

Abstract
The 26S proteasome is the macromolecular machine responsible for ATP/ubiquitin dependent degradation. As aberration in proteasomal degradation has been implicated in many human diseases, structural analysis of the human 26S proteasome complex is essential to advance our understanding of its action and regulation mechanisms. In recent years, cross-linking mass spectrometry (XL-MS) has emerged as a powerful tool for elucidating structural topologies of large protein assemblies, with its unique capability of studying protein complexes in cells. To facilitate the identification of cross-linked peptides, we have previously developed a robust amine reactive sulfoxide-containing MS-cleavable cross-linker, disuccinimidyl sulfoxide (DSSO). To better understand the structure and regulation of the human 26S proteasome, we have established new DSSO-based in vivo and in vitro XL-MS workflows by coupling with HB-tag based affinity purification to comprehensively examine protein-protein interactions within the 26S proteasome. In total, we have identified 447 unique lysine-to-lysine linkages delineating 67 interprotein and 26 intraprotein interactions, representing the largest cross-link dataset for proteasome complexes. In combination with EM maps and computational modeling, the architecture of the 26S proteasome was determined to infer its structural dynamics. In particular, three proteasome subunits Rpn1, Rpn6, and Rpt6 displayed multiple conformations that have not been previously reported. Additionally, cross-links between proteasome subunits and 15 proteasome interacting proteins including 9 known and 6 novel ones have been determined to demonstrate their physical interactions at the amino acid level. Our results have provided new insights on the dynamics of the 26S human proteasome and the methodologies presented here can be applied to study other protein complexes.
The ubiquitin-proteasome system (UPS)1 represents the major intracellular pathway for selective removal of regulatory, misfolded, and damaged proteins in eukaryotic cells. Aberrant UPS regulation can result in irregular protein turnover and accumulation of dysfunctional proteins, thus leading to various human diseases. The 26S proteasome is the macromolecular machine in the UPS that is responsible for controlled degradation of ubiquitinated substrates (1). It is composed of at least 33 subunits, which assemble into two subcomplexes: the 20S core particle (CP) and the 19S regulatory particle (RP). The 20S CP is responsible for various proteolytic activities, and has a highly conserved “barrel”-like structure arranged into four heptameric rings stacked in the order of α7β7β7α7 (2, 3). In contrast to the highly ordered and stable structure of the 20S CP, the 19S RP appears to be much more flexible and dynamic (4–6). The 19S RP is responsible for diverse functions including substrate recognition, deubiquitination, protein unfolding, and substrate translocation to the 20S CP for degradation. The 19S RP consists of 19 subunits that assemble into the lid and base subcomplexes. The base is composed of six ATPases (Rpt1–6), and four non-ATPase subunits (Rpn1, 2, 10, and 13). The remaining nine subunits (Rpn3, 5–9, 11, 12, and Rpn15/Sem1) comprise the lid structure. The binding of ubiquitinated substrates to proteasomes is facilitated through intrinsic ubiquitin receptors Rpn10, Rpn13, and Rpn1 of the base (7–11), whereas deubiquitination of bound substrates occurs through the action of the intrinsic deubiquitinase Rpn11 (12–15). The unfolding and translocation of substrates is ATP-driven and executed by the six ATPases, which directly interact with the 20S CP and modulate its gate opening (16).
It has been an extremely challenging task to resolve the high-resolution structure of the 26S proteasome holocomplex because of compositional and conformational heterogeneity of the RPs. Recently, a series of Cryo-EM studies combined with X-ray crystallography and other biochemical experiments have revealed the molecular architectures of the yeast (4–6) and human 26S proteasomes (17). Most of the studies actually focused on the yeast proteasomes, whereas reports on the human 26S proteasome have been sparse. Only very recently, two high-resolution Cryo-EM structures (3.9 and 3.5 Å) of the human 26S proteasome were reported (18, 19), indicating that the overall architecture of the 26S holocomplex is highly conserved from yeast to human. The six Rpt subunits of the 19S RP form a hexameric ring to associate with the cylinder ends of the 20S CP, and are surrounded by a shell of Rpn subunits (4–6, 18, 19). However, different assignments were proposed for the multiple geometries of human proteasomal subunits, contradicting previous structural studies of yeast proteasome in the localizations of Rpn8, Rpn11, and Rpn12 (17). Subsequent studies revealed that limited number of particles and overestimated resolution led to the incorrect assignment of these subunits (4, 5), and that the subunit arrangement in the human proteasome is indeed identical to that in yeast (18). Because of its structural dynamics, the proteasome exhibits a number of three-dimensional arrangements. Cryo-EM studies conducted in the presence of either ATPγS or ubiquitinated model substrates, along with a deep classification of a very large data set led us to identify coexisting conformational states and to define the conformational landscape of the 26S proteasome (20–22). These conformational changes were largely observed in the base and lid complexes but not in the 20S CP. Peripheral subunits such as Rpn1, Rpn10, and Rpn13 displayed a large degree of structural flexibility compared with the static 20S CP, resulting in a lower resolution structure (4). These subunits are known to be ubiquitin receptors in vivo and in vitro (7, 23). In addition, Rpn1 serves as a platform for deubiquitinating enzyme Ubp6 and the shuttle factors Rad23 and Dsk2 (24, 25).
In recent years, cross-linking mass spectrometry (XL-MS) has become an effective and powerful strategy to probe protein-protein interactions and define the architectures of macromolecular protein complexes (6, 26–30). In contrast to conventional structural tools such as X-ray crystallography or NMR techniques, XL-MS approaches have significantly fewer restrictions on sample preparation, and are capable of dissecting static and dynamic structural states of protein complexes. In addition to residue-specific protein interconnectivity, cross-links can be utilized as distance constraints to drive novel structural models and/or provide complimentary information to corroborate existing structures (26, 28, 29). Moreover, XL-MS approaches can be employed to probe protein-protein interactions at a large-scale in living cells (31–34), which cannot be easily assessed by other structural tools. Despite advantages of XL-MS technologies, inherent challenges remain regarding unambiguous identification of cross-linked peptides because of complex fragmentation profiles of cross-linked peptides when conventional (i.e. noncleavable) cross-linkers are used. Each cross-linked peptide contains two covalently linked peptides, whose sequences have to be determined based on convoluted MS/MS spectra containing the fragments from the two linked sequences. In addition, the two linked peptides often yield inequitable numbers of sequence ions, thus preventing accurate identification of both peptides. Moreover, specialized database searching tools are required to properly determine cross-linked peptide sequences. Although new developments in bioinformatics tools have proven effective in identifying noncleavable cross-linked peptides (35–38), database searching is limited to restricted protein databases because of quadratic expansion of computational search space required for increasingly large protein databases (39). Therefore, further improvement is still needed to make them as robust as conventional database searching tools (e.g. Protein Prospector or SEQUEST) for determination of single peptide sequences. To circumvent such problems, MS-cleavable cross-linkers appear to be the most attractive alternative because of their unique capability of simplifying MS identification of cross-linked peptides. To this end, we have previously developed a suite of new MS-cleavable cross-linkers containing sulfoxide(s) groups within their spacer regions (e.g. disuccinimidyl sulfoxide (DSSO)) (34, 40–42). These MS-cleavable reagents contain symmetric MS-labile C-S bonds (adjacent to the sulfoxide group) that can be selectively and preferentially fragmented prior to peptide backbone cleavage during collision induced dissociation (CID) (34, 40–42). Such fragmentation has proven robust and predictable, occurring independently of cross-linking types, peptide charges, and sequences, thus enabling simplified and accurate identification of sulfoxide-containing cross-linked peptides by MSn analysis and conventional database searching tools. DSSO is one of the amine-reactive sulfoxide-containing MS-cleavable cross-linkers that has been successfully applied for in vitro studies of purified protein complexes (27, 40, 43) and cell lysates (39). In this work, we have extended the application of DSSO linker by establishing new DSSO-based in vivo and in vitro XL-MS workflows to obtain a comprehensive protein-protein interaction connectivity map within the human 26S proteasome complex and its interacting proteins. In combination with cryo-EM and integrative modeling, we have obtained new structural insights to help us further uncover the details of human proteasomal architecture and dynamics.
EXPERIMENTAL PROCEDURES
Chemical Reagents
Regular Dulbecco's modified Eagle's medium (DMEM), ImmunoPure streptavidin, horseradish peroxidase-conjugated antibody, Super Signal West Pico chemiluminescent substrate and TurboFect transfection reagent were obtained from Thermo Fisher Scientific (San Jose, CA). Antibodies against human Rpt6 and HRP-conjugated streptavidin were from Biomol International. Endoproteinase Lys-C was from WAKO chemicals (Irvine, CA). Sequencing-grade trypsin was purchased from Promega Corp (Madison, WI). All other general chemicals for buffers and culture media were purchased from Thermo Fisher Scientific or VWR International (Radnor, PA).
Plasmids and Cloning
pQCXIP-Rpn11-HTBH and pQCXIP-Rpn1-HTBH were made as previously described (44, 45). pQCXIP-HBTH-Rpn13/ADRM1 was made as follows: The Rpn13/ADRM1 fragment containing a PacI site at the 5′end and an EcoR1 site at the 3′ was removed from pQCXIP-hisFlag-ADRM1 (46) and replaced the CSN5 fragment with the same restriction sites in pQCXIP-HBTH-CSN (47) to form pQCXIP-HBTH-Rpn13/ADRM1. To make pQCXIP-HBTH-Rpt6 plasmid, Rpt6 was PCR amplified using pCDNA3-Flag-Rpt6 as template with the following primers: forward, TTAATTAA CGCGCTTGACGGACCAGAGCAGATGGAG; and reverse, GAATTCTCACTTCCATAATTTCTTGATGGACATG. The Rpt6 DNA fragment containing a PacI site at the 5′end and an EcoR1 site at the 3′ end replaced the Rpn13/ADRM1 fragment with the same restriction sites in pQCXIP-HBTH-ADRM1 to form pQCXIP-HBTH-Rpt6. To make pQCXIP-Pre10-HTBH plasmid, Pre10 was PCR amplified using human cDNA library as template with the following primers: forward, ATAAGAATGCGGCCGCATGAGCTCAATCGGCACTGGGTATGAC; and reverse, CCTTAATTAACATATTATCATCATCTGATTCATCTTCTTCC. The Pre10 DNA fragment containing a NotI site at the 5′end and a Pac1 site at the 3′ end replaced the Rpn11 fragment with the same restriction sites in pQCXIP-Rpn11-HTBH (44) to form pQCXIP-Pre10-HTBH construct. To make pQCXIP-HBTH-Rpn10 plasmid, the Rpn10 fragment containing a PacI site at the 5′end and an EcoR1 site at the 3′ was removed from pQCXIP-his Flag-Rpn10 (46) and replaced the Rpn13/ADRM1 fragment with the same restriction sites in pQCXIP-HBTH-Rpn13/ADRM1 to form pQCXIP-HBTH-Rpn10.
pQCXIP-HBTH-SCOC and pQCXIP-SSNA1-HTBH plasmids were made in the following way: SCOC was PCR amplified using pANT7_cGST-human SCOC (DNASU plasmid repository, Plasmid #HsCD00303652) as template with the following primers: forward, TTAATTAACGACGGGTCCAGGAAAGAGGAGGAGG; and reverse, GAATTCTTACTTTCTTTTGCT TTTTGTGTCAGTTG. The SCOC DNA fragment containing a PacI site at the 5′end and a BamH1 site at the 3′ end replace the Rpn13/ADRM1 fragment with the same restriction sites in pQCXIP-HBTH-ADRM1 to form pQCXIP-SSNA1-HTBH construct. SSNA1 was PCR amplified using pLDNT7_nFLAG_human SSNA1 (DNASU plasmid repository, Plasmid #HsCD00616884) as template with the following primers: forward, GCGGCCGCATGACCCAGCAGGGCGCGGCGCTG; and reverse, TTAATTAAGCTGTCCCTGCCGCCGCTACTTTTC. The SSNA1 DNA fragment containing a NotI site at the 5′end and a PacI site at the 3′ end replace the Rpn11 fragment with the same restriction sites in pQCXIP-Rpn11-HTBH (44) to form pQCXIP-SSNA1-HTBH construct.
Experimental Design and Statistical Rationale
Five 293 stable cell lines (293Rpn11-HTBH, 293HBTH-Rpn13/ADRM1, 293HBTH-Rpt6, 293Pre10-HTBH, and 293HBTH-Rpn10) were generated using retrovirus as previously described (44). Briefly, a 293 GP2 cell line was cotransfected with HB tagged constructs and a plasmid expressing VSV-G. The medium containing the retrovirus was used to transduce 293 cells, which were subsequently selected with puromycin to establish the stable cell lines expressing each HB tagged-bait. The details on retroviral gene transfer can be found at (http://www.Clontech.com/US/Products/Viral_Transduction/Retroviral_Vector_Systems/ibcGetAttachment.jsp?cItemId=17555&fileId=6684076&sitex=10020:22372:US).
In vitro and In vivo DSSO Cross-Linking of the Human 26S Proteasome
For in vitro cross-linking analysis, the human 26S proteasome was purified by binding to Streptavidin beads as previously described (44), and then on-bead cross-linked with 0.5 mm DSSO in PBS buffer for 1 h at 37 °C. For in vivo cross-linking analysis, intact cells were cross-linked with 2 mm DSSO for one hour at 37 °C in PBS buffer and lysed in fully denaturing buffer as previously described (34). In vivo cross-linked proteasome complexes were tandem affinity purified under fully denaturing conditions by binding to Ni2+ Sepharose resin, followed by binding to Streptavidin beads. Both in vitro and in vivo cross-linked proteasome complexes that remained bound on Streptavidin beads were reduced/alkylated and digested by trypsin prior to LC MSn analysis (34). More than two biological replicates were performed for each cell line in both in vitro and in vivo XL-MS experiments to assess reproducibility.
LC MSn Analysis
LC MSn analysis was carried out using LTQ-Orbitrap XL MS (Thermo Fisher, San Jose, CA) coupled on-line to an Easy-nLC 1000 (Thermo Fisher) as previously described (40, 41). To obtain more comprehensive data, later samples were also analyzed using Orbitrap Elite or Fusion Tribrid MS instruments because of their significantly better sensitivity and speed. The LC setup and gradient were similar for all instruments, utilizing the Easy-nLC 1000 system. For Orbitrap XL MS, each MSn experiment consists of one MS scan in FT mode (350–1400 m/z, resolution of 60,000) followed by two data-dependent MS2 scans in FT mode (resolution of 7500) with normalized collision energy at 20% on the top two MS peaks with charges at 4+ and up, and three MS3 scans in the LTQ with normalized collision energy at 35% on the top three peaks from each MS2. MSn acquisitions performed on the Orbitrap Elite consisted of a single MS scan in FT mode (350–1600 m/z, resolution of 60,000), followed by two data-dependent MS2 scans in FT mode (resolution 15,000) with normalized collision energy at 20% on the top two MS peaks with charges 4+ and up, and three subsequent MS3 scans in the LTQ with normalized collision energy of 35% on the top three peaks from each MS2. Orbitrap Fusion MSn acquisitions were comprised of a single MS scan in FT mode (350–1800 m/z, resolution of 120,000), followed by three data-dependent MS2 scans in FT mode (resolution 30,000) with normalized collision energy at 20% on the top three MS peaks with charge selection 4+ to 8+. For each MS2 scan, three MS3 scans were performed in the LTQ on the most intense MS2 peaks using HCD with activation energy of 35%.
Data Analysis and Identification of DSSO Cross-linked Peptides
Monoisotopic masses of parent ions and corresponding fragment ions, parent ion charge states, and ion intensities from LC MSn spectra were first extracted based on the Raw_Extract script from Xcalibur v2.4 as described (34, 40, 41). MS3 data was subjected to a developmental version of Protein Prospector (v. 5.17.0) for database searching, using Batch-Tag against a decoy database consisting of a normal Swissprot database concatenated with its randomized version (SwissProt.2014.12.4.random.concat with total 20,294 protein entries). Homo sapiens was set as the taxonomy, whereas mass tolerances for parent ions and fragment ions were set as ± 20 ppm and 0.6 Da respectively. Trypsin was set as the enzyme with three maximum missed cleavages allowed. Cysteine carbamidomethylation was set as the fixed modification. A maximum of three variable modifications were also allowed, including protein N-terminal acetylation, methionine oxidation, and N-terminal conversion of glutamine to pyroglutamic acid. In addition, three defined modifications on uncleaved lysines and free protein N termini were also selected: alkene (A: C3H2O, +54 Da), sulfenic acid (S: C3H4O2S, +104 Da), and unsaturated thiol (T: C3H2OS, +86 Da) modifications, because of DSSO remnant moieties. It is noted that the sulfenic acid moiety often undergoes dehydration to become a more stable and dominant unsaturated thiol moiety as previously described (34, 40, 41). Peptides were identified from MS3 data with a FDR at 2%. Then MSn data and MS3 database searching results were integrated in xl-Discoverer (an in-house script) to automatically generate and summarize identified cross-linked peptide pairs (40, 41). The final FDR of interlinked peptide identification was determined as ∼0.1%, which was calculated based on the total number of false interlink hits (containing at least one false sequence resulting from decoy databases) out of the total cross-link entries identified, in a way similar to previous publications (35, 36, 38). The reduction in FDR for the identification of cross-linked peptides was presumably because of the integration of MSn data. Raw data has been deposited at the MassIVE repository FTP site: ftp://MSV000080313@massive.ucsd.edu (Password: LH26SXL110416).
Cryo-EM and Image Analysis
Data acquisition and image processing was done as previously described (18, 48). A data set was collected on a Titan Krios with a Falcon II camera using the FEI EPU software. Images were acquired at a pixel size of 1.4 Å at the specimen level, a total dose of 45 electrons distributed over seven frames with a nominal defocus varying between −1.8 to −3 μm. The acquired data were processed, reconstructed and classified in an in-house modified version of XMIPP, which allows us to restrict the in-plane rotation and to focus the analysis on one of the RPs (22). Additional classification using small masks were performed as described Bohn et al. (49).
Purification and Cross-linking of Reconstituted UBLCP1-hRpn1Complex
The human UBLCP1 protein used in this study was purified as described (45). Purification of human Rpn1 was based on the method of Rpn2 purification (50) with modifications. Briefly, full-length human Rpn1 was cloned into the pQE30 vector as a His-TEV fusion and expressed in the M15(pREP4) strain of E. coli. The cells were induced at room temperature overnight with 0.4 mm IPTG and purified using Ni-NTA resin (Thermo). Eluted protein was further purified through a Superdex200 size exclusion column (GE). The UBLCP1-Rpn1 complex was in vitro constituted for cross-linking analysis.
Structural Modeling and Analyses
Comparative and integrative modeling was carried out to elucidate the architectures of the human 26S proteasome and proteasome-PIP complexes (Supplemental Methods).
Biochemical Validation of the Selected Proteasome Interactors
The 293 cells transiently expressing HBTH-SCOC or stably expressing SSNA1-HTBH were used for biochemical validation (Supplemental Methods). A single-step affinity purification of the human SCOC or SSNA1 containing complexes was carried out by binding to Streptavidin resins (44). The purified protein complexes were analyzed by immunoblotting using specific antibodies. In vitro DSSO cross-linking of SCOC and SSNA1 complexes was carried out similarly as described above.
RESULTS
In Vitro XL-MS Analysis of Human 26S Proteasome Complexes
In order to elucidate the human 26S proteasome architecture, we first performed in vitro DSSO XL-MS studies as illustrated in Fig. 1A. Human 26S proteasome complex was affinity purified from 293 cells that stably express an HB-tagged proteasome subunit (e.g. Rpn11-HB) (44). The HB tag is a versatile tandem tag that contains hexahistidine and biotin tags suited for affinity purification under both native and denaturing conditions (44, 51). To define subunit interaction contacts, affinity purified proteasome complexes were cross-linked by DSSO in vitro. Unambiguous identification of DSSO cross-linked peptides was accomplished through LC MSn analysis using three lines of evidence: MS1 mass matching, MS2 fragmentation, and MS3 sequencing (40) (Fig. 1C). The identified cross-linked peptides were then subjected to structural mapping and modeling (29, 52). To expand the coverage of protein interactions, we selected 5 subunits located in different regions of the proteasome as baits (i.e. Rpn11, Rpn10, Rpt6, Rpn13/ADRM1, and α7/Pre10) and generated their respective HB-tagged stable cell lines. In total, we identified 1606 unique interlinked DSSO peptides (supplemental Table S1A), describing 157 unique Lys-Lys linkages for 63 intersubunit interactions, and 191 unique Lys-Lys linkages for 26 intrasubunit interactions (supplemental Table S2). Among the interprotein interactions, we have determined 21 as base-base, 17 base-lid, 12 lid-lid, 8 19S-20S and 5 20S-20S pair-wise interactions.
Fig. 1.

The general DSSO based XL-MS strategies to study protein complexes. (A) in vitro and (B) in vivo cross-linking workflow schemes. C, LC MSn analysis for the identification of DSSO cross-linked peptides.
In Vivo XL-MS Analysis of Human 26S Proteasome Complexes
In order to examine proteasome interactions as they occur in living cells, we next performed in vivo DSSO cross-linking on intact cells stably expressing a HB-tagged proteasome subunit (Fig. 1B). Cross-linked cells were then lysed in fully denaturing buffer (i.e. 8 m urea), and in vivo cross-linked proteasomes were isolated by two-step HB-tag based tandem affinity purification, digested and analyzed by LC MSn (34). Immunoblotting analysis showed that in vivo DSSO cross-linking and subsequent affinity purification of proteasome complexes were effective (supplemental Fig. S1), similar to our previous report using Azide-A-DSBSO (34). The five stable cell lines generated for in vitro studies were utilized to obtain a more comprehensive in vivo cross-link map of human proteasome complexes. In total, we identified 1320 DSSO cross-linked peptides, representing a total of 313 unique Lys-Lys linkages of interlinked peptides of proteasome subunits, and describing 54 pair-wise intersubunit interactions and 22 intrasubunit interactions (supplemental Tables S1B and S2). Among the pair-wise interactions, we have determined 17 base-base, 13 base-lid, 11 lid-lid, 7 19S-20S and 6 20S-20S intersubunit interactions.
The Human 26S Proteasome Interaction Network Topology
To further explore the connectivity of the 26S proteasome subunits, we generated an experimentally derived proteasome interaction network topology map based on a total of 67 pair-wise intersubunit interactions determined in this work (Fig. 2). To the best of our knowledge, this represents the most comprehensive XL-MS data derived subunit-subunit connectivity map of the human proteasome, which encompasses all 19 canonical subunits of the 19S RP and 10 subunits of the 20S CP. Extensive interactions within the 19S lid (Rpn3, 5–9, 11, 12, and Rpn15), the 19S base (Rpt1–6, Rpn1, Rpn2, Rpn10, and Rpn13), and 20S CP have been defined. In addition, proximal interactions of the 19S base with the 19S lid and the 20S have been revealed. Although the amount of cross-linking data obtained and reported here significantly surpasses that of our previous studies of the yeast 19S RP (27), the residue specific interactions identified in the human 19S RP are very similar, confirming resemblances in the overall architectures of the yeast and human complexes as recently suggested (18, 19).
Fig. 2.
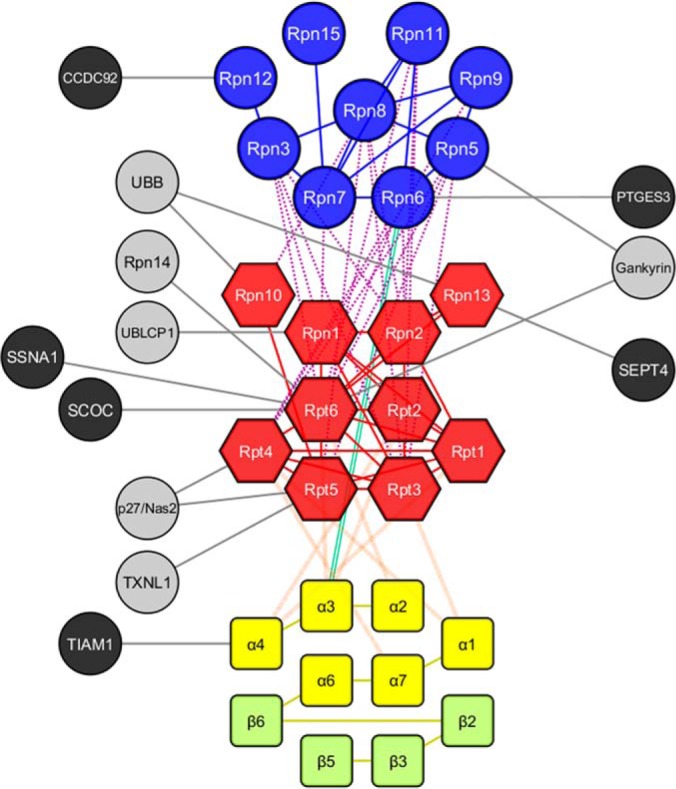
Interaction topology map of the human 26S proteasome. Nodes represent individual proteins whereas edges between nodes indicate identified cross-links within connected nodes. 26S proteasome subunits are categorized by primary color based on their subcomplex structures, i.e. 19S lid (blue), 19S base (red), 20S (yellow and light green, α and β, respectively). Corresponding primary color edges represent intermolecular linkages within the same subcomplex structure. Secondary color edges represent intermolecular linkages between different subcomplex structures, i.e. orange (19S base - 20S), purple (19S lid - 19S base), green (19S lid - 20S). Gray nodes represent known proteasome-interacting proteins, whereas black nodes indicate novel PIPs. Interactions (edges) between proteasome subunits and PIPs are shown in black.
Refining the Structure of the Human 26S Proteasome
The human 26S proteasome was purified from erythrocytes similarly as previously reported (53) for cryo-EM analysis, which resulted in 32,000 micrographs. We selected ∼250,000 particles with the same conformation for reconstruction, thus yielding a 3D density map of the human 26S proteasome at 6.8 Å (FSC = 0.143) resolution (Fig. 3). To elucidate the structure of the human 26S proteasome, we first generated a comparative model with the yeast 26S proteasome structure as a template (PDB ID: 4CR2; 53% average sequence identity) using MODELLER (54), followed by a refinement of the initial model based on the human EM density map determined in this work using MDFF (55). During refinement, the cross-correlation coefficient increased from 0.55 to 0.75. Most regions of the structure could be localized unambiguously. We estimated model precision by quantifying the variation in the model and defined the precision of each Cα position as the root-mean-square fluctuation (RMSF) from the mean position of the ensemble of models computed by MDFF. As expected, the highest RMSF values were located in loop regions and Rpn1, and appear to reflect the limited map resolution (supplemental Fig. S2). During the preparation of our manuscript, two high-resolution EM structures of human proteasome complexes were reported (i.e. PDB IDs: 5L4G, and 5GJR) (18, 19). In order to make certain that structural details in our refined model using the 6.8 Å (lower-resolution) EM map are similar to those using the two newly reported 3.5 Å and 3.9 Å (higher-resolution) EM maps, we relaxed our model in both 5L4G and 5GJR using MDFF. We saw a cross-correlation of our model with 5GJR and 5L4G as 0.7 and 0.66, respectively, showing high overlap between our model and new structures. The global RMSDs of 1.3 and 0.9 observed between our model to 5GJR and 5L4G, respectively, indicate minimal structural differences, which were further illustrated by the superposition of the human 26S models based on the three EM maps (supplemental Fig. S3).
Fig. 3.

6.8 Å resolution EM single particle reconstruction of the human 26S proteasome without imposed symmetry. The locally filtered density is displayed as an isosurface (left), as a mesh representation with an overlay isosurface in orange highlighting the main variances which indicates conformational variability (left middle), and colored according to the local resolution as indicated by the color key (right middle). Fit of a human homology model into the map of 6.8 Å (right).
Mapping In vitro and In vivo Cross-links onto the Structure of the 26S Proteasome
We next mapped the identified cross-links onto the human 26S structural model generated in this work. Considering the spacer length of DSSO (10.1 Å) and lysine side chains, as well as backbone dynamics, we considered lysine residues within Cα-Cα distance < 35 Å to be preferentially cross-linked by DSSO. To examine the distance constraints of identified cross-links, we plotted the distance distributions of our proteasome cross-link data sets using one of the high-resolution EM structures (5L4G) (18) (Fig. 4A). As a result, we were able to map 230 in vitro and 216 in vivo cross-links onto the human 26S EM structure. 189 (82.2%) and 173 (80.1%) of in vitro and in vivo cross-links, respectively, spanned distances shorter than 35 Å (Fig. 4B). For those outside the expected distance constraints (>35 Å), we grouped them as “violating” cross-links. Notably, the majority of violating in vitro (27/41 (65.9%)) and in vivo (32/43 (74.4%)) cross-links mapped to and within the Rpt6 subunit (Fig. 4C), suggesting that the conformation, if not also the position of this subunit, is dynamic. However, it is plausible that the identified violating cross-links may be attributed to the heterogeneity of cross-links that resulted from the presence of diverse forms of proteasome complexes in our XL-MS experiments.
Fig. 4.

Mapping of the cross-link dataset onto the 3. 9 Å human 26S structure (5L4G). A, Euclidean Cα-Cα distance distributions of all measured in vitro and in vivo cross-links mapped onto the human 26S model. The y axis provides the number of cross-links that were mapped onto the structural model. The dashed red line denotes the expected maximum reach of a cross-link. B, Matrix of all cross-links between and within the 26S subunits mapped onto the structural model. Satisfied (distance < 35 Å) and violating (distance > 35 Å) cross-links are colored in green and red, respectively. Observed cross-links from in vitro (bottom-right) and in vivo (upper-left) datasets on the structural 26S model. Larger circles represent unique cross-links. C, A detailed view of the Rpt6 subunit; color-coding of cross-links is the same as in B.
Among a total of 447 unique Lys-Lys linkages identified from the combined in vitro and in vivo datasets, 293 were mapped to the structural model (supplemental Table S2). The majority of these linkages correlated well onto the 26S human structural model within the expected spatial distance, suggesting that the core structure of the 26S proteasome remains similar under both experimental conditions. Interestingly, it is noted that the average cross-link distances identified in vitro and in vivo were similar (18.1 ± 7.4 Å and 18.5 ± 7.2 Å excluding “violating” cross-links, or 24.7 ± 18.0 Å and 25.5 ± 17.2 Å using the entirety of cross-linking data, respectively) (Fig. 4B). In addition, the two cross-linking strategies yielded significantly overlapping cross-link identifications (∼48%), thus further confirming the validity of our results. However, it is important to note that each method enabled the capture of unique interactions, most likely because of differences in sample preparation.
Dynamics of Rpn1, Rpn6, and Rpt6
In our EM structure (Fig. 3), the majority of subunits clearly show secondary structure elements, whereas subunits such as Rpn1 and Rpn6 are less resolved than the others. Because of its high variability, Rpn1 exhibits a resolution of 8.0∼8.5 Å. Although the variance at Rpn6 is not as dominant as in the case for Rpn1, the EM density of the N-terminal part of Rpn6 was smeared out after averaging (Fig. 3, bottom right). To obtain further insights into the various conformational states of the proteasome, we grouped the EM particles into different classes based on their structural variation of Rpn1 and Rpn6 (Fig. 5). The focused classifications of Rpn1 (Fig. 5, left side) revealed that Rpn1 has two distinct positions on the 19S RP. In the first, which is defined as the up-class, Rpn1 does not come into contact with the ATPases of the 19S base. This significantly differs from its position in the down-class, where we observed its direct interaction with the ATPases. The majority of the particles were found in the down-class (∼55%), with one third of the down-class particles containing an extra density connecting Rpn1 to Rpn2. We also observed a class in which Rpn1 is not detectable, which may be because of the transient binding of Rpn1. Rpn6 classification (Fig. 5, right side) resulted in three major classes. The first shows Rpn6 in the s1-conformation (i.e. substrate recruitment conformation), turning toward Rpn5. The majority of the particles grouped into the second class, “No Rpn6”, in which the N-terminal density for Rpn6 was missing but the remaining C-terminal regions were included in the horseshoe of the lid. The third class was a mix of the first and second classes, observed because of the flexibility of the subunits being averaged out in the EM map.
Fig. 5.
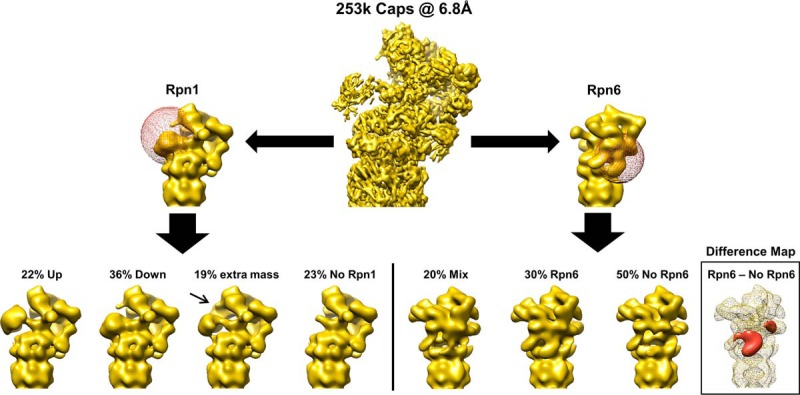
Classification of human 26S proteasomes reveals variations in the Rpn1 subunit (left) as well as in the Rpn6 subunit (right). Difference map between Rpn6-containing and non-Rpn6-containing 26S proteasomes is included to highlight differences in isosurfaces. UCSF Chimera was used to visualize the models.
Because our cryo-EM density maps suggest potential alternative states for Rpn1 and Rpn6 (Fig. 5), we next tested whether our cross-linking data supports the dynamics of these subunits. It has been reported that at least three distinct conformational states of the 19S RP (i.e. s1, s2, and s3) associate with the three crucial steps of the proteasomal degradation respectively: substrate recruitment (s1), irreversible commitment (s2), and substrate processing (s3) (20–22). We fitted the model of Rpn1 into the density maps with Rpn1 in “down” and “up” positions and placed it according to the yeast s1, s2, and s3 states. For each state of Rpn1, we optimized the positions of flexible loops and termini in the entire 26S proteasome (represented as beads, Methods) to minimize the distances between the cross-linked particles (supplemental Fig. S4A). Although there was no violating Rpn6 containing cross-links detected in our experiments, only 17 and 18 out of 63 cross-links between Rpn1 and other proteasome subunits were satisfied with Rpn1 in the “down” and “up” states, respectively (supplemental Fig. S4B). Similarly, Rpn1 superimposed onto yeast s1, s2, and s3 states satisfied 16, 20, and 21 of the cross-links, respectively. In total, these different states satisfy 43 (68%) unique cross-links (supplemental Fig. S4B). Again, these results suggest that the conformation, if not also the position of the Rpn1 subunit, is dynamic—and that potentially multiple subcomplexes exist because of sample heterogeneity.
Although cryo-EM analysis did not suggest any alternative conformations of Rpt6, the majority of the violating cross-links (Fig. 4C) include at least one end in the Rpt6 subunit, suggesting that the conformation and perhaps the position of this subunit may be dynamic. There was a total of 113 unique Lys-Lys linkages derived from the identified Rpt6 intraprotein and interprotein cross-linked peptides, representing the interactions within Rpt6 itself and with other proteasome subunits (supplemental Table S2). For intra-Rpt6 cross-links, 67 unique Lys-Lys linkages were identified with 53 from in vivo analysis and 45 from in vitro analysis. Because of missing residues in the structure model, only 55 out of 67 intra-Rpt6 cross-links could be mapped to the human 26S model, with only 52.6% (30/57) of them corresponding to Cα-Cα distances < 35 Å (supplemental Table S2). Interestingly, other than Rpt6, almost all proteasome subunits that can be mapped to the model do not carry intraprotein cross-links exceeding the required distance range. In addition, almost all (∼90%) of intra-Rpt6 violating cross-links were identified in in vivo XL-MS analysis, with in vitro analysis contributing only a few uniquely identified violating cross-links (supplemental Fig. S5). Similarly, for Rpt6 intersubunit cross-links, we have identified 45 unique intersubunit Lys-Lys linkages with 36 from in vivo and 33 from in vitro analyses, representing 13 pair-wise interactions between Rpt6 with proteasome subunits Rpn1–3, Rpn5–6, Rpn11, Rpt1–5, and α2–3 respectively. Based on structural mapping, 24/36 cross-links were measured within the expected Cα-Cα distance (< 35 Å), suggesting that the interactions of Rpt6 with Rpn11, Rpn6, Rpt2, Rpt3, α2, and α3 fit well with our proteasome model. However, 12 intersubunit cross-links were determined beyond the expected range (up to 109 Å), of which 4 are attributed to Rpt5-Rpt6 linkages and 5 to Rpn2-Rpt6 linkages. The hexametric ring structure of the six ATPase subunits is organized as Rpt3-Rpt6-Rpt2-Rpt1-Rpt5-Rpt4 in the proteasome (56). The close interactions of Rpt6 with Rpt2 and Rpt3 have been confirmed by 7 Rpt6-Rpt2 and 9 Rpt6-Rpt3 cross-links, respectively. Although Rpt5 is not in close proximity to Rpt6, five Rpt5-Rpt6 cross-links were identified with four corresponding to Cα-Cα distances > 35 Å. In contrast to Rpt6, all intra-Rpt5 cross-links fit perfectly well with the structure without exceeding expected distance. Therefore, the formation of these violating intersubunit cross-links is most likely because of the structural flexibility and dynamic movement of Rpt6, implying that Rpt6 is much more dynamic than anticipated. Potentially the Rpt6 violating cross-links may result from heterogeneous Rpt6-containing subcomplexes and/or tagging/overexpression of Rpt6 as most of these cross-links were identified only from Rpt6 purifications. It is noted that 10 cross-links between Rpt6 and other subunits cannot be mapped on the human 26S model because of insufficient atomic structural information, including the interactions between Rpt6 and Rpn1. Therefore, future studies would be needed to further explore structural dynamics of Rpt6 subunit in the 26S holocomplex.
Identification of Proteasome Interacting Proteins (PIPs)
In addition to the intra-26S cross-links, a total of 15 PIPs were identified with 36 cross-links describing intraprotein and interprotein cross-links with the 26S proteasome (supplemental Tables S1 and S2). Although in vitro XL-MS experiments identified 22 cross-links of PIPs, in vivo XL-MS experiments determined 26 PIP cross-links. Among the PIPs identified with cross-links, 9 are known PIPs including UBLCP1 (ubiquitin-like domain-containing CTD phosphatase 1), UCH37, proteasome assembly chaperones (i.e. Gankyrin/Nas6; PAAF1/Rpn14 and p27/Nas2), proteasome activators (PA200 and PA28β), Ubiquitin, and TXNL1 (Supplemental Table 2). In addition, we have found 6 unknown PIPs, including PTGES3, SEPT4, CCDC92, TIAM1, SCOC and SSNA1. Twelve out of the 15 PIPs were mapped to the 26S proteasome network (Fig. 2), and their residue specific interactions with proteasomes have not been reported before. To allow a more detailed characterization of proteasome function, we selected a proteasome phosphatase UBLCP1 as the PIP for integrative structural modeling to identify its potential binding sites at the proteasome.
UBLCP1 is the only phosphatase in human that contains a UBL domain, which is located at the N terminus of the protein followed by a flexible linker region and a C-terminal phosphatase domain (45). In vitro binding assays demonstrated that UBLCP1 selectively binds Rpn1 among all 19S subunits, consistent with the role of Rpn1 as a UBL receptor (10). UBLCP1 has been shown to regulate proteasome activity in the nucleus in a phosphatase-dependent manner (45), but how exactly it binds and dephosphorylates the 26S proteasome remains unknown. To provide insights into these questions, we carried out in vitro DSSO cross-linking experiments on reconstituted UBLCP1-Rpn1 complex, which identified 29 cross-links between UBLCP1 and Rpn1 (supplemental Table S4). 27 of the cross-links mapped to the phosphatase domain (residues 133–294) of UBLCP1, whereas 2 mapped to the linker region (residues 82–132). From the Rpn1 perspective, 27 cross-links mapped to the residues in proximity of the two binding sites for ubiquitin and ubiquitin-like domains (i.e. the T1 and T2 sites) (10) and one cross-link mapped to a distant residue, Lys66. This cross-linking data, as well as our previous finding that Lys44 located in the UBL domain is critical for Rpn1-UBLCP1 interaction (45), suggests that UBLCP1 uses its UBL domain to interact with one or both of the two known ubiquitin-binding sites on Rpn1.
To dissect the interaction between the 26S proteasome and UBLCP1 in more detail, we superimposed the cross-links of the reconstituted UBLCP1-Rpn1 complex onto existing structural models of the 26S proteasome and UBLCP1 (Supplemental Methods, Fig. 6). The UBLCP1 model was calculated based on the structure of its homolog from D. melanogaster (60% sequence identity). The template UBLCP1 structure was likely solved in the auto-inhibited conformation, because a part of the linker between the two domains binds and blocks the access to its active site. Moreover, placing the UBLCP1 comparative model onto T1 or T2 site of Rpn1 according to the structure of the Rpn1-diubiquitin complex positions the phosphatase domain away from any of the 26S subunits. Therefore, large changes in the relative arrangement of the two domains through conformational changes in the linker region are likely required not only for UBLCP1 activation but also for its phosphatase domain to access the target sites on the proteasome. Alternatively, UBLCP1 might bind to a different site on Rpn1 (or a different subunit), but this scenario is less likely given the absence of cross-links to alternative binding sites.
Fig. 6.

Structural model of the human 26S proteasome in complex with proteasome-interacting protein UBLCP1. Proposed structural model of the human 26S proteasome (gray and brown Rpn1) in complex with UBLCP1 (orange and red densities for models bound to T1 and T2, respectively). Putative binding sites for coiled-coil proteins SCOC and SSNA1 on Rpt6 are depicted with cyan spheres.
To uncover the potential active conformation(s) of UBLCP1 associated with the 26S proteasome, we turned to integrative modeling (Supplemental Methods, Fig. 6). We started by representing UBLCP1 as a set of two rigid bodies for the two domains, connected by a flexible linker. The ubiquitin-like domain was docked onto the T1 or T2 site of Rpn1 (10), according to the structure of the Rpn1-diubiquitin complex, whereas the phosphatase domain and the flexible linker were placed randomly. The flexible linker, the rigid phosphatase domain, and parts of the proteasome without structural information were the only segments allowed to move in our simulations. An ensemble of models that maximally satisfies the cross-linking and excluded volume restraints was calculated using Monte Carlo sampling with simulated annealing (supplemental Fig. S6). Although the resulting solutions are relatively precise (5.1 and 9.2 Å for models based on the T1 and T2 site, respectively), they only respectively satisfy 64 and 61% of the cross-links, suggesting that the position and conformation of UBLCP1 are dynamic. Coincidentally, the dynamic nature of UBLCP1 interaction with the human 26S proteasome has been previously elaborated through MAP-SILAC and PAM-SILAC based affinity purification and mass spectrometry approaches (57). Based on the dynamic nature of UBLCP1 and its long linker region, we hypothesize that the phosphatase domain of UBLCP1 may reach up to ∼150 Å away (considering fully extended conformation of the 50-amino-acid-long intervening linker region) from the T1/T2 binding sites in the proteasome. Clearly, future studies would be needed to test such hypothesis to fully understand the regulation of the human 26S proteasome by UBLCP1.
Identification and Validation of the 26S Interaction with Two Novel PIPs
The two selected novel PIPs for further analysis are SCOC (Short coil-coil protein) and SSNA1 (Sjoegren syndrome nuclear autoantigen 1). SCOC was recently identified as a positive regulator of starvation-induced autophagy (58, 59), presumably via its interaction with FEZ1 (fasciculation and elongation protein zeta 1), an inhibitor of the autophagy induction. SSNA1 is a putative coiled-coil protein and is involved in regulating cell division and cytokinesis as well as adult axonal development, presumably by interacting and modulating spastin, a microtubule-severing AAA ATPase (60). SCOC and SNNA1 were determined to interact with the 26S proteasome directly through a single cross-link with the coiled-coil region of Rpt6 respectively, i.e. SCOC:Lys122-Rpt6:Lys38 and SSNA1:Lys40-Rpt6:Lys55 (Supplemental Table 1). To confirm these interactions, we have performed biochemical validations. First, we generated HB-tagged SCOC and SSNA1 constructs and expressed them in 293 cells. Reciprocal HB-tag based purification was carried out using either SCOC-HB or SSNA1-HB as the bait. Immunoblotting analysis of the respective SCOC and SSNA1 copurified proteins revealed that they both captured the selected proteasome subunits Rpt6, Rpt1, and α4 and SCOC also captured α7/Pre10 (supplemental Fig. S7), suggesting that they interact with proteasome complexes. However, it seems that the amount of copurified Rpt6 appears to be significantly more abundant in comparison to the other three proteasome subunits, suggesting that Rpt6 is the major interactor for the two novel PIPs. These results correlate well with our XL-MS studies as SCOC and SSNA1 were only identified from Rpt6 purified samples, and not from other four proteasome baits. To further confirm the physical interactions of SCOC and SSNA1 with proteasomes, we performed in vitro DSSO cross-linking of affinity purified SCOC and SSNA1, respectively. MSn analysis has determined that Lys40 of SSNA1 cross-links with Lys55 of Rpt6, and that Lys122 of SCOC cross-links with Lys38 of Rpt6 (supplemental Table S5). These cross-links are identical to those previously identified from Rpt6 purified samples as described above (supplemental Table S2). This represents that protein interactions can be validated by both biochemical approaches and cross-linking experiments.
DISCUSSION
In this work, we have explored in vitro and in vivo DSSO cross-linking strategies to obtain an interaction topology map of the human 26S proteasomes containing a total of 67 intersubunit pair-wise interactions. Our results represent the first and most comprehensive cross-link dataset for the human 26S proteasome to date, as previous reports have mainly focused on yeast proteasomes (6, 27, 40, 61). In addition, this work describes both in vitro and in vivo cross-linking studies of proteasome complexes, which resulted in a comparable number of cross-links and a significant overlap of pair-wise intersubunit interactions. This is not entirely unexpected because the core structures of the 26S proteasomes are known to be stable. Interestingly, some specific lysine-lysine linkages within a given intersubunit interaction vary with experimental approaches (supplemental Table S2). Combination of the two approaches significantly increases the coverage of XL-MS experiments, thus yielding a comprehensive interaction network topology of the human 26S proteasome. However, in vitro analysis often yields more cross-link data than in vivo analysis, mainly because of differences in sample preparation and subsequent recovery of resulting proteasome complexes. For in vitro XL-MS analysis, human proteasomes were purified with one step under native conditions prior to cross-linking, whereas for in vivo studies, 2-step denaturing purification of proteasomes was performed after cross-linking of intact cells. However, in vivo cross-linking has proven to be more advantageous in capturing dynamic, weak and transient interactions (46, 62–64), and identification of in vivo cross-linked peptides can help unravel protein interaction topology and architecture of protein complexes as they occur in cells (34). Although it remain technically challenging to characterize in vivo cross-linked protein complexes because of low abundance, we have demonstrated that it is feasible to identify protein interactions of proteasome complexes from cross-linked cells without enrichment of cross-linked peptides. Our work further exhibits the effectiveness of the DSSO based XL-MS workflow that can be employed not only for in vitro, but also in vivo cross-linking studies, ultimately expanding its usage for probing protein-protein interactions in general.
Correlation analysis has revealed that our 26S model based on the lower-resolution EM map shares high similarity with those obtained from the two new higher-resolution EM structures. Importantly, our work has elucidated the dynamics of the three 19S subunits Rpn1, Rpn6 and Rpt6 for the first time by EM analysis and/or XL-MS data. The variance map of the human proteasome illustrates the degree of flexibility of Rpn1 and Rpn6 compared with other proteasomal subunits. Rpn1 flexibility was observed to be much more dominant in human 26S than seen in yeast (4) through its interaction with the coiled-coils of the ATPases Rpt1 and Rpt2. Rpn1 movement may be governed by the cycle of ATP hydrolysis, functioning as a transient docking station for diverse PIPs such as shuttling factors and deubiquitinating enzymes. The up- and down- conformations of Rpn1 may have an advantage in recruiting those PIPs. Indeed, it is also possible that the conformational change helps recruit substrates to one of the intrinsic proteasome ubiquitin receptors, Rpn10 or Rpn13. In addition, the focused classification of Rpn6 revealed an unexpected flexibility at its N-terminal region. Recent EM analyses showed that the N-terminal α-solenoid domain of Rpn6 undergoes a prominent conformational rearrangement to be incorporated into the holocomplex (65, 66). Thus, the flexibility of Rpn6 plays an important role to regulate the proteasome function by hinging the CP and the RP.
In addition to Rpn1 and Rpn6, XL-MS experiments have revealed the dynamics of Rpt6 in the 26S proteasome structure based on violating cross-links found in both intraprotein and interprotein interactions. Interestingly, more out-of-range cross-links involving Rpt6 were identified from the in vivo XL-MS strategy compared with in vitro analysis. This suggests that Rpt6 and its interacting proteasome subunits may adopt a wider range of possible conformations in cells, which would be better preserved and captured via in vivo cross-linking. The differences in conformational sampling could also be induced by proteasome interacting proteins and posttranslational modifications. Although much more complex samples are analyzed, in vivo XL-MS experiments clearly can provide unique structural information of protein complexes in their native environment. The DSSO-based in vivo XL-MS workflow established here will enable us to further explore structural dynamics of protein complexes under different physiological conditions in cells.
Rpn13 functions as a proteasome ubiquitin receptor, and the identification of multiple cross-links between Rpn13 and ubiquitin (Ub) (supplemental Table S2) has provided direct physical contacts to validate their close relationship. The common and dominant interactions identified in both in vitro and in vivo XL-MS experiments are the two interlinks: [Rpn13:Lys34-Ub:Lys48] and [Rpn13:Lys99-Ub:Lys6]. In comparison to in vitro experiments, two additional linkages between Rpn13 and Ub were identified in in vivo experiments, describing additional interactions of Lys42 of Rpn13 with Lys48 of Ub and Lys97 of Rpn13 with Lys63 of Ub. These results suggest that the N terminus of Rpn13 may be positioned nearby to both proximal and distal Ub components in ubiquitin chains, although it has been suggested that Rpn13 prefers binding to K48-linked chains based on its structure with monoubiquitin (67). It has also been hypothesized that Rpn13 can work cooperatively with proteasome ubiquitin receptor Rpn10 to bind polyubiquitin chains, facilitating the proper docking of ubiquitinated substrates to proteasomes prior to their degradation (68). In this work, we have identified a total of 5 interlinks between Rpn10 and Ub, in which in vitro studies captured two unique linkages between Lys106 of Rpn10 to Lys6 and Lys48 of Ub, and in vivo experiments identified interactions between K48 of Ub and Lys74, Lys81, and Lys103 of Rpn10, respectively (supplemental Table S2). Interestingly, the three lysines in Rpn10 that were found to interact with Ub are located at the VWA domain (AA 5–188) and not from its UIM1 (AA 211–230) or UIM2 (AA 287–291) domains. It has been suggested that the VWA domain of Rpn10 is involved in maintaining 19S RP stability, and extensively associates with proteasome subunits in the 19S lid (5, 6, 69). Our work has identified several cross-links of Rpn10 VWA domain with Rpn8 and Rpt5 respectively. The close contact between Rpn10 VWA domain and Rpn8 is expected given that both Rpn10 and Rpn8 directly interact with Rpn11. In addition, the spatial distances of Rpn10-Rpn8 cross-links are well within expected cross-linking distance (supplemental Table S2). Because of the positioning of Rpn10 between the lid and base structures of the 19S RP (5, 6), the Rpn10-Rpt5 interaction is not completely unexpected. However, previous EM analysis has revealed that yeast Rpn10 UIM domain projects internally toward Rpt4/Rpt5 (68). The distances between Rpn10 and Rpt5 cross-linked lysine residues are ∼50 Å (supplemental Table S2), beyond the expected range (35 Å). This implies that the N terminus of Rpn10 may be more flexible than anticipated, capable of assuming multiple positions within the 26S holocomplex. The lack of cross-links between Rpn10 UIMs with Ub is more likely attributed to the sparse distribution of lysines around their interaction interfaces and/or their cross-linkability. Nonetheless, our results provide new insights on how Rpn13 and Rpn10 may interact with various Ub chains across multiple sites. It is noted that most interaction interfaces involving ubiquitin chains are composed of hydrophobic cores, resulting in a low number of surrounding lysine residues available for cross-linking. Therefore, cross-linking reagents targeting other amino acid residues such as acidic residues (42) would be desired for future studies to further explore the binding of ubiquitin receptors with ubiquitin chains in cells.
Apart from proteasome itself, we have identified 11 PIPs with residue specific interactions with proteasome subunits for the first time. Although some of the identified PIPs have known functions, their detailed mechanisms of action at the proteasome were thus far unknown. For example, it remains unclear how UBLCP1 modulates the phosphorylation status of proteasome complexes to affect proteasome function. Based on our cross-linking data and structural modeling, we propose a model of the UBLCP1 regulation of the proteasome in which binding of free and inactive UBLCP1 to Rpn1 via its ubiquitin-like domain causes large conformational changes in its structure, in turn activating its phosphatase domain. The long and flexible linker between the UBLCP1 domains as well as multiple binding sites on Rpn1 may allow for modification of several residues in the 26S subunits. Dephosphorylation of the proteasome by UBLCP1 (and perhaps also UBLCP1 binding itself) may impair proteasomal activity by disassociating the 19S and 20S particles as reported before (45), by causing unproductive conformational changes in the 26S proteasome (e.g. changes in position of Rpn1 observed by cryo-EM), or by affecting the association of other cellular factors.
In this work, we identified 6 novel PIPs (i.e. SCOC, SSNA1, PTGES3, SEPT4, CCDC92, and TIAM1) with unknown biological implications of their interactions with proteasomes. Whether these proteins modulate proteasomal activity, recruit specific substrate proteins to the proteasome, or are simply proteasome substrates with specific binding sites on the proteasome remains to be seen. Among them, we have further validated the interactions of SCOC and SSNA1 with proteasome via biochemical approaches and reciprocal XL-MS experiments. Perfect agreement was observed in the identified cross-links between SCOC and SSNA1 with Rpt6, demonstrating the effectiveness of XL-MS studies in uncovering protein-protein interactions. Identification of cross-links between a pair of proteins could potentially eliminate the need for further biochemical validation of interacting proteins, which has been essential for conventional AP-MS experiments.
Structural prediction analysis using PairCoil2 (70) determined that c-terminal amino acids 79–142 of SCOC and the c-terminal amino acids 14–71 of SNNA1 adopt a coiled-coil domain. It is interesting to note that CCDC92 contains a coiled-coil domain as well. All Rpt subunits also contain a coiled-coil domain at their N termini, which extend away from the 19S base toward the lid subcomplex and have been suggested to be critical in maintaining the defined order of the ATPase ring. These domains are likely to be further involved in substrate recognition and maintaining the interaction between lid and base subcomplexes (71). Deletion of the N-terminal 40 AAs of Rpt5 or 50AAs of Rpt6 in yeast was sufficient to impair yeast growth, demonstrating the importance of N-terminal coiled-coil regions of Rpt subunits for normal proteasome function (71). Interestingly, SCOC and SSNA1 were found to interact with Lys38 and Lys55 of Rpt6 respectively, right at the coiled-coil region of Rpt6. The amino acids in SCOC and SSNA1 (amino acid 122 and 40, respectively), which form interactions with Rpt6, lie in the predicted coiled-coil region as well. In reciprocal XL-MS experiments using tagged SCOC and SSNA1 as baits, we also identified cross-links between SCOC and SSNA1 to Hsp70A. Hsp70 has been shown to bind to the 19S RP and play a role in maintaining the 26S proteasome assembly upon oxidative stress (72). Moreover, SSNA1 can pull down SCOC by AP-MS as shown in our work and another report (73). Collectively, we suspect that SCOC and SSNA1 may play an important role in assisting the structure and function of the 19S RP. However, whether they work redundantly or cohesively requires further elucidation. In comparison to SSNA1 and SCOC, CCDC92 was determined to directly interact with Rpn12, which confirms a recent large-scale AP-MS report that indicates that CCDC92 copurified with proteasomal components (73). Rpn12 is an essential proteasomal subunit that is crucial for the complete assembly of the 19S lid and its subsequent incorporation with the base to form the 26S holocomplex (74). Because of the importance of coiled-coil structure in proteasome assembly and function, the binding of an additional coiled-coil motif to the proteasome may disrupt its activity, as suggested by the experiments with the Rpt coiled-coil peptide mimetics that inhibit the proteasome (71). Nonetheless, further studies are needed to clarify the link of the known functions of these coiled-coil PIPs with the proteasome modulation.
CONCLUSIONS
We have established new DSSO-based in vitro and in vivo XL-MS workflows by coupling with HB-tag based affinity purification strategies, which have been successfully employed to dissect the interaction and structure of the human 26S proteasome. In comparison, both XL-MS approaches contributed significantly to the elucidation of proteasome architectures because of its stable core structure. However, in vivo analysis enabled the capture of diverse protein conformations in cells to reveal protein structural dynamics. Our results allowed us to assemble the largest subunit connectivity map of the human 26S proteasome. In combination with EM and structural modeling, we have defined the dynamics of three proteasome subunits attributed to interaction and/or conformational dynamics. The identification of known and novel PIPs with specific linkages to proteasome subunits help us understand their potential functions and their action mechanisms at the proteasome. This study has established a solid foundation for future studies to define structural dynamics of the human 26S proteasome under different physiological conditions. Given the recent commercialization of DSSO, the XL-MS strategies presented here will have a broad impact on cross-linking studies and can be directly applied to probe other protein complexes in vitro and in living cells.
DATA AVAILABILITY
Raw data has been deposited at the MassIVE repository FTP site: ftp://MSV000080313@massive.ucsd.edu (Password: LH26SXL110416).
Supplementary Material
Acknowledgments
We thank members of the Huang laboratory, especially David Vong, as well as Antje Aufderheide and Till Rudack for their help during this study. We would like to thank Prof. A.L. Burlingame and Robert Chalkley at UCSF for their support of the development version of Protein Prospector.
Footnotes
Author contributions: L.H. conceived the project. X.W., P.C., C.Y., E.S., X.G., W.B., Andrej S., and L.H. designed the experiments. X.W. and Y.Y. generated constructs, stable cell lines and performed in vitro and in vivo XL-MS experiments of proteasomes. X.W. performed biochemical validation. C.Y. and L.H. performed LC MSn analysis. C.Y. performed some in vitro XL-MS experiments. C.Y., A.S.H., X.W., Y.Y., and L.H. analyzed XL-MS data. P.C., N.C., and C.G. performed integrative modeling and structural mapping. A.S., F.B., and E.S. performed EM analysis and data interpretation. X.G. and J.E. prepared UBLCP1-related samples for XL-MS experiments. E.J.N. and S.D.R. contributed chemical reagents. A.L. and P.N. assisted in MS data acquisition. A.K. contributed to initial data processing. J.D. and R.A. provided analytical tools for the project. X.W., P.C., C.Y., N.C., E.S., and L.H. contributed to writing the manuscript. All authors contributed to editing the manuscript.
* This work was supported by National Institutes of Health grants RO1GM074830 to L.H., R01GM106003 to L.H. and S. R., R01GM083960 and P41GM109824 to Andrej S. and DK018849-36 to J.E.D. E.J.N. was supported by an institutional Chemical and Structural Biology Training Grant predoctoral fellowship (T32-GM10856). X.G. was supported by a Susan G. Komen postdoctoral fellowship (KG111280). R.A. was supported by ERC AdG “Proteomics v.3.0” (grant 233226) and AdG 679821 “Proteome in context”. E.S. and W.B. were supported by the German Science Foundation Excellence Cluster Center for Integrated Protein Science Munich and Collaborative Research Center (SFB)-1035/Project A01. E. S. is supported by Marie Curie CIG (proteAmics). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- UPS
- ubiquitin proteasome system
- XL-MS
- cross-linking mass spectrometry
- CP
- core particle
- RP
- regulatory particle
- UBL
- ubiquitin-like domain
- UBA
- ubiquitin associating domain
- AP-MS
- affinity purification mass spectrometry
- PIP
- proteasome interacting proteins
- HB
- histidine and biotin tag
- LC MSn
- liquid chromatography multistage tandem mass spectrometry.
REFERENCES
- 1. Voges D., Zwickl P., and Baumeister W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 [DOI] [PubMed] [Google Scholar]
- 2. Löwe J., Stock D., Jap B., Zwickl P., Baumeister W., and Huber R. (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science 268, 533–539 [DOI] [PubMed] [Google Scholar]
- 3. Groll M., Ditzel L., Lowe J., Stock D., Bochtler M., Bartunik H. D., and Huber R. (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386, 463–471 [DOI] [PubMed] [Google Scholar]
- 4. Beck F., Unverdorben P., Bohn S., Schweitzer A., Pfeifer G., Sakata E., Nickell S., Plitzko J. M., Villa E., Baumeister W., and Forster F. (2012) Near-atomic resolution structural model of the yeast 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 109, 14870–14875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lander G. C., Estrin E., Matyskiela M. E., Bashore C., Nogales E., and Martin A. (2012) Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lasker K., Forster F., Bohn S., Walzthoeni T., Villa E., Unverdorben P., Beck F., Aebersold R., Sali A., and Baumeister W. (2012) Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Husnjak K., Elsasser S., Zhang N., Chen X., Randles L., Shi Y., Hofmann K., Walters K. J., Finley D., and Dikic I. (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X., Lee B. H., Finley D., and Walters K. J. (2010) Structure of proteasome ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Mol. Cell. 38, 404–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elsasser S., Chandler-Militello D., Muller B., Hanna J., and Finley D. (2004) Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822 [DOI] [PubMed] [Google Scholar]
- 10. Shi Y., Chen X., Elsasser S., Stocks B. B., Tian G., Lee B. H., Zhang N., de Poot S. A., Tuebing F., Sun S., Vannoy J., Tarasov S. G., Engen J. R., Finley D., and Walters K. J. (2016) Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351, pii: aad9421. doi: 10.1126/science.aad9421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verma R., Aravind L., Oania R., McDonald W. H., Yates J. R. I., Koonin E. V., and Deshaies R. J. (2002) Role of Rpn11 Metalloprotease in Deubiquitination and Degradation by the 26S Proteasome. Science 298, 611–615 [DOI] [PubMed] [Google Scholar]
- 13. Yao T., and Cohen R. E. (2002) A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 419, 403–407 [DOI] [PubMed] [Google Scholar]
- 14. Pathare G. R., Nagy I., Sledz P., Anderson D. J., Zhou H. J., Pardon E., Steyaert J., Forster F., Bracher A., and Baumeister W. (2014) Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc. Natl. Acad. Sci. U.S.A. 111, 2984–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Worden E. J., Padovani C., and Martin A. (2014) Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat Struct Mol Biol. 21, 220–227 [DOI] [PubMed] [Google Scholar]
- 16. Zhang F., Hu M., Tian G., Zhang P., Finley D., Jeffrey P. D., and Shi Y. (2009) Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 34, 473–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. da Fonseca P. C., He J., and Morris E. P. (2012) Molecular model of the human 26S proteasome. Mol Cell. 46, 54–66 [DOI] [PubMed] [Google Scholar]
- 18. Schweitzer A., Aufderheide A., Rudack T., Beck F., Pfeifer G., Plitzko J. M., Sakata E., Schulten K., Forster F., and Baumeister W. (2016) Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. U.S.A 113, 7816–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang X., Luan B., Wu J., and Shi Y. (2016) An atomic structure of the human 26S proteasome. Nat Struct Mol Biol 23, 778–785 [DOI] [PubMed] [Google Scholar]
- 20. Matyskiela M. E., Lander G. C., and Martin A. (2013) Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol. 20, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forster F., Unverdorben P., Sledz P., and Baumeister W. (2013) Unveiling the long-held secrets of the 26S proteasome. Structure 21, 1551–1562 [DOI] [PubMed] [Google Scholar]
- 22. Unverdorben P., Beck F., Sledz P., Schweitzer A., Pfeifer G., Plitzko J. M., Baumeister W., and Forster F. (2014) Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 111, 5544–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deveraux Q., Ustrell V., Pickart C., and Rechsteiner M. (1994) A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 269, 7058–7061 [PubMed] [Google Scholar]
- 24. Elsasser S., Gali R. R., Schwickart M., Larsen C. N., Leggett D. S., Muller B., Feng M. T., Tubing F., Dittmar G. A., and Finley D. (2002) Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol. 4, 725–730 [DOI] [PubMed] [Google Scholar]
- 25. Leggett D. S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R. T., Walz T., Ploegh H., and Finley D. (2002) Multiple associated proteins regulate proteasome structure and function. Mol. Cell. 10, 495–507 [DOI] [PubMed] [Google Scholar]
- 26. Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F. U., Ban N., Malmstrom L., and Aebersold R. (2012) Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352 [DOI] [PubMed] [Google Scholar]
- 27. Kao A., Randall A., Yang Y., Patel V. R., Kandur W., Guan S., Rychnovsky S. D., Baldi P., and Huang L. (2012) Mapping the structural topology of the yeast 19S proteasomal regulatory particle using chemical cross-linking and probabilistic modeling. Mol. Cell. Proteomics 11, 1566–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi Y., Fernandez-Martinez J., Tjioe E., Pellarin R., Kim S. J., Williams R., Schneidman D., Sali A., Rout M. P., and Chait B. T. (2014) Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol. Cell. Proteomics 13, 2927–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erzberger J. P., Stengel F., Pellarin R., Zhang S., Schaefer T., Aylett C. H., Cimermancic P., Boehringer D., Sali A., Aebersold R., and Ban N. (2014) Molecular architecture of the 40SeIF1eIF3 translation initiation complex. Cell 158, 1123–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi Y., Pellarin R., Fridy P. C., Fernandez-Martinez J., Thompson M. K., Li Y., Wang Q. J., Sali A., Rout M. P., and Chait B. T. (2015) A strategy for dissecting the architectures of native macromolecular assemblies. Nat. Methods 12, 1135–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chavez J. D., Schweppe D. K., Eng J. K., Zheng C., Taipale A., Zhang Y., Takara K., and Bruce J. E. (2015) Quantitative interactome analysis reveals a chemoresistant edgotype. Nat. Commun. 6, 7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng C., Yang L., Hoopmann M. R., Eng J. K., Tang X., Weisbrod C. R., and Bruce J. E. (2011) Cross-linking measurements of in vivo protein complex topologies. Mol. Cell. Proteomics 10, M110 006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chavez J. D., Weisbrod C. R., Zheng C., Eng J. K., and Bruce J. E. (2013) Protein interactions, post-translational modifications and topologies in human cells. Mol. Cell. Proteomics 12, 1451–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaake R. M., Wang X., Burke A., Yu C., Kandur W., Yang Y., Novtisky E. J., Second T., Duan J., Kao A., Guan S., Vellucci D., Rychnovsky S. D., and Huang L. (2014) A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol. Cell. Proteomics 13, 3533–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Forster F., Beck M., and Aebersold R. (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods. 9, 901–903 [DOI] [PubMed] [Google Scholar]
- 36. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., and Dong M. Q. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods. 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 37. Chu F., Baker P. R., Burlingame A. L., and Chalkley R. J. (2009) Finding Chimeras: A bioinformatic strategy for identification of cross-linked peptides. Mol. Cell. Proteomics 9, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A. L., and Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu F., Rijkers D. T., Post H., and Heck A. J. (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods. 12, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 40. Kao A., Chiu C. L., Vellucci D., Yang Y., Patel V. R., Guan S., Randall A., Baldi P., Rychnovsky S. D., and Huang L. (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics 10, M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu C., Kandur W., Kao A., Rychnovsky S., and Huang L. (2014) Developing new isotope-coded mass spectrometry-cleavable cross-linkers for elucidating protein structures. Anal Chem. 86, 2099–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gutierrez C. B., Yu C., Novitsky E. J., Huszagh A. S., Rychnovsky S. D., and Huang L. (2016) Developing an acidic residue reactive and sulfoxide-containing MS-cleavable homobifunctional cross-linker for probing protein-protein interactions. Anal. Chem. 88, 8315–8322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu J., Yu C., Hu X., Kim J. K., Bierma J. C., Jun H. I., Rychnovsky S. D., Huang L., and Qiao F. (2015) Dissecting Fission Yeast Shelterin Interactions via MICro-MS Links Disruption of Shelterin Bridge to Tumorigenesis. Cell Rep. 12, 2169–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X., Chen C. F., Baker P. R., Chen P. L., Kaiser P., and Huang L. (2007) Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565 [DOI] [PubMed] [Google Scholar]
- 45. Guo X., Engel J. L., Xiao J., Tagliabracci V. S., Wang X., Huang L., and Dixon J. E. (2011) UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc. Natl. Acad. Sci. U.S.A. 108, 18649–18654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu C., Yang Y., Wang X., Guan S., Fang L., Liu F., Walters K. J., Kaiser P., and Huang L. (2016) Characterization of dynamic UbR-proteasome subcomplexes by in vivo cross-linking (X) assisted bimolecular tandem affinity purification (XBAP) and label-free quantitation. Mol. Cell. Proteomics 15, 2279–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fang L., Wang X., Yamoah K., Chen P. L., Pan Z. Q., and Huang L. (2008) Characterization of the human COP9 signalosome complex using affinity purification and mass spectrometry. J. Proteome Res. 7, 4914–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aufderheide A., Beck F., Stengel F., Hartwig M., Schweitzer A., Pfeifer G., Goldberg A. L., Sakata E., Baumeister W., and Forster F. (2015) Structural characterization of the interaction of Ubp6 with the 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 112, 8626–8631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bohn S., Beck F., Sakata E., Walzthoeni T., Beck M., Aebersold R., Forster F., Baumeister W., and Nickell S. (2010) Structure of the 26S proteasome from Schizosaccharomyces pombe at subnanometer resolution. Proc. Natl. Acad. Sci. U.S.A. 107, 20992–20997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He J., Kulkarni K., da Fonseca P. C., Krutauz D., Glickman M. H., Barford D., and Morris E. P. (2012) The structure of the 26S proteasome subunit Rpn2 reveals its PC repeat domain as a closed toroid of two concentric alpha-helical rings. Structure 20, 513–521 [DOI] [PubMed] [Google Scholar]
- 51. Guerrero C., Tagwerker C., Kaiser P., and Huang L. (2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell. Proteomics 5, 366–378 [DOI] [PubMed] [Google Scholar]
- 52. Luo J., Cimermancic P., Viswanath S., Ebmeier C. C., Kim B., Dehecq M., Raman V., Greenberg C. H., Pellarin R., Sali A., Taatjes D. J., Hahn S., and Ranish J. (2015) Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol Cell. 59, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu C. W., Li X., Thompson D., Wooding K., Chang T. L., Tang Z., Yu H., Thomas P. J., and DeMartino G. N. (2006) ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell. 24, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 55. Trabuco L. G., Villa E., Mitra K., Frank J., and Schulten K. (2008) Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tomko R. J. Jr, Funakoshi M., Schneider K., Wang J., and Hochstrasser M. (2010) Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol. Cell. 38, 393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang X., and Huang L. (2008) Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell. Proteomics 7, 46–57 [DOI] [PubMed] [Google Scholar]
- 58. Joachim J., Wirth M., McKnight N. C., and Tooze S. A. (2012) Coiling up with SCOC and WAC: two new regulators of starvation-induced autophagy. Autophagy 8, 1397–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McKnight N. C., Jefferies H. B., Alemu E. A., Saunders R. E., Howell M., Johansen T., and Tooze S. A. (2012) Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J. 31, 1931–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goyal U., Renvoise B., Chang J., and Blackstone C. (2014) Spastin-interacting protein NA14/SSNA1 functions in cytokinesis and axon development. PLoS ONE 9, e112428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Politis A., Stengel F., Hall Z., Hernandez H., Leitner A., Walzthoeni T., Robinson C. V., and Aebersold R. (2014) A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods. 11, 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guerrero C., Milenkovic T., Przulj N., Kaiser P., and Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 13333–13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kaake R. M., Milenkovic T., Przulj N., Kaiser P., and Huang L. (2010) Characterization of cell cycle specific protein interaction networks of the yeast 26S proteasome complex by the QTAX strategy. J. Proteome Res. 9, 2016–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fang L., Kaake R. M., Patel V. R., Yang Y., Baldi P., and Huang L. (2012) Mapping the protein interaction network of the human COP9 signalosome complex using a label-free QTAX strategy. Mol. Cell. Proteomics 11, 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luan B., Huang X., Wu J., Mei Z., Wang Y., Xue X., Yan C., Wang J., Finley D. J., Shi Y., and Wang F. (2016) Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proc. Natl. Acad. Sci. U.S.A. 113, 2642–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tomko R. J. Jr, Taylor D. W., Chen Z. A., Wang H. W., Rappsilber J., and Hochstrasser M. (2015) A single alpha helix drives extensive remodeling of the proteasome lid and completion of regulatory particle assembly. Cell 163, 432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schreiner P., Chen X., Husnjak K., Randles L., Zhang N., Elsasser S., Finley D., Dikic I., Walters K. J., and Groll M. (2008) Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453, 548–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sakata E., Bohn S., Mihalache O., Kiss P., Beck F., Nagy I., Nickell S., Tanaka K., Saeki Y., Forster F., and Baumeister W. (2012) Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc. Natl. Acad. Sci. U.S.A. 109, 1479–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tomko R. J. Jr., and Hochstrasser M. (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 82, 415–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McDonnell A. V., Jiang T., Keating A. E., and Berger B. (2006) Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22, 356–358 [DOI] [PubMed] [Google Scholar]
- 71. Inobe T., and Genmei R. (2015) Inhibition of the 26S proteasome by peptide mimics of the coiled-coil region of its ATPase subunits. Biochem. Biophys. Res. Commun. 468, 143–150 [DOI] [PubMed] [Google Scholar]
- 72. Grune T., Catalgol B., Licht A., Ermak G., Pickering A. M., Ngo J. K., and Davies K. J. (2011) HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic Biol Med. 51, 1355–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huttlin E. L., Ting L., Bruckner R. J., Gebreab F., Gygi M. P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K., Dong R., Guarani V., Vaites L. P., Ordureau A., Rad R., Erickson B. K., Wuhr M., Chick J., Zhai B., Kolippakkam D., Mintseris J., Obar R. A., Harris T., Artavanis-Tsakonas S., Sowa M. E., De Camilli P., Paulo J. A., Harper J. W., and Gygi S. P. (2015) The BioPlex Network: A systematic exploration of the human interactome. Cell. 162, 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tomko R. J. Jr., and Hochstrasser M. (2011) Incorporation of the Rpn12 subunit couples completion of proteasome regulatory particle lid assembly to lid-base joining. Mol. Cell. 44, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data has been deposited at the MassIVE repository FTP site: ftp://MSV000080313@massive.ucsd.edu (Password: LH26SXL110416).