

Abstract
Influenza A virus (IAV) infections are a major cause for respiratory disease in humans, which affects all age groups and contributes substantially to global morbidity and mortality. IAV have a large natural host reservoir in avian species. However, many avian IAV strains lack adaptation to other hosts and hardly propagate in humans. While seasonal or pandemic IAV strains replicate efficiently in permissive human cells, many avian IAV cause abortive nonproductive infections in these hosts despite successful cell entry. However, the precise reasons for these differential outcomes are poorly defined. We hypothesized that the distinct course of an IAV infection with a given virus strain is determined by the differential interplay between specific host and viral factors. By using Spike-in SILAC mass spectrometry-based quantitative proteomics we characterized sets of cellular factors whose abundance is specifically up- or downregulated in the course of permissive versus nonpermissive IAV infection, respectively. This approach allowed for the definition and quantitative comparison of about 3500 proteins in human lung epithelial cells in response to seasonal or low-pathogenic avian H3N2 IAV. Many identified proteins were similarly regulated by both virus strains, but also 16 candidates with distinct changes in permissive versus nonpermissive infection were found. RNAi-mediated knockdown of these differentially regulated host factors identified Vpr binding protein (VprBP) as proviral host factor because its downregulation inhibited efficient propagation of seasonal IAV whereas overexpression increased viral replication of both seasonal and avian IAV. These results not only show that there are similar differences in the overall changes during permissive and nonpermissive influenza virus infections, but also provide a basis to evaluate VprBP as novel anti-IAV drug target.
Influenza viruses are a major cause for waves of respiratory disease, which affects all age groups and can occur repeatedly in any particular individual. These infections have a strong socio-economic impact as they are responsible for about 3 to 5 million cases of severe illness annually and about 250,000 to 500,000 deaths, worldwide (1). Furthermore, influenza pandemics that are caused by novel virus strains originating from animal host reservoirs of influenza A virus (IAV)1 as well as the ongoing highly lethal zoonotic infections with avian H5N1 and H7N9 subtype strains remain a constant threat for the human population (2). Human influenza virus was first isolated more than 80 years ago (3). Therefore, we have a fairly good understanding of its structures, genetics and principal modes of replication. In contrast, influenza virus host interactions have only partially been explored mainly because many analyses examined isolated properties such as activation of a single signaling pathway or the contribution of one gene product to virus replication (4–8). Despite the accumulated knowledge, we have also only incomplete understanding of the cellular factors that determine species specificity or the molecular basis for high virulence of certain zoonotic strains. Still, knowledge of these topics is crucial for an improved risk assessment of seasonal and emerging influenza virus strains.
Viral infection leads to perturbations of many cellular functions such as metabolism or DNA/protein synthesis and often triggers an inflammatory/immune response (9). One major question that arises from the increased detection of zoonotic inter-species transmissions in recent years (10) concerns the cellular factors that determine the success of a viral infection in a given host cell in terms of generating high levels of progeny viruses. A permissive host cell supports virus replication, gives rise to high levels of progeny viruses and will eventually enter a lytic phase resulting in the host cell's death. If the host cell is nonpermissive, the virus may be internalized, but will not efficiently produce viruses (11, 12). Only a few early studies have addressed differences and similarities between permissive and nonpermissive IAV infections by biochemical and cell biological approaches (11, 13), but systematic investigations of this topic are lacking. Previous holistic analyses of IAV focused on the cellular responses to seasonal, pandemic or mouse-adapted influenza strains at early or late time-points of infection (14–20), or identified host factors required for efficient IAV replication by genome-wide RNAi screens (21–24). Simon and colleagues, for example, detected more profound changes in the global proteome of the human lung epithelial cell line A549 due to novel H7N9 and highly pathogenic H5N1 infection compared with infection with low-pathogenic H1N1 virus at early time points post infection (14). Permissive influenza virus infection depends on the virus' ability to suppress the anti-viral host cell response, as well as on adaptations within the viral genome that determine efficient viral entry or polymerase activity. However, protein signatures within the host cell proteome typical for permissive or nonpermissive course of IAV infection were not identified, so far.
Based on metabolic SILAC labeling, we quantitatively compared the proteome signatures in human lung epithelial cells during the entire course of infection with either highly productive seasonal IAV of the H3N2 subtype or an avian H3N2 strain causing abortive infection. The majority of the 3500 quantified proteins per sample were similarly regulated by both virus strains, but also candidates with distinct changes in permissive versus nonpermissive infection were found. One example was the Vpr binding protein (VprBP), also known as DCAF-1, which was significantly downregulated selectively during nonpermissive infection. Functional validation by RNAi-mediated knockdown identified VprBP as a novel host factor of influenza virus propagation. Downregulation of VprBP inhibited efficient propagation of seasonal IAV and reduced cellular as well as viral gene expression. In contrast, overexpression of VprBP increased viral protein expression and replication of both, seasonal and avian IAV. Thus, our comprehensive proteomic screen elucidated for the first time both, similarities and differences in the host cell response to permissive and nonpermissive infection over time and identified VprBP as a promising host cell factor that might be suitable as a novel anti-IAV drug target.
EXPERIMENTAL PROCEDURES
Cells and Viruses
HEK 293T and A549 cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% (v/v) FCS, 2 mm l-glutamine, and antibiotics. MDCK type II cells were grown in minimal essential medium (MEM) supplemented with the same additives. Vero cells were cultivated in serum free OPTI PROTM medium supplemented with 1.6 mm l-glutamine. All cells were maintained at 37 °C and 5% CO2. Stocks of the influenza viruses A/Mallard/439/2004 (H3N2) (Mal) (GISAID accession numbers EPI859640-EPI859647) and A/Turkey/Italy/472/1999 (H7N1) (Turk/It) were grown in the allantoic cavities of 10-day-old embryonated chicken eggs for 3 days at 37 °C. Virus stocks of A/Panama/2007/1999 (H3N2) (Pan) (NCBI accession numbers: DQ487333-DQ487340) and A/Udorn/307/1972 (H3N2) (Udorn) (NCBI accession numbers: DQ508926-DQ508933) were propagated in MDCK type II cells for 2 days at 37 °C. Pan ΔNS1 virus was grown in Vero cells for 2 days at 37 °C. To analyze viral replication, confluent A549 or Vero cell cultures were infected at an MOI of 0.01 and incubated for 48 or 72 h at 37 °C in culture medium containing 0.2% (w/v) BSA and trypsin. Virus titers were determined on MDCK type II cells by standard plaque assay (25).
Plasmids and Antibodies
FLAG-tagged construct DCAF1-iso1 (Gen-Bank accession number NP_055518) was kindly provided by Florence Margottin-Goguet (26). The following antibodies were used in this study: anti-CrkL (Merck Millipore), anti-VprBP (Abcam), anti-Rsl1D1 (Sigma-Aldrich), anti-SAMHD1 (Sigma-Aldrich), anti-NP (Serotec), anti-M1 (Serotec), anti-NS1 (27), anti-HA (abcam), anti-PA (GeneTex), anti-Actin (Sigma-Aldrich).
Spike in SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture)-based mass spectrometry to measure host proteome response to IAV infection
A549 cells were grown in stable isotope-labeled DMEM (SILAC-DMEM, PAA) containing either light (R0K0: R = 12C6, 14N4, K = 12C6, 14N2) or heavy (R10K8: R = 13C6, 15N4, K = 13C6, 15N2) arginine and lysine (Cambridge Isotope Laboratories, Tewksbury, MA), 10% dialyzed FBS (Invitrogen), 2 mm l-glutamine and antibiotics for at least six cell doublings prior to infection. Successful incorporation of heavy amino acids was verified by MS with incorporation rates of 98.7% for lysine and 97.1% for arginine. Heavy-labeled cells were mock infected to generate heavy labeled reference proteins, while light-labeled cells were infected with Pan or Mal virus at an MOI of 3. At indicated time points cell extracts were prepared using 1% SDS lysis buffer (20 mm Tris-HCl [pH7.5], 150 mm NaCl, 1% SDS). Protein concentration of each lysate was determined by BCA protein assay (Thermo Fisher Scientific), and each light cell lysate was spiked at a 1:1 ratio with heavy-labeled reference proteins.
Experimental Design and Statistical Rationale
Each sample for mass spectrometry (MS) analysis was generated by combining two individual SILAC-labeled cell populations. In total 20 samples, two biological replicates for each time point post infection (p.i.) (0 h, 4 h, 8 h, 16 h, 24 h) with Pan or Mal virus were analyzed (Fig. 2). R0K0-labeled, mock-infected A549 cells served as internal standard control. The different time points p.i. were chosen to cover the entire course of infection. Scatter plot analyses and calculation of Pearson correlation were performed to ensure reproducibility of biological replicates. Statistical tests were performed by volcano plot analysis (t test, both sides, FDR 0.05) using Perseus (V. 1.5.0.31).
Fig. 2.

Schematic representation of “spike-in SILAC” approach. R10K8-labeled A549 cells were mock infected generating the “SILAC standard.” R0K0 cells were infected with either Pan or Mal virus (MOI 2) for the indicated time points. Same amounts of proteins were mixed and prepared for measurement by a Q Exactive™ Orbitrap (Thermo Scientific™) equipped with a Nano-LC. The ratio of peak intensities from the heavy and light peptides display the abundance of the corresponding proteins. MS data were acquired using Xcalibur software and searched using the Andromeda algorithm in Maxquant 1.5.1.2 against the Homo Sapiens (ncbi) database (89,601 entries). Spiking of light cell populations with “SILAC standard” enables indirect comparison of light samples; n = 2.
Sample Preparation
After lysis samples were reduced by adding DTT to a final concentration of 0.1 m and incubation for 5 min at 95 °C. Sulfhydryl groups were alkylated by adding iodoacetamide to a final concentration of 0.25 m and incubation for 20 min in the dark at room temperature. Proteins were precipitated according to Wessel and Fluegge (28), resuspended in 6 m urea/2 M thiourea and digested into peptides with C-terminal lysine or arginine using Lys-C (3 h) and Trypsin (overnight, diluted 4× with 50 mm ABC). Enzyme activity was quenched by acidification of the samples with trifluoroacetic acid. The peptides were desalted with C18 Stage Tips (29) prior to nanoLC-MS/MS analysis.
LC-MS/MS Analysis
Peptides were separated on a 2-m monolithic column (MonoCap C18 High Resolution 2000 (GL Sciences Einhoven, The Netherlands), 100 μm i.d. × 2000 mm at a flow rate of 300 nL/min with a 5 to 45% acetonitrile gradient on an EASY-nLC II system (Thermo Fisher Scientific) with 480 min gradient. A Q Exactive plus instrument (Thermo Fisher Scientific) was operated in the data dependent mode with a full scan in the Orbitrap followed by top 10 MS/MS scans using higher-energy collision dissociation (HCD). The full scans were performed with a resolution of 70,000, a target value of 3 × 106 ions and a maximum injection time of 20 ms. The MS/MS scans were performed with a 17,500 resolution, a 1 × 106 target value and a 60 ms maximum injection time.
Peptide and Protein Identification
Peak lists were generated from raw data files using MaxQuant software version 1.5.1.2 according to the standard workflow (30), which corrects for systematic inaccuracies of measured peptide masses and corresponding retention times of extracted peptides (30, 31). Proteins were identified by searching against the recent Uniprot human database (2013–4; 89,601 protein sequences) as well as 12 viral protein sequences of Pan and Mal virus. Proteins were identified using the integrated Andromeda search engine and the following search parameters: carbamidomethylation of cysteine as a fixed modification, oxidation of methionine, acetylation of protein N terminus, deamidation of asparagine and glutamine (32) and appropriate SILAC labels as variable modifications; tryptic digestion with a maximum of two missed cleavages; a peptide precursor mass tolerance of 20 ppm and a fragment mass tolerance of 4.5 ppm. Known contaminants were included for protein identification (2014–11; contaminant.fasta, MaxQuant). Intensity threshold was set to 500 (default setting). The decoy database search option was enabled and all peptide spectrum matches (PSMs) and proteins were filtered with a maximum false discovery rate (FDR) of 0.01 by using the MaxQuant target-decoy approach. Annotated spectra can be visualized using the ProteinProspector MS-viewer (33) with the following search key: jt7cvdk2q3. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD005825.
Protein Quantification
The MaxQuant output file proteinGroups.txt was used to determine relative protein abundance by means of Perseus software version 1.5.0.31. This table contains information on the identified proteins, sequence coverage, number of unique peptides and their H/L ratios in the processed raw-files. Each single row contains the group of proteins that could be reconstructed from a set of peptides. Protein quantification was performed with at least 2 unique and razor (i.e. peptides shared by different proteins of a group) peptides by means of the measured peak intensities. The relative abundance of a protein was derived from its H/L ratio in the differently labeled cell populations. An H/L ratio of 0.5 for a given protein was considered to signal increased abundance in the infected R0K0 cell population. Scatter plot analyses and calculation of Pearson correlation were performed to ensure reproducibility of biological replicates. Thereby, PWP1 protein was excluded as outlier in the Pan_4h data set, because of strong variation between the two replicates (Rep1: −80.50; Rep2: −0.15). Because data sets have normal distribution, significant differences between data points were determined by t test using volcano plot analysis. A p value < 0.05 was considered significant. Principle component analysis (PCA) (cutoff method: Benjamini-Hochberg; FDR 0.05) and IDAS 2.1 (34) hierarchical clustering (Euclidian distance) were used to display structure and variance of significant proteins having valid values in each sample. Classification of proteins with significant log2(fold change) >1 or <-1 upon infection with Pan or Mal virus was performed using PANTHER (Protein analysis through evolutionary relationships) Classification System Version 10.0 according to their gene ontology terms “biological process” (GOBP).
Immunofluorescence Microscopy
IAV infected A549 cells (MOI 1 and 0.5, respectively, as indicated) were used for indirect immunofluorescence staining (35). Viral NP and cellular SAMHD1 proteins were detected with primary antibodies as well as suitable secondary anti-mouse IgG conjugated with Alexa Fluor-594 dye and anti-rabbit IgG conjugated with Alexa Fluor-488 dye (Life Technologies GmbH, Darmstadt, Germany), respectively. Images of cells were captured using Axio Observer.D1 (Zeiss, Jena, Germany) or LSM510 Meta confocal laser scanning microscope with a C-Apochromat 63/1.2 water objective lens (Zeiss) and a pinhole setting of 1, as indicated. Data were analyzed and processed with the Zeiss ZEN 2012 (blue edition) and Adobe Photoshop CS6 software packages. For quantification of SAMHD1 positive and negative cells, values below a threshold of 52 were classified as SAMHD1 negative by means of the “Histo” function of Zeiss ZEN 2012 software package.
Immunoblot Analysis
To analyze protein expression levels, A549 cells were mock-treated or infected with indicated viruses at an MOI of 2 or 3, respectively. Cell extracts were prepared at indicated time points in 1% SDS lysis buffer and subjected to SDS-PAGE and immunoblot analysis. Proteins were detected with specific primary and suitable secondary antibodies together with an enhanced chemiluminescence protocol (Thermo Fisher Scientific).
Minigenome Reporter Assay
Cellular and viral gene expression was analyzed by a luciferase reporter minigenome assay (36). Briefly, 293T cells were transiently transfected with the pHW-Pan expression plasmids coding for PB1, PA, PB2 (50 ng of each), and NP (100 ng) as well as pPol-NS-Luc (50 ng) encoding a firefly luciferase cDNA (Luc) of negative polarity flanked by the noncoding regions of the viral NS segment. The constitutive expression vector pTK-RL (5 ng) (Promega, Mannheim, Germany) was used to detect changes in cellular gene expression levels by means of constitutive renilla luciferase (RL) expression. The effect of VprBP on cellular and viral gene expression was analyzed by co-transfection of the expression construct (500 ng) or 50 pmol Flexitube siRNA (Qiagen, Hilden, Germany), respectively.
IFN-β ELISA
Cell culture supernatant samples from infected cell cultures (MOI 2, 24 h p.i.) were stored at −80 °C, thawed at room temperature and IFN-β concentrations were measured using a human IFN-β ELISA Kit (Fujirebio Inc./Invitrogen, Karlsruhe, Germany).
SiRNA-mediated Knockdown Experiments
All siRNAs (Silencer® Select) were purchased from Thermo Fisher. A549 cells were reversely transfected with siRNAs in 96-well plates at final siRNA concentrations of 20 nm. For this, 8000 cells were added to the siRNAs, which were complexed previously by adding HiPerfect (1.4 μl per well, Qiagen). The cells were incubated for an additional 48 h at 37 °C, 5% CO2. To assess the cytotoxicity of siRNAs, a WST-1 assay (Roche) was performed 48 h post transfection by adding WST-1 reagent to the cells, followed by incubation at 37 °C for 1.5 h. Absorbance was measured at 460 nm and at the reference wavelength 590 nm. Nontargeting siRNA and siPLK1 (target sequence: CACCATATGAATTGTACAGAA) were used as positive and negative control, respectively. The sequences of the siRNAs used can be obtained upon request.
Virus replication in siRNA-transfected cells was assessed 48 h post-transfection by washing the cells with PBS, followed by inoculation with either Pan or Mal viruses at MOI 0.02. After 1 h, infection medium (DMEM supplemented with 0.2% bovine serum albumin, 4 mm l-glutamine and antibiotics) containing 1 μg/ml TPCK-treated trypsin (Sigma-Aldrich) was added and cells kept under normal culture conditions for 39 h (Pan virus) or 48 h (Mal virus). To quantify the virus load upon siRNA knockdown, the supernatants were harvested and added to MDCK cells, seeded the day before at 10,000/well. To this end, MDCK cells were washed with PBS and inoculated with undiluted (Mal virus) or diluted (1:100, Pan virus) supernatants for 1 h. Virus was removed and MDCK cells were cultured for 6 h with infection medium. Cells were stained for nuclei (Hoechst, Sigma) and the viral NP using a monoclonal NP-specific antibody, and the infection rate (i.e. the percentage of NP positive cells) was determined as described previously (21). The final read-out of the assay was the virus titer, which was extrapolated from the infection rate using the following formula: multiplicity of infection = -ln(1-infection rate). Raw data were further analyzed using the R package cellHTS2 (37). Data normalization was performed by scaling the raw data relative to an inactive (nontargeting) and an active (siNP, target sequence: AAGGAUCUUAUUUCUUCGGAG) siRNA. Finally, a Z-score transformation was applied, and based on the calculated Z-scores, hits with Z-score >1.5 or <−1.5 were selected.
Statistical Analysis of Non-MS Data
Data were presented as mean and standard deviation. Using GraphPad Prism 5 software the Mann-Whitney U test was performed to delineate significant differences (p < 0.05 (*), p < 0.01 (**), p < 0.001 (***)) between data points if not indicated otherwise.
RESULTS
Replication of Low-pathogenic Avian H3N2 Influenza Virus is Restricted in Human Lung Epithelial Cells
Despite successful replication in birds, many avian IAV are not adapted to propagate efficiently in humans as they lack the necessary adaptation (38). This restriction is likely a multifactorial process, but can even be observed in cultured human cells that are largely nonpermissive for avian IAV (12, 39, 40). To gain novel insights into the cellular factors determining these differences, we chose to compare two viruses of the H3N2 subtype, seasonal A/Panama/2007/1999 (Pan) and the low-pathogenic avian A/Mallard/439/2004 (Mal) virus. Both viruses entered human A549 lung epithelial cells with similar efficiency, as demonstrated by immunofluorescence detection of the viral NP in infected cells (Fig. 1A). Immunoblot analysis showed that both viruses expressed comparable levels of viral proteins in A549 cells, although there were differences in the relative accumulation of the M1 and NS1 gene products (Fig. 1B). Interestingly, only the seasonal Pan virus, but not the avian Mal virus replicated efficiently in A549 cells to final titers of 107 PFU/ml in a multicyclic setting (Fig. 1C). In contrast, Mal virus titers were significantly decreased by 3 log steps at 72 h p.i. demonstrating that human lung epithelial cells are highly permissive for Pan but not for the avian Mal virus (Fig. 1C). In general, a permissive infection indicates that a virus avoids or suppresses activation of innate cellular defenses such as the antiviral type I interferon (IFN) system (41). To analyze whether the avian Mal virus was unable to suppress the innate IFN response, we determined the IFN-β levels in the supernatant of infected cells. However, Mal virus infection did not trigger increased IFN-β secretion whereas the seasonal Pan virus provoked only modest IFN-β levels most likely due to efficient suppression of IFN expression (35) (Fig. 1D). As a control, the recombinant influenza Pan ΔNS1 mutant virus was used to determine strong influenza virus-dependent IFN-β induction in the absence of antagonistic NS1 protein (27). Growth curve analyses in Vero cells that do not express intact IFN-α/β genes showed similar differences in the replication of the Pan and Mal viruses suggesting that the poor propagation of the avian virus in human cells is not caused by the type I IFN response (Fig. 1E).
Fig. 1.

Replication of low-pathogenic avian H3N2 influenza virus is restricted in human lung epithelial cells despite successful internalization. A, A549 cells were infected with Pan or Mal virus at MOI of 1 for 4 h. Cells were fixed, permeabilized, stained with DAPI and specific primary antibody against NP. Images were representative for five independent experiments. Scale bar: 10 μm. B, A549 cells were infected with Pan or Mal virus at an MOI of 2 for indicated time points. Cell lysates were prepared and subjected to immunoblot analyses; n = 3. C, A549 cells were infected with indicated viruses at an MOI of 0.01. Supernatants were collected at indicated time points and titrated; n = 3. D, A549 cells were infected with indicated viruses at MOI of 2 for 24 h. Supernatants were analyzed for IFN-β expression using IFN-β ELISA (Fujirebio), n = 5. E, VeroSF cells were infected with indicated viruses at an MOI of 0.01. Supernatants were collected at indicated time points and titrated; n = 3. C–E: Data represent means + S.E. *, p < 0.05; **, p < 0.01 (Mann-Whitney U test).
Human Proteome Response to Permissive versus Nonpermissive Influenza A Virus Infection
Here, we used a global approach to systematically analyze sets of cellular factors whose abundance is specifically up- or downregulated during the course of permissive versus nonpermissive IAV infection using the two H3N2 strains. Based on metabolic SILAC labeling of two cell populations that enables the relative quantification and comparison of cellular proteomes (42), we generated a mock-infected cell population labeled with heavy arginine and lysine. Lysate of this cell population was used as internal standard and spiked into each sample of a second cell population being cultivated in medium with light arginine and lysine. The cultures of light-labeled cells were infected with seasonal or avian H3N2 viruses for 0, 4, 8, 16, or 24 h, respectively, in two experimental repeats (Fig. 2). After the MS measurement, SILAC-spiking enabled the direct comparison of all samples in a quantitative manner (Fig. 2).
Data evaluation was performed with MaxQuant, which calculates protein heavy to light ratios as the median of all peptide ratios assigned to a distinct protein or protein group (30, 31). Only unique and razor peptides were used for protein quantification with a minimum ratio count of two and a FDR of 1%. The MS measurement resulted in identification of 4000 protein groups and quantification of about 3500 protein groups per sample (Fig. 3A, supplemental Table S1). We did not attempt to unambiguously identify which member of a protein group was present; for clarity, “protein groups” will be referred to as “proteins” hereafter. Accession numbers of all proteins that were combined into a protein group are provided in each of the supplementary Tables S1–S4. Peptide sequences used for protein identification are listed in supplemental Table S2, whereas supplemental Table S3 indicates those peptides unique for a protein group and a specific protein, respectively. Quantification values were specified as log2 (light to heavy ratios) (L/H) to display changes in protein abundance upon infection. Reproducibility of both biological replicates was verified by scatter plot analysis and calculation of Pearson correlation (Fig. 3B, supplemental Fig. S1). Depending on time point and virus strain, the Pearson correlation ranged from 0.67 to 0.76 (Mal) and 0.28 to 0.81 (Pan) (Fig. 3B, supplemental Fig. S1). Statistically significant differences of protein expression levels in two different samples were calculated by t test using a cutoff of 1 and −1 (log2(L/H)), respectively, and a p value < 0.05, meaning that protein abundances changed at least 2-fold upon infection (supplemental Table S4). Interestingly, analysis of data structure and variance using PCA as well as IDAS 2.1 hierarchical cluster analysis (34) revealed that the 20 generated samples assembled according to the infection period rather than the used viruses (Fig. 3C–3D), indicating that similar cellular processes occur during both, permissive and nonpermissive, IAV infection. Fig. 3C–3D clearly shows increasing variation between the samples during the course of viral infection.
Fig. 3.
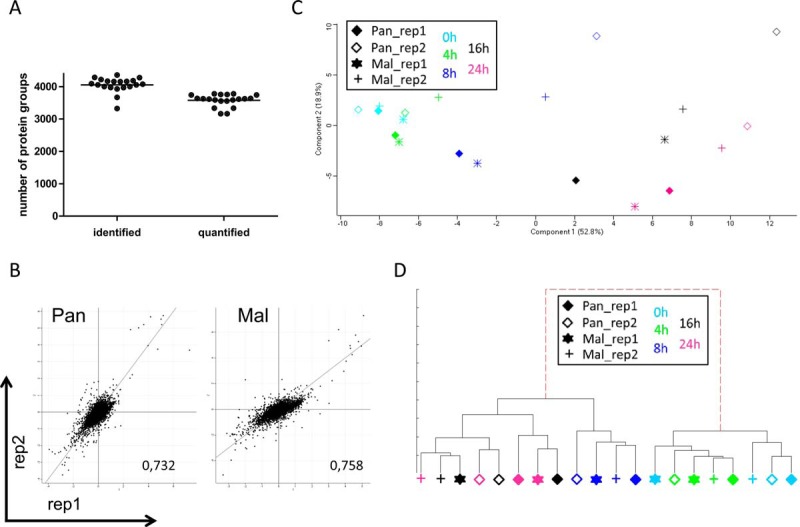
MS data evaluation revealed quantification of about 3,500 proteins per sample with good correlation between replicates and time points. A, MS data were acquired using Xcalibur software and searched using the Andromeda algorithm in Maxquant 1.5.1.2 against the Homo Sapiens (ncbi) database (2014–4; 89,601 protein sequences). Evaluation in Perseus 1.5.0.31 identified about 4000 protein groups per sample of which 3500 were used for protein quantification. B, Scatter plot analysis of biological replicates at 24 h p.i. Displayed are log2(L/H) values of Pan (left panel) and Mal (right panel) infected cells and the corresponding Pearson correlation; dots represent protein groups. C, Principle component analysis using Perseus 1.5.0.31 and 944 significant proteins (p < 0.05) having valid values in each sample to determine structure and variance of MS data. D, Hierarchical cluster analysis by means of IDAS 2.1 (34) using 85 proteins having significant log2(fold changes) >1 or <−1 and valid values in each sample. C–D: Each symbol represents one sample: filled diamond - Pan infected cells, replicate 1; diamond - Pan infected cells, replicate 2; star - Mal infected cells, replicate 1; cross - Mal infected cells, replicate 2; light blue - 0 h p.i.; green - 4 h p.i.; dark blue - 8 h p.i.; black - 16 h p.i.; pink - 24 h p.i.
Changes in protein abundance at early and late time points post infection in comparison to the 0 h time point were displayed by volcano plot analyses (Fig. 4A–4B). Proteins with p values > -log(0.05) are displayed in blue, while red dots represent significantly regulated cellular proteins beyond the cutoff of 1 and −1, respectively. These evaluations revealed that changes in protein expression levels increase during viral infection independent of the virus strain used (Fig. 4A–4B). Thereby, most proteins were significantly downregulated compared with 0 h p.i., which likely reflects the global host cell shut off known for IAV infected cells (43). In total, we identified 135 significantly regulated proteins upon Pan or Mal infection which were involved in comparable biological processes, such as metabolic process, biological regulation, biological adhesion, response to stimulus and apoptotic process (Fig. 4C, supplemental Table S4). Additionally, we generated a hierarchical cluster analysis as well as corresponding profile plots (Fig. 5). This scrutiny demonstrated that the vast majority of the significantly regulated cellular proteins were downregulated upon infection with both viruses (e.g. Cluster c), but with variations in the extent of regulation (Fig. 5). Cluster d, for instance, shows proteins with strong downregulation upon Pan infection, whereas Mal infection decreased protein levels only moderately (Fig. 5B).
Fig. 4.

Statistical evaluation of proteins with significant log2(fold changes) >1 or <−1 upon Mal or Pan IAV infection. A, B, Volcano plot analyses were used to display the changes in protein abundance at early and late time points post infection with Mal (A) and Pan (B) virus, respectively, in comparison to the 0 h time point: differences between L/H ratio of two samples were plotted against -log(p value); blue: log2(fold change) <1 or >−1, p value<0.05; red: log2(fold change) >1 or <−1, p value<0.05; t test, n = 2. C, Classification of proteins with significant log2(fold change) >1 or <−1 upon infection with Pan or Mal virus according to their gene ontology terms “biological process” (GOBP) using PANTHER (Protein analysis through evolutionary relationships) Classification System Version 10.0.
Fig. 5.

Cluster analysis of protein expression pattern during the course of IAV infection. A, Hierarchical cluster analysis based on protein expression pattern over time generated by means of Perseus software version 1.5.0.31. Used were the Z-scores of 135 identified proteins with significant log2(fold changes) >1 or <−1 (p value< 0.05) and a Euclidean distance threshold of 1.9. B, Profile plots a-f illustrate the expression patterns of distinct clusters in A.
The focus of our study was the identification of protein signatures discriminating between permissive and nonpermissive IAV infections. Therefore, we directly compared protein expression levels in Pan and Mal infected cells at each single time point post infection and discovered 16 proteins with at least 2-fold difference (p < 0.05) (Fig. 6A, Table I). These proteins are described to be functionally involved in biological processes including apoptosis, immune system process, developmental process, cellular and metabolic process (supplemental Fig. S2). Differences in protein expression levels detected by the MS measurement were evaluated and confirmed for CrkL, Rsl1D1 (both more abundant in Mal infected cells compared with Pan infected cells (Fig. 6C)) and VrpBP (decreased expression levels in Mal infected cells compared with Pan infected cells (Fig. 6C)) for the Pan and Mal viruses (Fig. 6B–6C), as well as for additional human and avian strains by immunoblot analysis (Fig. 6B–6D). Infection with human A/Udorn/307/1972 (Udorn) virus, which also replicates efficiently in A549 cells (44), did not affect protein expression levels of VprBP and CrkL, but strongly decreased abundance of Rsl1D1 at 24 h p.i. similar to Pan virus (Fig. 6B, 6D). In contrast, the influenza A/Turkey/Italy/472/1999 (Turk/It) virus, another representative of low-pathogenic avian IAV (45) led to decreased abundance of VprBP and increased CrkL protein levels at 24 h p.i., whereas Rsl1D1 expression was not affected (Fig. 6B–6D). Taken together, our approach identified significant changes in the host cell proteome in response to IAV infection. Most of the 135 proteins were similarly regulated by both virus strains, but also 16 candidates with distinct changes in permissive versus nonpermissive infection were discovered.
Fig. 6.

Identification of protein signatures during permissive versus nonpermissive IAV infection. A, Volcano plot analyses display the changes in protein abundance due to permissive (Pan) versus nonpermissive (Mal) IAV infection at the indicated time points: differences between L/H ratio of two samples were plotted against -log(p value); blue: log2(fold change) <1 or >−1, p value<0.05; red: log2(fold change) >1 or <−1, p value<0.05; t test, n = 2. B, A549 cells were infected with indicated viruses at an MOI of 2 (VprBP) or 3 (CrkL, Rsl1D1), respectively. At 4 h and 24 h p.i. cell lysates were prepared in 1% SDS buffer. Protein expression levels were analyzed by SDS-PAGE and immunoblot. Data from one representative experiment of at least three are shown. C, Band intensities of VprBP, Rsl1D1 and CrkL proteins at the 24 h time point post Mal virus or Turk/It virus infection were quantified, normalized to actin levels and displayed in relation to the protein expression levels at 24 h Pan virus infection; n ≥ 3. D, Band intensities of VprBP, Rsl1D1 and CrkL proteins at 24 h p.i. with the indicated viruses were quantified, normalized to actin levels and displayed in relation to the protein expression levels at 4 h p.i.; n ≥ 3.
Table I. Differentially regulated proteins in Pan vs. Mal infected A549 cells. Listed are proteins with significant differences upon infection with Pan and Mal virus at indicated time points calculated by volcano plot analysis (log2(fold change) >1 or < −1, p value <0.05; t-test, n = 2). Values represent log2(L/H ratio) of the samples, (+) indicates p value <0.05.
| Gene names | 4 h p.i. |
8 h p.i. |
16 h p.i. |
24 h p.i. |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pan | Mal | sign. | Pan | Mal | sign. | Pan | Mal | sign. | Pan | Mal | sign. | |
| ASPSCR1 | 1.054 | 1.517 | 0.725 | 0.810 | −0.118 | 0.979 | −0.041 | 1.135 | + | |||
| CHTOP | −0.035 | −0.054 | 0.035 | 0.025 | −1.311 | −0.310 | −1.266 | −0.169 | + | |||
| CRKL | 0.513 | 0.794 | 0.280 | 1.069 | 0.741 | 1.602 | 0.350 | 1.582 | + | |||
| CYP1B1 | −0.207 | −0.574 | −0.400 | −0.380 | −0.223 | −0.442 | −0.527 | −1.769 | + | |||
| GRB2 | 0.221 | 0.381 | −0.214 | 0.437 | −0.794 | 0.674 | −0.678 | 0.729 | + | |||
| HERC2 | 0.151 | −0.870 | + | −0.623 | −0.345 | −0.742 | −0.608 | −0.620 | −0.850 | |||
| KIAA0101 | 0.522 | 0.064 | −0.821 | −0.526 | −2.082 | NaN | −2.614 | −1.366 | + | |||
| MUC5AC | −1.313 | −1.524 | −1.138 | −1.066 | −1.756 | −0.411 | −1.509 | −0.481 | + | |||
| NRCAM | −0.688 | −0.506 | −1.047 | −2.062 | + | −2.624 | −3.085 | −3.031 | −2.502 | |||
| POLDIP3 | −0.095 | 0.102 | −0.138 | −0.106 | −1.437 | 0.083 | + | −2.934 | −0.139 | + | ||
| RSL1D1 | −0.042 | 0.029 | 0.155 | 0.055 | −0.428 | 0.261 | −0.989 | 0.095 | + | |||
| TMBIM6 | −0.201 | 0.214 | −0.118 | 0.165 | −0.845 | 0.324 | −1.732 | −0.012 | + | |||
| TRAF2 | 0.120 | 0.680 | −1.191 | 0.658 | + | −0.794 | NaN | −0.101 | NaN | |||
| TRIM47 | 0.628 | 0.704 | −0.276 | 0.545 | −1.385 | 0.001 | −1.805 | 0.248 | + | |||
| USP36 | −0.506 | −0.358 | −0.475 | 0.308 | −0.666 | −0.514 | −1.787 | −0.357 | + | |||
| VPRBP | −0.013 | −0.397 | 0.003 | −0.545 | −0.181 | −1.284 | −0.493 | −2.149 | + | |||
Functional Validation of Significant Protein Signatures by siRNA-mediated Knockdown
To examine the functional importance of the 16 differentially regulated proteins in IAV infection, we conducted a limited RNAi-mediated screen by transfection of three different siRNAs per gene separately in A549 cells for 48 h. Knockdown of target genes did not affect overall cell viability (supplemental Fig. S3). After siRNA-mediated knockdown, cells were infected with Pan or Mal virus and viral titers were determined at 39 h or 48 h p.i., respectively. Genes were classified as affecting virus replication, if the median of the three different siRNAs as well as at least two out of three values fell outside the cutoff of 1.5 (Fig. 7). The described MS measurements had detected Trim47, CrkL and Grb2 to be more abundant in Mal infected cells at 24 h p.i., whereas VprBP expression was decreased in Mal infected cells compared with Pan (Fig. 6, Table I). The RNAi-mediated screen revealed that knockdown of Trim47 resulted in decreased viral titers of both Pan and Mal virus compared with negative control, while siRNA-mediated knockdown of CrkL, Grb2 and VprBP only affected Pan virus replication (Fig. 7). Based on these findings we suggest that well-balanced expression levels of CrkL, Grb2 and Trim47 are important for their correct function during permissive IAV infection. Moreover, we hypothesized that VprBP enhances viral replication during permissive IAV infection.
Fig. 7.

Functional validation of significant protein signatures by siRNA-mediated knockdown. Three siRNAs per gene (Thermo Fisher) were used to transfect A549 cells for 48 h. Then, cells were infected with Pan or Mal virus (MOI 0.02) for 39 and 48 h, respectively. Supernatants were titrated by infection of MDCKII cells for 7 h and NP staining. Normalized values were displayed as Z-score in relation to NP (positive control) and Allstars siRNA (negative control); n = 3. Cut-off: ± 1.5.
VprBP Supports IAV Replication
VprBP is known to bind and stabilize HIV Vpr/Vpx proteins, inducing cell cycle arrest in G2 and to direct antiviral host factors such as SAM domain and HD domain-containing protein 1 (SAMHD1) to proteasomal degradation (26, 46–50). Furthermore, VprBP can increase replication and protein expression of hepatitis C virus (HCV) (51). We first performed a viral mini-genome luciferase assay to analyze if VprBP knockdown has an influence on IAV gene expression (36). Transfection of Pan vRNP genes PB1, PB2, PA and NP together with four siRNAs directed against VprBP mRNA strongly reduced VprBP levels and decreased viral gene expression significantly to about 62% compared with negative control siRNA (Fig. 8A). Although cell viability was not affected by VprBP knockdown (supplemental Fig. S3), cellular gene expression as measured by Renilla luciferase activity expressed from a constitutive promoter was impaired to nearly the same extent (∼70%), suggesting a rather general role of VprBP as positive regulator for cellular and viral gene expression (Fig. 8A). This conclusion was supported by the findings that VprBP overexpression significantly elevated cellular gene expression (Fig. 8B) and increased viral titers for both the Pan and Mal viruses (Fig. 8C), accompanied by enhanced accumulation of viral proteins compared with vector-transfected cells (Fig. 8C). These findings suggest that VprBP positively regulates cellular as well as viral gene expression, thereby supporting IAV replication.
Fig. 8.

VprBP supports IAV replication. A, B, 293T cells were transiently transfected with the pHW expression plasmids coding for PB1, PA, PB2, NP and pPol-A/NS-Luc encoding a firefly luciferase cDNA of negative polarity flanked by the noncoding regions of the viral A/NS segment. The effect of VprBP was analyzed by co-transfection of 50 pmol Flexitube siRNA directed against VprBP (Qiagen, Hilden, Germany) (A) and by co-transfection of the expression construct (500 ng) (B). Luciferase activity of cells transfected with AllStars Control siRNA or empty plasmid was set to 100%. Cells not expressing PB1 were used as negative control. Data are means + S.E. (A: n = 3; B, n = 6; biological duplicates). p values were calculated using Wilcoxon signed rank test. C, A549 cells were transfected with 500 ng VprBP expression plasmid for 24 h. Then, cells were infected with Pan or Mal virus at an MOI of 1. At 48 h p.i. supernatants were collected and cell lysates were prepared in 1% SDS buffer. Titers were determined using standard plaque assay and protein content of infected cells was analyzed by immunoblot. Cells transfected with an empty plasmid were set to 100%. p values were determined by Mann Whitney U test. Data are means + S.E. (n = 4; biological duplicates). A–C: *, p < 0.05; **, p < 0.01; ***, p < 0.001. D: A549 cells were infected with Pan or Mal virus at an MOI of 0.5. At 4 h and 16 h p.i. cells were fixed, permeabilized, stained with specific primary antibody against NP, SAMHD1 and with DAPI. White arrows indicate NP positive cells with low SAMHD1 expression levels, yellow arrows mark infected cells with high levels of SAMHD1. Images were representative for three independent experiments. Scale bar: 10 μm. Infected cells (white NP signal) were classified according to their SAMHD1 expression levels. Cells with SAMHD1 signal below the threshold of 52 (green signal) were classified as SAMHD1 negative, while cells with SAMHD1 expression levels above the threshold of 52 were ranked as SAMHD1 positive (red signal). Data are means + S.E. (n = 3).
VprBP has been described to facilitate proteasomal degradation of SAMHD1, a known restriction factor for several human pathogens including hepatitis B virus, herpes simplex virus, vaccinia virus and retroviruses (52–58). To investigate the hypothesis that SAMHD1 is regulated in influenza virus infection we compared its expression in human cells infected with the Pan or Mal viruses, respectively, at 4 h and 16 h p.i. in a microscopy approach. Interestingly, this analysis showed strong reduction of SAMHD1 signals in Pan, but not Mal infected cells at the late time point (Fig. 8D, supplemental Fig. S4). Less than 10% of the Pan infected cells were SAMHD1 positive, whereas approx. 60% of the Mal infected cells showed an SAMHD1 signal above the threshold (Fig. 8D). Based on this observation we hypothesize that SAMHD1 is subject to VprBP-dependent downregulation during permissive influenza virus infection.
In summary, we identified protein signatures of permissive and nonpermissive IAV infection in human lung epithelial cells. Moreover, our data suggest that VprBP is beneficial for viral gene expression and supports viral propagation in permissive IAV infection.
DISCUSSION
Cellular factors that determine the course of avian influenza virus infection in human cells are poorly defined. Previous quantitative proteome studies of IAV infections focused mainly on responses in epithelial cells or macrophages to viruses with different pathogenicity in humans, such as representatives of the H1N1 and zoonotic H5N1 or H7N9 subtypes, respectively (14–20). In contrast, our study aimed to gain a systematic understanding of the differences in the proteomes during the courses of permissive and nonpermissive IAV infections and to identify differentially regulated key factors, as they may be part of the poorly defined species barrier that prevents regular transmission of avian strains to humans (59). Our experimental setup investigated the responses to permissive versus nonpermissive IAV infection using two H3N2 subtype viruses throughout the course of infection up to 24 h p.i. Several polymorphisms in the viral genomes have been associated with host range restrictions of IAV (10). For instance, a lysine present at position 627 within the polymerase basic protein 2 (PB2) in most human isolates confers high-level polymerase activity in human cells compared with the glutamic acid found at this position in the Mal virus and many other avian IAV isolates (60, 61). The HA protein of the Mal strain also has the characteristic Gln226/Gly228 amino acid constellation that confers preferential binding to α2,3-linked sialic acid receptor determinants, whereas human strains such as Pan preferentially recognize α2,6-linked sialic acids (62). However, similar efficiencies of virus uptake (Fig. 1A) and cumulative protein production demonstrated that other determinants play an important role in restricting Mal virus replication in human cells (Fig. 1B). We initially eliminated the possibility that the avian Mal virus was restricted by a strong innate IFN response in human cells as Mal infected cells secreted only minimal IFN-β levels (Fig. 1D) and Mal virus was similarly attenuated on Vero cells that lack functional type I IFN genes (63) (Fig. 1E).
We used a spike-in SILAC approach that enabled the comparison of different states of infection as well as the proteome responses to different viruses. For each infection state, we quantified about 3500 proteins with a strong Pearson correlation of 0.7 to 0.8 between the two replicates (Fig. 2A–2B, supplemental Fig. S1). Exceptions were the eight and 16-hour time points of Pan infected cells. Possibly because of high dynamics of this infection period (Fig. 1C) and a consequent nonsynchronous infection, cell populations had only a weak correlation coefficient of 0.3 (supplemental Fig. S1). However, despite the low Pearson correlation, we identified similar numbers of significantly changed proteins at these time points compared with Mal virus infected cells (Supplemental Table S4). To enable the identification of the most interesting proteins regulated during IAV infection, we applied stringent filters to the entire data set. Only proteins with a minimum fold change of two and a p value < 0.05 were considered for further analyses. Interestingly, the 20 generated samples clustered according to the infection period rather than the used viruses (Fig. 3C–3D), indicating that many changes in cellular processes occur during both, permissive and nonpermissive IAV infection. In general, we detected more profound changes in the proteome response at later time points of infection (Fig. 3C–3D, 4A–4B). Although Pan virus infection resulted in 1, 20, 33, and 72 proteins with significantly changed abundance, Mal virus infection led to altered expression levels of 2, 14, 45, and 58 cellular factors at 4 h, 8 h, 16 h, and 24 h p.i., respectively (supplemental Table S4). Those proteins were not only involved in similar biological processes, e.g. biological regulation, biological adhesion, response to stimulus and apoptotic process (Fig. 4C), but also similarly regulated independent of the virus strain, as revealed by hierarchical cluster analysis of protein expression levels (Fig. 5). These observations indicate that the distinct outcomes of IAV infections in human cells might be determined by only a few differentially regulated proteins.
To identify protein signatures characteristic for permissive IAV infection in human cells, we compared protein expression levels in Pan and Mal infected cells at each single time point post infection. The 16 proteins identified by at least 2-fold differential expression (p < 0.05) were described to be functionally involved in biological processes such as apoptosis, immune system process, developmental process, cellular and metabolic process (Fig. 6A, Fig. S2, Table I). By immunoblot analysis, differences in protein expression levels were confirmed for CrkL, Rsl1D1 and VrpBP (Fig. 6B-C). Furthermore, we demonstrated that the effect on protein abundance was not only restricted to Pan and Mal viruses, but matched also for the permissive seasonal Udorn strain and the low-pathogenic avian influenza Turk/It virus (Fig. 6B, 6D). Thus, we confirmed distinct general effects on CrkL, Rsl1D1 and VprBP protein levels during permissive and nonpermissive infections, respectively.
To further narrow down which of the 16 differentially regulated factors influence replication efficiency in human cells, we performed a limited siRNA screen involving targeted siRNA-mediated knockdown in A549 cells. Thereby, it was found that knockdown of VprBP, Trim47, CrkL and Grb2 reduced virus replication of Pan, whereas low Mal virus propagation was even further diminished only by Trim47 knockdown (Fig. 7). Trim47 protein levels were downregulated upon Pan virus infection, but not affected by Mal virus infection (Table I). Trim47 has a similar structure as Trim25 known to be involved in Rig-I activation upon IAV infection (64). Similar to Trim25, Trim47 can activate the IFN-β promoter, as well as nuclear factor κB (NFκB)- and IFN-dependent gene expression (65). However, its role in influenza virus replication has not been investigated, so far. Our results indicate that a well-balanced regulation of Trim47 expression levels might be important in infected human cells, as Trim47 is downregulated during permissive IAV infection (Table I), whereas its knockdown negatively affects IAV replication (Fig. 7).
Grb2 is a cellular adapter protein that provides a critical link between cell surface growth factor receptors and the Raf signaling pathway (66). Inhibition of Raf signaling results in nuclear retention of influenza virus ribonucleoprotein complexes, impaired function of the viral nuclear-export protein and concomitant inhibition of virus production (67). Hemagglutinin accumulation of seasonal H3N2 viruses at the cell surface was reported to trigger Raf signaling (68). Therefore, Grb2 might be a possible link for HA induced stimulation of Raf signaling in Pan virus infected cells, while Grb2 knockdown had only minor effects for Mal virus replication (Fig. 7). Recently, it was reported that Grb2 is incorporated in virions of human cytomegalovirus (HCMV), acts as proviral factor for HCMV replication and is upregulated during infection (69). In case of IAV, Grb2 is slightly downregulated during permissive infection and faintly upregulated during nonpermissive infection (Table I). While isoform 1 is a positive regulator for Raf signaling, isoform 2 acts as a dominant negative protein over Grb2 and suppresses proliferative signals (70). However, in our experimental setup it is not possible to discriminate the different isoforms of Grb2, because isoform 2 only lacks 40 amino acids of isoform 1 (70). Hence, further mechanistical investigation is needed to define how Grb2 isoforms affect IAV infection.
The SH3 domain protein CrkL belongs together with CrkI and CrkII to the family of Crk adaptor proteins, which are ubiquitously expressed and function as scaffolds for the formation of protein complexes involved in numerous cellular signaling processes (71, 72). Previously, it was reported that interactions of the NS1 proteins of avian IAV and of influenza B virus with Crk adaptor proteins inhibit influenza virus-mediated activation of c-jun N-terminal kinase (JNK), which results in suppressed virus-induced apoptosis, whereas reduction of cellular Crk protein levels led to slightly decreased influenza virus propagation (73, 74). CrkL knockdown was also reported to significantly suppress cell viability and phosphorylation of ERK, which is a downstream kinase of the Raf signaling pathway (75). Reduced Pan virus replication due to knockdown of CrkL seems plausible in light of the finding that inhibition of Raf signaling decreases IAV propagation (76) (Fig. 7). Mal virus infection led to upregulation of CrkL expression levels (Table I), but the effects of this increased protein abundance on Raf signaling and avian IAV infection remain to be determined. Knockdown of both CrkL and Grb2 indicate a role for the Raf signaling cascade on permissive influenza virus infection (Table I), but these findings have to be further investigated to clarify the roles of Grb2 and CrkL upstream of the Raf signaling cascade in IAV infected human cells.
VprBP has been described to interact with HIV Vpr and to support HIV as well as HCV replication (26, 46–51). In this study, we observed that VprBP has a stimulatory effect on cellular gene expression, and supports IAV protein accumulation and replication (Fig. 8). SiRNA-mediated reduction of VprBP levels resulted in decreased viral titers of Pan virus (Fig. 7), which is in confirmation of the finding from a previous study that detected impaired propagation of influenza WSN/33 virus (H1N1) upon VprBP knockdown (21, 24). In line with our own results, VprBP protein expression was also not changed in permissive infections with H7N9, H5N1 and seasonal H1N1 subtype influenza viruses (14). In contrast, VprBP levels were reduced in Mal virus infected cells and thereby compromised in their ability to promote virus replication (Table I, Fig. 6), raising the question for its mode of action. Our data support a model in which the positive effects of VprBP for cellular and viral gene expression in infected cells are at least partially exerted via degradation of its binding partner SAMHD1, which is known to deplete cellular dNTP levels and to degrade viral RNA (56, 58, 77). Indeed, we detected decreased SAMHD1 expression levels during permissive Pan infection (Fig. 8D), in analogy to HIV and SIV infected cells (78). IAV with its single stranded RNA genome that is transcribed in viral mRNA might be susceptible to the RNase activity of unaltered SAMHD1 expression during nonpermissive infection, but this hypothesis remains to be investigated in future analyses.
In conclusion, by the use of SILAC-based quantitative proteomics, our study identified several interesting factors that characterize or determine the outcome of an IAV infection in human cells. Further analyses are warranted to elucidate the precise roles of these cellular proteins for the success of influenza virus infections in human host cells. Especially, the exact role of proviral acting VprBP for influenza virus propagation as well as its suitability as anti-influenza drug target should be unraveled.
DATA AVAILABILITY
Annotated spectra can be visualized using the ProteinProspector MS-viewer (33) with the following search key: jt7cvdk2q3. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD005825.
Supplementary Material
Acknowledgments
We thank Florence Margottin-Goguet (Paris, France) for kindly providing us the plasmid DCAF1-iso1. We also thank Gudrun Heins as well as Isabella Gravenstein for their excellent technical support, Koshi Imami for setting up the nLC system using monolithic columns and Peter R. Baker for uploading the data set to MS-viewer.
Footnotes
Author contributions: A.S., A.H., T.F.M., M.S., and T.W. designed research; A.S., K.P., K.E., B.B., S.S., M.B., M.L., and A.K. performed research; A.S., K.H., A.K., and T.W. analyzed data; A.S. and T.W. wrote the paper.
* This work was supported by the German Ministry of Education and Research (Virosign (grant 0316180D)) and the German Research Foundation (grant WO 554/4–1).
 This article contains supplemental material.
This article contains supplemental material.
Conflict of interest: There is no conflict of interests within this article.
1 The abbreviations used are:
- IAV
- influenza A virus(es)
- ABC
- Ammonium bicarbonate
- BCA
- bicinchoninic acid
- DAPI
- 4′,6-Diamidin-2-phenylindol
- DMEM
- Dulbecco modified Eagle medium
- DNA
- deoxyribonucleic acid
- FBS
- fetal bovine serum
- FDR
- false discovery rate
- h
- hours
- H
- hemagglutinin
- H/L
- heavy/light ratio
- HCMV
- human cytomegalovirus
- HCV
- hepatitis C virus
- HIV
- human immunodeficiency virus
- L/H
- light/heavy ratio
- IFN
- Interferon
- JNK
- c-jun N-terminal kinase
- K
- lysine
- Mal
- A/Mallard/439/2004 (H3N2) virus
- MEM
- minimal essential medium
- MOI
- multiplicity of infection
- N
- neuraminidase
- NCBI
- National Center for Biotechnology Information
- NS1
- nonstructural protein 1
- NP
- Nucleoprotein
- PA
- polymerase acidic protein
- Pan
- A/Panama/2007/1999 (H3N2) virus
- PB1
- polymerase basic protein 1
- PB2
- polymerase basic protein 2
- PCA
- principle component analysis
- PEP
- posterior error probability
- PFU
- plaque forming units
- p.i.
- post infection
- ppm
- parts per million
- PSM
- peptide spectrum match
- R.
- arginine
- RNA
- ribonucleic acid
- SAMHD1
- SAM domain and HD domain-containing protein 1
- SH3
- Src homology 3
- SILAC
- stable isotope labelling by amino acids in cell culture
- siRNA
- small interfering RNA
- Turk/It
- A/Turkey/Italy/472/1999 (H7N1) virus
- Udorn
- A/Udorn/307/1972 (H3N2) virus
- VprBP
- Vpr binding protein
- vRNP
- viral ribonucleoprotein
- WST
- water soluble tetrazolium.
REFERENCES
- 1. WHO. (2014) Influenza (Seasonal), Fact sheet N°211 [Google Scholar]
- 2. Donatelli I., Castrucci M. R., De Marco M. A., Delogu M., and Webster R. G. (2016) Human-animal interface: The case for influenza interspecies transmission. Adv. Exp. Med. Biol. 1–17 [DOI] [PubMed] [Google Scholar]
- 3. Smith W., Andrewes C. H., and Laidlaw P. P. (1933) Originally published as Volume 2, Issue 5732A Virus Obtained From Influenza Patients. Lancet 222, 66–68 [Google Scholar]
- 4. Zheng W., Cao S., Chen C., Li J., Zhang S., Jiang J., Niu Y., Fan W., Li Y., Bi Y., Sun L., Gao G. F., and Liu W. (2016) Threonine 80 phosphorylation of nonstructural protein NS1 regulates the replication of influenza A virus by reducing the binding affinity with RIG-I. Cell Microbiol. 19, e12643. [DOI] [PubMed] [Google Scholar]
- 5. Hu J., Mo Y., Gao Z., Wang X., Gu M., Liang Y., Cheng X., Hu S., Liu W., Liu H., Chen S., Liu X., Peng D., and Liu X. (2016) PA-X-associated early alleviation of the acute lung injury contributes to the attenuation of a highly pathogenic H5N1 avian influenza virus in mice. Med. Microbiol. Immunol. 205, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gotz V., Magar L., Dornfeld D., Giese S., Pohlmann A., Hoper D., Kong B. W., Jans D. A., Beer M., Haller O., and Schwemmle M. (2016) Influenza A viruses escape from MxA restriction at the expense of efficient nuclear vRNP import. Sci. Rep. 6, 23138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weber-Gerlach M., and Weber F. (2016) Standing on three legs: antiviral activities of RIG-I against influenza viruses. Curr. Opinion Immunol. 42, 71–75 [DOI] [PubMed] [Google Scholar]
- 8. Dudek S. E., Nitzsche K., Ludwig S., and Ehrhardt C. (2016) Influenza A viruses suppress cyclooxygenase-2 expression by affecting its mRNA stability. Sci. Rep. 6, 27275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hale B. G., Albrecht R. A., and Garcia-Sastre A. (2010) Innate immune evasion strategies of influenza viruses. Future Microbiol. 5, 23–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reperant L. A., Kuiken T., and Osterhaus A. D. (2012) Adaptive pathways of zoonotic influenza viruses: from exposure to establishment in humans. Vaccine 30, 4419–4434 [DOI] [PubMed] [Google Scholar]
- 11. Lau S. C., and Scholtissek C. (1995) Abortive infection of Vero cells by an influenza A virus (FPV). Virology 212, 225–231 [DOI] [PubMed] [Google Scholar]
- 12. Portincasa P., Conti G., and Chezzi C. (1990) Abortive replication of influenza A viruses in HeLa 229 cells. Virus Res. 18, 29–40 [DOI] [PubMed] [Google Scholar]
- 13. Gujuluva C. N., Kundu A., Murti K. G., and Nayak D. P. (1994) Abortive replication of influenza virus A/WSN/33 in HeLa229 cells: defective viral entry and budding processes. Virology 204, 491–505 [DOI] [PubMed] [Google Scholar]
- 14. Simon P. F., McCorrister S., Hu P., Chong P., Silaghi A., Westmacott G., Coombs K. M., and Kobasa D. (2015) Highly Pathogenic H5N1 and Novel H7N9 Influenza a viruses induce more profound proteomic host responses than seasonal and pandemic H1N1 strains. J. Proteome Res. 14, 4511–4523 [DOI] [PubMed] [Google Scholar]
- 15. Liu L., Zhou J., Wang Y., Mason R. J., Funk C. J., and Du Y. (2012) Proteome alterations in primary human alveolar macrophages in response to influenza A virus infection. J Proteome Res 11, 4091–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kroeker A. L., Ezzati P., Halayko A. J., and Coombs K. M. (2012) Response of primary human airway epithelial cells to influenza infection: a quantitative proteomic study. J. Proteome Res. 11, 4132–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu X., Wang S., Yu Y., Zhang J., Sun Z., Yan Y., and Zhou J. (2013) Subcellular proteomic analysis of human host cells infected with H3N2 swine influenza virus. Proteomics 13, 3309–3326 [DOI] [PubMed] [Google Scholar]
- 18. Ding X., Lu J., Yu R., Wang X., Wang T., Dong F., Peng B., Wu W., Liu H., Geng Y., Zhang R., Ma H., Cheng J., Yu M., and Fang S. (2016) Preliminary proteomic analysis of A549 cells infected with avian influenza virus H7N9 and influenza A virus H1N1. PLoS ONE 11, e0156017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soderholm S., Fu Y., Gaelings L., Belanov S., Yetukuri L., Berlinkov M., Cheltsov A. V., Anders S., Aittokallio T., Nyman T. A., Matikainen S., and Kainov D. E. (2016) Multi-omics studies towards novel modulators of influenza A virus-host interaction. Viruses 8, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soderholm S., Kainov D. E., Ohman T., Denisova O. V., Schepens B., Kulesskiy E., Imanishi S. Y., Corthals G., Hintsanen P., Aittokallio T., Saelens X., Matikainen S., and Nyman T. A. (2016) Phosphoproteomics to characterize host response during influenza A virus infection of human macrophages. Mol. Cell. Proteomics 15, 3203–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karlas A., Machuy N., Shin Y., Pleissner K. P., Artarini A., Heuer D., Becker D., Khalil H., Ogilvie L. A., Hess S., Maurer A. P., Muller E., Wolff T., Rudel T., and Meyer T. F. (2010) Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463, 818–822 [DOI] [PubMed] [Google Scholar]
- 22. Watanabe T., Kawakami E., Shoemaker J. E., Lopes T. J., Matsuoka Y., Tomita Y., Kozuka-Hata H., Gorai T., Kuwahara T., Takeda E., Nagata A., Takano R., Kiso M., Yamashita M., Sakai-Tagawa Y., Katsura H., Nonaka N., Fujii H., Fujii K., Sugita Y., Noda T., Goto H., Fukuyama S., Watanabe S., Neumann G., Oyama M., Kitano H., and Kawaoka Y. (2014) Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe 16, 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konig R., and Stertz S. (2015) Recent strategies and progress in identifying host factors involved in virus replication. Curr. Opin. Microbiol. 26, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tripathi S., Pohl M. O., Zhou Y., Rodriguez-Frandsen A., Wang G., Stein D. A., Moulton H. M., DeJesus P., Che J., Mulder L. C., Yanguez E., Andenmatten D., Pache L., Manicassamy B., Albrecht R. A., Gonzalez M. G., Nguyen Q., Brass A., Elledge S., White M., Shapira S., Hacohen N., Karlas A., Meyer T. F., Shales M., Gatorano A., Johnson J. R., Jang G., Johnson T., Verschueren E., Sanders D., Krogan N., Shaw M., Konig R., Stertz S., Garcia-Sastre A., and Chanda S. K. (2015) Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zielecki F., Semmler I., Kalthoff D., Voss D., Mauel S., Gruber A. D., Beer M., and Wolff T. (2010) Virulence determinants of avian H5N1 influenza A virus in mammalian and avian hosts: role of the C-terminal ESEV motif in the viral NS1 protein. J. Virol. 84, 10708–10718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Rouzic E., Morel M., Ayinde D., Belaidouni N., Letienne J., Transy C., and Margottin-Goguet F. (2008) Assembly with the Cul4A-DDB1DCAF1 ubiquitin ligase protects HIV-1 Vpr from proteasomal degradation. J. Biol. Chem. 283, 21686–21692 [DOI] [PubMed] [Google Scholar]
- 27. Matthaei M., Budt M., and Wolff T. (2013) Highly pathogenic H5N1 influenza A virus strains provoke heterogeneous IFN-α/β responses that distinctively affect viral propagation in human cells. PLoS ONE 8, e56659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wessel D., and Flugge U. I. (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 [DOI] [PubMed] [Google Scholar]
- 29. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 30. Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J. V., and Mann M. (2009) A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 [DOI] [PubMed] [Google Scholar]
- 31. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 32. Bogdanow B. Z., H; Selbach M. (2016) Systematic Errors in Peptide and Protein Identification and Quantification by Modified Peptides Mol. Cell. Proteomics 15, 2791–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baker P. R., and Chalkley R. J. (2014) MS-viewer: a web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bondarenko I., Treiger B., Van Grieken R., and Van Espen P. (1996) IDAS: a Windows based software package for cluster analysis. Spectrochimica Acta Part B: Atomic Spectroscopy 51, 441–456 [Google Scholar]
- 35. von Recum-Knepper J., Sadewasser A., Weinheimer V. K., and Wolff T. (2015) Fluorescence-activated cell sorting-based analysis reveals an asymmetric induction of interferon-stimulated genes in response to seasonal influenza A virus. J. Virol. 89, 6982–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wunderlich K., Juozapaitis M., Manz B., Mayer D., Gotz V., Zohner A., Wolff T., Schwemmle M., and Martin A. (2010) Limited compatibility of polymerase subunit interactions in influenza A and B viruses. J. Biol. Chem. 285, 16704–16712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boutros M., Bras L. P., and Huber W. (2006) Analysis of cell-based RNAi screens. Genome Biol. 7, R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beare A. S., and Webster R. G. (1991) Replication of avian influenza viruses in humans. Arch. Virol. 119, 37–42 [DOI] [PubMed] [Google Scholar]
- 39. Hayman A., Comely S., Lackenby A., Hartgroves L. C., Goodbourn S., McCauley J. W., and Barclay W. S. (2007) NS1 proteins of avian influenza A viruses can act as antagonists of the human alpha/beta interferon response. J. Virol. 81, 2318–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lohmeyer J., Talens L. T., and Klenk H. D. (1979) Biosynthesis of the influenza virus envelope in abortive infection. J. Gen. Virol. 42, 73–88 [DOI] [PubMed] [Google Scholar]
- 41. Randall R. E., and Goodbourn S. (2008) Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89, 1–47 [DOI] [PubMed] [Google Scholar]
- 42. Meissner F., and Mann M. (2014) Quantitative shotgun proteomics: considerations for a high-quality workflow in immunology. Nat. Immunol. 15, 112–117 [DOI] [PubMed] [Google Scholar]
- 43. Rivas H. G., Schmaling S. K., and Gaglia M. M. (2016) Shutoff of host gene expression in influenza A virus and herpesviruses: similar mechanisms and common themes. Viruses 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Min J.-Y., and Krug R. M. (2006) The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knepper J., Schierhorn K. L., Becher A., Budt M., Tonnies M., Bauer T. T., Schneider P., Neudecker J., Ruckert J. C., Gruber A. D., Suttorp N., Schweiger B., Hippenstiel S., Hocke A. C., and Wolff T. (2013) The novel human influenza A(H7N9) virus is naturally adapted to efficient growth in human lung tissue. MBio 4, e00601–e00613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cassiday P. A., DePaula-Silva A. B., Chumley J., Ward J., Barker E., and Planelles V. (2015) Understanding the molecular manipulation of DCAF1 by the lentiviral accessory proteins Vpr and Vpx. Virology 476, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Le Rouzic E., Belaidouni N., Estrabaud E., Morel M., Rain J. C., Transy C., and Margottin-Goguet F. (2007) HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6, 182–188 [DOI] [PubMed] [Google Scholar]
- 48. Romani B., Baygloo N. S., Hamidi-Fard M., Aghasadeghi M. R., and Allahbakhshi E. (2016) HIV-1 Vpr Protein Induces Proteasomal Degradation of Chromatin-associated Class I HDACs to Overcome Latent Infection of Macrophages. J. Biol. Chem. 291, 2696–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Romani B., Shaykh Baygloo N., Aghasadeghi M. R., and Allahbakhshi E. (2015) HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J. Biol. Chem. 290, 17380–17389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou D., Wang Y., Tokunaga K., Huang F., Sun B., and Yang R. (2015) The HIV-1 accessory protein Vpr induces the degradation of the anti-HIV-1 agent APOBEC3G through a VprBP-mediated proteasomal pathway. Virus Res. 195, 25–34 [DOI] [PubMed] [Google Scholar]
- 51. Yan Y., Huang F., Yuan T., Sun B., and Yang R. (2016) HIV-1 Vpr increases HCV replication through VprBP in cell culture. Virus Res. 223, 153–60 [DOI] [PubMed] [Google Scholar]
- 52. Berger A., Sommer A. F., Zwarg J., Hamdorf M., Welzel K., Esly N., Panitz S., Reuter A., Ramos I., Jatiani A., Mulder L. C., Fernandez-Sesma A., Rutsch F., Simon V., Konig R., and Flory E. (2011) SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 7, e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Z., Zhu M., Pan X., Zhu Y., Yan H., Jiang T., Shen Y., Dong X., Zheng N., Lu J., Ying S., and Shen Y. (2014) Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun. 450, 1462–1468 [DOI] [PubMed] [Google Scholar]
- 54. Hollenbaugh J. A., Gee P., Baker J., Daly M. B., Amie S. M., Tate J., Kasai N., Kanemura Y., Kim D. H., Ward B. M., Koyanagi Y., and Kim B. (2013) Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog. 9, e1003481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hrecka K., Hao C., Gierszewska M., Swanson S. K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M. P., and Skowronski J. (2011) Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim E. T., White T. E., Brandariz-Nunez A., Diaz-Griffero F., and Weitzman M. D. (2013) SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J. Virol. 87, 12949–12956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Segeral E., Yatim A., Emiliani S., Schwartz O., and Benkirane M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sommer A. F. R., Rivière L., Qu B., Schott K., Riess M., Ni Y., Shepard C., Schnellbächer E., Finkernagel M., Himmelsbach K., Welzel K., Kettern N., Donnerhak C., Münk C., Flory E., Liese J., Kim B., Urban S., and König R. (2016) Restrictive influence of SAMHD1 on Hepatitis B Virus life cycle. Sci. Reports 6, 26616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Short K. R., Richard M., Verhagen J. H., van Riel D., Schrauwen E. J., van den Brand J. M., Manz B., Bodewes R., and Herfst S. (2015) One health, multiple challenges: The inter-species transmission of influenza A virus. One Health 1, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kirui J., Bucci M. D., Poole D. S., and Mehle A. (2014) Conserved features of the PB2 627 domain impact influenza virus polymerase function and replication. J. Virol. 88, 5977–5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Steel J., Lowen A. C., Mubareka S., and Palese P. (2009) Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog. 5, e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stencel-Baerenwald J. E., Reiss K., Reiter D. M., Stehle T., and Dermody T. S. (2014) The sweet spot: defining virus-sialic acid interactions. Nat. Rev. Micro. 12, 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Diaz M. O., Ziemin S., Le Beau M. M., Pitha P., Smith S. D., Chilcote R. R., and Rowley J. D. (1988) Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. U.S.A. 85, 5259–5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gack M. U., Shin Y. C., Joo C. H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., and Jung J. U. (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 [DOI] [PubMed] [Google Scholar]
- 65. Versteeg G. A., Rajsbaum R., Sanchez-Aparicio M. T., Maestre A. M., Valdiviezo J., Shi M., Inn K. S., Fernandez-Sesma A., Jung J., and Garcia-Sastre A. (2013) The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38, 384–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lowenstein E. J., Daly R. J., Batzer A. G., Li W., Margolis B., Lammers R., Ullrich A., Skolnik E. Y., Bar-Sagi D., and Schlessinger J. (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442 [DOI] [PubMed] [Google Scholar]
- 67. Pleschka S., Wolff T., Ehrhardt C., Hobom G., Planz O., Rapp U. R., and Ludwig S. (2001) Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3, 301–305 [DOI] [PubMed] [Google Scholar]
- 68. Marjuki H., Yen H. L., Franks J., Webster R. G., Pleschka S., and Hoffmann E. (2007) Higher polymerase activity of a human influenza virus enhances activation of the hemagglutinin-induced Raf/MEK/ERK signal cascade. Virol. J. 4, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cavignac Y., Lieber D., Laib Sampaio K., Madlung J., Lamkemeyer T., Jahn G., Nordheim A., and Sinzger C. (2015) The cellular proteins Grb2 and DDX3 are increased upon human cytomegalovirus infection and act in a proviral fashion. PLoS ONE 10, e0131614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fath I., Schweighoffer F., Rey I., Multon M. C., Boiziau J., Duchesne M., and Tocque B. (1994) Cloning of a Grb2 isoform with apoptotic properties. Science 264, 971–974 [DOI] [PubMed] [Google Scholar]
- 71. Liu D. (2014) The adaptor protein Crk in immune response. Immunol. Cell Biol. 92, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Birge R. B., Kalodimos C., Inagaki F., and Tanaka S. (2009) Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun. Signal. 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hrincius E. R., Wixler V., Wolff T., Wagner R., Ludwig S., and Ehrhardt C. (2010) CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell Microbiol. 12, 831–843 [DOI] [PubMed] [Google Scholar]
- 74. Sadewasser A., Saenger S., Paki K., Schwecke T., and Wolff T. (2016) Disruption of Src homology 3-binding motif within non-structural protein 1 of influenza B virus unexpectedly enhances viral replication in human cells. J. Gen. Virol. 97, 2856–2867 [DOI] [PubMed] [Google Scholar]
- 75. Miyazaki M., Nishihara H., Hasegawa H., Tashiro M., Wang L., Kimura T., Tanino M., Tsuda M., and Tanaka S. (2013) NS1-binding protein abrogates the elevation of cell viability by the influenza A virus NS1 protein in association with CRKL. Biochem. Biophys. Res. Commun. 441, 953–957 [DOI] [PubMed] [Google Scholar]
- 76. Droebner K., Pleschka S., Ludwig S., and Planz O. (2011) Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res. 92, 195–203 [DOI] [PubMed] [Google Scholar]
- 77. Choi J., Ryoo J., Oh C., Hwang S., and Ahn K. (2015) SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology 12, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ahn J., Hao C., Yan J., DeLucia M., Mehrens J., Wang C., Gronenborn A. M., and Skowronski J. (2012) HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 287, 12550–12558 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Annotated spectra can be visualized using the ProteinProspector MS-viewer (33) with the following search key: jt7cvdk2q3. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD005825.