

Abstract
Mass spectrometry allows quantification of tens of thousands of phosphorylation sites from minute amounts of cellular material. Despite this wealth of information, our understanding of phosphorylation-based signaling is limited, in part because it is not possible to deconvolute substrate phosphorylation that is directly mediated by a particular kinase versus phosphorylation that is mediated by downstream kinases. Here, we describe a framework for assignment of direct in vivo kinase substrates using a combination of selective chemical inhibition, quantitative phosphoproteomics, and machine learning techniques. Our workflow allows classification of phosphorylation events following inhibition of an analog-sensitive kinase into kinase-independent effects of the inhibitor, direct effects on cognate substrates, and indirect effects mediated by downstream kinases or phosphatases. We applied this method to identify many direct targets of Cdc28 and Snf1 kinases in the budding yeast Saccharomyces cerevisiae. Global phosphoproteome analysis of acute time-series demonstrated that dephosphorylation of direct kinase substrates occurs more rapidly compared with indirect substrates, both after inhibitor treatment and under a physiological nutrient shift in wt cells. Mutagenesis experiments revealed a high proportion of functionally relevant phosphorylation sites on Snf1 targets. For example, Snf1 itself was inhibited through autophosphorylation on Ser391 and new phosphosites were discovered that modulate the activity of the Reg1 regulatory subunit of the Glc7 phosphatase and the Gal83 β-subunit of SNF1 complex. This methodology applies to any kinase for which a functional analog sensitive version can be constructed to facilitate the dissection of the global phosphorylation network.
Virtually all cellular behavior is influenced either directly or indirectly by protein phosphorylation (1). The extensive phosphorylation-based signaling network of the cell (2, 3) is governed by a highly interconnected network of protein kinases, phosphatases, and phospho-dependent recognition modules. A central problem in modern biology and drug discovery is therefore to identify the direct in vivo targets of protein kinases. Recent advances in mass spectrometry (MS) have allowed the profiling of 10,000s of phosphosites from few milligrams of cell lysate (4, 5). However, the diversity of kinases in the cell and the organization of kinases into complex regulatory networks precludes the effective deconvolution of large-scale phosphoproteomic datasets into defined signaling hierarchies (6). A particular difficulty arises in isolating the effects of a specific kinase from downstream kinases or phosphatases that form a local signaling network. This situation is further complicated by the propensity of kinases to form a globally connected network (7). Thus, even highly specific perturbation of a given kinase may trigger many ancillary events not associated with the primary set of target substrates. In addition, many phosphorylation events may arise from nonspecific kinase activity, which may contribute substantial nonfunctional noise to the phosphoproteome (8, 9).
Protein kinases must correctly discriminate their cognate substrates and phosphorylation sites within these substrates from a nonspecific background of ∼700,000 potentially phosphorylatable residues in the proteome. There are multiple mechanisms responsible for this exquisite specificity, including the structure of the catalytic site, local and distal interactions between the kinase and substrate, the formation of complexes with scaffolding and adaptor proteins that spatially regulate the kinase, systems-level competition between substrates, and error-correction mechanisms (10). The responsibility for the recognition of substrates by protein kinases appears to be distributed among a large number of independent, imperfect specificity mechanisms, many of which are not fully recapitulated by in vitro assays. Conversely, the interpretation of in vivo data is confounded by the complexity and cross-connections of the global signaling network, in which perturbation of a single kinase can alter phosphorylation not only of cognate direct substrates but also through indirect effects on many other substrates that may reside in distant regions of the network. Due to these difficulties, the identification of direct kinase substrates requires both in vivo and in vitro studies with multiple levels of cross-validation that is time consuming and often not comprehensive (11, 12).
Genetic approaches enable the manipulation of kinase activities in vivo through either simple gene deletion or the targeted introduction of inactivating or activating point mutations. However, the interpretation of the effects of such mutations on the phosphoproteome is heavily confounded by compensating effects in the network (13). In a more elegant approach, it is possible to engineer many protein kinases to accept cell-permeable bulky ATP analogs that are not able to bind or inhibit wild-type (wt) kinases (14). These engineered kinases, which are referred to as analog-sensitive (as) alleles provide several key advantages over other genetic techniques for kinase inactivation. In particular, the chemical inhibitor can be applied on short timescales to avoid compensation mechanisms that would otherwise buffer the loss of the target kinase. Both tyrosine and serine/threonine kinases are amenable to this approach, which has been used for selective inhibition of mutant Src-family kinases (14), Abl-family kinases (15), mitogen-activated kinases (16), cyclin-dependent kinases (CDKs) (16), p21-activated kinases (17), and Ca2+/calmodulin-dependent kinases. Despite the power of the as-allele approach, the functional integrity of the mutant kinase and the actual target specificity of the inhibitor are crucial considerations. It has been reported that the catalytic activity of some protein kinases is affected by as mutations (15) and unintended off-target effects of the inhibitor on other enzymes (18).
Given that kinase inactivation will shift the equilibrium of cognate substrates to the dephosphorylated state, due to the action of constitutive phosphatases, a combination of quantitative phosphoproteomics and selective chemical inhibition can in principle be used to discover in vivo kinase targets on a large-scale. This approach has for example been used to identify Cdc28 substrates in S. cerevisiae (19). Based on criteria of at least twofold dephosphorylation in response to kinase inhibition and the presence of a full CDK consensus motif, a total of 547 phosphorylation sites have been assigned to 308 candidate Cdc28 substrates (19). Unexpectedly, a significant proportion of the quantified sites on these substrates were up-regulated following Cdc28 inhibition, which suggests the influence of feedback loops that may activate additional kinases and/or inactivate phosphatases. Notably, some of these up-regulated phosphosites were even located within the CDK consensus sequence. Additional criteria are thus needed to resolve the spectrum of direct in vivo substrates for any given kinase.
To address the above issues with kinase substrate identification, we set out to develop a method for unbiased large-scale identification of direct in vivo kinase substrates based on a combination of specific kinase inhibition, large-scale phosphoproteomics, and machine learning approaches (Fig. 1a). The point of departure for our strategy is the selective inhibition of an as-form of a kinase of interest by the chemical inhibitor 1NM-PP1 (20). Upon treatment with the inhibitor, we expect changes in phosphorylation in response to the inhibitor itself (kinase independent) and changes that result from inhibition of the as kinase (kinase dependent). The latter may arise from either dephosphorylation of direct substrates or phosphorylation/dephosphorylation of indirect substrates. The latter substrates would represent downstream targets that are not directly phosphorylated by a kinase, for instance when the targeted kinase modulates the activity of another kinase or phosphatase, which in turn affects the phosphorylation of other proteins. To discriminate between kinase-independent, direct, and indirect substrate classes, we developed and validated a machine learning strategy that uses the support vector machine (SVM) (21) and ideas from positive and unlabeled learning (22). A proof of principle using the well-studied Cdc28 kinase in yeast (23) allowed us to identify many novel substrates and obtain a deeper understanding of kinase–substrate relationships. To demonstrate applicability to other less-characterized kinases, we then use our approach to identify direct targets of Snf1, a kinase that is required for transcription of glucose-repressed genes, thermotolerance, sporulation, and peroxisome biogenesis in yeast (24, 25). Our integrated approach should enable the identification of direct substrates for any kinase for which a functional as allele can be engineered. The systematic application of this method should help reveal the true structure of the global phosphorylation-based network and provide insights into biological regulation.
Fig. 1.
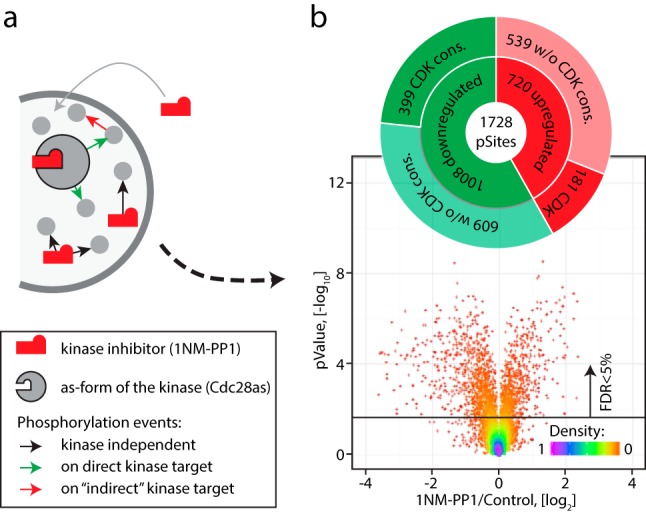
Global effects of Cdc28-as inhibition. (a) Addition of inhibitor to cells bearing as-form of a kinase can result in three types of phosphorylation events represented by colored arrows. The inhibitor itself can affect cellular homeostasis leading to changes in the phosphoproteome (kinase-independent). Upon inhibition of the target kinase its direct substrates will be dephosphorylated, which in turn can affect phosphorylation of other downstream targets, termed indirect substrates). (b) Yeast cells containing the cdc28-as allele were treated with vehicle or 10 μm 1NM-PP1 for 15 min. Experiments were performed in six replicates and only phosphopeptides quantified by MS in at least four replicates were considered for subsequent analysis. Significant (FDR<5%) changes were observed for 1,728 phosphosites without a clear prevalence of dephosphorylation (1,008) over phosphorylation (720) but with enrichment of CDK consensus motif among dephosphorylated phosphosites.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
Inhibition of CDK11 and SNF1 with 1NM-PP1 was performed as six independent treatments, every time using reverse triple SILAC labeling (see supplemental Fig. S1). To find significantly affected phosphosites, t test pValue threshold was chosen based on false discovery rate (FDR) <5% using permutation-based correction. For all dynamic (time course) experiments, only one replicate was used for analysis and significance was calculated based on the shape of the entire kinetic profile rather than individual measurements (26).
Yeast Strain Construction
The S. cerevisiae strains used in this study were isogenic with the S288C background. Single deletion strains, in which open reading frames were replaced by the kanMX cassette, were obtained from H. Bussey (McGill University, Montreal, Canada), Research Genetics and EUROSCARF. For SILAC labeling, the wt strain YAL6B (MATa his3Δ leu2Δ met15Δ ura3Δ lys1::KanMX6 arg4::KanMX4) was provided by Ole Jensen (University of Southern Odense, Denmark). To construct the cdc28-as1 strain used for SILAC analysis, a mutation resulting in an F88G substitution was introduced at the CDC28 locus in strain YAL6B by PCR amplification from a CDC28::Tadh1::HIS3MX6 selection cassette (27). The presence of the F88G mutation and absence of secondary mutations were confirmed by sequencing the entire CDC28 open reading frame. The snf1-as strain for SILAC labeling was constructed by integration of an snf1(I132G)-LEUMX cassette into an snf1::kanMX deletion strain (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) followed by crossing to the YAL6B strain and selection for a spore clone that was unable to grow on -Lys or -Arg medium. The snf1-as mutation and flanking regions of genomic DNA were sequence verified. Phosphorylation site mutations were introduced at the genomic loci by homologous recombination-mediated seamless integration in a two-step acceptor/donor protocol. First, an acceptor strain was generated by transformation of a linear DNA fragment that contained 45 bp upstream and 45 bp downstream of the mutation site flanking the URA3MX cassette into parental strain BY4741 and selection on -Ura medium. Second, the acceptor strain was transformed with a donor DNA fragment that contained 150 bp upstream and 150 bp downstream of the mutation site, which substituted the natural codon for an alanine codon, followed by selection on 5-FOA medium. All phosphorylation site mutant strains were verified by sequencing of at least 200 bp upstream and downstream of the mutation site. Three independent isolates of each strain were assessed for mutation-associated phenotypes. An SNF1(S391A) snf4Δ double mutant strain was constructed by PCR-based disruption of the SNF4 locus with an His3MX cassette in the parental SNF1(Ser391Ala) strain.
Cell Culture
Yeast cells were grown in synthetic dextrose (S.D., 0.17% yeast nitrogen base without amino acids, 0.5% ammonium sulfate, and appropriate amino acids) supplemented with either light (Lys0/Arg0), medium (Lys4/Arg6), or heavy (Lys8/Arg10) lysine (30 mg/l) and arginine (20 mg/l) (Cambridge Isotope Laboratories). For experiments with the cdc28-as allele, cultures were grown in S.D. in the presence of 2% glucose. For experiments with snf1-as, cultures were grown in S.D. media containing 2% ethanol. All cultures were grown to a optical density of OD600 = 0.7. Kinase inhibition was performed by adding the 1NM-PP1 to a final concentration of 10 μm, whereas control cultures were treated with vehicle (DMSO). After 15 min, TCA was added to final concentration of 10%, cells were collected by centrifugation a 2,000 × g for 10 min, washed with ice-cold PBS, and the SILAC channels were combined before cell lysis. For nutrient shift experiments, snf1-wt strain was grown in S.D. media containing 2% EtOH, concentrated glucose solution was added to light SILAC culture to final 2% (w/w), whereas heavy SILAC culture was used as a control. Probes from both SILAC channels were taken every 2 min for a time course of 30 min. For growth assays, mutant strains were cultured in the S.D. medium containing the appropriate carbon source (2% glucose or 2% galactose or 3% glycerol) or XY medium (2% bactopeptone, 1% yeast extract, 0.01% adenine, 0.02% tryptophan) containing 3% glycerol, as indicated. For size analysis, strains were inoculated from fresh YPD plates into indicated media and cell size profiles determined after 8, 16, 24, and 40 h on a Beckman–Coulter Z2 particle sizer. Daughter cell size was defined as the half maximal peak on the left-hand side of the cell size distribution, and mode size was defined as the value at maximum peak height. For growth curve measurements, cultures were seeded from an overnight culture in 2% glucose medium at density 2 × 106 cells per ml in 100 μl of media in 96-well plates. Culture growth was measured in triplicate on a Tecan Sunrise automated shaker/reader and analyzed using Magellan V7.1 software. Growth parameters were calculated using custom cell growth curve software (http://sysbiolab.bio.ed.ac.uk:3838/my-app/GCA.shiny/).
Protein Extraction and Enzymatic Digestion
Cells were lysed by bead beating for two cycles of 5 min each in lysis buffer (8 m urea, 50 mm Tris, pH 8.0, supplemented with HALT phosphatase inhibitor mixture, Pierce). Samples were centrifuged at 40,000 × g for 10 min, and the supernatants were transferred into clean tubes prior to determination of protein concentrations by bicinchoninic acid assay (Thermo Fisher Scientific). Disulfide bonds were reduced by adding dithiothreitol to a final concentration of 5 mm, and samples were incubated at 56 °C for 30 min. Reduced cysteines were alkylated by adding iodoacetamide to 15 mm and incubating for 30 min in the dark at room temperature. Alkylation was quenched with 5 mm dithiothreitol for 15 min. Samples were diluted sixfold with 20 mm TRIS, pH 8, containing 1 mm CaCl2 prior to overnight digestion at 37 °C with trypsin (Sigma-Aldrich) using an enzyme to substrate ratio of 1:50 (w/w). Tryptic digests were acidified with 1% formic acid (FA), centrifuged (20,000 × g 10 min) and desalted on Oasis HLB cartridges (Waters) according to manufacturer instructions. Peptide eluates were snap-frozen in liquid nitrogen, lyophilized in a speedvac centrifuge, and stored at −80 °C.
Phosphopeptide Isolation and Fractionation
Tryptic digests were subjected to enrichment on TiO2 beads as described previously (28). Sample loading, washing, and elution steps were performed using custom StageTips (29, 30) made from 200 μl pipette tips containing an SDB-XC membrane (3 m) frit and filled with TiO2 beads. We equilibrated TiO2 material in 250 mm lactic acid 70% acetonitrile 3% TFA; the same buffer was used for sample loading. After extensive washing steps, retained phosphopeptides were displaced from TiO2 with 500 mm phosphate buffer at pH = 7. Peptides were desalted in 50 μl of 1% FA directly on SDB-XC frits and subsequently eluted using 50 μl of 50% acetonitrile 1% FA. Eluates were dried in a speedvac and stored at −80 °C. To increase phosphoproteome coverage prior to MS analysis, phosphopeptides were fractionated offline by strong cation exchange chromatography. Peptides were solubilized in 100 μl of loading buffer (0.2% FA 10% acetonitrile) and loaded on StageTips containing 10 mg of poly-sulfoethyl-A strong cation exchange phase (5 μm 300A, Canada Life Science). Columns were washed with 50 μl of loading buffer, and peptides were eluted in five separate 100 μl salt steps of 25, 50, 80, 150, and 250 mm NaCl. All fractions (including flow-through) were collected, dried in a speedvac, and resuspended in 10 μl of 4% FA prior to MS analysis.
Mass Spectrometry
Enriched phosphopeptide extracts were analyzed by LC-MS/MS using a Proxeon nanoflow HPLC system coupled to a tribrid fusion mass spectrometer (Thermo Fisher Scientific). Each sample was loaded and separated on a reverse-phase analytical column (18 cm length, 150 μm inner diameter) (Jupiter C18, 3 μm, 300 Å, Phenomenex) packed manually. LC separations were performed at a flow rate of 0.6 μl/min using a linear gradient of 5–30% aqueous acetonitrile (0.2% FA) in 106 min. MS spectra were acquired with a resolution of 60,000. The TopSpeed (maximum number of sequencing events within 5 s window) method was used for data-dependent scans on the most intense ions using high energy dissociation. AGC target values for MS and MS/MS scans were set to 5e5 (max fill time 200 ms) and 5e4 (max fill time 200 ms), respectively. The precursor isolation window was set to m/z 1.6 with a high energy dissociation normalized collision energy of 25. The dynamic exclusion window was set to 30 s.
Data Processing and Analysis
MS data were analyzed using MaxQuant (31, 32) software version 1.3.0.3 and searched against the SwissProt subset of the S. cerevisiae uniprot database (http://www.uniprot.org/) containing 6,630 entries (November 2013). A list of 248 common laboratory contaminants included in MaxQuant was also added to the database as well as reversed versions of all sequences. The enzyme specificity was set to trypsin with a maximum number of missed cleavages set to 2. Peptide identification was performed with an allowed initial precursor mass deviation up to 7 ppm and an allowed fragment mass deviation of 20 ppm with subsequent nonlinear mass recalibration (33). Phosphorylation of serine, threonine, and tyrosine residues was searched as variable modification; carbamidomethylation of cysteines was searched as a fixed modification. The FDR for peptide, protein, and site identification was set to 1% and was calculated using decoy database approach. The minimum peptide length was set to 6, and the “peptide requantification” function was enabled. The option match between runs (1-min time tolerance) was enabled to correlate identification and quantitation results across different runs. In addition to an FDR of 1% set for peptide, protein, and phosphosite identification levels, we considered only phosphosites for which localization confidence was higher than 75%. Relative quantification of the peptides against their heavy-labeled counterparts was performed with MaxQuant using area under 3D peptide peak shapes (31, 32).
Detailed information on peptide and protein identifications is available in MaxQuant output tables provided along with the raw data at http://www.peptideatlas.org/ (identifier PASS00888, password: ZD2377ro). Annotated MS/MS spectra can be accessed through MS-Viewer (34) (http://msviewer.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer) with the following search keys: noj5x15byw (Ccd28 inhibition time course), hqgdudqu8g (nutrient shift time course), teobkhfobl (Cdc28 15-min inhibition), and jri18xoti1 (Snf1 15-min inhibition).
Machine Learning
We devised an algorithm to allow the learning of a molecular recognition model despite the lack of validated substrates. We let x be a phosphosite and y be a binary label equal to 1 if x is a direct kinase target (positive example) and 0 otherwise (negative example). Additionally, let s = 1 if x is a known direct target (or not), and s = 0 if it is unclear. From our experimental strategy, only negative examples are bona fide, meaning that y = 0 is certain when s = 1, but y can either be 0 or 1 when s = 0. Formally, we have that P(s = 1|x, y = 1) = 0 and we want to learn a model function f(x) that approximates P(y = 1|x) as closely as possible. This learning paradigm is known as negative and unlabeled learning; its twin problem, positive and unlabeled learning, has been studied previously (27). The data generating distribution, f(x) can be derived from g(x) = P(s = 1|x). This implies that it is sufficient to learn a model that discriminates down-regulated Cdc28-related phosphosites from the negative set to derive a model that can discriminate direct Cdc28 phosphosites. The binary learning set for g(x) was thus composed of up-regulated phosphosites (s = 1, certainly negative) and down-regulated Cdc28-related phosphosites (s = 0, unclear if positive or negative). To approximate g(x), we choose the SVM (21) for its ability to produce probabilistic outputs, its effectiveness at handling noisy data, and its compatibility with kernels. In proteomics, examples are in the form of peptide sequences while virtually all learning algorithms need examples in the form of real valued feature vectors. String kernels have been specifically designed to address this limitation (35). Such kernels can implicitly map strings of amino acids to an arbitrary feature vector to allow the computation of the dot product between any two strings. Kernel methods, such as the SVM, rely solely on the dot product, in contrast with the actual vectors. This approach is well suited for learning models from peptides and other biomolecules. In this study, phosphosites were represented using a fixed window of 15 amino acids centered on the phosphorylated residue. Residues with overlapping windows were considered separately. In addition, the generic string (GS) kernel (36) was chosen as kernel for the amino acids windows because it can account for the properties of amino acids and the flexibility of the substrates. In this study, the Blocks Substitution Matrix was used to compare amino acids in the GS kernel. The GS kernel has been shown to produce models with improved accuracies for a variety of peptide-related learning problems. In particular, this kernel has produced models that are more complex and accurate than position-specific weight matrix and motifs (37). However, this improved accuracy comes at the cost of producing models that are very hard to manually interpret. 10-fold cross-validation and the F1-score (harmonic mean of precision and recall) were used to determine the optimal parameters for the SVM and the GS kernel. With these parameters, a model was learned using all but one phosphosite, and the probability that this left-out site was a direct substrate was predicted. This leave-one-out prediction scheme was repeated for each phosphosite. Following Lemma 1 from (27), f(x) = P(y = 1|x) = g(x)/c was obtained by applying a constant transformation to g(x) = P(s = 1|x), effectively predicting the probability that a site x is directly phosphorylated by the kinase of interest. A more detailed description of the machine learning algorithm is available as a Supplemental Note.
NetworKIN, GO, and PPI Network Analyses
Prediction of kinase–substrate relationships were achieved using KinomeXplorer (http://networkin.info/) with default thresholding criteria (38). Gene ontology enrichment analyses were performed against whole S. cerevisiae proteome as background using the Database for Annotation, Visualization and Integrated Discovery bioinformatics resources (39, 40). All protein–protein interaction networks were built in STRING (41, 42). All interaction predictions were based on experimental evidences with the minimal confidence score of 0.9, which is considered as the highest confidence filter in STRING. Results were visualized in Cytoscape (43, 44, 45).
Data Availability
The mass spectrometry raw data from this publication along with MaxQuant output files and fasta files have been submitted to the Peptide Atlas database (http://www.peptideatlas.org/) and assigned the identifier PASS00888 (Password: ZD2377ro).
RESULTS
Inhibition of Cdc28-as Results in Phosphorylation and Dephosphorylation of a Large Number of Substrates
We used quantitative SILAC-based phosphoproteomics to study the global effect of inhibition of an as-form of Cdc28 (Cdc28-as) upon treatment with 1NM-PP1. Cells were grown at 30 °C in media containing light (0Lys, 0Arg), medium (4Lys, 6Arg), or heavy (8Lys, 10Arg) isotopic forms of lysine and arginine then treated with 10 μm 1NM-PP1 or vehicle (DMSO) for 15 min. Cells were rapidly harvested by addition of TCA (46) and the SILAC channels for each culture combined before lysis in denaturing urea buffer (supplemental Fig. S1a). Extracted proteins were digested into peptides with trypsin and phosphopeptides were concentrated on TiO2 affinity resin with subsequent offline strong cation exchange fractionation and LC-MS/MS analyses on an LTQ-Orbitrap Fusion instrument (supplemental Fig. S1b). We performed six independent treatments and only phosphosites quantified in at least four replicas were considered for subsequent analysis (supplemental Table S1, Fig. 1b). As a result, we obtained 1,008 down-regulated and 720 up-regulated phosphosites, corresponding to ∼28% of all quantified sites (FDR<5%). The observed ratio between phosphorylated and dephosphorylated phosphosites in our dataset was consistent with a previous study (19). As expected, we found a relative enrichment of the CDK consensus motif among dephosphorylated phosphosites (Fig. 1b). The large fraction of up-regulated sites indicated significant effects due to 1NM-PP1 off-target inhibition and/or the occurrence indirect substrates as a result of regulation of other kinases/phosphatases by Cdc28. We thus sought to improve our experimental and data analysis workflow to filter out contributions from these nonspecific or indirect effects.
1NM-PP1 Itself Triggers Many Phosphorylation Events Independent of Cdc28-as Activity
To evaluate the effect of 1NM-PP1 on the phosphoproteome, we repeated the inhibitor and control treatment with a wt strain that did not contain the cdc28-as mutation (Fig. 2a). Surprisingly, we observed significant changes in the abundance of 757 phosphosites (∼13% of the total quantified phosphosites) with an approximately equal number of up-regulated and down-regulated sites. This result suggests that 1NM-PP1 itself caused substantial changes in the phosphoproteome in the absence of any as-kinase allele. To understand the biological basis for the effect of 1NM-PP1 alone, we extracted sequence motifs from affected phosphosites. Up-regulated phosphosites did not share any obvious motifs, whereas down-regulated sites were enriched for basophilic kinase motifs containing basic amino acids (R/K) in the -2/-3 positions (Fig. 2b). We attempted to link these dephosphorylated sites with putative kinases using KinomeXplorer (38), a platform that integrates an improved NetworKIN (47) algorithm for modeling kinase signaling networks by combining linear motif analysis with protein–protein interactions data. We found that the majority of dephosphorylated sites were associated with PKA activity (Fig. 2b). This result suggested that 1NM-PP1 either directly inhibits PKA catalytic subunits or decreases PKA activity through an indirect mechanism. Notably, each PKA catalytic subunit in yeast (Tpk1, Tpk2, Tpk3) contains a methionine residue in the gatekeeper position, and hence, their ATP-binding pocket cannot accommodate the bulky 1NM-PP1 moiety, suggesting that 1NM-PP1 does not directly inhibit endogenous PKA activity. It is possible that 1NM-PP1 produces a stress response that causes a subsequent decrease in PKA activity (26, 48, 49). Consistent with this possibility, gene ontology analysis of the phosphosites altered in response to 1NM-PP1 revealed an enrichment for proteins involved in the response to osmotic stress (such as Sln1, Ssk1, Fps1) along with amino acid transporters (Avt1/3/4, Tat1) that might help to remove 1NM-PP1 from the cell (supplemental Fig. S2). Regardless of the precise mechanism, the significant off-target effect of the inhibitor required reinterpretation of the as-inhibition data.
Fig. 2.

1NM-PP1 causes off-target phosphorylation events. (a) In order to estimate the effect of 1NM-PP1 treatment alone a wild type (non-as) strain was treated with 10 μm 1NM-PP1 for 15 min and compared with DMSO control treatment. Significant changes were observed on ∼13% of detected phosphosites. (b) MotifX analysis revealed basophilic kinase motifs among the dephosphorylated sites in a 1NM-PP1 treated wt strain. NetworKIN analysis also suggested that ∼60% of dephosphorylated sites were predicted substrates of the basophilic kinase PKA (Tpk1, Tpk2, and Tpk3).
Normalization of 1NM-PP1 Off-Target Phosphorylation Allows Definition of Cdc28-Dependent Phosphorylation
As a next step, we compared inhibition of both cdc28-as and wt strains to select phosphosites that were significantly (FDR<5%) regulated in as- compared with wt cells. This subtractive approach yielded a list of phosphorylation sites that were definitively associated with the loss of Cdc28 activity (Fig. 3a). In this refined dataset, the majority of the affected phosphosites were dephosphorylated (523 down-regulated sites versus 207 up-regulated sites), as expected for bona fide Cdc28 substrates. Importantly, removal of the nonspecific 1NM-PP1-dependent events lowered the total number of regulated sites (and complexity of the dataset) by ∼2.5-fold compared with the initial dataset. This normalization also substantially increased the enrichment CDK consensus motifs and known Cdc28 substrates reported in the PhosphoGRID database (http://www.phosphogrid.org/) (50, 51) among the dephosphorylated targets (Figs. 3b and 3c). However, the observed increase in phosphorylation on a substantial number of phosphosites also indicated the presence of indirect Cdc28 substrates in the dataset. As such indirect substrates might become either phosphorylated or dephosphorylated on Cdc28 inhibition, we inferred that the dephosphorylated sites were a mixture of direct and indirect substrates of Cdc28.
Fig. 3.

Cdc28-specific phosphorylation sites. (a) Subtraction of phosphosites due to 1NM-PP1 alone from a cdc28-as inhibition experiment revealed targets whose abundance was affected by the change in Cdc28 activity. In total ∼12% of all identified phosphosites were regulated by Cdc28 with strong preference for dephosphorylation (523 dephosphorylated versus 207 phosphorylated sites). The increase in phosphorylation on 207 substrates indicates existence of indirect downstream effectors of Cdc28 inhibition. (b) Removal of 1NM-PP1 off-target phosphorylation events (1NM-PP1) from the Cdc28as results (global) improved the quality of the dataset as indicated by dramatic increase of the ratio of known Cdc28 substrates and sites with a CDK consensus motif between down-regulated and up-regulated peptides (Cdc28). (c) NetworKIN analysis revealed enrichment of CDK substrates among dephosphorylated phosphosites.
Machine Learning Separates Direct and Indirect Cdc28-Dependent Phosphorylation Events
As it was not possible to design a purely experimental strategy to filter out indirect substrates from actual down-regulated Cdc28-regulated targets, we used an SVM approach to computationally partition phosphosites into direct and indirect classes. In our setting, a typical supervised learning problem consists of learning a binary classification model from a set of validated substrates and negative controls. Such a dataset would allow the learning of a statistical model capable of differentiating direct from indirect substrates. However, de novo studies like ours do not contain validated training examples needed to learn a conventional model. We circumvent this problem by using a binary learning approach similar to positive and unlabeled learning (22).
To filter out indirect substrates from down-regulated by Cdc28 phosphosites, we used a mixture of direct and indirect Cdc28 targets (dephosphorylated phosphosites) as potential positive examples along with negative controls (up-regulated phosphosites across all experiments) to build a model (see Experimental Procedures for details). Accuracy was estimated by 10-fold cross-validation on the negative control group (Fig. 4a) with a value of 93.2% that suggests a good separation between indirect and direct Cdc28 substrates. Notably, despite the model having no prior information on the CDK consensus sequence and known Cdc28 targets (inferred from www.phosphogrid.org), phosphosites predicted as direct substrates were enriched for both of these independent criteria. To further confirm that the machine learning approach made biologically relevant predictions, we used the Yeast Kinase Interaction Database (52) (KID, http://www.moseslab.csb.utoronto.ca/KID/), which contains high- and low-throughput data relevant to Cdc28. To independently benchmark our results against other experimental studies, we extracted all Cdc28 interacting partners from the KID database and computed the overlap with our data. This analysis revealed a much better correlation between KID and our direct Cdc28 target set compared with the indirect target set (Fig. 4b). We stress that our model did not use any prior information from KID or any other previous datasets.
Fig. 4.

A machine learning algorithm separates direct from indirect Cdc28 targets. (a) Correction for the 1NM-PP1 off-target effect yielded a list of dephosphorylated sites that correspond to both direct and indirect Cdc28 targets. A mixture of direct and indirect Cdc28 targets (dephosphorylated phosphosites) along with part of the negative control set of up-regulated phosphosites across all experiments was used to build a support vector machine (SVM) model. Accuracy was estimated by cross-validation on the remaining part of the negative control group. Known CDK substrates as well as CDK motifs were enriched among direct targets predicted by the model. (b) The KID was used to extract known Cdc28 partners from literature-based studies, which were compared with direct and indirect Cdc28 sites predicted by the SVM model.
Among 260 phosphosites predicted to be directly phosphorylated by Cdc28, we found a number of known substrates. Notable examples included components of the SBF and MBF G1/S transcription complexes (supplemental Fig. S3) and a phosphorylation site on Ser429 of the phosphatase Cdc14 involved in feedback regulation of Cdc28 activity (53). Our data also revealed some Cdc28 targets implicated in cytoskeletal dynamics (supplemental Fig. S4). Interestingly, on many Cdc28 targets, we identified multiple phosphosites that corresponded to the direct and indirect classes. These substrates may represent examples of complex regulation whereby a substrate is first directly phosphorylated by Cdc28, followed by indirect phosphorylation events mediated by downstream kinases/phosphatases (supplemental Fig. S4a). In this regard, we note that inhibition of Cdc28 triggers changes in the phosphorylation status of 33 kinases and phosphatases, which may explain the dramatic contribution (∼50%) of indirect phosphorylation events to our dataset (supplemental Fig. S5).
Direct Cdc28 Sites Are Dephosphorylated Faster Than Indirect Sites
We investigated possible correlations between experimentally measured parameters and the probability of a phosphosite being a direct Cdc28 substrate. We found a dependence between the score generated by the model, that is, the probability that the phosphosite is direct, and the observed fold change in phosphosite abundance between control and inhibitor-treated conditions (Pearson r = 0.49, Fig. 5a). Direct Cdc28 sites were thus significantly more down-regulated compared with indirect sites (Wilcoxon test p = 1 × 10−8, Fig. 5b). It is important to highlight that no quantitative information was used to train the model (see Supplemental Note for additional information on the machine learning workflow).
Fig. 5.
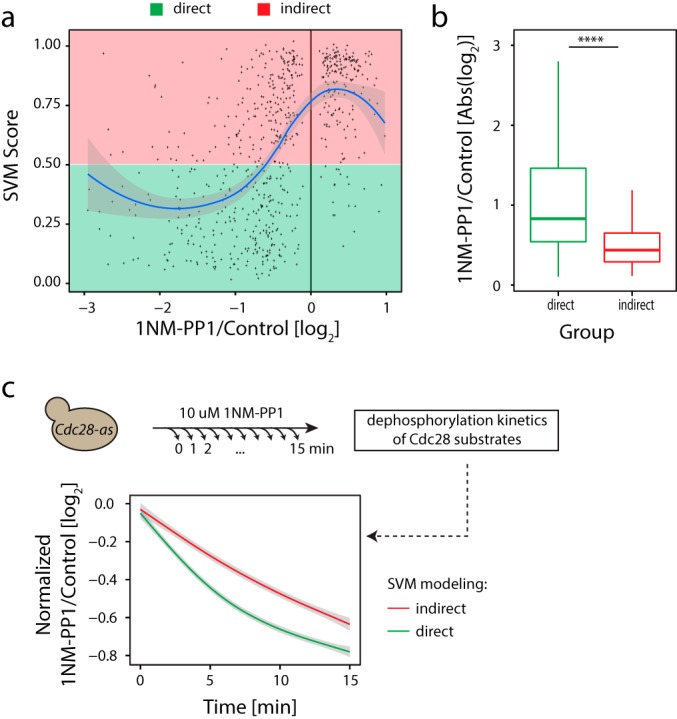
Predicted direct Cdc28 substrates are dephosphorylated more rapidly than predicted indirect substrates. (a) Correlation between extent of site dephosphorylation and SVM score. (b) Extent of dephosphorylation was larger for direct versus indirect substrates (pValue = 1e-8, Wilcoxon test). (c) Kinetic measurements of phosphorylation changes upon Cdc28 inhibition over a 15-min time course at 1 min intervals revealed differential dephosphorylation dynamics between direct versus indirect Cdc28 targets.
Although multiple parameters underlie the kinetics of phosphorylation/dephosphorylation (54, 55), we tested the simple hypothesis that direct substrates might be dephosphorylated more rapidly compared with indirect targets, which would result in higher absolute fold change values upon kinase inhibition. To test this notion, we performed kinetic experiments by determining global phosphorylation changes every 1 min following 1NM-PP1 treatment for a total duration of 15 min (Fig. 5c and supplemental Table S1). We obtained a 95% overlap between this dynamic dataset and our original single time point datasets. We assigned high-quality kinetic profiles, defined as sites being quantified in at least 14 of 16 measurements across the 15-min time series, for a majority of the Cdc28-associated direct and indirect phosphosites. We used this kinetic data to plot averaged profiles for dephosphorylated direct (n = 237) and indirect (n = 213) Cdc28 targets (Fig. 5c). These results confirmed the predicted faster dynamics of dephosphorylation for direct compared with indirect targets. We did not observe an obvious temporal delay between up-regulated phosphorylation events, in agreement with previous high-resolution dynamic studies (26, 56, 57).
Identification of Direct Snf1 Targets
To test our approach on a less well-characterized kinase, we applied the same workflow to study the Snf1 kinase, which regulates cell growth and metabolism under nutrient-limiting conditions. Snf1 is the founding member of the SNF1/AMPK family of protein kinases, which is highly conserved in eukaryotes and required for energy homeostasis in mammals, plants, and fungi (58). In S. cerevisiae, Snf1 allows cells to respond to declining levels of glucose by promoting respiratory metabolism, glycogen accumulation, gluconeogenesis, autophagy, the glyoxylate cycle, and peroxisome biogenesis (59, 60, 61, 62).
We constructed strain bearing an snf1-as allele and repeated inhibition with 1NM-PP1 under the same conditions as for the cdc28-as experiment (10 μm 1NM-PP1 for 15 min). To maintain the activity of Snf1, we used culture medium with ethanol as a carbon source. In total, this experiment yielded 408 down-regulated and 442 up-regulated phosphosites (supplemental Table S2). In this Snf1 experiment, we noticed a much smaller off-target effect of 1NM-PP1 as only 3% of the quantified sites were affected by 1NM-PP1 alone compared with 13% in the Cdc28 experiment. This difference may be explained by the use of ethanol versus glucose as a carbon source since the major effect of 1NM-PP1 in glucose medium appears to be mediated by an indirect decrease in PKA activity (Fig. 2b). In the absence of glucose, PKA activity would be significantly reduced, and therefore, an additional inhibition with 1NM-PP1 would produce a smaller relative change in the phosphoproteome. As for Cdc28, correction for the nonspecific effects of 1NM-PP1 on the phosphoproteome allowed us to identify Snf1-specific events (Fig. 6a) that were represented by both phosphorylation (73 sites) and dephosphorylation (225 sites) events. Machine learning was used to separate between direct (134) and indirect (78) Snf1 substrates among the dephosphorylated sites with an estimated cross-validation accuracy of 93%. Notably, we found the same correlation between the fold change decrease in phosphorylation and the probability generated by the model as for Cdc28 (Pearson r = 0.54, supplemental Figs. S6a and S6b). Snf1 direct substrates were also dephosphorylated more rapidly than indirect substrates. We were not able to find a consensus linear motif among the indirect Snf1 substrates, whereas for direct Snf1 targets we identified a motif in which the phosphorylated residue was followed by a leucine residue at the +4 position. Gene ontology analysis of the predicted direct substrates revealed an enrichment for proteins implicated in sugar metabolism and growth regulation (Fig. 6b), consistent with the known role of Snf1. Finally, we detected phosphorylation on many proteins previously associated with Snf1-mediated signaling events (Fig. 6c). Of particular note, we identified direct phosphorylation sites on the well-known Snf1 targets Mig1 (63, 64), Hxk1/2 (65, 66), and Cat8 (67, 68), including previously undocumented sites. We also observed phosphorylation on S241 of adenylate cyclase, which has recently been reported to be a direct Snf1 substrate (69, 70). We found both direct (Sip1) and indirect (Sip1, Gal83, and Sip2) phosphorylation on all β-subunits of SNF1 complex, which play a role in determining Snf1 substrate specificity and subcellular localization (24). In an interesting new regulatory connection, we identified Ser66, Ser75, and Ser925 as direct Snf1 sites on the Reg1 regulatory subunit of the Glc7 (PP1) phosphatase that inactivates Snf1, suggesting a potential feedback regulatory mechanism. Finally, we identified a new direct autophosphorylation site on Ser391 of Snf1 itself, suggestive of yet another regulatory mechanism.
Fig. 6.

Identification of direct Snf1 substrates. (a) Application of global phosphorylation profiles and machine learning to identify direct targets of the Snf1 kinase. After removal of 1NM-PP1 inhibitor off-target phosphorylation events, significant regulation was observed for ∼ 5% of all detected sites with a strong prevalence of dephosphorylation. SVM model allowed separation of direct (134) and indirect (78) Snf1 targets among all dephosphorylated targets. (b) MotifX analysis revealed enrichment of a sequence motif where the phosphorylated residue was followed by leucine in the +4 position for direct Snf1 substrates. No significantly enriched motifs were obtained in a similar analysis of indirect Snf1 targets. Gene ontology analysis revealed enrichment of terms associated with sugar metabolism and regulation of cell growth. (c) Many known components of the local Snf1 signaling network were identified as either direct or indirect targets.
Physiological Regulation of Direct Snf1 Sites
To demonstrate that the Snf1 sites were also affected under physiological conditions, we undertook a large-scale phosphoproteomic study of a nutrient shift that would alter endogenous Snf1 activity. A wt non-as strain was grown in the presence of ethanol as a carbon source and then transferred to the glucose-containing medium to cause the physiological inactivation of Snf1. Changes in the phosphoproteome were monitored every 2 min over a 30-min time course. In total, we were able to monitor the dynamic changes on ∼4,000 phosphosites with a minimum representation of nine measurements across the 16-point time series (supplemental Table S3). In this physiological nutrient shift experiment, we observed changes on 42 phosphosites out of the 134 that were identified as direct Snf1 substrates. All of these sites exhibited dephosphorylation upon the nutrient shift, consistent with the loss of Snf1 activity. We also observed dynamic changes for 30 phosphosites out of 78 that were classified as indirect Snf1 substrates. When we averaged normalized kinetic profiles for direct and indirect sites in the nutrient response, we observed the same trend as for Cdc28 inhibition, namely that dephosphorylation of direct targets was faster than on indirect direct targets (supplemental Fig. S6b). This result in the context of a physiological response provided support for the machine-learning-based classification of phosphorylation sites.
Mutational Analysis of Snf1 Phosphosites
We evaluated the functional importance of a number of both direct and indirect Snf1 phosphorylation sites by mutational analysis. Nonphosphorylatable alanine mutations were introduced into the endogenous loci for a total of 16 phosphosites in 8 Snf1 substrates within the immediate Snf1 signaling network. We assessed the effect of either single or combined mutations on culture doubling time in log phase, lag time for re-entry into log phase from stationary phase, and cell size on glucose, galactose, or glycerol medium. Strikingly, we were able to detect one or more growth-related phenotypes for each phosphosite mutant generated, suggesting that both direct and indirect phosphorylation sites contribute to the function of the Snf1 signaling network (supplemental Table S4). For example, mutation of phosphosites on each of the β-subunits of the SNF1 complex (Gal83, Sip1, and Sip2) caused phenotypes of differing severities. Gal83 is the most abundant β-subunit and targets SNF1 into the nucleus where it activates transcription factors Sip4 and Cat8 and deactivates Mig1. Mutation of the S44 site on Gal83 caused a decrease in growth rate and cell size on glycerol medium when compared with the wt control strain but without any difference when the strains were grown on glucose medium (supplemental Figs. S7a and S7b). This result illustrates the functional importance of an indirect Snf1 phosphosite. Mutation of other mapped sites on the Sip1 and Sip2 β-subunits caused a more modest effect on the lag time for log phase re-entry without a clear effect on doubling time or cell size (supplemental Table S4). In another example, mutation of the Ser925 and Ser66/Ser75 direct phosphosites on Reg1 both caused a significant delay in growth rate and a smaller cell size on glycerol medium. Interestingly, these phenotypes on the nonfermentable carbon source were even more severe than in the corresponding reg1Δ deletion strain (supplemental Table S4). A particularly notable direct phosphosite was mapped to S391 on Snf1 itself. Although mutation of this previously unknown site did not cause an obvious phenotype in isolation, the S391A mutation partially suppressed the loss of Snf4, which is an essential activating subunit of the SNF1 kinase complex (supplemental Fig. S7c). The SNF1(S391A) mutation thus bypassed the pronounced size defect of an snf4Δ deletion strain on both glucose and glycerol medium. In contrast, an snf1Δ snf4Δ double mutant grows as poorly on a nonfermentable carbon source as either single deletion (71). From these results, we postulate that the SNF1(S391A) allele encodes a hyperactive form of the kinase that is partially Snf4 independent and that autophosphorylation of Ser391 might be a feedback mechanism to limit Snf1 activity.
DISCUSSION
Our study addresses a straightforward but important biological question, namely, why does selective inhibition of a particular kinase result in both an increase and decrease in phosphorylation on a large number of proteins? To explain this phenomenon, we show that the global phosphorylation response to selective kinase inhibitors arises from three distinct contributions: nonspecific off-target effects of the inhibitor, direct substrates of the kinase, and indirect signaling events that arise downstream of the primary kinase. We developed a new method to disentangle these different contributions and performed a benchmark analysis on an as allele of the Cdc28 cell cycle regulatory kinase in S. cerevisiae. This analysis revealed an unexpectedly large effect of the 1NM-PP1 inhibitor on the phosphoproteome. Correction for these off-target events then allowed us to obtain a set of Cdc28-specific phosphorylation sites. Many of these sites were inferred to be indirect consequences of Cdc28 inhibition because a significant number exhibited increased phosphorylation upon Cdc28 inhibition, indicative downstream signaling effects. To deconvolute direct from indirect phosphorylation sites, we developed a machine learning model with strong discriminatory power, as assessed against independent datasets. Consistently, we demonstrated higher experimental fold changes and faster dephosphorylation kinetics for direct substrates compared with indirect substrates. Finally, we successfully applied our workflow to identify direct substrates of the less-well-characterized kinase Snf1 in S. cerevisiae. We identified many new direct and indirect phosphorylation sites on Snf1 substrates, for which a significant fraction of the direct sites exhibited faster dephosphorylation during a physiological nutrient shift that inactivates Snf1. Mutational analysis of a number of both direct and indirect Snf1 sites revealed new regulatory features of the Snf1 signaling network, including a potential new autoinhibition mechanism on Snf1 itself. This combined methodology based on specific kinase inhibition, appropriately controlled global phosphoproteome profiles, and machine learning may in principle be applied to any kinase for which an analog sensitive form can be engineered. The identification of direct in vivo kinase substrates in an unbiased manner will provide the foundation for deconvolution of the labyrinthine phosphoregulatory architecture of the cell.
Supplementary Material
Acknowledgments
The Institute for Research in Immunology and Cancer receives infrastructure support from the Canadian Center of Excellence in Commercialization and Research, the Canadian Foundation for Innovation, and the Fonds de Recherche du Quebec-Sante (FRQS). Proteomics analyses were performed at the Center for Advanced Proteomic Analyses, a Node of the Canadian Genomic Innovation Network supported by the Canadian Government through Genome Canada.
Footnotes
Author contributions: E.K., P.T., and M.T. designed the research; E.K., S.G., and J.C. performed the research; E.K., S.G., M.D.T., and P.T. analyzed data; and E.K., S.G., M.T., and P.T. wrote the paper.
* This work was supported by Canada Research Chairs in Proteomics and bioanalytical mass spectrometry (PT) and in Systems and Synthetic Biology (MT) and by Genome Canada Bioinformatics grant (MT, PT), the National Science and Engineering Research Council (311598, PT); the Canadian Institutes for Health Research (MOP 126129 to MT, and MOP 366608 to MT and PT), the National Institutes of Health (R01RR024031 to MT), the Ministère de l'enseignement supérieur, de la recherche, de la science et de la technologie du Québec through Génome Québec (MT), and the Quebec Consortium for Drug Discovery (MT).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CDK1
- cyclin-dependent kinase
- Cdc28
- cyclin-dependent kinase 1
- as
- analog-sensitive alleles
- 1NM-PP1
- 1-(1,1-dimethylethyl)-3-(1-naphthalenylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- OD
- optical density
- SILAC
- stable isotope labeling with amino acids in culture
- TRIS
- tris(hydroxymethyl)aminomethane
- FDR
- false discovery rate
- SVM
- support vector machine
- GS
- generic string
- YPD
- yeast extract peptone dextrose
- AGC
- automatic gain control
- PPI
- protein-protein pnteraction(s)
- GO
- gene ontology
- STRING
- (Search Tool for the Retrieval of Interacting; Genes/Proteins)
- SCB-binding factor
- SBF
- MCB-binding factor
- MBF
- SCB
- Swi4/6 cell cycle box
- MCB
- MluI cell cycle box
- SNF1
- Sucrose NonFermenting 1.
REFERENCES
- 1. Pawson T., and Scott J. D. (2005) Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 30, 286–290 [DOI] [PubMed] [Google Scholar]
- 2. Hunter T. (2000) Signaling—2000 and beyond. Cell 100, 113–127 [DOI] [PubMed] [Google Scholar]
- 3. Hunter T., and Plowman G. D. (1997) The protein kinases of budding yeast: Six score and more. Trends Biochem. Sci. 22, 18–22 [DOI] [PubMed] [Google Scholar]
- 4. Macek B., Mann M., and Olsen J. V. (2009) Global and site-specific quantitative phosphoproteomics: Principles and applications. Annu. Rev. Pharmacol. Toxicol. 49, 199–221 [DOI] [PubMed] [Google Scholar]
- 5. Rigbolt K. T., Prokhorova T. A., Akimov V., Henningsen J., Johansen P. T., Kratchmarova I., Kassem M., Mann M., Olsen J. V., and Blagoev B. (2011) System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 4, rs3. [DOI] [PubMed] [Google Scholar]
- 6. Levy E. D., Landry C. R., and Michnick S. W. (2010) Cell signaling. Signaling through cooperation. Science 328, 983–984 [DOI] [PubMed] [Google Scholar]
- 7. Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z. Y., Breitkreutz B. J., Stark C., Liu G., Ahn J., Dewar-Darch D., Reguly T., Tang X., Almeida R., Qin Z. S., Pawson T., Gingras A. C., Nesvizhskii A. I., and Tyers M. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Landry C. R., Levy E. D., and Michnick S. W. (2009) Weak functional constraints on phosphoproteomes. Trends Genetics 25, 193–197 [DOI] [PubMed] [Google Scholar]
- 9. Levy E. D., Michnick S. W., and Landry C. R. (2012) Protein abundance is key to distinguish promiscuous from functional phosphorylation based on evolutionary information. Phil. Trans. Royal Soc. London B Biol. Sci. 367, 2594–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nature Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 11. Galan J. A., Geraghty K. M., Lavoie G., Kanshin E., Tcherkezian J., Calabrese V., Jeschke G. R., Turk B. E., Ballif B. A., Blenis J., Thibault P., and Roux P. P. (2014) Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14–3-3. Proc. Natl. Acad. Sci. U.S.A. 111, E2918–E2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeffery D. C., Kakusho N., You Z., Gharib M., Wyse B., Drury E., Weinreich M., Thibault P., Verreault A., Masai H., and Yankulov K. (2015) CDC28 phosphorylates Cac1p and regulates the association of chromatin assembly factor I with chromatin. Cell Cycle 14, 74–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bodenmiller B., Wanka S., Kraft C., Urban J., Campbell D., Pedrioli P. G., Gerrits B., Picotti P., Lam H., Vitek O., Brusniak M. Y., Roschitzki B., Zhang C., Shokat K. M., Schlapbach R., Colman-Lerner A., Nolan G. P., Nesvizhskii A. I., Peter M., Loewith R., von Mering C., and Aebersold R. (2010) Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 3, rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bishop A. C., Shah K., Liu Y., Witucki L., Kung C., and Shokat K. M. (1998) Design of allele-specific inhibitors to probe protein kinase signaling. Current Biol. 8, 257–266 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y., Witucki L. A., Shah K., Bishop A. C., and Shokat K. M. (2000) Src-Abl tyrosine kinase chimeras: Replacement of the adenine binding pocket of c-Abl with v-Src to swap nucleotide and inhibitor specificities. Biochemistry 39, 14400–14408 [DOI] [PubMed] [Google Scholar]
- 16. Bishop A. C., Ubersax J. A., Petsch D. T., Matheos D. P., Gray N. S., Blethrow J., Shimizu E., Tsien J. Z., Schultz P. G., Rose M. D., Wood J. L., Morgan D. O., and Shokat K. M. (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 [DOI] [PubMed] [Google Scholar]
- 17. Weiss E. L., Bishop A. C., Shokat K. M., and Drubin D. G. (2000) Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nature Cell Biol. 2, 677–685 [DOI] [PubMed] [Google Scholar]
- 18. Bishop A. C., Buzko O., and Shokat K. M. (2001) Magic bullets for protein kinases. Trends Cell Biol. 11, 167–172 [DOI] [PubMed] [Google Scholar]
- 19. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., and Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papa F. R., Zhang C., Shokat K., and Walter P. (2003) Bypassing a kinase activity with an ATP-competitive drug. Science 302, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 21. Cortes C., and Vapnik Vladimir (1995) Support-vector networks. Machine Learning, 20, 273–297 [Google Scholar]
- 22. Elkan C., and Noto K. (2008) Learning classifiers from only positive and unlabeled data. Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining [Google Scholar]
- 23. Malumbres M. (2014) Cyclin-dependent kinases. Genome Biol. 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conrad M., Schothorst J., Kankipati H. N., Van Zeebroeck G., Rubio-Texeira M., and Thevelein J. M. (2014) Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 38, 254–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kayikci O., Nielsen J. (2015) Glucose repression in Saccharomyces cerevisiae. FEMS Yeast Res. 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanshin E., Kubiniok P., Thattikota Y., D'Amours D., and Thibault P. (2015) Phosphoproteome dynamics of Saccharomyces cerevisiae under heat shock and cold stress. Mol. Syst. Biol. 11, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chylek L. A., Akimov V., Dengjel J., Rigbolt K. T., Hu B., Hlavacek W. S., and Blagoev B. (2014) Phosphorylation site dynamics of early T-cell receptor signaling. PloS One 9, e104240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanshin E., Michnick S. W., and Thibault P. (2013) Displacement of N/Q-rich peptides on TiO2 beads enhances the depth and coverage of yeast phosphoproteome analyses. J. Proteome Res. 12, 2905–2913 [DOI] [PubMed] [Google Scholar]
- 29. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 30. Ishihama Y., Rappsilber J., and Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 31. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 32. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 33. Cox J., Michalski A., and Mann M. (2011) Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass Spectrom. 22, 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baker P. R., and Chalkley R. J. (2014) MS-viewer: A web-based spectral viewer for proteomics results. Mol. Cell Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leslie C. S., Eskin E., and Noble W. S., The spectrum kernel: A string kernel for SVM protein classification. Pacific Symp. Biocomput. 2002, 7. [PubMed] [Google Scholar]
- 36. Giguère S., Marchand M., Laviolette F., Drouin A., and Corbeil J. (2013) Learning a peptide–protein binding affinity predictor with kernel ridge regression. BMC Bioinformatics 14, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giguère S., Laviolette F., Marchand M., Tremblay D., Moineau S., Liang X., Biron É., and Corbeil J. (2015) Machine learning assisted design of highly active peptides for drug discovery. PLoS Comput. Biol. 11, e1004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horn H., Schoof E. M., Kim J., Robin X., Miller M. L., Diella F., Palma A., Cesareni G., Jensen L. J., and Linding R. (2014) KinomeXplorer: An integrated platform for kinome biology studies. Nature Methods, 11, 603–604 [DOI] [PubMed] [Google Scholar]
- 39. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 40. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., and von Mering C. (2011) The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. von Mering C., Huynen M., Jaeggi D., Schmidt S., Bork P., and Snel B. (2003) STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cline M. S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., Hanspers K., Isserlin R., Kelley R., Killcoyne S., Lotia S., Maere S., Morris J., Ono K., Pavlovic V., Pico A. R., Vailaya A., Wang P. L., Adler A., Conklin B. R., Hood L., Kuiper M., Sander C., Schmulevich I., Schwikowski B., Warner G. J., Ideker T., and Bader G. D. (2007) Integration of biological networks and gene expression data using Cytoscape. Nature Protocols 2, 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., and Ideker T. (2011) Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kanshin E., Tyers M., and Thibault P. (2015) Method bias effects in quantitative phosphoproteomics. J. Proteome Res. 14, 2998–3004 [DOI] [PubMed] [Google Scholar]
- 47. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A., Jørgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., Samson L. D., Woodgett J. R., Russell R. B., Bork P., Yaffe M. B., and Pawson T. (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garrett S., and Broach J. (1989) Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAKI, whose product may act downstream of the cAMP-dependent protein kinase. Genes Dev. 3, 1336–1348 [DOI] [PubMed] [Google Scholar]
- 49. Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D., and Brown P. O. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sadowski I., Breitkreutz B. J., Stark C., Su T. C., Dahabieh M., Raithatha S., Bernhard W., Oughtred R., Dolinski K., Barreto K., and Tyers M. (2013) The PhosphoGRID Saccharomyces cerevisiae protein phosphorylation site database: version 2.0 update. Database, 2013, bat026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stark C., Su T. C., Breitkreutz A., Lourenco P., Dahabieh M., Breitkreutz B. J., Tyers M., and Sadowski I. (2010) PhosphoGRID: A database of experimentally verified in vivo protein phosphorylation sites from the budding yeast Saccharomyces cerevisiae. Database, bap026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharifpoor S., Nguyen Ba A. N., Youn J. Y., van Dyk D., Friesen H., Douglas A. C., Kurat C. F., Chong Y. T., Founk K., Moses A. M., and Andrews B. J. (2011) A quantitative literature-curated gold standard for kinase-substrate pairs. Genome Biol. 12, R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Y., Cross F. R., and Chait B. T. (2014) Method for identifying phosphorylated substrates of specific cyclin/cyclin-dependent kinase complexes. Proc. Natl. Acad. Sci. U.S.A. 111, 11323–11328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salazar C., and Hofer T. (2006) Competition effects shape the response sensitivity and kinetics of phosphorylation cycles in cell signaling. Ann. N.Y. Acad. Sci. 1091, 517–530 [DOI] [PubMed] [Google Scholar]
- 55. Salazar C., and Höfer T. (2006) Kinetic models of phosphorylation cycles: A systematic approach using the rapid-equilibrium approximation for protein–protein interactions. Bio Systems 83, 195–206 [DOI] [PubMed] [Google Scholar]
- 56. Kanshin E., Bergeron-Sandoval L. P., Isik S. S., Thibault P., and Michnick S. W. (2015) A cell-signaling network temporally resolves specific versus promiscuous phosphorylation. Cell Reports 10, 1202–1214 [DOI] [PubMed] [Google Scholar]
- 57. Humphrey S. J., Azimifar S. B., and Mann M., High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nature Biotechnol. 2015 [DOI] [PubMed] [Google Scholar]
- 58. Hedbacker K., and Carlson M. (2006) Regulation of the nucleocytoplasmic distribution of Snf1-Gal83 protein kinase. Eukaryotic Cell 5, 1950–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ashrafi K., Lin S. S., Manchester J. K., and Gordon J. I. (2000) Sip2p and its partner snf1p kinase affect aging in S. cerevisiae. Genes Dev. 14, 1872–1885 [PMC free article] [PubMed] [Google Scholar]
- 60. Lin S. S., Manchester J. K., and Gordon J. I. (2003) Sip2, an N-myristoylated beta subunit of Snf1 kinase, regulates aging in Saccharomyces cerevisiae by affecting cellular histone kinase activity, recombination at rDNA loci, and silencing. J. Biol. Chem. 278, 13390–13397 [DOI] [PubMed] [Google Scholar]
- 61. Hedbacker K., and Carlson M. (2008) SNF1/AMPK pathways in yeast. Front. Biosci. 13, 2408–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Usaite R., Jewett M. C., Oliveira A. P., Yates J. R. 3rd, Olsson L., Nielsen J. (2009) Reconstruction of the yeast Snf1 kinase regulatory network reveals its role as a global energy regulator. Mol. Syst. Biol. 5, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Treitel M. A., Kuchin S., andCarlson M. (1998) Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 18, 6273–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Karunanithi S., and Cullen P. J. (2012) The filamentous growth MAPK pathway responds to glucose starvation through the Mig1/2 transcriptional repressors in Saccharomyces cerevisiae. Genetics 192, 869–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fernandez-Garcia P., Pelaez R., Herrero P., and Moreno F. (2012) Phosphorylation of yeast hexokinase 2 regulates its nucleocytoplasmic shuttling. J. Biol. Chem. 287, 42151–42164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ahuatzi D., Herrero P., de la Cera T., and Moreno F. (2004) The glucose-regulated nuclear localization of hexokinase 2 in Saccharomyces cerevisiae is Mig1-dependent. J. Biol. Chem. 279, 14440–14446 [DOI] [PubMed] [Google Scholar]
- 67. Roth S., Kumme J., and Schüller H. J. (2004) Transcriptional activators Cat8 and Sip4 discriminate between sequence variants of the carbon source-responsive promoter element in the yeast Saccharomyces cerevisiae. Current Genetics 45, 121–128 [DOI] [PubMed] [Google Scholar]
- 68. Vincent O., and Carlson M. (1999) Gal83 mediates the interaction of the Snf1 kinase complex with the transcription activator Sip4. EMBO J. 18, 6672–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Busnelli S., Tripodi F., Nicastro R., Cirulli C., Tedeschi G., Pagliarin R., Alberghina L., and Coccetti P. (2013) Snf1/AMPK promotes SBF and MBF-dependent transcription in budding yeast. Biochim. Biophys. Acta 1833, 3254–3264 [DOI] [PubMed] [Google Scholar]
- 70. Nicastro R., Tripodi F., Gaggini M., Castoldi A., Reghellin V., Nonnis S., Tedeschi G., and Coccetti P. (2015) Snf1 phosphorylates adenylate cyclase and negatively regulates protein kinase A-dependent transcription in Saccharomyces cerevisiae. J. Biol. Chem. 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Usaite R., Nielsen J., and Olsson L. (2008) Physiological characterization of glucose repression in the strains with SNF1 and SNF4 genes deleted. J. Biotechnol. 133, 73–81 [DOI] [PubMed] [Google Scholar]
- 72. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER Suite: protein structure and function prediction. Nature Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Roy A., Kucukural A., and Zhang Y. (2010) I-TASSER: A unified platform for automated protein structure and function prediction. Nature Protocols 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry raw data from this publication along with MaxQuant output files and fasta files have been submitted to the Peptide Atlas database (http://www.peptideatlas.org/) and assigned the identifier PASS00888 (Password: ZD2377ro).