF in BMDMs were therefore well matched and offered an excellent opportunity to assess the relative roles of class I PI3K isoforms in mediating GPCR- and RTK-dependent responses, singly and in combination, within the same cell. The amount of PIP3 generated in response to C5a (after 30-s stimulation) in BMDMs lacking p110γ (γ-KO) was similar to that found in unstimulated wild-type cells; however, neither the loss of p110β (β-KO) nor the expression of a kinase-inactive p110δ (δ-KI) reduced the PIP3 responses of those cells (Fig. 1B). These results suggest that p110γ plays a dominant, nonredundant role in the generation of PIP3 in response to C5a. In contrast, no single class I PI3K isoform played a similarly dominant role in the generation of PIP3 in response to M-CSF (Fig. 1D), at least at 30 s.
Fig. 1. Characterization of RTK- and GPCR-driven PIP3 responses in BMDMs: Roles of class I PI3Ks.

(A to D) Serum-starved BMDMs (1.2 × 106 cells) from wild-type (WT) mice were stimulated with 30 nM C5a (A) or M-CSF (30 ng/ml) (C) for the indicated times, whereas BMDMs (1.2 × 106) from WT (closed bars), β-KO (open bars), δ-KI (dark gray bars), or γ-KO (light gray bars) mice were serum-starved overnight before being stimulated for 30 s with either 30 nM C5a (B) or M-CSF (30 ng/ml) (D) or vehicle control (B and D). Where indicated (hatched bars, D), the p110α-selective inhibitor BYL719 (1 μM final) was added to WT BMDMs 5 min before the cells were stimulated. (E) BMDMs (1.2 × 106 cells) from WT or δ-KI mice were serum-starved and incubated with the indicated concentrations of TGX221 (WT, black diamonds; δ-KI, black squares) or 1 μM IC87114 (WT, gray diamonds; δ-KI, gray squares) for 5 min before being stimulated for 30 s with M-CSF (30 ng/ml). As a control, WT BMDMs were also left unstimulated (gray triangle). (F) BMDMs (1.2 × 106 cells) from WT (black diamonds) or δ-KI (black squares) mice were serum-starved and incubated with vehicle (WT) or 1 μM IC87114 (δ-KI) and the indicated concentrations of BYL719 for 5 min before being stimulated for 30 s with M-CSF (30 ng/ml). As a control, WT BMDMs were also left unstimulated (gray triangle). (A to F) Reactions were quenched, lipids were extracted, and the amounts of PIP3 and PI were quantitated by mass spectrometry ( MS) as described in Materials and Methods. Data are means ± SEM of at least three experiments, each performed in duplicate [except for the WT control in (E), where n = 1], and are expressed as the ratio of the abundance of PIP3 to that of PI (PIP3/PI) to account for any variations in cell input. The following statistical analyses were performed. (B) For the comparisons shown: NS, not significant; *P < 0.05, **P < 0.01, by one-way analysis of variance (ANOVA) with Holm-Sidak post hoc test. (E) For M-CSF + 0 nM TGX221 versus M-CSF + IC87114, P = 0.0156 (WT), P = 0.7417 (δ-KI); for M-CSF + 0nMTGX221 versus M-CSF + TGX221, P <0.05 (WT), P < 0.05 (δ-KI), by ratio paired t test with Holm-Sidak correction for multiple comparisons and independent t test. (F) For comparison to the 0 μM BYL719 control, *P < 0.05, **P < 0.01, and ***P < 0.005 by repeated-measures ANOVA with Holm-Sidak post hoc test, with Holm-Sidak correction for multiple comparisons.
To interrogate further the potential roles of class Ia PI3Ks in the PIP3 response to M-CSF, we investigated the effect of inhibiting different combinations of isoforms. First, p110β and p110δ were inhibited in experiments with a range of concentrations of TGX221 and both wild-type and δ-KI BMDMs. TGX221 has predicted IC50 (median inhibitory concentration) values for p110β and p110δ of 7 and 100 nM, respectively (17, 38). We also investigated the effect of a single high concentration of the p110δ-selective inhibitor IC87114, which has a predicted IC50 for p110δ of 500 nM (35, 38). Either pharmacological or genetic inhibition of p110δ or a combination of the two inhibited the M-CSF–dependent generation of PIP3 by ~40% compared to that in wild-type cells treated with dimethyl sulfoxide (DMSO) (Fig. 1E). Inhibition of p110β by low concentrations of TGX221 (up to 40 nM) also reduced the amount of PIP3 generated by ~40%, whereas combined inhibition of p110β and p110δ (by treating δ-KI BMDMs with 40 nM TGX221) reduced the amount of PIP3 generated in response to M-CSF by about 65% (Fig. 1E).
Next, the effects of increasing concentrations of the p110α-specific inhibitor BYL719, which has a predicted IC50 in cells of ~100 nM (39), were investigated in both wild-type and δ-KI BMDMs pretreated with 40 nM TGX221 (Fig. 1F). Relatively low concentrations of BYL719 resulted in a ~25% further reduction in the amount of PIP3 generated in response to M-CSF in δ-KI BMDMs pretreated with TGX221, resulting in an overall inhibition of ~90% (Fig. 1F). Together, these data suggest that all three class Ia PI3K isoforms (p110α, p110β, and p110δ) are engaged downstream of the M-CSF receptor and play substantial roles in supporting PIP3 production. Our best estimate of the individual relative contributions suggests that each isoform plays a largely additive role (40% δ, 30% β, and 30% α), but these estimates are subject to significant error. The larger effect of pharmacological versus genetic inhibition of p110β is assumed to result from partial compensation for the loss of p110β protein by other class Ia isoforms; that is, heterodimers containing either p110α or p110δ can occupy more activating phosphotyrosines in the absence of p110β.
Coincident activation of macrophages by C5a and M-CSF results in a synergistic PIP3 response that requires Gβγ to bind to p110β
Simultaneous stimulation of wild-type BMDMs with a combination of both C5a and M-CSF resulted in a synergistic PIP3 response (Fig. 2A). The amount of PIP3 produced in response to this combined stimulation was ~85% greater than that predicted by the sum of the individual responses (costimulation/additive ratio; Fig. 2A, right). This synergistic PIP3 response was changed to an additive response by the loss of p110β but not by the loss of p110γ or the expression of kinase-inactive p110δ (Fig. 2B). The lack of involvement of p110γ in the synergistic PIP3 response to the combination of C5a and M-CSF was particularly striking because there was no measurable PIP3 generated in response to C5a alone in γ-KO BMDMs (Fig. 2B, left). The synergistic PIP3 response to coincident stimulation by C5a and M-CSF was also reduced to an additive response in β-Gβγ BMDMs, which essentially phenocopied β-KO cells (Fig. 2C). However, substantial synergy was still seen in β-RBD macrophages (Fig. 2C). These results suggest that the direct binding of Gβγ to p110β is required for the p110β-dependent production of PIP3 in response to stimulation by both C5a and M-CSF.
Fig. 2. Synergistic PIP3 production by BMDMs in response to costimulation of RTKs and GPCRs is dependent on p110β and requires binding to Gβγ proteins.

(A) Left: BMDMs (1.2 × 106 cells) from WT mice were serum-starved overnight before being stimulated for 30 s with 30 nM C5a or M-CSF (30 ng/ml) alone or in combination, as indicated. PIP3 was quantified by MS as described for Fig. 1. Data are means ± SEM of at least 10 experiments, each performed in duplicate, and are expressed as a percentage of the response to M-CSF. Right: Ratios for costimulation (the PIP3 response to simultaneous addition of C5a and M-CSF) divided by additive (the sum of the individual PIP3 responses to C5a and M-CSF) for the same experiments were calculated as described in Materials and Methods. (B) Left: WT (closed bars), β-KO (open bars), δ-KI (dark gray bars), and γ-KO (light gray bars) BMDMs (1.2 × 106) were left unstimulated (control) or were stimulated with the indicated agonists, and PIP3/PI ratios were quantitated as described in (A). Data are means ± SEM of at least three experiments, each performed in duplicate, and are expressed as a percentage of the PIP3 abundance of WT macrophages in response to M-CSF. Data for the unstimulated and singly stimulated cells include data from the experiments shown in (A) and Fig. 1(B and D). Right: Ratios (costimulation/additive) for the same experiments were calculated as described in (A). (C) Left: BMDMs from WT (closed bars), β-RBD (hatched bars), β-Gβγ (striped bars), and β-KO (open bars) mice were prepared and treated as described in (A). Data are means ± SEM of at least three experiments, each performed in duplicate, and are expressed as a percentage of the response of WT cells to M-CSF. Right:Ratios (costimulation/additive) for the same experiments were calculated as described in (A). *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.0001 by ratio paired t test (A, right) with Holm-Sidak correction for multiple comparisons (B and C, right).
Both the C5a- and fMLP-stimulated generation of PIP3 in neutrophils require p110γ
We and others previously showed that p110γ plays a major role in neutrophil activation by various Gi-coupled GPCRs (33, 38, 40–42). Here, we reevaluated the relative involvement of p110β and p110γ in GPCR-dependent PIP3 generation elicited by either 100 nM C5a or 10 μM f MLP (N-formyl-Met-Leu-Phe). Both C5a and f MLP stimulated very rapid and substantial generation of PIP3 by wild-type BMNs, with an initial maximal accumulation of PIP3 at 10 s, whereas f MLP also induced a second peak at 60 s (Fig. 3, A and B). The amount of PIP3 generated by wild-type BMNs in response to C5a was greater than that in response to fMLP (about threefold different) and was greater than the amount of PIP3 generated by wild-type BMDMs in response to C5a (about sixfold), but the kinetics of PIP3 production were similar. The amount of PIP3 that accumulated in γ-KO BMNs in response to either f MLP or C5a was reduced by ≥90% compared to that in wild-type BMNs (Fig. 3, C and D). In contrast, the amounts of PIP3 generated by wild-type BMNs in response to either fMLP or C5a were not statistically significantly affected by inhibition of p110β with 40 nM TGX221, nor were they substantially reduced in β-KO neutrophils (Fig. 3, C and D).
Fig. 3. PIP3 generation in BMNs in response to the GPCR agonists f MLP and C5a requires p110γ but not p110β.

(A and B) WT BMNs (0.5 × 106) were stimulated in suspension with 10 μM fMLP (A) or 100 nM C5a (B) for the indicated times. The reactions were quenched, and lipids were extracted, derivatized, and quantitated by MS as described earlier. Data are means ± SEM of the PIP3/PI ratio of at least three experiments, each performed in at least duplicate. (C and D) BMNs (0.5 × 106) from WT, β-KO, and γ-KO mice were preincubated with 40 nM TGX221 (hatched bars) or 0.05% DMSO (vehicle, closed bars) before being stimulated for 10 s with fMLP (C) or C5a (D). Reactions were then stopped, and lipids were extracted and quantitated as described earlier. The PIP3/PI ratio in unstimulated WT BMNs (WT-CON) is also shown. Data are means ± SEM of the PIP3/PI ratio of at least three experiments, each performed in at least duplicate. NS, not significant. ***P < 0.005 by one-way ANOVA with Holm-Sidak post hoc test comparisons as indicated (C and D).
However, TGX221 reduced the mean amount of PIP3 generated in γ-KO cells in response to either f MLP or C5a by ~5 to 10%, which suggests that p110β might make a very small contribution to basal or GPCR-stimulated PIP3 formation. Consistent with the lack of a major role for p110β in the PIP3 responses to C5a and f MLP, the PI3K-dependent formation of ROS in response to these agonists was also unaffected by 40 nM TGX221 (fig. S5A).
Both intact Gβγ- and Rac/Cdc42-binding domains are required to support p110β-dependent neutrophil activation by FcγRs and β2 integrins
We previously described an essential role for p110β in the responses of mouse neutrophils to immune complexes and adhesive surfaces (31). These ligands are recognized initially by FcγRs and integrins, respectively, which then drive a highly cooperative and complex sequence of “outside-in” and “inside-out” signaling events that regulate increased binding avidity, spreading, granule secretion, and NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase (NOX)–dependent formation of ROS (43, 44). Furthermore, other observations suggest that these two receptor systems share several common signaling elements, including phosphorylation of the γ chain of FcRs (Fc receptors) by an Src family kinase and the consequent recruitment and activation of Syk (45).
Our previous studies suggested that a paracrine loop involving the inflammatory mediator leukotriene B4 (LTB4) and its GPCR BLT1 is required downstream of FcγRs for the efficient activation of p110β and NOX (31). Therefore, we were interested in exploring the potential involvement of the direct binding of Gβγ to p110β in this response. The addition of wild-type BMNs to immobilized immunoglobulin G (IgG)–bovine serum albumin (BSA) induced cell adhesion, spreading, and the production of ROS, which was maximal at around 10 min and was maintained for over 40 min (Fig. 4, A to E). Pharmacological (40 nM TGX221) or genetic (β-KO) inhibition of p110β reduced each of these responses by about 70% (Fig. 4, C to E), confirming a central role for p110β. Cell adhesion and the production of ROS, and to a lesser extent cell spreading, were substantially reduced in β-Gβγ BMNs compared to those in wild-type BMNs (Fig. 4, C to E). Substantial reductions in adhesion and ROS formation were also seen with β-RBD BMNs (Fig. 4, C to E). The addition of 40 nM TGX221 still resulted in small reductions in the ROS responses of β-Gβγ and β-RBD neutrophils (Fig. 4E), which suggests that preventing the direct binding of Gβγ, Rac, or Cdc42 to p110β results in the substantial, but incomplete, elimination of the role of p110β in this response.
Fig. 4. Adhesion, spreading, and ROS production in BMNs in response to immune complexes and adhesive proteins are p110β-dependent responses.

(A) BMNs from WT, β-Gβγ, β-RBD, and β-KO mice were incubated for 10 min with either DMSO or 40 nM TGX221 before being applied to glass coverslips coated with poly-RGD+. Cells were incubated at 37°C for 20 min, and adherent cells were fixed in 3.7% paraformaldehyde, washed, and mounted on glass microslides before being visualized with a Nikon Ti-E Live Cell Imager inverted microscope with a 20× objective lens. Images of a 1:15 field of view of adherent spread (S) and nonspread (NS) cells are representative of three experiments performed in duplicate (except for cells from β-KO mice, for which n = 2). (B) WT BMNs (0.5 × 106) were preincubated with horseradish peroxidase (HRP) and luminol before being added to a 96-well plate coated with IgG-BSA (triangles), FGN (diamonds), orpoly-RGD+ (circles), or to a plate containing IgG-opsonized SRBCs (squares). ROS generation was measured in triplicate for each condition by chemiluminescence and recorded with a Berthold MicroLumat Plus luminometer and are expressed as relative light units (RLUs) per second. Data are from one experiment and are representative of at least three experiments. (C and D) BMNs from the indicated mice were pretreated for 10 min with either DMSO or 40 nM TGX221 before being incubated on duplicate coverslips coated with IgG-BSA, FGN, or poly-RGD+, as indicated, and were processed as described for (A). Images were analyzed for cell adhesion and spreading with ImageJ software (Fiji), selecting for size and brightness as described in Materials and Methods. Data are means ± SEM of the number of adherent cells per field of view (C) and of the percentage of spread cells (D) from three experiments performed in duplicate (except for cells from β-KO mice, for which n = 2). (E) BMNs (0.5 × 106) from WT, β-RBD, β-Gβγ, and β-KO mice were preincubated with HRP/luminol in the presence of 40 nM TGX221 (hatched bars) or DMSO (0.05%, closed bars) before being added to wells precoated with IgG-BSA, FGN, or poly-RGD+, as indicated. ROS was then measured as described earlier. Data are means ± SEM for accumulated light emission (RLU) over a 20-min recording of at least three experiments performed in at least duplicate and are expressed as a percentage of the ROS generated in DMSO-treated WT BMNs. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001 by t test with Holm-Sidak corrections for multiple comparisons.
We extended these observations by evaluating the role of p110β in the generation of ROS in response to the FcγR-dependent phagocytosis of IgG-opsonized sheep red blood cells (SRBCs) by BMNs primed with tumor necrosis factor–α (TNF-α) and granulocyte M-CSF (GM-CSF) [nonprimed cells do not generate ROS in response to IgG-SRBCs (46)]. The addition of wild-type BMNs to IgG-SRBCs elicited the generation of less but still a substantial amount of ROS than did their addition to IgG-BSA, which was maximal at around 7 min and lasted for more than 40 min (Fig. 4B). Similar to the IgG-BSA response described earlier, production of ROS in response to the phagocytosis of IgG-SRBCs was highly dependent on p110β activity, being reduced by 70% in β-KO BMNs or by the addition of TGX221 towild-type BMNs (fig. S5B). The amounts of ROS produced in response to IgG-SRBCs were also greatly reduced in β-Gβγ or β-RBD neutrophils (fig. S5B), with 40 nM TGX221 reducing the response further only in β-Gβγ cells, which suggests that the RBD plays a particularly important role in the regulation of p110β in this context.
Neutrophils also adhere, spread, and generate ROS in response to the engagement of β2 integrins with extracellular ligands (47), although the ROS response in our experiments did not require BLT1 (fig. S6). Adhesion of BMNs to the physiological Mac1 ligand fibrinogen (FGN) in the presence of the proinflammatory cytokine TNF-α (Fig. 4C) or onto the synthetic multivalent integrin ligand poly–Arg-Gly-Asp+ (poly-RGD+) (Fig. 4C) was largely unaffected by the inhibition of p110β. However, cell spreading and the production of ROS in response to both stimuli were substantially reduced in either β-KO BMNs or TGX221-treated wild-type BMNs (Fig. 4, D and E). Furthermore, ROS production and, to a lesser extent, cell spreading were reduced in β-Gβγ BMNs compared to those in wild-type BMNs (Fig. 4, D and E), whereas β-RBD BMNs displayed reduced ROS production but had unaffected spreading (Fig. 4, D and E). As before, preincubation with 40 nM TGX221 elicited further small reductions in ROS generation by β-Gβγ and β-RBD BMNs; however, this did not reach statistical significance (Fig. 4E).
Together, these data suggest that both an intact RBD and the direct binding of Gβγ subunits substantially contribute to the regulation of p110β in FcγR- and β2 integrin–mediated signaling pathways in neutrophils. The extent to which p110β activity controls various downstream elements, such as receptor clustering and activation, cytoskeletal rearrangement, or oxidase activation, is difficult to discern; however, because the relationships between these pathways are still incompletely understood. From our data, it appears that adhesion, spreading, and ROS formation in response to IgG-BSA are similarly dependent on p110β; however, ROS formation in response to integrin engagement is much more dependent on p110β than is cell adhesion.
Ideally, we would have wished to measure PIP3 formation in response to these agonists as a more direct readout of p110β activity. Unfortunately, the sizes of the PIP3 responses elicited in response to TNF-α–FGN, IgG-BSA, or IgG-SRBCs were too small to measure with enough precision to enable a useful dissection of potential regulatory inputs into p110β. This is possibly because of the imprecise time at which the neutrophils first encountered the stimuli in these types of assay. However, a statistically significant 50% increase in PIP3 abundance above that in unstimulated cells was measured 10 min after BMNs were plated onto poly-RGD+ (Fig. 5), and this was similarly reduced in β-KO, β-Gβγ, and β-RBD BMNs or upon the addition of 40 nM TGX221 to wild-type BMNs (Fig. 5). These data suggest that the RBD and Gβγ binding are required for p110β stimulation in response to integrin activation. Furthermore, the basal amounts of PIP3 in BMNs in suspension were reduced further by the addition of the pan-PI3K inhibitor wortmannin (100 nM), which suggests that they were generated throughalargelyp110β-independentroute (Fig. 5).
Fig. 5. The p110β-dependent generation of PIP3 in BMNs in response to adhesion to poly-RGD+ requires both an intact RBD and binding to Gβγ proteins.
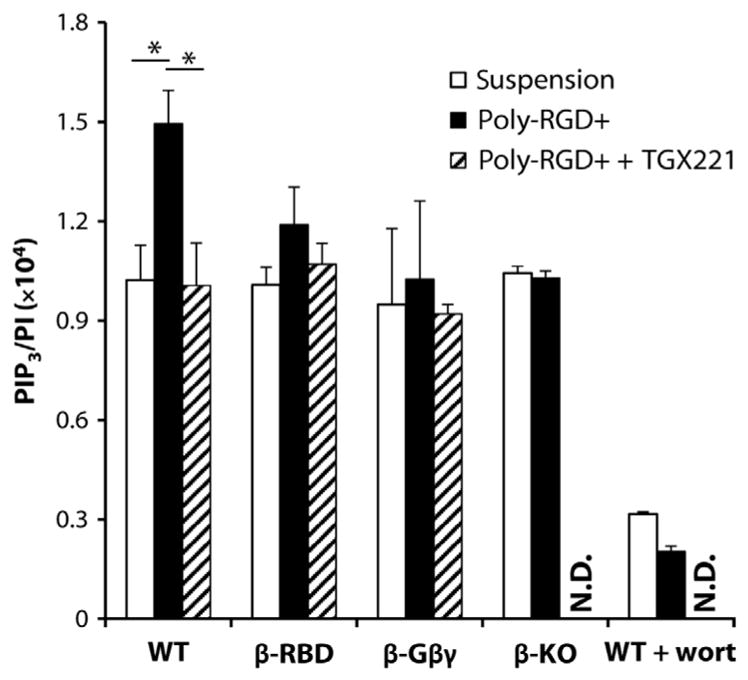
BMNs from WT, β-RBD, β-Gβγ, and β-KO mice were preincubated with 40 nM TGX221 or vehicle control before being added to glass coverslips coated with poly-RGD+ or were left in suspension. Where indicated, WT BMNs were additionally preincubated with 100 nM wortmannin (wort). Cells were allowed to adhere and spread for 10 min at 37°C and then harvested for PIP3 measurement by MS as described earlier. Data are means ± SEM of PIP3/PI ratios from three experiments (except for data from wortmannin-treated cells, for which n = 2), each performed in duplicate. N.D., not determined. *P < 0.05 by one-sample t test with Holm-Sidak correction for multiple comparisons.
DISCUSSION
There is steadily accumulating evidence of selective roles for p110β in individual cell responses and animal physiology (1, 3, 32). p110β is also implicated in driving pathology, such as thrombosis (48), lung fibrosis (9), autoimmune inflammation (18, 39), and cancer (49). Thus, p110β has been suggested to be a potentially useful therapeutic target (31, 50, 51). There has been substantial progress in defining some of the molecular properties of p85-p110β dimers that may enable p110β to play such selective roles (24, 30, 52), but the extent to which they are individually or cooperatively responsible in a given context in vivo remains uncertain. In particular, there is debate over the extent to which p110β is a direct transducer of GPCR signals. p85-p110β heterodimers were initially characterized as responding to direct activation by Gβγ subunits in vitro, though the extent of this activation was modest in the absence of costimulation by tyrosine-phosphorylated peptides (24, 25). Furthermore, pharmacological or genetic inhibition of p110β suggested that p85-p110β plays a widespread role downstream of several GPCRs (for example, receptors for lysophosphatidic acid, CXCL12, or sphingosine 1-phosphate) and acts redundantly with p101-p110γ heterodimers in cells in which both heterodimers are present (for example, cells of hematopoietic origin) (27, 28). In contrast, p110β is generally accepted to play a confusingly minor role downstream of many RTKs, particularly those responding to growth factors, such as PDGF, EGF, or insulin (27, 28, 53), the exception being in circumstances in which p110α activity is low, for example, after p110α-selective pharmacological inhibition (54, 55) or when p110α signaling is inhibited by a PTEN-PIP3–mediated negative feedback loop (56). Furthermore, a study described an alternative, indirect route for the Gβγ-dependent activation of p110β through the Gβγ-dependent regulation of guanine nucleotide exchange on Rac and the subsequent binding of GTP-Rac to the RBD of p110β (9).
The identification of the Gβγ-binding site on p110β enabled the generation of point mutants (KK532/533DD) that selectively ablate the binding of Gβγ but have no effect on basal p110β activity, its interaction with Rab5, or the activation of p85-p110β heterodimers by phosphotyrosine peptides (24). This enabled us to generate a knock-in mouse (β-Gβγ) expressing a mutant p110β to which Gβγ binding was selectively blocked, which then provided us with an opportunity to investigate the role of this interaction in vivo. The abundance of p110β-KK526/527DD was normal (comparable to that of the protein in wild-type cells) in primary macrophages and neutrophils, and homozygous knock-in mice were born at normal Mendelian ratios with no overt phenotype. This contrasts with the growth and developmental defects seen in p110β KO or p110β kinase–inactive knock-in mice, which indicates that the β-Gβγ mice retain some p110β function (26–28).
We chose to assess the effect of the KK526/527DD mutation on the GPCR-stimulated formation of PIP3 in macrophages and neutrophils, because these cells contain all four class I PI3K isoforms and exhibit both GPCR and RTK-dependent PI3K signaling. We measured the accumulation of PIP3 formation at early times as the most linear readout of class I PI3K activity available. Individual stimulation of BMDMs by C5a or of BMNs by C5a or fMLP resulted in the rapid synthesis of PIP3 in a manner that was almost entirely dependent on p110γ and largely independent of p110β. Previous work suggested that the C5a-stimulated phosphorylation of Akt in macrophages is substantially dependent on both p110β and p110γ (28). At present, we have no explanation for the difference between these results and ours; however, the primary role of p110γ downstream of GPCR signaling in neutrophils agrees with most previous studies and concurs with the much more sensitive and potent Gβγ-dependent activation of p101-p110γ than of p85-p110β in vitro (24, 57, 58).
In BMDMs, the generation of PIP3 in response to M-CSF enabled a direct comparison of the involvement of different PI3K isoforms in similarly sized RTK and GPCR responses. The M-CSF–stimulated generation of PIP3 at 30 s involved cooperation between all three class Ia PI3Ks, with similar contributions from p110α, p110β, and p110δ. In principle, the widely held assumption that class Ia p110 isoforms share a common pool of phosphotyrosine-binding regulatory subunits provides a natural mechanism for redundancy downstream of RTKs, although previous work implied a predominant role for either p110α or p110δ in the responses of macrophages to M-CSF (59–61). It seems likely that the explanation for these apparently disparate results lies in the different assays, time scales, and cell preparations used. It is also possible that the level of redundancy among class Ia isoforms has been generally underestimated in the past because of the use of cellular preparations exhibiting widely differing relative amounts of p110 isoforms, poor quantitation of class I PI3K activity, or both.
Coincident stimulation of BMDMs by C5a and M-CSF resulted in the synergistic production of PIP3 in amounts that were about twice those that were expected on the basis of a purely additive effect. Furthermore, loss of p110β selectively removed this synergistic component of the response, whereas inhibition of p110α, p110γ, or p110δ had no effect on the degree of synergy. In addition, β-Gβγ macrophages also failed to support the synergistic production of PIP3 in response to C5a and M-CSF, which suggests that this role for p110β is entirely dependent on its direct interaction with Gβγ. The role of the RBD in regulating p110β activity in these responses was less clear; the mean amount of PIP3 produced by β-RBD BMNs in response to both C5a and M-CSF was ~20% less than that produced by wild-type BMNs, but this did not reach statistical significance.
These results suggest that PI3Kβ does indeed have a direct role in transducing GPCR signals but only in the context of parallel activation of an RTK. Thus, Gβγ binding to a p85-p110β heterodimer alone is insufficient to elicit substantial PIP3 production; however, in the context of simultaneous p85-phosphotyrosine docking (as occurs during parallel RTK activation), the binding of Gβγ to p110β supports additional PI3Kβ activation, giving rise to a synergistic increase in PIP3 formation. These conclusions align closely with studies describing the direct and synergistic activation of p85-p110β heterodimers by Gβγ subunits and phosphotyrosine peptides in vitro, and also our current mechanistic understanding of how Gβγ and phosphotyrosine docking might cause enzyme activation (by increasing membrane dwell time and allosteric activation, respectively) (6, 24, 49).
We sought to extend our understanding of the role of Gβγ in the regulation of p110β by investigating ROS production in BMNs in response to several stimuli. We and others previously established that the stimulation of mouse neutrophils with C5a and fMLP results in the rapid and transient production of ROS, which is dependent on p110γ (38). We also described a role for p110β in the more slowly developing and sustained production of ROS in response to FcγR-mediated recognition of immune complexes and β2 integrin–mediated adhesion (31). Here, we confirmed that BMN ROS responses to immobilized IgG-BSA or adhesion to FGN or poly-RGD+ are all dependent on p110β activity. Furthermore, we extended our studies to demonstrate that ROS generation in response to the FcγR-mediated phagocytosis of IgG-opsonized SRBCs was also highly dependent on p110β, implicating p110β activity in an important role downstream of the activation of FcγR and supporting previous observations that proposed a common mechanism used by both FcγR and integrins to activate NOX2 in neutrophils. ROS responses to IgG-BSA, adhesion, or IgG-SRBCs were also substantially reduced in β-Gβγ and β-RBD BMNs compared to those in wild-type BMNs. The relative scale of these reductions compared to those seen in β-KO cells and the extent of additional TGX221-dependent inhibition suggest that both direct Gβγ binding to p110β and input through the RBD play important and cooperative roles in p110β regulation. The input through the RBD is most important in ROS production in response to IgG-SRBCs and poly-RGD+, and this may correlate with the central role that GTP-Rac plays in these responses (62).
These results indicate that Gβγ subunits are involved in the activation of p110β downstream of FcγRs and β2 integrins, which directly implicates GPCRs in these responses. BLT1, the GPCR for LTB4, has been suggested to play a role in FcγR-mediated phagocytosis by macrophages (63) and in ROS production by neutrophils in response to immobilized immune complexes (31). Neutrophils can generate substantial amounts of LTB4 in response to FcγR activation, and thus, there is the potential to set up a paracrine loop in vitro that involves coincident signaling through GPCR and tyrosine kinase signaling; FcγRs initiate signaling through non-RTKs of the Src and Syk families (64). However, BLT1 did not appear to be involved in the generation of ROS in response to β2 integrin stimulation (fig. S6), which suggests that GPCR signaling plays a previously uncharacterized role in these responses.
Our results suggest that the direct binding of Gβγ subunits to p110β cooperates with the binding of Rac and Cdc42 to the p110β-RBD and also the binding of the p85 SH2 domain to phosphotyrosines to enable p85-p110β to play a unique role downstream of coincident GPCR and RTK signaling. However, the extent to which the interaction with Gβγ can operate independently of the other regulatory inputs to transduce isolated GPCR signals (for example, in the absence of the more dominant p101-p110γ isoform) is still difficult to assess. No mutations in p110β have yet been defined that selectively ablate allosteric activation through SH2-phosphotyrosine docking, and almost all cells in their “basal” state probably receive survival signals through low-level, phosphotyrosine-mediated activation of class I PI3Ks, for example, through integrin-mediated adhesion (65). Nevertheless, with or without coincident RTK signaling, there is a clear case for searching for previously uncharacterized GPCR inputs where p110β plays a major role in physiology or pathology.
MATERIALS AND METHODS
Reagents
Bovine FGN, poly-RGD+, fMLP, luminol, TMS-diazomethane, wortmannin, fatty acid–free BSA, and HRP were from Sigma-Aldrich. Murine GM-CSF and M-CSF were from PeproTech, whereas murine TNF-α and recombinant human C5a were from R&D Systems. SRBCs in Alsever’s were from TCS Biosciences. Hanks’ balanced salt solution and Dulbecco’s phosphate-buffered saline (dPBS) with Ca2+ and Mg2+ were from Sigma. The isoform-selective PI3K inhibitors IC87114 and TGX221 were from Calbiochem, whereas BYL719 was from Active Biochem. All tissue culture products were from Invitrogen. All buffer components were from Sigma-Aldrich and were endotoxin-free or low-endotoxin, as available. Internal standards for lipid analysis, 1-heptadecanoyl-2-hexadecanoyl-sn-glycero-3-(phosphoinositol 3,4,5-trisphosphate) (C17:0/C16:0-PIP3, as a hepta sodium salt), and C17:0/C16:0-PI were synthesized at the Babraham Institute. All chemicals and solutions were of analytical reagent grade.
Mouse strains
Mice lacking p110γ (γ-KO) (33) or p110β (β-KO) (31) and mice expressing a kinase-inactive p110δ (δ-KI, p110δ-D910A) (34) or point mutations in the RBD of p110β (β-RBD; Jackson Laboratory, JAX strain 025930) (9) have been previously described. We generated knock-in mice expressing a point-mutated PI3Kβ that is deficient in binding to Gβγ dimers (described below and in the Supplementary Materials). In all experiments, mice were compared with appropriate age- and strain-matched wild-type controls. Mice were housed in the Biological Support Unit at the Babraham Institute under specific pathogen–free conditions. All work was performed under Home Office Project license PPL 70/8100.
Generation of knock-in mice expressing Gβγ-insensitive p110β
The amino acid residues that bind to and enable activation of p110β by Gβγ dimers, K532 and K533, have previously been identified in the human sequence (24). Through sequence alignment of human and mouse p110β, we identified K526 and K527 as the corresponding residues in mouse p110β (fig. S1A). The Pik3cbKK526,527DD targeting construct was generated with a recombineering-based methodology as previously described (66). This method uses the homologous recombination functions of the λ phage Red genes to subclone and retrieve DNA from bacterial artificial chromosomes into higher-copy plasmids and enables insertions, mutations, or deletions to be made in the retrieved gene of interest. For full details about generation of the targeting construct and resultant knock-in mice expressing Gβγ-insensitive p110β, see the Supplementary Materials.
Preparation and stimulation of BMDMs
BMDMs were prepared essentially as previously described (60). Briefly, flushed bone marrow was resuspended in complete medium [RPMI 1640 medium containing L-glutamine supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS), 1% penicillin and streptomycin, 1 mM sodium pyruvate, 1% nonessential amino acids, 0.5 μM β-mercaptoethanol, and M-CSF (20 ng/ml)]. Cells were distributed to 10-cm2 tissue culture plates (10 × 106 cells per plate) and cultured for 72 hours at 37°C, 5% CO2. BMDMs (nonadherent cells) were harvested and stored frozen in 90% HI-FBS and 10% DMSO under liquid nitrogen until use. Before experiments were performed, BMDMs were thawed and grown in complete medium on nontissue culture dishes (1.2 × 106 cells per dish) for a further 3 days before detaching (with versene) and replating on 10-cm2 tissue culture dishes (at 1.2 × 106 cells per dish). Flow cytometric analysis of cells (to detect CD11b and F4/80) revealed that >95% of the cells were macrophages. Before stimulation, the BMDMs were starved overnight in medium in the absence of HI-FBS and M-CSF. Where indicated in the figure legends, cells were pretreated with inhibitors (for 10 min at 37°C, 5% CO2) before being stimulated for the times indicated in the figure legends (at 37°C, 5% CO2). Reactions were terminated by aspiration of the buffer and the addition of 1 ml of ice-cold 1 M HCl. Adherent cells, on ice, were harvested by scraping and transferred to 2-ml safe-lock Eppendorf tubes on ice for centrifugation at 15,000g at 4°C and removal of the supernatant. At this point, cell pellets could be snap-frozen in liquid nitrogen and stored at −80°C before proceeding with lipid extraction.
Preparation of BMNs
For ROS assays and soluble agonist lipid analysis, mature mouse neutrophils were isolated from bone marrow at room temperature on a discontinuous Percoll+ gradient as described previously (38). Purity was determined by cytospin and Reastain QUICK-DIFF (Reagena) staining, and preparations were at least 70 to 85% pure. After washing, BMNs were resuspended in dPBS with Ca2+ and Mg2+, glucose (1 g/liter), and 4 mM sodium bicarbonate (dPBS+). For phospholipid analysis of adherent cells, BMNs were prepared at 4°C as described previously (67). Cells (60 to 80% pure, as assessed by cytospin) were resuspended at 5 × 106 cells/ml in dPBS+ in the presence of 0.05% endotoxin and essentially fatty acid–free BSA (dPBS++). Cell suspensions were allowed to equilibrate for 5 min at room temperature and then for 10 min at 37°C in the presence or absence of 40 nM TGX221, 100 nM wortmannin, or DMSO (vehicle control), as indicated, before adhesion.
Preparation of adhesive surfaces
Immune complexes were immobilized by coating luminometer plate wells (Berthold Technologies; for ROS assays) or glass coverslips (for adhesion assays) overnight at 4°C with PBS containing endotoxin-free and essentially fatty acid–free BSA (100 μg/ml). After washing in PBS, wells or coverslips were blocked with 1% fat-free milk in PBS for 1 hour at room temperature. After further washing, immune complexes were formed by the addition of a 1:10,000 dilution of monoclonal anti-BSA antibody (Sigma, B2901) in dPBS+ for 1 hour at room temperature, followed by washing to remove unbound antibodies. Bovine FGN (150 μg/ml) was adsorbed onto luminometer wells (for ROS assays) or coverslips (for adhesion assays) overnight at 4°C, and surfaces were washed with dPBS before assays were performed. BMNs were preincubated with murine TNF-α (20 ng/ml) for 10 min at 37°C in the absence or presence of inhibitor before being added to FGN-coated surfaces. Poly-RGD+ in dPBS+ was adsorbed onto luminometer plates (at 20 μg/ml for ROS assays) or glass coverslips (at 20 μg/ml for adhesion assays or at 100 μg/ml for phospholipid analysis) for 3 hours at room temperature and were washed with dPBS+ before the BMNs were added.
Preparation of IgG-opsonized SRBCs
IgG-opsonized SRBCs were prepared for ROS assays as previously described (46). Briefly, 10 μl of SRBCs in Alsever’s (TCS Biosciences) was washed, centrifuged at 1500g for 4 min, and resuspended in 1 ml of dPBS++. SRBCs were opsonized with a 1:1000 dilution of IgG anti-SRBC (rabbit, MP Biomedicals), rotating end over end at room temperature for 20 min. SRBCs were then washed twice and resuspended in 800 μl of dPBS++.
Measurement of ROS production
Rate kinetics of ROS production were measured by chemiluminescence with a luminol-based assay in polystyrene 96-well plates (Berthold Technologies) as described previously (68). Cells (0.5 × 106) were incubated with 150 μM luminol and HRP (18.75 U/ml) for 10 min at 37°C in the presence or absence of 40 nM TGX221 (or DMSO vehicle control, 0.05%) as indicated in the figure legends. BMNs were added manually to wells precoated with FGN (150 μg/ml), poly-RGD+ (20 μg/ml), or immobilized immune complexes (1:10,000 anti-BSA), which were prepared as described earlier. For IgG-opsonized SRBC-generated ROS assays, BMNs were primed for 1 hour at 37°C in the presence of mouse TNF-α (1000 U/ml) and GM-CSF (100 ng/ml). For all assays, measurements were started immediately, and light emission was recorded by a Berthold MicroLumat Plus luminometer (Berthold Technologies). Data output was in RLUs per second or total RLUs integrated over the indicated measured time periods.
BMN adhesion and spreading assays
BMNs (0.5 × 106 cells for poly-RGD+ and IgG-BSA; 1 × 106 cells for FGN) from wild-type, p110β Gβγ–insensitive (β-Gβγ), p110β RBD–insensitive (β-RBD), or p110β knockout (β-KO) mice were applied in duplicate to 15-mm glass coverslips in 24-well tissue culture plates pre-coated with poly-RGD+ (20 μg/ml), IgG-BSA (1:10,000 anti-BSA), or FGN (150 μg/ml), as described earlier, after a 10-min pretreatment at 37°C with 40 nM TGX221 or DMSO vehicle. Cells were incubated at 37°C for 20 min, nonadherent cells were aspirated, and adherent cells were fixed in 3.7% paraformaldehyde in dPBS (pH 7.4) for 15 min at room temperature. After being washed three times in dPBS, the coverslips were rinsed in MilliQ double-distilled water before being mounted on glass microslides with Aqua-Poly/Mount antifading solution (Polysciences). Cells were visualized on wide field by differential interference contrast, and images were captured with a Nikon Ti-E Live Cell Imager inverted microscope with an integrated camera, using a 20× objective lens. Images were analyzed for cell adhesion and spreading in Fiji X64 for 10 to 15 (poly-RGD+), 15 to 20 (IgG-BSA), or 25 to 30 (FGN) random fields of view per coverslip, selecting for cell size and cell brightness.
BMN adhesion assays: Preparation for lipid analysis
Isolated BMNs (1 × 106 cells in 200 μl) were added at 37°C to 32-mm glass coverslips within 35-mm culture dishes precoated with poly-RGD+, as described earlier, containing 800 μl of dPBS++ (in the presence of 40 nM TGX221, 100 nM wortmannin, or 0.05% DMSO as required) and were allowed to adhere for 10 min at 37°C. Reactions were terminated by aspirating the buffer, adding 0.5 ml of ice-cold 1 M HCl, and placing the dishes on ice. Adherent cells were scraped from coverslips and transferred to 2-ml safe-lock Eppendorf tubes on ice. For suspension controls, 1 × 106 BMNs were incubated at 37°C for 10 min in the absence or presence of 40 nM TGX221, 100 nM wortmannin, or 0.05% DMSO as required. Reactions were stopped by the addition of 1.5 ml of ice-cold 1 M HCl, and the tubes were placed on ice. Cells from adhesion and suspension samples were pelleted by centrifugation (18,000g for 10 min at 4°C), and the supernatant was removed. Cells pellets were then processed for lipid analysis.
Lipid analysis
BMN cell pellets from adhesion assays or BMDM cell pellets prepared as described earlier were resuspended in 920 μl of primary extraction solution [CHCl3/MeOH/1 M HCl (484/242/23.22)/H2O, 750:170 (v/v)]. The internal standards C17:0/C16:0-PI (100 ng) and C17:0/C16:0-PIP3 (10 ng) were added to the initial extract resuspensions. The lipids were extracted and derivatized with trimethylsilyl-diazomethane, and PIP3 and PI were analyzed by MS, using neutral loss of derivatized head groups with an AB Sciex QTRAP 4000 connected to a Waters Acquity UPLC system as described by Clark et al. (69), with the exception that final samples were dried in a rotary SpeedVac evaporator rather than under nitrogen. For lipid measurements in BMNs exposed to soluble agonists, 0.5 × 106 BMNs in 150 μlwere equilibrated to 37°C for 3 min before stimulation at 37°C with 10 μM fMLP or 100 nM C5a for the times indicated in the figure legends. Where indicated in the figure legends, BMNs were preincubated for 10 min at 37°C with 40 nM TGX221 or 0.05% DMSO before being stimulated. Reactions were terminated by the addition of 750 μl of ice-cold CHCl3/MeOH/1 M HCl (484/242/23.22), and samples were placed on ice until extraction (which occurred within 10 min). C17:0/C16:0-PIP3 and C17:0/C16:0-PI internal standards were added, and the samples were processed as described earlier. Data were generated as response ratios, calculated by normalizing the multiple reaction monitoring–targeted, lipid-integrated response area to that of a known amount of relevant internal standard. Where indicated, PIP3 amounts were divided by PI amounts to correct for any cell input variability between experiments and are presented as nominal PIP3/PI ratios. For the BMDM PIP3 data presented in Fig. 2, for each separate experiment, the “additive response” was calculated by adding the individual C5a-mediated PIP3 response above control (control being the amount of PIP3 in unstimulated cells) to the individual M-CSF–mediated response above control, whereas the “costimulation response” represents the actual response to both agonists added simultaneously (C5a and M-CSF) above control (that is, a single subtraction for the amount of PIP3 in unstimulated cells was performed). A ratio of costimulation response divided by the additive response was then calculated for each separate experiment, and the collected data were subjected to statistical analysis.
Statistics
Data are means ± SEM of at least three experiments performed with at least duplicate samples (unless otherwise indicated). BMNs and BMDMs were isolated from at least two pooled mice of each genotype for each experiment. One-way ANOVA followed by Holm-Sidak post hoc tests were performed on all BMN data (PIP3 and ROS) and for BMDM PIP3 responses, except for the use of ratio paired t tests with Holm-Sidak post hoc test to compare costimulation and additive PIP3 responses to C5a and M-CSF. For each test, *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.0001.
Supplementary Material
Fig. S1. Targeting of Pik3cbKK526,527DD.
Fig. S2. Confirmation of the correct targeting of Pik3cbKK526,527DD.
Fig. S3. p110β quantities in BMDMs and BMNs are unaffected by the introduction of the RBD- and Gβγ-insensitive mutations.
Fig. S4. Dose responses of GPCR (C5a) and RTK (M-CSF) agonists on PIP3 production by BMDMs.
Fig. S5. ROS generation in BMNs in response to the phagocytosis of IgG-SRBCs, but not soluble GPCR agonists, requires p110β activity.
Fig. S6. ROS generation in BMNs in response to adhesion to poly-RGD+, but not immune complexes, is independent of BLT1 activation. Reference (70)
Acknowledgments
We would like to thank members of the Small Animal Breeding Unit at the Babraham Institute for animal husbandry; R. Williams (Medical Research Council Laboratory of Molecular Biology, Cambridge) for communicating essential information about the direct interaction between Gβγ and p110β; J. Clark for help with the MS analysis of lipids; A. Segonds-Pichon for statistical analysis; S. Walker and H. Okkenhaug (Babraham Institute Imaging Facility) for assistance with image analysis; and S. Suire and D. Gyori for helpful discussions.
Funding: This work was supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) UK (BB/J004456/1 to K.E.A., L.R.S., and P.T.H.), the Welcome Trust (WT085889MA to T.C. and S.K.), the NIH (GM112524 to J.M.B.), and the European Research Council (RASTARGET to J.D.). D.M.H. is a recipient of a BBSRC PhD studentship.
Footnotes
Author contributions: K.E.A., D.M.H., S.K., and T.C. performed experiments; K.E.A., D.H., P.T.H., and L.R.S. analyzed data; R.F., J.D., and J.M.B. supplied important resources and information; P.T.H., K.E.A., and L.S. designed research; and P.T.H., K.E.A., T.C., and L.R.S. wrote the article.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Use of the p110β KO and p110δ D910A-KI mice requires a material transfer agreement from the Ludwig Institute for Cancer Research.
References
- 1.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta. 2015;1851:882–897. doi: 10.1016/j.bbalip.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Salamon RS, Backer JM. Phosphatidylinositol-3,4,5-trisphosphate: Tool of choice for class I PI 3-kinases. Bioessays. 2013;35:602–611. doi: 10.1002/bies.201200176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammond GRV, Balla T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim Biophys Acta. 2015;1851:746–758. doi: 10.1016/j.bbalip.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 6.Burke JE, Williams RL. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS) Adv Biol Regul. 2013;53:97–110. doi: 10.1016/j.jbior.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115–127. doi: 10.1016/j.tibs.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 9.Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, Downward J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell. 2013;153:1050–1063. doi: 10.1016/j.cell.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. 2013;31:675–704. doi: 10.1146/annurev-immunol-032712-095946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghigo A, Morello F, Perino A, Hirsch E. Phosphoinositide 3-kinases in health and disease. Subcell Biochem. 2012;58:183–213. doi: 10.1007/978-94-007-3012-0_6. [DOI] [PubMed] [Google Scholar]
- 12.Wymann M. PI3Ks—Drug targets in inflammation and cancer. Subcell Biochem. 2012;58:111–181. doi: 10.1007/978-94-007-3012-0_5. [DOI] [PubMed] [Google Scholar]
- 13.Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJH, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B. Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–666. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 14.Vogt PK, Hart JR, Gymnopoulos M, Jiang H, Kang S, Bader AG, Zhao L, Denley A. Phosphatidylinositol 3-kinase: The oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foukas LC, Bilanges B, Bettedi L, Pearce W, Ali K, Sancho S, Withers DJ, Vanhaesebroeck B. Long-term p110α PI3K inactivation exerts a beneficial effect on metabolism. EMBO Mol Med. 2013;5:563–571. doi: 10.1002/emmm.201201953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurent PA, Séverin S, Hechler B, Vanhaesebroeck B, Payrastre B, Gratacap MP. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood. 2015;125:881–888. doi: 10.1182/blood-2014-07-588335. [DOI] [PubMed] [Google Scholar]
- 17.Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, Sturgeon SA, Prabaharan H, Thompson PE, Smith GD, Shepherd PR, Daniele N, Kulkarni S, Abbott B, Saylik D, Jones C, Lu L, Giuliano S, Hughan SC, Angus JA, Robertson AD, Salem HH. PI 3-kinase p110β: A new target for antithrombotic therapy. Nat Med. 2005;11:507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 18.Gyõri D, Csete D, Benkõ S, Kulkarni S, Mandl P, Dobó-Nagy C, Vanhaesebroeck B, Stephens L, Hawkins PT, Mócsai A. The phosphoinositide 3-kinase isoform PI3Kβ regulates osteoclast-mediated bone resorption in humans and mice. Arthritis Rheumatol. 2014;66:2210–2221. doi: 10.1002/art.38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillermet-Guibert J, Smith LB, Halet G, Whitehead MA, Pearce W, Rebourcet D, León K, Crépieux P, Nock G, Strömstedt M, Enerback M, Chelala C, Graupera M, Carroll J, Cosulich S, Saunders PTK, Huhtaniemi I, Vanhaesebroeck B. Novel role for p110β PI 3-kinase in male fertility through regulation of androgen receptor activity in Sertoli cells. PLOS Genet. 2015;11:e1005304. doi: 10.1371/journal.pgen.1005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciraolo E, Morello F, Hobbs RM, Wolf F, Marone R, Iezzi M, Lu X, Mengozzi G, Altruda F, Sorba G, Guan K, Pandolfi PP, Wymann MP, Hirsch E. Essential role of the p110β subunit of phosphoinositide 3-OH kinase in male fertility. Mol Biol Cell. 2010;21:704–711. doi: 10.1091/mbc.E09-08-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuzugullu H, Baitsch L, Von T, Steiner A, Tong H, Ni J, Clayton LK, Bronson R, Roberts TM, Gritsman K, Zhao JJ. A PI3K p110β–Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat Commun. 2015;6:8501. doi: 10.1038/ncomms9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA) Proc Natl Acad Sci USA. 2012;109:15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dbouk HA, Vadas O, Shymanets A, Burke JE, Salamon RS, Khalil BD, Barrett MO, Waldo GL, Surve C, Hsueh C, Perisic O, Harteneck C, Shepherd PR, Harden TK, Smrcka AV, Taussig R, Bresnick AR, Nürnberg B, Williams RL, Backer JM. G protein–coupled receptor–mediated activation of p110β by Gβγ is required for cellular transformation and invasiveness. Sci Signal. 2012;5:ra89. doi: 10.1126/scisignal.2003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katada T, Kurosu H, Okada T, Suzuki T, Tsujimoto N, Takasuga S, Kontani K, Hazeki O, Ui M. Synergistic activation of a family of phosphoinositide 3-kinase via G-protein coupled and tyrosine kinase-related receptors. Chem Phys Lipids. 1999;98:79–86. doi: 10.1016/s0009-3084(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 26.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110β in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, Dastrù W, Martin EL, Silengo L, Altruda F, Turco E, Lanzetti L, Musiani P, Rückle T, Rommel C, Backer JM, Forni G, Wymann MP, Hirsch E. Phosphoinositide 3-kinase p110β activity: Key role in metabolism and mammary gland cancer but not development. Sci Signal. 2008;1:ra3. doi: 10.1126/scisignal.1161577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJH, Okkenhaug K, Vanhaesebroeck B. The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc Natl Acad Sci USA. 2008;105:8292–8297. doi: 10.1073/pnas.0707761105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurosu H, Katada T. Association of phosphatidylinositol 3-kinase composed of p110β-catalytic and p85-regulatory subunits with the small GTPase Rab5. J Biochem. 2001;130:73–78. doi: 10.1093/oxfordjournals.jbchem.a002964. [DOI] [PubMed] [Google Scholar]
- 30.Dou Z, Pan JA, Dbouk HA, Ballou LM, DeLeon JL, Fan Y, Chen JS, Liang Z, Li G, Backer JM, Lin RZ, Zong WX. Class IA PI3K p110β subunit promotes autophagy through Rab5 small GTPase in response to growth factor limitation. Mol Cell. 2013;50:29–42. doi: 10.1016/j.molcel.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, Hirose M, Juss J, Oxley D, Chessa TAM, Ramadani F, Guillou H, Segonds-Pichon A, Fritsch A, Jarvis GE, Okkenhaug K, Ludwig R, Zillikens D, Mocsai A, Vanhaesebroeck B, Stephens LR, Hawkins PT. PI3Kβ plays a critical role in neutrophil activation by immune complexes. Sci Signal. 2011;4:ra23. doi: 10.1126/scisignal.2001617. [DOI] [PubMed] [Google Scholar]
- 32.Dbouk HA. PI3King the right partner: Unique interactions and signaling by p110β. Postdoc J. 2015;3:71–87. doi: 10.14304/surya.jpr.v3n6.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 34.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJH, Vanhaesebroeck B. Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science. 2002;297:1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 35.Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. Essential role of phosphoinositide 3-kinase δ in neutrophil directional movement. J Immunol. 2003;170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- 36.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, Hamon J, Fabbro D, Caravatti G. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Jones GE, Prigmore E, Calvez R, Hogan C, Dunn GA, Hirsch E, Wymann MP, Ridley AJ. Requirement for PI 3-kinase γ in macrophage migration to MCP-1 and CSF-1. Exp Cell Res. 2003;290:120–131. doi: 10.1016/s0014-4827(03)00318-5. [DOI] [PubMed] [Google Scholar]
- 38.Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M, Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- 39.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira SM, Wilson C, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hofmann F, Sellers WR. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13:1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 40.Deladeriere A, Gambardella L, Pan D, Anderson KE, Hawkins PT, Stephens LR. The regulatory subunits of PI3Kγ control distinct neutrophil responses. Sci Signal. 2015;8:ra8. doi: 10.1126/scisignal.2005564. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 42.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 43.Brandsma AM, Jacobino SR, Meyer S, ten Broeke T, Leusen JHW. Fc receptor inside-out signaling and possible impact on antibody therapy. Immunol Rev. 2015;268:74–87. doi: 10.1111/imr.12332. [DOI] [PubMed] [Google Scholar]
- 44.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside–outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012;24:600–606. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson KE, Chessa TAM, Davidson K, Henderson RB, Walker S, Tolmachova T, Grys K, Rausch O, Seabra MC, Tybulewicz VLJ, Stephens LR, Hawkins PT. PtdIns3P and Rac direct the assembly of the NADPH oxidase on a novel, pre-phagosomal compartment during FcR-mediated phagocytosis in primary mouse neutrophils. Blood. 2010;116:4978–4989. doi: 10.1182/blood-2010-03-275602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan SR, Berton G. Regulation of Src family tyrosine kinase activities in adherent human neutrophils. Evidence that reactive oxygen intermediates produced by adherent neutrophils increase the activity of the p58c-fgr and p53/56lyn tyrosine kinases. J Biol Chem. 1996;271:23464–23471. doi: 10.1074/jbc.271.38.23464. [DOI] [PubMed] [Google Scholar]
- 48.Martin V, Guillermet-Guibert J, Chicanne G, Cabou C, Jandrot-Perrus M, Plantavid M, Vanhaesebroeck B, Payrastre B, Gratacap MP. Deletion of the p110β isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood. 2010;115:2008–2013. doi: 10.1182/blood-2009-04-217224. [DOI] [PubMed] [Google Scholar]
- 49.Dbouk HA, Vadas O, Williams RL, Backer JM. PI3Kβ downstream of GPCRs–crucial partners in oncogenesis. Oncotarget. 2012;3:1485–1486. doi: 10.18632/oncotarget.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nylander S, Kull B, Björkman JA, Ulvinge JC, Oakes N, Emanuelsson BM, Andersson M, Skärby T, Inghardt T, Fjellström O, Gustafsson D. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J Thromb Haemost. 2012;10:2127–2136. doi: 10.1111/j.1538-7836.2012.04898.x. [DOI] [PubMed] [Google Scholar]
- 51.Shuttleworth SJ, Silva FA, Cecil ARL, Tomassi CD, Hill TJ, Raynaud FI, Clarke PA, Workman P. Progress in the preclinical discovery and clinical development of class I and dual class I/IV phosphoinositide 3-kinase (PI3K) inhibitors. Curr Med Chem. 2011;18:2686–2714. doi: 10.2174/092986711796011229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Backer JM. The regulation of class IA PI 3-kinases by inter-subunit interactions. Curr Top Microbiol Immunol. 2010;346:87–114. doi: 10.1007/82_2010_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, Rikova K, Zhao J, Hirsch E, Benes CH, Engelman JA. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell. 2015;27:97–108. doi: 10.1016/j.ccell.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, Shah RH, Huynh T, Mino-Kenudson M, Sgroi D, Isakoff S, Thabet A, Elamine L, Solit DB, Lowe SW, Quadt C, Peters M, Derti A, Schegel R, Huang A, Mardis ER, Berger MF, Baselga J, Scaltriti M. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature. 2015;518:240–244. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, Scher HI, Barry ST, Sawyers CL, Chandarlapaty S, Rosen N. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell. 2015;27:109–122. doi: 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The Gβγ sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 58.Vadas O, Dbouk HA, Shymanets A, Perisic O, Burke JE, Abi Saab WF, Khalil BD, Harteneck C, Bresnick AR, Nürnberg B, Backer JM, Williams RL. Molecular determinants of PI3Kγ-mediated activation downstream of G-protein–coupled receptors (GPCRs) Proc Natl Acad Sci USA. 2013;110:18862–18867. doi: 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mouchemore KA, Sampaio NG, Murrey MW, Stanley ER, Lannutti BJ, Pixley FJ. Specific inhibition of PI3K p110δ inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J. 2013;280:5228–5236. doi: 10.1111/febs.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papakonstanti EA, Zwaenepoel O, Bilancio A, Burns E, Nock GE, Houseman B, Shokat K, Ridley AJ, Vanhaesebroeck B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci. 2008;121:4124–4133. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 61.Vanhaesebroeck B, Jones GE, Allen WE, Zicha D, Hooshmand-Rad R, Sawyer C, Wells C, Waterfield MD, Ridley AJ. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat Cell Biol. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- 62.Costa C, Germena G, Hirsch E. Dissection of the interplay between class I PI3Ks and Rac signaling in phagocytic functions. Scientific World Journal. 2010;10:1826–1839. doi: 10.1100/tsw.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okamoto F, Saeki K, Sumimoto H, Yamasaki S, Yokomizo T. Leukotriene B4 augments and restores FcγRs-dependent phagocytosis in macrophages. J Biol Chem. 2010;285:41113–41121. doi: 10.1074/jbc.M110.175497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fodor S, Jakus Z, Mócsai A. ITAM-based signaling beyond the adaptive immune response. Immunol Lett. 2006;104:29–37. doi: 10.1016/j.imlet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 65.Khwaja A, Rodriguez-Viciana P, Wennström S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Damoulakis G, Gambardella L, Rossman KL, Lawson CD, Anderson KE, Fukui Y, Welch HC, Der CJ, Stephens LR, Hawkins PT. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci. 2014;127:2589–2600. doi: 10.1242/jcs.153049. [DOI] [PubMed] [Google Scholar]
- 68.Anderson KE, Boyle KB, Davidson K, Chessa TAM, Kulkarni S, Jarvis GE, Sindrilaru A, Scharffetter-Kochanek K, Rausch O, Stephens LR, Hawkins PT. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. 2008;112:5202–5211. doi: 10.1182/blood-2008-04-149450. [DOI] [PubMed] [Google Scholar]
- 69.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJO, Stephens LR, Hawkins PT. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods. 2011;8:267–272. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR. Targeting genes for self excision in the germ line. Genes Dev. 1999;13:1524–1528. doi: 10.1101/gad.13.12.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Targeting of Pik3cbKK526,527DD.
Fig. S2. Confirmation of the correct targeting of Pik3cbKK526,527DD.
Fig. S3. p110β quantities in BMDMs and BMNs are unaffected by the introduction of the RBD- and Gβγ-insensitive mutations.
Fig. S4. Dose responses of GPCR (C5a) and RTK (M-CSF) agonists on PIP3 production by BMDMs.
Fig. S5. ROS generation in BMNs in response to the phagocytosis of IgG-SRBCs, but not soluble GPCR agonists, requires p110β activity.
Fig. S6. ROS generation in BMNs in response to adhesion to poly-RGD+, but not immune complexes, is independent of BLT1 activation. Reference (70)