

Summary
Roles for cell death in development, homeostasis, and the control of infections and cancer have long been recognized. Whereas excessive cell damage results in passive necrosis, cells can be triggered to engage molecular programs that result in cell death. Such triggers include cellular stress, oncogenic signals that engage tumor suppressor mechanisms, pathogen insults, and immune mechanisms. The best-known forms of programmed cell death are apoptosis and a recently recognized regulated necrosis termed necroptosis. Of the two best understood pathways of apoptosis, the extrinsic and intrinsic (mitochondrial) pathways, the former is induced by the ligation of death receptors, a subset of the TNF receptor (TNFR) superfamily. Ligation of these death receptors can also induce necroptosis. The extrinsic apoptosis and necroptosis pathways regulate each other and their balance determines whether cells live. Integral in the regulation and initiation of death receptor-mediatedactivation of programmed cell death is the aspartate-specific cysteine protease (caspase)-8. This review describes the role of caspase-8 in the initiation of extrinsic apoptosis execution and the mechanism by which caspase-8 inhibits necroptosis. The importance of caspase-8 in development and homeostasis and the way that dysfunctional caspase-8 may contribute to the development of malignancies in mice and humans are also explored.
Keywords: Caspase-8, apoptosis, necroptosis, inflammation, development, hematopoiesis
Introduction
The earliest studies on apoptosis defined this as a “programmed” cell death, one that is controlled by molecular pathways. More recently, the realization that necrosis can also be a regulated form of cell death, has led to the identification of several forms of “programmed” necrosis. These include necroptosis, pyroptosis, ferroptosis, mitotic catastrophy and autophagic cell death, among others. While it has been demonstrated that each of these newly identified forms of programmed cell death have their own important functions and consequences in their own respective settings, it has become clear that necroptosis has a broad impact on biology, similar to apoptosis.
Deregulation of the apoptotic mechanism has been implicated in a variety of disease settings. Indeed, blockade or evasion of apoptosis was listed as one of the original “hallmarks” of cancer (1). However, the physiological function of necroptosis upon similar perturbations in homeostasis has not been as well defined. Recently, multiple studies have suggested there are roles for necroptosis in development (2–5) and disease (reviewed in (6–8)), such as viral infection (9), atherosclerosis (10, 11), renal ischaemic reperfusion injury (12), and cancer (13–15).
The extrinsic pathway of apoptosis is initiated by ligation of death receptors, a subset of the tumor necrosis factor superfamily (TNFRSF) that includes tumor necrosis factor (TNF) receptor-1 (TNFR1; TNFRSF1a), CD95 (TNFRSF6, Apo-1, and Fas), TNF-related apoptosis-inducing ligand (TRAIL) receptor-1 and -2 (TRAIL-R1/2; DR4/5, and TNFRSF10a/b), DR3 (TNFRSF25), and DR6 (TNFRSF21). Ligation of these receptors by their cognate ligands or by agonistic antibodies can promote apoptosis.
It has become evident that there is a bridge between apoptosis and necroptosis in homeostasis and disease (3–5). The extrinsic apoptotic and necroptotic pathways are tightly intertwined and depend on multiple proteins that regulate whether a cell (or animal) lives or dies. Integral to the initiation and regulation of death receptor-mediatedactivation of apoptosis and necroptosis is a complex consisting of the proteins Fas associated via death domain (FADD), aspartate-specific cysteine protease (caspase)-8 (formerly known as FLICE) or caspase-10 (FLICE2) and FLICE-like inhibitory protein (cFLIP; encoded by casp8 and FADD-like apoptosis regulator, cflar). As such, this complex has been shown to determine cell fate upon extrinsic death receptor signaling.
The importance of cell death regulation by FADD-caspase-8-cFLIP extends well beyond the individual cell. If the balance skews towards apoptosis, cells reduce to apoptotic bodies that are removed by resident phagocytic cells and surrounding cells are not “alarmed”, instead they produce signals that prevent inflammation. However, when necroptosis is engaged, cells die by rupture of the plasma membrane and the cellular contents, including damage-associated molecular patterns (DAMPs), pro-inflammatory cytokines, and other alarmins, are released into the microenvironment and surrounding cells respond in a pro-inflammatory manner. Thus, whereas apoptosis is generally anti-inflammatory, necroptosis induces inflammation.
This review describes the functions and regulation of the FADD-caspase-8-cFLIP complex in cell death and non-cell death pathways with a particular focus on caspase-8.
Caspase-8 in executing extrinsic apoptosis
The term apoptosis (greek “falling off”) was first proposed by Kerr, Wyllie and Currie in 1972 to distinguish between classical necrosis and the observation of structures that are now known to be apoptotic bodies observed in ischemic liver injury (16, 17). Since then, it has been well established that two forms of apoptosis exist; intrinsic apoptosis and extrinsic apoptosis. Both intrinsic and extrinsic apoptosis are processes that rely on activation of members of the aspartate-specific cysteine protease (caspase) family. In the intrinsic, or mitochondrial pathway of apoptosis, effector caspases are activated after a signaling cascade involving mitochondrial outer membrane permeabilization (MOMP), causing the release of proteins of the intermembrane space. These include cytochrome c and second mitochondria-derived activator of caspases (SMAC), which promote the formation of the so-called “apoptosome”, and the activation of caspases that orchestrate the death of the cell (18). In the extrinsic, or death receptor pathway of apoptosis, ligation of death receptors on the cell surface leads to caspase activation. In both forms of apoptosis cells die by effector caspase-induced cell blebbing, DNA fragmentation, phosphatidylserine externalization and formation of apoptotic bodies, the characteristics of apoptosis described by Kerr et al.
Extrinsic apoptosis relies on the formation of a death-inducing signaling complex (DISC), which always includes FADD and caspase-8. In the case of ligation of TNFR1, another adapter, tumor necrosis factor receptor type 1-associated DEATH domain (TRADD), is first engaged, which in turn recruits FADD, and TRADD is required for apoptosis induced by TNF, but not for that induced by other death receptor ligands. Other death receptors engage FADD directly.
FADD is an adapter protein that consists of a C-terminal death domain (DD) and a N-terminal death effector domain (DED). FADD is recruited to the intracellular death domains of trimerized death receptors (DR) through homotypic DD interactions and recent stoichiometric studies show that the ratio between DR and FADD is 3:1 (19). FADD binding to the trimerized receptor induces a conformational change in the protein that exposes the DED to bind caspase-8. In humans, caspase-10 (which is not present in rodents) can also be recruited to FADD, but its role in apoptosis is less clear. Both procaspase-8 and -10 are expressed as a zymogen that comprises a N-terminal pro-domain that includes two DEDs (tandem DED), a large protease subunit containing the catalytic site (p20/p18), a short linker region, and a small subunit (p12/p10).
Procaspase-8 binds to the exposed DED of death receptor-associated FADD through a pocket in its DED1, forming the DISC (19, 20) (Figure 1). Additional procaspase-8 molecules are then recruited and bind through a second DED1 pocket to a motif in DED2 of the prior procaspase-8 by dominant hydrophobic interactions (20). This results in DED-mediated procaspase-8 oligomerization. Binding of procaspase-8 to FADD thus prompts recruitment of additional procaspase-8 molecules that leads to the formation of a filament assembled in a unidirectional manner (20). Stoichiometric studies show that on average six procaspase-8 molecules bind to a single FADD protein (19). Filament formation is important, because it allows the proteolytic domains of the interacting procaspase-8 molecules to homodimerize. Within the homodimer, autoproteolytic cleavage of an aspartate residue between the linker and small subunit (D384 human, D387 mouse) then partially activates the protein. Subsequent cleavage of an aspartate connecting DED2 and the large subunit releases the now fully matured enzyme and allows initiation of apoptosis.
Figure 1. cFLIP regulates death receptor-mediated apoptosis.
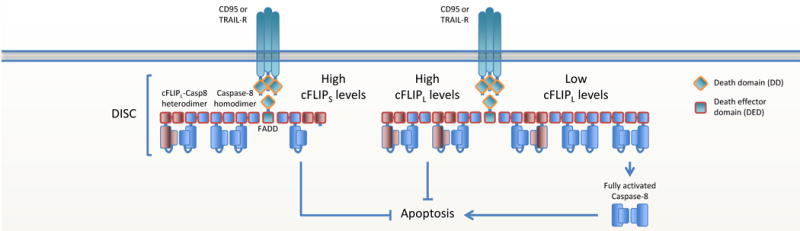
Ligated CD95 or TRAILR recruits FADD via homotypic DD interactions. FADD then recruits caspase-8 via DED interactions, forming the DISC. When cFLIP levels are low, caspase-8 homodimerizes and activates itself by autoproteolytic cleavage, resulting in the release of activated caspase-8 and the initiation of apoptosis. cFLIPS blocks apoptosis by abrogating caspase-8 filament formation. cFLIPL blocks apoptosis by inhibiting the full maturation and release of caspase-8.
The effector caspases, caspase-3 and caspase-7, exist as dimeric zymogens that are activated by cleavage between their large and small subunits. Fully matured caspase-8 cleaves these zymogens and thus activates them. However, in some cases the activity of the effector caspases is subsequently inhibited by X-linked Inhibitor of Apoptosis Protein (XIAP). Caspase-8 can circumvent this inhibition as follows. Caspase-8 can also cleave the protein BID and cleaved BID (cBID) then activates the effector proteins BAX and BAK, which mediate MOMP. Upon MOMP, proteins that antagonize XIAP (such as SMAC) are released from the mitochondrial intermembrane space, allowing executioner caspases to promote apoptosis. This process was demonstrated most strikingly through a series of elegant in vivo experiments. Administration of the ligand for the death receptor CD95 results in rapid liver damage and lethality in mice dependent on the presence of caspase-8 in the liver. Animals lacking BID are resistant to this lethal effect (19, 21). However, animals in which both BID and XIAP are ablated are fully sensitive to the lethal effects of CD95 ligation (22).
The apoptotic function of caspase-8 can be modulated by phosphorylation of specific residues on the protein. Human procaspase-8b can be phosphorylated on tyrosines 273, 293, 380, 448 and serines 287, 305 and 347 (23–25). Human procaspase-8 is a substrate for Polo-like kinase 3 (Plk3) that interacts with CD95, FADD, and caspase-8. Upon DISC formation, Plk3 phosphorylates residue T273 of procaspase-8 that promotes the pro-apoptotic function of procaspase-8 (23). Phosphorylation of Y380 also controls apoptosis and is regulated by the kinases Fyn, Lyn and Src and phosphatase SHP1 (26, 27). Procaspase-8 Y380 phosphorylation does not impair recruitment to the DISC, nor does it interfere with DED chain assembly and D384 processing. It does, however, block apoptosis by hampering further autoproteolytic cleavage and hence release from the DED, possibly by the interaction with unknown factors that hinder maturation in a steric way (28). Furthermore, Y380 phosphorylation induces translocation to the plasma membrane where caspase-8 can apparently promote migration (in an unknown manner) and interact with the p85a subunit of PI3K to promote endosome maturation (29, 30). However, mice lack a tyrosine in the linker region between the large and small subunits, and therefore these regulatory effects are not evident.
cFLIP regulates extrinsic apoptosis
The presence of the FLICE-like inhibitory protein (cFLIP) in the FADD-caspase-8-cFLIP complex determines if and how cells die. As such, cFLIP is a switch that determines cell fate. cFLIP comes in two major isoforms, depending on post-transcriptional mRNA splicing (31, 32). cFLIP long (cFLIPL) is a procaspase-8-like protein with the major difference that it lacks proteolytic activity due to the absence of a catalytic cysteine in the large subunit. cFLIP short (cFLIPS) is a truncated form of procaspase-8 that only contains two tandem DEDs and a short C-terminal tail. The cFLIPS protein is an inhibitor of caspase-8 and blocks DISC-dependent procaspase-8 activation by disrupting DED-mediated procaspase-8 oligomer assembly. Concordantly, equal numbers of FADD, procaspase-8 and cFLIPS are found in the DISC (19, 32–35). The function of cFLIPL is more complex as it regulates the extent of activation and possibly substrate specificity of procaspase-8 (4, 36). Low levels of cFLIPL can enhance apoptotic signaling, whereas apoptosis is inhibited when cFLIPL levels are high. It is not completely clear how cFLIPL mechanistically inhibits apoptosis. A classical view portrays cFLIP directly binding to FADD (37), inhibiting apoptosis by directly competing with procaspase-8 for binding to FADD (31, 37–39). However, this dogma is now challenged by recent studies that show that cFLIP is only weakly able to bind FADD, but that upon FADD-procaspase-8 interaction, it is procaspase-8 that recruits cFLIP to the DISC in a co-operative and hierarchical manner (19, 20, 40). How exactly the DED domains of procaspase-8 and cFLIPL interact remains to be elucidated, but it is clear that the procaspase-8 catalytic domain prefers heterodimerization to the cFLIPL caspase-like domain over homodimerization with catalytic domains of other procaspase-8 molecules (41, 42). Heterodimerization to c-FLIPL rearranges the catalytic site of procaspase-8, producing a conformation that renders the heterodimer highly active even in the absence of proteolytic processing of either caspase-8 or cFLIPL (42–44). A non-cleavable procaspase-8 mutant (mouse; D387A), that in the absence of cFLIP is unable to initiate apoptosis, becomes catalytically active when heterodimerized with cFLIPL (4, 36). Therefore the current paradigm is that cFLIP regulates procaspase-8 filament formation. The cFLIPS protein abrogates the formation of procaspase-8 filaments. Low levels of cFLIPL enhance the specific activity of the heterodimer and procaspase-8 homodimer activation without disrupting the filament. High levels of cFLIPL reduces the activity of procaspase-8, possibly by disrupting procaspase-8 homodimer filaments. Thus, the cFLIP isoforms and their expression levels are critical determinants whether cells live or die.
CD95 and TRAIL-R ligation induces apoptosis by direct recruitment of FADD-caspase-8 in a complex called the death inducing signaling complex (DISC). As described above, isoform levels of cFLIP then determine whether apoptosis is blocked or engaged (Figure 1).
TNFR1-induced apoptosis activation is more complex (Figure 2). Ligation of TNFR1 can both induce caspase-8-mediated apoptosis as well as block apoptosis via the NF-κB-induced expression of cFLIP. Receptor interacting serine/threonine kinases 1 (RIPK1) is key in regulating TNFR1-induced FADD-caspase-8-mediated apoptosis. RIPK1 consists of a N-terminal kinase domain (KD), an intermediate domain (ID), which includes a RIP homotypic interaction motif (RHIM), and a C-terminal DD. Furthermore, the KD and ID contain caspase-8 cleavage sites. RIPK1 is highly regulated post-translationally, mainly by phosphorylation and ubiquitylation and the balance thereof determines whether cells resist or engage death. Following ligation of TNFR1, RIPK1 is recruited to the DD of the adaptor protein TRADD and is then heavily ubiquitylated by the E3 ligases cellular inhibitor of apoptosis (cIAP) 1 and 2. This gives RIPK1 a scaffolding function that contributes to the activation of NF-κB and MAPK pathways (45), resulting in expression of both pro-inflammatory cytokines and pro-survival genes, including cFLIP. Conversely, deubiquitylated RIPK1, mediated by cIAP inhibition or the deubiquitylases A20 or CYLD, induces apoptosis by associating with FADD and procaspase-8 (called complex IIa). RIPK1 ubiquitylation thus regulates whether cells survive or die by apoptosis following TNFR1 ligation.
Figure 2. Regulation of TNFR1-mediated cell death.
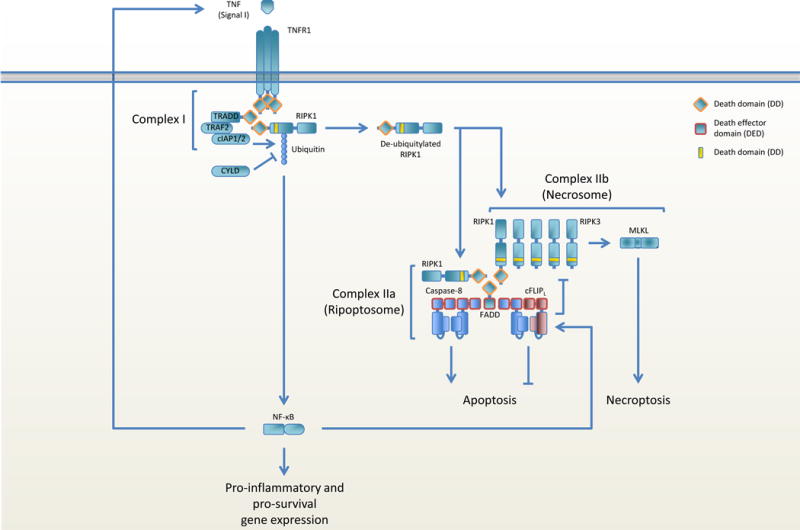
TNFR1 ligation leads to the recruitment of TRADD, TRAF2, cIAP1/2 and RIPK1 (complex I). RIPK1 ubiquitylation by cIAP1/2 mediates activation of NF-κB and the production of pro-inflammatory and pro-survival gene expression. RIPK1 is deubiquitylated by CYLD and leaves complex I to recruit FADD via homotypic DD interactions and RIPK3 through homotypic RHIM domain interactions, thereby forming the ripoptosome. Homodimerization and activation of caspase-8 on FADD induces apoptosis, whereas RIPK3 ameloid formation and MLKL activation engages necroptosis. One of the expressed pro-survival genes, cFLIPL, heterodimerizes with caspase-8, resulting in inhibition of caspase-8 activation and apoptosis. Caspase-8 retains its catalytic activity and is able to cleave RIPK1 and RIPK3 in the apoptosome, thereby blocking necroptosis.
As discussed above, TNF treatment of cells can induce apoptosis through the activation of caspase-8 in the RIPK1-FADD-caspase-8 complex (complex IIa). Surprisingly, it was observed that cells die if FADD or caspase-8 are absent or inhibited by pharmacological, viral, or bacterial factors, indicating a role for these proteins in a new form of non-apoptotic cell death. Close examination revealed that these cells do not display apoptotic features; rather they die by swelling and rupture of the cell membrane similar to death of necrotic cells. TNF thus engages a second form of programmed cell death with phenotypical similarities of necrosis. This form of death has been termed necroptosis (6).
The FADD-procaspase-8-cFLIPL complex inhibits necroptosis
Necroptosis is a form of programmed necrosis that is both morphologically and mechanistically distinct from apoptosis. Whereas extrinsic apoptosis depends on initiator and effector caspase activation, necroptosis relies on the kinase activity of receptor interacting serine/threonine kinases (RIPK) 1 and 3 to activate pseudokinase mixed lineage kinase domain-like (MLKL). The observation that necroptosis ensues in cells lacking FADD or caspase-8 pointed to a necroptosis-inhibitory role for the FADD-procaspase-8-cFLIPL complex (Figure 2).
TNFR1 ligation-induced necroptosis depends on binding of deubiquitylated RIPK1 to RIPK3 through homotypic RHIM domain interactions of both proteins (Figure 2). This leads to RIPK3 oligomerization, activation and the subsequent engagement of MLKL, the effector protein that once activated translocates to the plasma membrane, where it induces rupture and subsequent cell death.
As described above, deubiquitylated RIPK1 can interact with the FADD-procaspase-8-cFLIPL complex through homotypic DD interaction between FADD and RIPK1. RIPK1 can also recruit RIPK3 to this complex via homotypic interaction of their RHIM domains. The resulting complex that is formed around RIPK1 is called the ripoptosome. RIPK1 (46) and RIPK3 (47) each contain one or more caspase-8 cleavage sites and are substrates for the procaspase-8-cFLIPL heterodimer. Inhibition of caspase-8, for instance by the pharmacological inhibitor zVAD-fmk or viral cFLIPS mimetics, abrogates its ability to cleave RIPK1 and RIPK3 and leads to RIPK1-dependent TNF-induced RIPK3 activation and necroptosis. This is dependent on the kinase activity of RIPK1 since RIPK1 inhibition by the pharmacological inhibitor necrostatin-1s (Nec-1s) rescues TNF/zVAD-induced necroptosis (2). Hence, the FADD-procaspase-8-cFLIPL complex inhibits necroptosis by proteolytic cleavage of RIPK1 and RIPK3 in the ripoptosome.
Necroptosis can also be initiated independently of RIPK1 by so-called ‘signal II’ stimuli (2) (Figure 3). In those cases, RIPK3 is activated following ligation of TLR3 and TLR4 (2, 48), the interferon receptors IFNAR and IFNγR (2), DAI (9, 49, 50), or recognition of oxidized LDL through an as yet unknown mechanism (51). The DNA sensor DAI and the adaptor protein TRIF, activated by TLR3/4, each contain RHIM domains through which they can bind to and activate RIPK3. How necroptosis is induced by type I and II Interferons and oxidized LDL remains unclear, though IFNAR and IFNγR seem to signal via PKR (52). Either absence of RIPK1 or inhibition of caspase-8 is sufficient to promote signal II-induced necroptosis in the presence of TLR ligands or interferons (2, 52, 53), implying that the FADD-procaspase-8-cFLIPL-mediated abolishment of signal II-induced necroptosis is facilitated by RIPK1. Through homotypic RHIM domain interactions between RIPK1 and RIPK3, the RIPK1-FADD-procaspase-8-cFLIPL complex can associate with TRIF or DAI and form a complex that has great similarities with the TNFR-induced ripoptosome. Since this complex contains additional interacting proteins and its formation is initiated through signal II receptors we classify this complex as the non-canonical ripoptosome.
Figure 3. Regulation of signal II-mediated cell death.
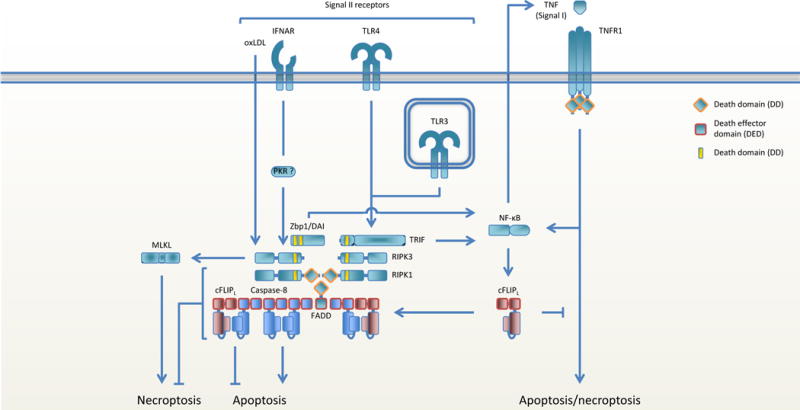
Ligation of signal II receptors induces pro-inflammatory and pro-survival gene expression and activates RIPK3 to engage necroptosis. RIPK1 links the inhibitory FADD-caspase-8-cFLIPL complex to RIPK3 via homotypic RHIM domain interactions between RIPK1 and RIPK3 and homotypic DD interactions between RIPK1 and FADD. When cFLIPL levels are low, RIPK3 induces necroptosis, but when MLKL is absent or blocked RIPK3 induces caspase-8 mediated necroptosis. When cFLIPL levels are high, the caspase-8-cFLIPL heterodimer blocks both apoptosis and necroptosis.
The way RIPK1 blocks signal II-induced necroptosis seems twofold. First, RIPK1 blocks signal II-induced necroptosis by recruiting the FADD-procaspase-8-cFLIPL complex (2). Second, as Nec-1s treatment protects cells from signal II-induced necroptosis without the need for caspase-8 activity (2), RIPK1 itself may block signal II-induced RIPK3 activation in an as yet unknown way. Thus, RIPK1 has a dual role in necroptosis that reflects how RIPK3 is activated. Following TNFR1 ligation, RIPK3 is activated by RIPK1 and the function of RIPK1 is pro-necroptotic. Type II signals, however, activate RIPK3 independently of RIPK1 and in this setting RIPK1 acts in an anti-necroptotic fashion (2). In both the canonical (TNFR-induced) and non-canonical ripoptosomes, the procaspase-8-cFLIPL heterodimer can cleave RIPK1 and RIPK3 and block necroptosis.
Activated RIPK3 kills cells by engaging MLKL-dependent necroptosis. However, the observation that MLKL-deficient cells also die following RIPK3 activation (54, 55) indicates that RIPK3 can also induce FADD-caspase-8-mediated apoptosis. The kinase activity of RIPK3 is essential for necroptosis, but dispensable for apoptosis (54–58) and a fully intact RHIM domain may be expendable for apoptosis to occur as well (55). Thus, activated RIPK3 can have a pro-apoptotic role by inducing RIPK1-FADD-caspase-8-mediated apoptosis independent of its catalytic activity through retrograde signaling in the non-canonical ripoptosome.
IFNγ can induce cell death through RIPK3 and co-ablation of caspase-8 and MLKL is sufficient to block IFNγ-induced death in mouse cells (2). RIPK1 deubiquitylation can be achieved through the inhibition of cIAP1/2, for instance following SMAC release upon MOMP or by pharmacological SMAC mimetics. The absence of caspase-8 and RIPK3 suffices to block death in mouse cells even in the presence of MLKL following IFNγ and SMAC mimetic treatment (59). However, human caspase-8 and RIPK3 double-deficient HT29 cells die following IFNγ and SMAC mimetic treatment (59). Death was attributed to IFNγ-induced gene expression, RIPK1 and caspase-10 in human HT29 cells, but independent of TNFR1/2, Fas, TRAILR, TWEAKR, IFNAR and ATG5, a protein essential for autophagy. These data hint at the existence of a yet unknown intrinsic signal that mediates death through RIPK1.
Caspase-8 in development
It was long believed that a non-apoptotic role of caspase-8 was necessary during development. Fadd (60), casp8 (61) or cFLIP (cflar) (62) ablation in mice yields embryonic lethality at E10.5. The embryos have a pale appearance and show defects in heart muscle development, erythrocyte accumulation, neural tube development and yolk sac vasculature. The cause of death in utero is most likely due to defects in yolk sac development, since heart and neural tube deficiencies were not observed in whole E10.5 embryo ex vivo cultures (63). Furthermore, catalytically inactive caspase-8 mutant mice (casp8C382A/C362A) are embryonically lethal (64), suggesting embryogenesis is dependent on the catalytic activity of caspase-8. Conditional deletion of caspase-8 in brain, heart, or liver does not impact development (65–67), whereas conditional deletion of caspase-8 in the endothelial compartment (Tie1-Cre) phenocopies casp8−/− embryos (66). Conditional deletion of caspase-8 from the hematopoietic development system (Vav-Cre) also results in embryonic lethality (68), but more specific caspase-8 deletion in either T cells, B cells, macrophages or dendritic cells renders viable animals (64, 68–70), suggesting a role for caspase-8 in other hematopoietic compartments in early development.
To determine if the apoptotic function of caspase-8 is necessary during development, mice were generated in which the aspartate in the self-processing site at position D387 was mutated to alanine. This non-cleavable caspase-8 is impaired in inducing apoptosis, but remains able to block necroptosis (4, 36, 71). These casp8D387A/D387A mice survive and reach adulthood without any observed complications (71), showing that the apoptotic function of caspase-8 is not required for development. The finding that casp8D387A/D387A mice survive and reach adulthood suggests that the primary role for caspase-8 in development is to inhibit necroptosis. Indeed, casp8−/− mice in which either one of the necroptosis-mediating genes ripk3 or mlkl is co-ablated are fully viable and wean at Mendelian frequencies (3, 4, 72).
That casp8D387A/D387A mice live does not mean that extrinsic apoptosis is completely irrelevant for development. Catalytically inactive RIPK3 (ripk3D161N/D161N) mutant mice, in which RIPK3 is unable to induce necroptosis, die during embryogenesis with similar features as casp8−/−, fadd−/− and especially cflar−/− mice (58). The yolk-sac vasculature of these embryos is disrupted by caspase-dependent apoptosis. Ripk3D161N/+ and ripk3D161N/− animals are viable, suggesting either that this mutation is dominant negative or that threshold levels are required for the effects to take place. Co-ablation of casp8 or ripk1 fully rescues the development of ripk3D161N/D161N animals, showing that RIPK3 can induce apoptosis through RIPK1 and caspase-8 in a kinase-independent fashion and thus emphasizes the pro-apoptotic role of RIPK3 discussed above. In contrast, mutation of K51A also renders RIPK3 catalytically inactive, but yield viable animals that do not show a developmental phenotype (57). This suggests that it is not the inactivity of RIPK3 but another, yet unknown, effect of the D161N mutation that causes lethality. From the developmental point of view it thus seems that the inhibition of both apoptosis and necroptosis determines if the yolk sac vasculature develops normally and if the embryo survives this and other checkpoints of development (56).
Recent studies suggest how necroptosis is activated during development. The early lethality of casp8−/− embryos is at least partially dependent on TNFR1 as co-ablation of TNFRSF1A in caspase-8 deficient animals delays embryonic lethality to E16.5 (2). Yolk-sac vasculature seems intact in these embryos, thus it remains unclear why development of these tnfr−/−casp8−/− embryos stops at E16.5. Furthermore, co-ablation of ripk1 in caspase-8 deficient animals rescues embryonic lethality to birth with similar kinetics to ripk1−/− animals (2), suggesting that caspase-8 requires RIPK1 for the inhibition of necroptotic cell death at E10.5 and E16.5. Ripk1−/− animals die perinatally and show abundant apoptosis. Co-ablation of fadd, casp8 or tnfr1, however, does not prevent lethality, suggesting that these mice die by a TNF-independent non-apoptotic death pathway. Ripk3 or mlkl ablation only briefly extends the life span of ripk1−/− animals (2, 5, 73, 74), but the additional removal of fadd, caspase-8, or tnfr1 fully prevents perinatal lethality in ripk1−/−ripk3−/− mice (2), indicating that both apoptosis and necroptosis must be blocked for mice to reach adulthood. Ripk1 ablation is dispensable for this, since fadd−/− or casp8−/− co-ablation with ripk3−/− or mlkl−/− is sufficient for mouse survival (3, 4, 72). These data imply that necroptosis must be engaged through signals other than TNF shortly after birth. TRIF and DAI both contain RHIM domains that facilitate their binding to RIPK3. Co-ablation of trif and zbp1 in ripk1−/− mice prolongs life until weaning, but only co-ablation of caspase-8 fully rescues these trif−/−zbp1−/−ripk1−/− animals to adulthood (50). Trif ablation in ripk1−/−tnfr1−/− mice delays lethality, but most mice die around weaning (2), indicating that the lethal signal is not mediated through TRIF. DAI was shown to bind to RIPK1 and RIPK3 via RHIM interactions in vitro and recruitment of RIPK3 to DAI induced RIPK3 autophosphorylation (75). The weaning casp8−/−zbp1−/−ripk1−/− animals shows that DAI is the receptor that initiates necroptosis shortly after birth (50), albeit that this conclusion is based on only a few observed animals.
Recent work shows that the RHIM domain interaction between RIPK1 and RIPK3 is essential for blocking DAI-induced necroptosis. Mice in which the RHIM domain of RIPK1 is mutated (ripk1RHIM/RHIM mice) die perinatally, similar to ripk1−/− animals (49, 50). Of note is that RIPK1 does not rely on its RHIM domain to transduce TNFR signaling, implying that caspase-8-mediated apoptosis is still blocked in these animals. Ripk1RHIM/RHIM mice can be rescued by RIPK3 deficiency, MLKL deficiency, catalytically inactive RIPK3 (D161N), RHIM mutant RIPK3, or DAI deficiency. The observation that TRIF ablation does not rescue ripk1RHIM/RHIM (49) mice further delineates that DAI is the receptor that initiates necroptosis after birth. The RHIM domain of RIPK1 hampers the interaction between DAI and RIPK3 (50), but how it does so requires further in-depth investigation. It has been suggested that RIPK1 may directly compete with DAI for RIPK3 binding (49, 50) and RIPK1 itself was shown to bind to DAI in absence of RIPK3 (75). Alternatively, the RHIM domain may be needed to bind the necroptosis-inhibitory RIPK1-FADD-caspase-8-cFLIPL complex to RIPK3 and as such block necroptosis induced by the interaction between DAI and RIPK3.
It is of interest to examine whether DAI also plays an important role during early development or if TNFR signaling mediates necroptosis during embryogenesis and DAI is engaged only around the time of birth. The generation of casp8−/−zbp1−/−tnrf−/− and casp8−/−zbp1−/− animals may prove valuable to answering that question.
Caspase-8-associated pathology
Despite their normal development, casp8−/−ripk3−/−, casp8−/−mlkl−/−, fadd−/−ripk3−/−, fadd−/−mlkl−/− and casp8−/−trif−/−zbp1−/−ripk1−/− mice manifest a lymphoaccumulative syndrome with characteristics of Autoimmune LymphoProliferative Syndrome (ALPS) several weeks after birth (2–4, 50, 72). ALPS is characterized by splenomegaly and lymphadenopathy, autoimmunity, occasional lymphoma, and a massive accumulation of CD3+B220+CD4-CD8-TCR+ T cells (76), all displayed by these mice. ALPS develops from the T cell lineage, since RIPK3-deficient mice in which caspase-8 is conditionally deleted in T cells accumulate these CD3+B220+ cells (77). In mice and humans, ALPS is usually a consequence of mutations or deletions of CD95 or its ligand CD95L. Because apoptotic CD95 signaling depends on FADD-caspase-8 it is assumed that it is the loss of FADD-caspase-8-induced apoptosis, permitted by RIPK3 or MLKL ablation, that is responsible for ALPS. Alternatively, since T cells from the non-cleavable caspase-8 mutant mouse do not show functional defects and splenomegaly of the mouse was not reported (71, 78), the scaffolding functions of caspase-8, important for antigen and death receptor-induced gene expression (described below) (79, 80), could be required for proper T cell functioning and simultaneously hints to a role for necroptosis in ALPS. However, whereas ripk3D161N/D161N mice are asympomatic, co-ablation of caspase-8 induces development of ALPS in ripk3D161N/D161N animals, suggesting that it is the balance between apoptosis and necroptosis that keeps the T cells from developing their ALPS phenotype.
Splenomegaly, lymphadenopathy and mild systemic inflammatory disease develop in mice in which caspase-8 is conditionally deleted in myeloid cells (LysM-Cre) (81) or dendritic cells (CD11c; itgax) (64, 82) or upon conditional FADD deletion in myeloid cells (83). Unlike casp8−/−ripk3−/− mice, the phenotypes in these animals are not associated with ALPS, since the indicative CD3+B220+ cell population is absent. The increase in spleen size is associated with an increase in collagen content, but is independent of total splenocyte numbers, although stromal cell, TER119+ cell and some myeloid subset numbers are increased (64). Co-ablation of RIPK3 in casp8LysM-Cre animals completely prevents splenomegaly and lymphadenopathy. Co-ablation of RIPK3 in casp8CD11c-Cre mice induced ALPS, but in these mice caspase-8 was also deleted in T cells, concomitant with reports that CD11c can be expressed in T cells (64, 84). These studies point towards a role for caspase-8 and FADD in blocking necroptosis during erythropoiesis. Furthermore, splenomegaly and lymphadenopathy were completely prevented by treating casp8LysM-Cre mice with oral antibiotics, suggesting a role for microbiome-induced TLR signaling in the observed pathology (81).
Conditional loss of caspase-8 leads to necroptotic death of intestinal epithelial cells (IECs) in the adult mouse, resulting in weight loss and sometimes death of the animal, and these effects can be completely prevented by RIPK3 co-ablation (85). Similarly, conditional FADD deletion in the intestine induces inflammatory bowel disease, which can be prevented by deletion of RIPK3, CYLD, TNF or MyD88 (86). Shedding and death of cells in the villi upon engagement of TLR3 and TLR4 are controlled by caspase-8. Ligation of either TLR can induce necroptosis in intestinal epithelial cells, but in mechanistically different ways; TLR3 signals activate RIPK3 through TRIF whereas TLR4-mediated death appears to be dependent on TNFR1 signaling (87). Also in macrophages TLR4-induced necroptosis is dependent on TNFR1. TLR4 ligation induces TNF expression that then activates TNFR1. Caspase-8 blocks TLR4-induced necroptosis by mediating the degradation of CYLD in a TRIF and RIPK1-dependent manner (88) and as such prevents RIPK1 deubiquitylation and the formation of the necrosome. LPS-induced necroptosis is not increased in non-cleavable caspase-8 (casp8D387A/D387A) macrophages, again indicating that non-cleavable caspase-8 has catalytic activity. Interestingly, Yersinia infection of these macrophages induced RIPK3-dependent necroptosis, apparently due to the activity of the bacterial YopJ protein that inhibits the ability of the IKK complex to phosphorylate RIPK1 (78) and as such prevents RIPK1 from gaining its pro-survival function (89). In contrast to conventional macrophages, TLR3/4-induced necroptosis in microglia is entirely dependent on TRIF (90).
Tamoxifen-induced acute conditional deletion of FADD (91), caspase-8 (85, 92), or cFLIP (93) in the skin of adult mice induces local keratinocyte death, tissue disruption, and inflammation. Mice expressing a catalytically inactive caspase-8 mutant show the same phenotype, indicating that the enzymatic activity of caspase-8 is required for normal skin homeostasis (92). Caspase-8 loss was associated with necroptotic cell death and RIPK3 co-ablation rescued the phenotype (85). Concordantly, the skin of casp8−/−ripk3−/−, casp8−/−mlkl−/−, fadd−/−ripk3−/− or fadd−/−mlkl−/− double knock-out (DKO) mice is completely normal, further establishing that the severe skin inflammation observed in conditional FADD or caspase-8 knock-out animals is induced by necroptosis.
In contrast, cFLIP ablation was associated with apoptotic but not necroptotic cell death and RIPK3 co-ablation had no effect (93). TNFR signaling induced cell death and the resulting pathology in both cFLIP and caspase-8 deficient skin, since TNF neutralization abrogated disease formation in both cases (85, 93). However, the requirement for TNF in the skin pathology remains controversial, since another study showed that the disease caused by caspase-8 deficiency in keratinocytes is triggered independently of TNF, IL-1, TLR adapter proteins or macrophages, but rather is associated with constitutive signaling for IRF3 activation (92).
Conditional deletion of RIPK1 in keratinocytes (K14-Cre) also induces inflammation and necroptotoc cell death (94) and RIPK1 depends on its RHIM domain to block this phenotype, since mice that express the mutant RIPK1 RHIM only in keratinocytes show this pathology (49). Skin pathology of these animals evolves gradually and was suggested to depend on the presence of cytokines (IFNβ) within the tissue, resulting in the gradual increase of zbp1 expression. As in mouse development, skin inflammation and necroptosis were abrogated by mlkl or zbp1 deficiency, suggesting that the RIPK1 prevents DAI-induced necroptosis in the skin of these mice (49). As discussed above, RIPK1 may do so by direct competition with DAI for RIPK3 binding or may mediate DAI-RIPK3 disruption via the activity of the FADD-caspase-8-cFLIPL complex.
Caspase-8 in inflammasomes
Besides being induced via death receptor ligation, caspase-8-mediated apoptosis can be engaged by inflammasome components (detailed below). Caspase-8-mediated apoptosis is activated by the NLRC4 inflammasome through the interaction of SUG1 and FADD (95) and complex formation of NLRP3-ASC on mitochondria can activate caspase-8 in epithelial cells (96). In absence of caspase-1 in dendritic cells (97) or macrophages (98) inflammasomes can induce apoptosis, but in presence of caspase-1 and -11 inflammasomes can engage another form of programmed necrotic cell death; pyroptosis. In pyroptosis, inflammasome activation leads to the proteolytic processing of gasdermin D (GSDMD) by caspase-1/11 (reviewed in (99)). A recent study shows that the GSDMD-related protein deafness associated tumor suppressor 5 (DFNA5) can be activated by caspase-3 during apoptosis to induce secondary pyroptosis (100). Caspase-8 can cleave caspase-3 upon DR ligation and as such may regulate DFNA5-induced pyroptosis, but this remains to be formally shown. Since caspase-8 can cleave some substrates at the same site as caspase-1/11, it also remains to be determined whether caspase-8 can activate GSDMD, DFNA5 or other gasdermin family members directly.
Inflammasomes are cytosolic complexes that process the pro-inflammatory cytokines pro-IL1β and pro-IL18 into their biologically active forms. Similar to the DISC, inflammasomes consist of a receptor (NLR or PYHIN family or AIM2), the adapter protein apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1/11 that mediate the cleavage of pro-IL1β and pro-IL18. Similar to FADD, the ASC adapter protein contains a two interaction motifs, a caspase recruitment domain (CARD) and a pyrin domain (PYD), that mediate complex formation via homotypic interactions. Following receptor activation, ASC aggregates into a single ‘spek’ in the cell and recruits and activates caspase-1/11 to cleave its substrates. Pathogen detection by membrane bound or cytosolic innate immune receptors, such as TLRs, RIG-I or IFI16, induces the expression of the pro-inflammatory cytokines pro-IL1β and pro-IL18 as well as components of the inflammasome (priming). Expressed pro-IL1β and pro-IL18 need to be cleaved into their biologically active forms in order to be secreted and this cleavage is typically mediated by inflammasomes upon recognition of the invading pathogen (trigger).
Several studies show that, depending on the cell type, caspase-8 plays a role in both priming as well as in proteolytical cleavage of the substrates into their bioactive forms. Dectin-1 ligation on DCs induces pro-IL1β expression through the CARD9-Bcl-10-MALT1 scaffold (described below) and recruitment of caspase-8 and ASC into this scaffold is crucial for processing of pro-IL-1β by caspase-8 (101). DR3 signals through MALT1-TRADD-FADD-caspase-8 to mature pro-IL1β in monocyte-derived macrophages (102). Ligation of CD95 on bone marrow-derived macrophages induces pro-IL1β activation dependent on FADD and caspase-8 and independent of RIPK3 or inflammasome components (103). In macrophages (104) and bone marrow-derived dendritic cells (BMDCs) (105, 106) TLR4 ligation induces caspase-8 mediated IL1β maturation by formation of the non-canonical ripoptosome that mediates processing and secretion of pro-IL1β (105). In line with the observations described above, IL1β maturation is enhanced when RIPK3 kinase activity is blocked (105), suggesting that caspase-8 activation through non-canonical ripoptosome formation not only blocks necroptosis but also induces secretion of pro-inflammatory cytokines.
Caspase-8 is involved in inflammasome activity as well (reviewed in (107–109)) and can be activated by NLRP3, AIM2 and NLRC4 inflammasomes in macrophages (110, 111) and NLRP3 inflammasome in dendritic cells (97). The DED domain of caspase-8 can interact with the PYD domain of ASC and results in caspase-8 filament formation and subsequent activation on ASC (112). Dependent on the cell type, caspase-8 has both positive and negative roles in NLRP3 inflammasome activation (64, 113). In macrophages, caspase-8 and FADD facilitate both pro-IL1β production and activation of the canonical and non-canonical NLRP3 inflammasomes (113), whereas in DCs caspase-8 inhibits RIPK1-RIPK3-MLKL-mediated NLRP3 inflammasome activation (64). Furthermore, caspase-8 is able to regulate the NLRP3 inflammasome upstream of NLRP3 under certain conditions (114, 115). These observations show that caspase-8 can regulate inflammasome activation, the expression and maturation of pro-inflammatory cytokines IL1β and IL18 and as a consequence can regulate inflammation in ways independent of death receptor signaling.
Non-cell death roles of caspase-8
In various cell types death receptor ligation induces pro-inflammatory cytokine expression. The produced cytokines are secreted before the cell dies and function to attract immune cells to clear the apoptotic cell (116, 117). Ligation of CD95, TRAILR, antigen receptors, Fc receptors, or Toll-like receptor 4 in T, B, and natural killer cells can lead to NF-κB activation that may depend on caspase-8 (118). FADD, caspase-8 and cFLIPL are integral for mediating NF-κB activation following antigen receptor ligation in T and B cells. Activated T cell receptor recruits a complex consisting of PKCθ and CARMA-BCL-10-MALT1 (CBM) to the lipid raft of the immunological synapse. The FADD-caspase-8-cFLIPL complex then associates with CBM and this leads to processing of caspase-8 and cFLIPL to p43 (119) and the dissociation of FADD from the complex (118). RIPK1 and TRAF2 (119)), both known to bind to cFLIPL p43, and the IKK complex (IKKα, IKKβ, NEMO) bind to the complex and NF-κB activation follows (118, 119). The processing of cFLIPL indicates that the catalytic activity of caspase-8 may be needed for TCR-induced NF-κB activation. However, procaspase-8 auto-processing is not essential for BMDM or T cell activation. T cells expressing the non-cleavable caspase-8 D387A mutant are activated upon TCR stimulation (71) and BMDMs depend on the catalytic activity but not auto-processing of caspase-8 for optimal TLR-induced cytokine expression (78). Caspase-8 also associates with the IKK complex upon TLR4 ligation in B cells leading to enhanced RelA phosphorylation at serine 536 and subsequent NF-κB nuclear translocation (68). Concomitantly, NF-κB translocation to the nucleus is impaired in B, T and NK cells of human patients carrying a mutation that reduces the stability of caspase-8 and makes it catalytically inactive (casp8C248T) (118, 120). These experiments are confounded by the role of caspase-8 in controlling necroptosis, however, and T cells lacking caspase-8 display normal NF-κB activation and proliferation when RIPK1 is inhibited or RIPK3 is ablated (4, 77).
CD95 and TRAILR ligation also induces pro-inflammatory cytokine expression. The DISC rapidly forms on activated CD95 and TRAILR and initiates death, but a subsequent secondary complex, dubbed the FADDosome (80), was shown to form (Figure 4). This complex does not contain the receptor and ligand, but consists of FADD-caspase-8 in association with RIPK1, cIAP1/2, TRAF2 and NEMO, that can function to activate the NF-κB, JNK and p38 pathways (79, 80, 116, 117). The interaction between FADD and caspase-8 is integral, since TRAIL-induced cytokine expression is abrogated in cells lacking FADD or caspase-8, or cells that express a caspase-8 mutant that is unable to bind FADD (80, 117). ERK and p38 activation depend on the catalytic activity of caspase-8 at least to some extend, but this activity is totally dispensable for NF-κB activation (80, 117). These studies indicate that the FADD-caspase-8 complex, distinct from its role in cell death, has a scaffold role that is required for cytokine production following TRAILR ligation.
Figure 4. Caspase-8 regulates apoptosis, gene expression and necroptosis following CD95 and TRAILR ligation.
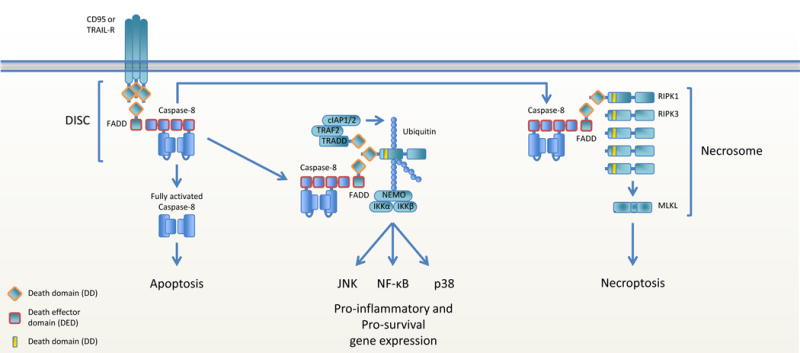
Ligation of CD95 or TRAILR induces the rapid association of FADD-caspase-8 with the intracellular domain of the receptor, forming the DISC. This can lead to caspase-8 homodimerization, activation and dissociation from the DISC to initiate apoptosis. The FADD-caspase-8 complex can also dissociate from the ligated receptor and recruit RIPK1. The additional association of TRADD, TRAF2 and cIAP1/2 leads to the formation of a complex similar to complex I in TNFR1 signaling and results in gene expression. Recruitment of RIPK1 and RIPK3 to the dissociated FADD-caspase-8 complex results in RIPK3 and MLKL activation and necroptosis.
The role of cFLIP in transcriptional regulation is less well defined, with both stimulatory and inhibitory effects on gene expression have been reported. cFLIPL was initially shown to have an inhibitory role in CD95-induced NF-κB activation (34, 121), but it was later demonstrated that it is the c-terminal domain of cFLIPL that inhibits the interaction between the DD of RIPK1 and the prodomain of caspase-8 that is required for CD95-induced NF-κB activation (122). Processing of full-length cFLIPL to p43/p22 by caspase-8 in the FADD-caspase-8-cFLIPL complex mediates NF-κB activation (123) by allowing cFLIPL p43 to interact with TRAF2 (124), RIPK1 and the IKK complex. Furthermore, the p22 fragment of processed cFLIPL was shown to be able to induce NF-κB activation by interacting with NEMO in the IKK complex (125). Depending on it’s processing by caspase-8 in the FADD-caspase-8-cFLIP complex, cFLIP thus can both negatively and positively regulate NF-κB activation.
Several studies show that caspase-8 can interfere with inflammatory signaling. Caspase-8 hampers pro-inflammatory gene expression induced by the cytosolic RNA sensor retinoic acid-inducible gene I (RIG-I) through cleavage of RIPK1, which is recruited to a complex of RIG-I and its adapter molecule mitochondrial antiviral signaling protein (MAVS) on mitochondria (126). Furthermore, caspase-8 can suppress an inflammatory pathway downstream of a cytosolic innate DNA receptor (92). Multiple cytosolic DNA sensors have been described, including DAI, cGAS, IFI16 and AIM2 (reviewed in (127)) and most cytosolic DNA sensors signal through the adapter protein STING, which is present on mitochondria. It may thus be that caspase-8 hampers signaling of STING or its downstream partners. It is now known that caspase-8 is connected to DAI through RIPK3-RIPK1-FADD, suggesting that caspase-8 might hamper DAI-induced pro-inflammatory signaling. Again, the role of caspase-8 in the control of necroptosis confuses the interpretation of these findings, and it will be important to examine the effects of ablation of MLKL in such settings. Studies in animals lacking caspase-8 and MLKL failed to show defects in the production of IFNs, although detailed studies of DNA sensing in the cells of such animals are currently lacking.
Caspase-8 in hematopoiesis
Hematopoiesis starts in the bone marrow (BM) where hematopoietic stem cells (HSC) rise to multipotent progenitor cells (MPP), which in turn produce more the committed common lymphoid progenitors (CLP) and common myeloid progenitors (CMP). All hematopoietic progenitor cells express the receptor c-kit (CD117). Several studies show that stem cell factor (SCF), the cognitive ligand for c-kit, prevents CD95-mediated cell death in hematopoietic progenitor cells (128–130). SCF inhibits CD95-mediated apoptosis by hampering caspase-3 and caspase-8 activation. SCF induces activation of Src-family kinases and these kinases were identified to be essential for both proliferation and resistance to CD95-mediated cell death of hematopoietic cells, since their inhibition induced apoptosis (130–132). This is in line with the observations that Src-kinase family members Lyn and Src phosphorylate caspase-8 at Y380 described above, but whether c-kit ligation leads to Src-mediated caspase-8 phosphorylation in hematopoietic progenitor cells remains to be examined.
Multiple studies show that FADD and caspase-8 are critical for hematopoietic progenitor cell proliferation (66, 133, 134). Casp8−/− embryos manifest a decrease in hematopoietic progenitor cells (61) and conditional caspase-8 deletion (Mx1-Cre) in bone marrow cells impairs hematopoietic progenitor cell differentiation (66). FADD and the catalytic activity of caspase-8 seem critical for cytokine-induced proliferation of hematopoietic progenitor cells (133). Caspase-8 is needed in the myeloid lineage, both at an early progenitor stage and at a more differentiated monocyte precursor stage, and in B and T lymphocyte progenitor cells (66). However, since casp8Lck-Cre mice have normal thymocyte development (135), FADD and caspase-8 are dispensable for lymphopoiesis after lineage commitment (134, 135). Monocytic bone marrow precursor cells of casp8LysM-Cre mice fail to differentiate into macrophages (66). Concomitantly, block of caspase-8 catalytic activity by zVAD-fmk blocks macrophage colony-stimulating factor (M-CSF)-induced differentiation of human peripheral blood monocytes into macrophages (136) and, concurrently, FADD overexpression has the opposite effect (137). This differentiation deficiency is specific for macrophages, as differentiation to dendritic cells (DC) or granulocytes is not affected (66, 137). M-CSF-induced monocyte to macrophage differentiation thus requires caspase-8 and is regulated by NF-κB. M-CSF induces the formation of the RIPK1-FADD-procaspase-8-cFLIPL complex, which appears to regulate NF-κB activity through cleavage of RIPK1 by procaspase-8-cFLIPL (137).
Taken together, it seems that FADD and caspase-8 regulate specific stages of hematopoiesis, especially during the early hematopoietic stages in the bone marrow, independent of their role in extrinsic apoptosis. What their exact role is remains to be elucidated, but because in the BM of conditional faddMx1 mice reduced HSC and progenitor-enriched populations are found (134), also in this setting it has to be taken into consideration that their function may be to inhibit non-apoptotic cell death (necroptosis). Alternatively, FADD-caspase-8 may function as a signal-transducing platform that regulates transcriptional processes involved in progenitor cell commitment. It will be important to examine the effects of (co-)ablation of RIPK3 or MLKL on hematopoietic progenitor cell proliferation in animal models.
Concluding remarks
Two conundrums in the field are why caspase-8 plays such a dual role in regulating cell death and why RIPK3 can be engaged by multiple receptors. It has been proposed that the necroptotic pathway evolved as a back-up mechanism to control infection when pathogens interfere with the apoptotic pathway. The dual nature of caspase-8 may lie exactly here and it has to be kept in mind that pathogen recognition by PRRs can trigger RIPK3-mediated necroptosis. Pathogenic impairment of apoptosis by targeting FADD, caspase-8 or cFLIP leads to the release of the FADD-procaspase-8-cFLIPL-mediated block on (signal II-induced) necroptosis. Conversely, targeting of RIPK3 or MLKL can trigger non-canonical ripoptosome-mediated apoptosis. Many pathogens have evolved strategies to block both apoptosis and necroptosis. For example, enteropathic Escherichia coli utilizes its encoded Non-LEE encoded effector proteins (Nle)B1 and NleF to inhibit CD95-induced cell death by targeting FADD, caspase-8 and RIPK1 (138) and EspL to cleave all four RHIM domain-containing proteins (139) to block both apoptosis and necroptosis.
Novel regulators of the apoptotic and necroptotic pathways are being identified and add to the complexity of the regulation of these processes. RIPK1 is shown to be ubiquitylated in the necrosome by LUBAC components HOIP and HOIL1 (140), in contrast with the current dogma that ubiquitylation of RIPK1 functions in a pro-survival way. RIPK3 ubiquitylation also regulates necroptosis. RIPK3 ubiquitylation at lysine 5 supports RIPK3-RIPK1 complex formation and is negatively regulated by the deubiquitylating enzyme A20 (141). Furthermore, RIPK1 and RIPK3 are targeted for lysosome-dependent degradation by the E3 ligase C-terminus HSC70 interacting protein (CHIP). Chip−/− mice are viable but die within several weeks of birth due to intestinal inflammation and necroptotic cell death and are rescued by co-ablation of ripk3 (142). Inhibition of the proteasome induces RIPK3-mediated necroptosis dependent on the RHIM domain, but without the need for caspase-8 inhibition (143). TAK1, a kinase required for IKK activation, also regulates apoptotic and necroptotic signaling by activating RIPK1 and caspase-8 (144). The regulation and balance of apoptosis and necroptosis is becoming more and more intricate with novel involved proteins and processes being regularly reported. How all these observations will shape the current model remains to be determined.
The FADD-procaspase-8 complex intricately regulates extrinsic cell death and the concomitant homeostasis of the surround tissue. Whereas engagement of apoptosis mainly results in the attraction of immune cells that ‘clean up’ the cell debris, necroptosis can have big inflammatory consequences due to the release of DAMPs, pro-inflammatory cytokines, and other alarmins. Under physiological conditions caspase-8 lies silent but once recruited to FADD it can function in a pleiotropic fashion depending on the stimulus and cell type. It remains to be determined whether caspase-8 always needs the adapter protein FADD for its various functions. The recent description of the intricate associations of FADD, caspase-8 and cFLIP allows us to think of new possibilities to study how these three proteins work, together or alone, in various biological settings including the immune response, cancer, and inflammatory disease. It has become evident that caspase-8 is integral for the initiation and regulation of death receptor-mediatedactivation of apoptosis, necroptosis and inflammation.
Acknowledgments
The work from our laboratory described herein was supported by grants from the US National Institutes of Health and from ALSAC.
Footnotes
The authors do not declare any conflict of interest.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Dillon CP, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaiser WJ, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 8.Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Thapa RJ, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe. 2016;20:674–681. doi: 10.1016/j.chom.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin J, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–210. doi: 10.1016/j.celrep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Meng L, Jin W, Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci U S A. 2015;112:11007–11012. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kers J, Leemans JC, Linkermann A. An Overview of Pathways of Regulated Necrosis in Acute Kidney Injury. Semin Nephrol. 2016;36:139–152. doi: 10.1016/j.semnephrol.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Hockendorf U, et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell. 2016;30:75–91. doi: 10.1016/j.ccell.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Hockendorf U, Yabal M, Jost PJ, Killing AML. RIPK3 leads the way. Cell Cycle. 2017;16:3–4. doi: 10.1080/15384101.2016.1232069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strilic B, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215–218. doi: 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
- 16.Kerr JF. History of the events leading to the formulation of the apoptosis concept. Toxicology. 2002;181–182:471–474. doi: 10.1016/s0300-483x(02)00457-2. [DOI] [PubMed] [Google Scholar]
- 17.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green DR. The cell’s dilemma, or the story of cell death: an entertainment in three acts. FEBS J. 2016;283:2568–2576. doi: 10.1111/febs.13658. [DOI] [PubMed] [Google Scholar]
- 19.Hughes MA, et al. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell. 2016;61:834–849. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.TM Fu, et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol Cell. 2016;64:236–250. doi: 10.1016/j.molcel.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin XM, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- 22.Jost PJ, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmke C, et al. Ligand stimulation of CD95 induces activation of Plk3 followed by phosphorylation of caspase-8. Cell Res. 2016;26:914–934. doi: 10.1038/cr.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandal R, Raab M, Matthess Y, Becker S, Knecht R, Strebhardt K. pERK 1/2 inhibit Caspase-8 induced apoptosis in cancer cells by phosphorylating it in a cell cycle specific manner. Mol Oncol. 2014;8:232–249. doi: 10.1016/j.molonc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthess Y, Raab M, Knecht R, Becker S, Strebhardt K. Sequential Cdk1 and Plk1 phosphorylation of caspase-8 triggers apoptotic cell death during mitosis. Mol Oncol. 2014;8:596–608. doi: 10.1016/j.molonc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia SH, Parodo J, Kapus A, Rotstein OD, Marshall JC. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol Chem. 2008;283:5402–5413. doi: 10.1074/jbc.M706462200. [DOI] [PubMed] [Google Scholar]
- 27.Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838–854. doi: 10.1016/j.cell.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Powley IR, Hughes MA, Cain K, MacFarlane M. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing procaspase-8 maturation and cycling within the complex. Oncogene. 2016;35:5629–5640. doi: 10.1038/onc.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senft J, Helfer B, Frisch SM. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 2007;67:11505–11509. doi: 10.1158/0008-5472.CAN-07-5755. [DOI] [PubMed] [Google Scholar]
- 30.Torres VA, Mielgo A, Barila D, Anderson DH, Stupack D. Caspase 8 promotes peripheral localization and activation of Rab5. J Biol Chem. 2008;283:36280–36289. doi: 10.1074/jbc.M805878200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irmler M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 32.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–1548. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 33.Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol. 2010;190:377–389. doi: 10.1083/jcb.201002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kavuri SM, et al. Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J Biol Chem. 2011;286:16631–16646. doi: 10.1074/jbc.M110.148585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 36.Pop C, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–457. doi: 10.1042/BJ20101738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majkut J, et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat Commun. 2014;5:3350. doi: 10.1038/ncomms4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasper DM, et al. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–288. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- 39.Yang JK, et al. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell. 2005;20:939–949. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schleich K, et al. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016;23:681–694. doi: 10.1038/cdd.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by FLIP(L) Biochem J. 2004;382:651–657. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu JW, Jeffrey PD, Shi Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci U S A. 2009;106:8169–8174. doi: 10.1073/pnas.0812453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang DW, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Micheau O, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 45.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 46.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng S, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin J, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540:124–128. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newton K, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–133. doi: 10.1038/nature20559. [DOI] [PubMed] [Google Scholar]
- 51.Karunakaran D, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224. doi: 10.1126/sciadv.1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thapa RJ, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110:E3109–3118. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orozco S, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–1521. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cook WD, et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014;21:1600–1612. doi: 10.1038/cdd.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Remijsen Q, et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014;5:e1004. doi: 10.1038/cddis.2013.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dillon CP, Tummers B, Baran K, Green DR. Developmental checkpoints guarded by regulated necrosis. Cell Mol Life Sci. 2016;73:2125–2136. doi: 10.1007/s00018-016-2188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mandal P, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newton K, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 59.Tanzer MC, et al. Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017 doi: 10.1038/cdd.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeh WC, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 61.Varfolomeev EE, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 62.Yeh WC, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 63.Sakamaki K, et al. Ex vivo whole-embryo culture of caspase-8-deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 2002;9:1196–1206. doi: 10.1038/sj.cdd.4401090. [DOI] [PubMed] [Google Scholar]
- 64.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 65.Dillon CP, et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang TB, et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 67.Krajewska M, et al. Neuronal deletion of caspase 8 protects against brain injury in mouse models of controlled cortical impact and kainic acid-induced excitotoxicity. PLoS One. 2011;6:e24341. doi: 10.1371/journal.pone.0024341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lemmers B, et al. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- 69.Beisner DR, Ch’en IL, Kolla RV, Hoffmann A, Hedrick SM. Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J Immunol. 2005;175:3469–3473. doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- 70.Salmena L, Hakem R. Caspase-8 deficiency in T cells leads to a lethal lymphoinfiltrative immune disorder. J Exp Med. 2005;202:727–732. doi: 10.1084/jem.20050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang TB, et al. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol. 2008;181:2522–2532. doi: 10.4049/jimmunol.181.4.2522. [DOI] [PubMed] [Google Scholar]
- 72.Alvarez-Diaz S, et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity. 2016;45:513–526. doi: 10.1016/j.immuni.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaiser WJ, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111:7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rickard JA, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 75.Rebsamen M, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10:916–922. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ. 2003;10:124–133. doi: 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- 77.Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Philip NH, et al. Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 2016;12:e1005910. doi: 10.1371/journal.ppat.1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hartwig T, et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol Cell. 2017;65:730–742 e735. doi: 10.1016/j.molcel.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Henry CM, Martin SJ. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol Cell. 2017;65:715–729 e715. doi: 10.1016/j.molcel.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 81.Cuda CM, et al. Conditional deletion of caspase-8 in macrophages alters macrophage activation in a RIPK-dependent manner. Arthritis Res Ther. 2015;17:291. doi: 10.1186/s13075-015-0794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuda CM, et al. Caspase-8 acts as a molecular rheostat to limit RIPK1- and MyD88-mediated dendritic cell activation. J Immunol. 2014;192:5548–5560. doi: 10.4049/jimmunol.1400122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schock SN, Young JA, He TH, Sun Y, Winoto A. Deletion of FADD in macrophages and granulocytes results in RIP3- and MyD88-dependent systemic inflammation. PLoS One. 2015;10:e0124391. doi: 10.1371/journal.pone.0124391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weinlich R, et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 2013;5:340–348. doi: 10.1016/j.celrep.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Welz PS, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 87.Gunther C, et al. Caspase-8 controls the gut response to microbial challenges by Tnf-alpha-dependent and independent pathways. Gut. 2015;64:601–610. doi: 10.1136/gutjnl-2014-307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Legarda D, et al. CYLD Proteolysis Protects Macrophages from TNF-Mediated Auto-necroptosis Induced by LPS and Licensed by Type I IFN. Cell Rep. 2016;15:2449–2461. doi: 10.1016/j.celrep.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dondelinger Y, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015;60:63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 90.Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bonnet MC, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 92.Kovalenko A, et al. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med. 2009;206:2161–2177. doi: 10.1084/jem.20090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Panayotova-Dimitrova D, et al. cFLIP regulates skin homeostasis and protects against TNF-induced keratinocyte apoptosis. Cell Rep. 2013;5:397–408. doi: 10.1016/j.celrep.2013.09.035. [DOI] [PubMed] [Google Scholar]
- 94.Dannappel M, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Raghawan AK, et al. A Disease-associated Mutant of NLRC4 Shows Enhanced Interaction with SUG1 Leading to Constitutive FADD-dependent Caspase-8 Activation and Cell Death. J Biol Chem. 2017;292:1218–1230. doi: 10.1074/jbc.M116.763979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chung H, et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 2016;23:1331–1346. doi: 10.1038/cdd.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Antonopoulos C, et al. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem. 2015;290:20167–20184. doi: 10.1074/jbc.M115.652321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pierini R, et al. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19:1709–1721. doi: 10.1038/cdd.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 100.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gringhuis SI, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012;13:246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 102.Hedl M, Abraham C. A TNFSF15 disease-risk polymorphism increases pattern-recognition receptor-induced signaling through caspase-8-induced IL-1. Proc Natl Acad Sci U S A. 2014;111:13451–13456. doi: 10.1073/pnas.1404178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bossaller L, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maelfait J, et al. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moriwaki K, Bertin J, Gough PJ, Chan FK. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol. 2015;194:1938–1944. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stammler D, et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J Immunol. 2015;195:5421–5431. doi: 10.4049/jimmunol.1501195. [DOI] [PubMed] [Google Scholar]
- 107.Gurung P, Kanneganti TD. Novel roles for caspase-8 in IL-1beta and inflammasome regulation. Am J Pathol. 2015;185:17–25. doi: 10.1016/j.ajpath.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Monie TP, Bryant CE. Caspase-8 functions as a key mediator of inflammation and pro-IL-1beta processing via both canonical and non-canonical pathways. Immunol Rev. 2015;265:181–193. doi: 10.1111/imr.12284. [DOI] [PubMed] [Google Scholar]
- 109.Vince JE, Silke J. The intersection of cell death and inflammasome activation. Cell Mol Life Sci. 2016;73:2349–2367. doi: 10.1007/s00018-016-2205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol. 2013;191:5239–5246. doi: 10.4049/jimmunol.1301581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sagulenko V, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vajjhala PR, et al. The Inflammasome Adaptor ASC Induces Procaspase-8 Death Effector Domain Filaments. J Biol Chem. 2015;290:29217–29230. doi: 10.1074/jbc.M115.687731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gurung P, et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kang S, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lawlor KE, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cullen SP, et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell. 2013;49:1034–1048. doi: 10.1016/j.molcel.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 117.Varfolomeev E, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem. 2005;280:40599–40608. doi: 10.1074/jbc.M509560200. [DOI] [PubMed] [Google Scholar]
- 118.Su H, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 119.Misra RS, et al. Caspase-8 and c-FLIPL associate in lipid rafts with NF-kappaB adaptors during T cell activation. J Biol Chem. 2007;282:19365–19374. doi: 10.1074/jbc.M610610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chun HJ, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- 121.Kreuz S, et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol. 2004;166:369–380. doi: 10.1083/jcb.200401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Matsuda I, Matsuo K, Matsushita Y, Haruna Y, Niwa M, Kataoka T. The C-terminal domain of the long form of cellular FLICE-inhibitory protein (c-FLIPL) inhibits the interaction of the caspase 8 prodomain with the receptor-interacting protein 1 (RIP1) death domain and regulates caspase 8-dependent nuclear factor kappaB (NF-kappaB) activation. J Biol Chem. 2014;289:3876–3887. doi: 10.1074/jbc.M113.506485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Koenig A, et al. The c-FLIPL cleavage product p43FLIP promotes activation of extracellular signal-regulated kinase (ERK), nuclear factor kappaB (NF-kappaB), and caspase-8 and T cell survival. J Biol Chem. 2014;289:1183–1191. doi: 10.1074/jbc.M113.506428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kataoka T, Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol. 2004;24:2627–2636. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Golks A, Brenner D, Krammer PH, Lavrik IN. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J Exp Med. 2006;203:1295–1305. doi: 10.1084/jem.20051556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rajput A, et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP-1 protein. Immunity. 2011;34:340–351. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 127.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Endo T, et al. Stem cell factor protects c-kit+ human primary erythroid cells from apoptosis. Exp Hematol. 2001;29:833–841. doi: 10.1016/s0301-472x(01)00660-9. [DOI] [PubMed] [Google Scholar]
- 129.Lee JW, Gersuk GM, Kiener PA, Beckham C, Ledbetter JA, Deeg HJ. HLA-DR-triggered inhibition of hemopoiesis involves Fas/Fas ligand interactions and is prevented by c-kit ligand. J Immunol. 1997;159:3211–3219. [PubMed] [Google Scholar]
- 130.Nishio M, et al. Stem cell factor prevents Fas-mediated apoptosis of human erythroid precursor cells with Src-family kinase dependency. Exp Hematol. 2001;29:19–29. doi: 10.1016/s0301-472x(00)00618-4. [DOI] [PubMed] [Google Scholar]
- 131.Corey SJ, Anderson SM. Src-related protein tyrosine kinases in hematopoiesis. Blood. 1999;93:1–14. [PubMed] [Google Scholar]
- 132.Linnekin D, DeBerry CS, Mou S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. 1997;272:27450–27455. doi: 10.1074/jbc.272.43.27450. [DOI] [PubMed] [Google Scholar]
- 133.Pellegrini M, et al. FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood. 2005;106:1581–1589. doi: 10.1182/blood-2005-01-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rosenberg S, Zhang H, Zhang J. FADD deficiency impairs early hematopoiesis in the bone marrow. J Immunol. 2011;186:203–213. doi: 10.4049/jimmunol.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Salmena L, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–895. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sordet O, et al. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood. 2002;100:4446–4453. doi: 10.1182/blood-2002-06-1778. [DOI] [PubMed] [Google Scholar]
- 137.Rebe C, et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood. 2007;109:1442–1450. doi: 10.1182/blood-2006-03-011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pollock GL, et al. Distinct roles of the anti-apoptotic effectors NleB and NleF from enteropathogenic Escherichia coli. Infect Immun. 2017 doi: 10.1128/IAI.01071-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pearson JS, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. 2017;2:16258. doi: 10.1038/nmicrobiol.2016.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.de Almagro MC, Goncharov T, Newton K, Vucic D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 2015;6:e1800. doi: 10.1038/cddis.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Onizawa M, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. 2015;16:618–627. doi: 10.1038/ni.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Seo J, et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat Cell Biol. 2016;18:291–302. doi: 10.1038/ncb3314. [DOI] [PubMed] [Google Scholar]
- 143.Moriwaki K, Chan FK. Regulation of RIPK3- and RHIM-dependent Necroptosis by the Proteasome. J Biol Chem. 2016;291:5948–5959. doi: 10.1074/jbc.M115.700997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guo X, Yin H, Chen Y, Li L, Li J, Liu Q. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 2016;7:e2381. doi: 10.1038/cddis.2016.294. [DOI] [PMC free article] [PubMed] [Google Scholar]