

Abstract
To date, Alzheimer's disease (AD) drug candidates have produced negative results in human trials and progress moving new targets out of the laboratory and into trials has been slow. However, based on three decades of previous work, there is reason to hope that amyloid-based and other novel therapies will move at a faster pace towards successful clinical trials. Here we highlight selected preclinical research topics that are rapidly advancing in the laboratory.
The amyloid cascade hypothesis
In 1907, experimental observations by Alois Alzheimer led to the identification of Aβ and tau pathologies, which are the main pathological constituents in Alzheimer's Disease (AD) (Alzheimer, 1987). Thereafter, familial studies identified mutations in APP, PSEN1 and PSEN2, that lead to misprocessing of Aβ and autosomal dominant AD (ADAD) that is typically of early onset 1,2. These advances led to the formulation of the amyloid cascade hypothesis proposed by Hardy and Higgins in 1992 3. According to this hypothesis, Aβ peptide, which originates from sequential proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase and the γ-secretase complex, is the causative agent of Alzheimer's pathology. In AD, Aβ42 is the more pathologic Aβ fragment (the predominant species being Aβ40), and more readily adopts misfolded conformers that are more prone to aggregate. Overproduction or reduced clearance of Aβ leads to the accumulation of different Aβ conformations i.e. oligomers, fibrils, that can block neuronal transmission and lead to synaptic loss and neuronal toxicity. Updated in 2002 to propose tau pathology as downstream of the original insult of Aβ accumulation 4, and then more recently in light of newly identified genetic risk variants 5, the amyloid hypothesis has been reevaluated to include apoE/cholesterol effects, inflammation, and endosomal recycling as additional cellular drivers downstream of initial Aβ deposition 6.
Though the evidence supporting different species of Aβ as causative of AD is strong, it is widely recognized that neurofibrillary tangle (NFT) pathology is a closer correlate of neuronal death and symptom manifestation in AD 7. Hyperphosphorylated tau is a major component of such NFTs and therefore how Aβ might influence tau phosphorylation is an important topic. Multiple lines of evidence support elaboration of the amyloid hypothesis suggesting that soluble Aβ oligomers cause subsequent tau toxicity. In recent reports, Aβ42 oligomers have been shown to activate AMP-activated kinase (AMPK), which is associated with tau accumulation in AD neurons 8. Using HeLa cell lines and in vivo mouse models, Mairet-Coello and colleagues demonstrated that AMPKα1 phosphorylates tau on Serine 262 in response to Aβ42 oligomers. Furthermore, investigators found that blocking tau phosphorylation at Serine 262 prevents neurons from Aβ42-mediated toxicity and subsequent dendritic spine loss induced by AMPKα1 phosphorylation. Similarly, Pooler and colleagues demonstrated that Aβ increases tau aggregation and propagation by utilizing immunofluorescence labeling, cell quantification, fluorescence in situ hybridization, and real-time PCR analysis methods in vivo in a mouse model (rTgTauEC × APP/PS1 mice) that expresses tau and Aβ. Their results showed that Aβ increases tau spreading in the cortex as well as tau-induced neuronal toxicity 9. The lack of tau pathology in most animal models has long been cited as a problem for the amyloid cascade hypothesis, but the current consensus view is that model systems with human rather than murine tau support this version of the amyloid hypothesis 6.
Synaptic loss and elaborations on the amyloid cascade hypothesis
Synaptic loss has long been associated with cognitive decline in AD10 and correlates strongly with soluble Aβ early in disease progression, differentiating high pathology control from AD cases 11,12. As the field has moved to accept a version of the amyloid hypothesis where the primary proximal toxin to neurons and synaptic function is soluble oligomeric Aβ 6, we and others have demonstrated accumulations of oligomeric Aβ within presynaptic boutons 13,14. Synaptic Aβ consists of multiple oligomeric species, both prefibrillar and fibrillar 12,14, and insoluble Aβ fibrils also accumulate in presynaptic terminals 15. The recent development of a PET ligand to quantify synaptic density in the living human brain should greatly advance AD molecular research as well as empower clinicians with a robust complementary diagnostic tool16.
Aβ deposits in plaques may in themselves be relatively benign, but are a potential source of soluble oligomeric Aβ, which directly causes neurotoxicity and neuronal death through post-synaptic interactions with glutamate receptors, cellular prion protein, neuroligin, Wnt, and insulin receptors 17,18. Complicating work in this area is the difficulty of accurate quantification of soluble amyloid oligomers, combined with the precarious structural stability that is the nature of oligomers. Recent studies have made use of an oligomer sandwich enzyme-linked immunosorbent assay (ELISA) using an anti-Aβ antibody as both capture and detection, which accurately quantifies Aβ oligomers with n > 2. With this assay, high pathology but nondemented control samples showed lower oligomer levels compared to AD samples 19, suggesting that cognition is impaired when soluble oligomers reach a threshold.
Consistent with this hypothesis and extending it into the synaptic compartment, recent experiments by Bilousova and colleagues used flow cytometry and confocal microscopy methods to measure Aβ and tau within synaptosomes of human cortex and rodent models to test the amyloid hypothesis 12. Consistent with a synaptic amyloid cascade, synaptic Aβ accumulation occurred early in AD prior to the appearance of phosphorylated tau. Synaptic phosphorylated tau was not prevalent until late stage AD compared to earlier stages in both human samples and rodent models. Most importantly, soluble Aβ oligomers in synaptic terminals were increased in early AD human synaptosomes compared with high pathology non-demented controls, suggesting that a threshold soluble synaptic oligomer level is associated with the onset of a dementia. Confocal images with conformation dependent monoclonal antibodies reveal that both prefibrillar (M55 antibody) and fibrillar Aβ oligomers accumulate in AD synapses (M116 antibody; Fig. 1).
Fig. 1.
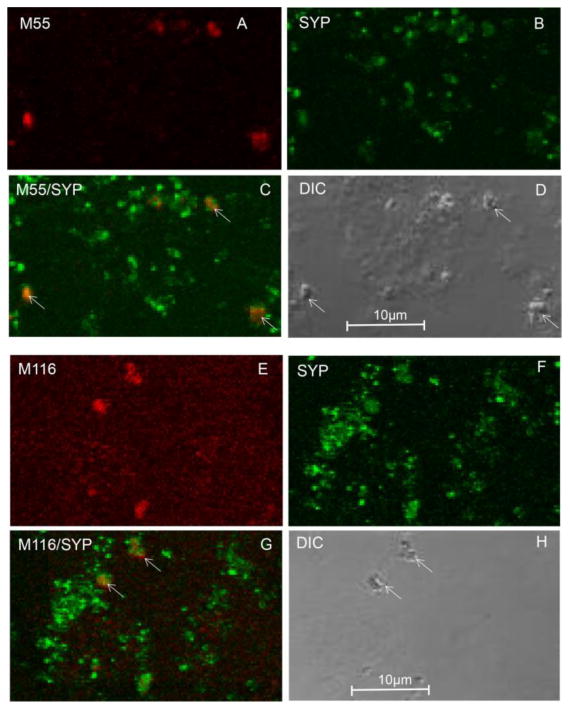
Conformation-dependent antibodies label Ab in synaptic terminals. (A–D) Synapto- somes were dual labeled with synaptophysin and the monoclonal antibody M55 directed against prefibrillar oligomers: (A) M55; (B) synaptophysin (SYP); (C) overlay image with ar- rows indicating colocalization; (D) differential interference contrast (DIC) image. (E–H) Synaptosomes dual labeled with synaptophysin and the monoclonal antibody M116 directed against fibrillar oligomers: (E) M116; (F) synaptophysin (SYP); (G) overlay image with arrows indicating colocalization; (H) DIC image.
Spread of tau pathology
A wide body of literature has shown that cytoskeletal protein tau accumulates in the brain leading to the formation of intracellular neurofibrillary tangles (NFTs), and it's long been known that tau pathology follows a progression suggestive of regional spread 20. More recent evidence has moved towards a model by which tau is released from neurons and may travel to surrounding neurons transsynaptically 21.
Recent data has greatly expanded understanding of specific cellular mechanisms driving the spread of tau pathology. For example, several lines of evidence demonstrate depolarization-dependent tau release. Pooler and colleagues utilized KCl-treated primary neuronal cultures to stimulate AMPA receptors to propagate action potentials and measured tau release with a sandwich ELISA. Investigators found that stimulation of healthy neurons causes tau release that is dependent on intracellular calcium and mediated by exosomes and membrane vesicle secretion22. Yamada and colleagues utilized in vivo microdialysis to measure tau from brain interstitial fluid of wild-type mice, demonstrating that presynaptic glutamate release drives tau release and neuronal activity increased extracellular tau 23. Experiments by our group expanded on the results from animal model systems into humans by measuring tau release from postmortem synaptosome preparations. KCl-mediated depoloarization markedly increased tau release from AD synaptosomes 24. Flow cytometry analysis also revealed the abundance of tau within presynaptic terminals, along with increased C-terminal truncated tau in AD. Similar experiments determining the exact tau peptide released in human AD will be important in guiding development of tau immunotherapies 25,26.
Exosomes are ∼100nm extracellular vesicles that are released by most cells, including those in the (CNS), and are thought to be derived from late endosomes termed multivesicular bodies 27. A good deal of evidence supports the hypothesis that extracellular and trans-synaptic tau spreading occurs via exosomes 28-31. Recent studies have also implicated microglia and microglial-derived exosomes in tau propagation 31,32. Alternatively, exosomes may also be a mechanism for sequestration of Aβ and misfolded proteins 33. More recently, exosome-associated total and p-tau in plasma have been suggested as predictors of incipient AD 34, suggesting promise for exosomes as circulating biomarkers of AD. Having a size in the low nanometer range means that there are significant technical barriers in the study of these particles; however, the potential impact on AD diagnosis and treatment has made this an active and dynamic areas of focus in the field of neurodegeneration.
ApoE and Cholesterol in AD
The APOE gene encodes an apolipoprotein that functions as a cholesterol carrier (apoE), and a good deal of evidence indicates that lipid metabolism modulates Aβ levels. More recently, a number of genes regulating lipid metabolism have been implicated in AD 35. Cholesterol levels in the brain are thought to play an important role in brain neuronal homeostasis with the distribution of cholesterol playing an important role 36. In the brain, cholesterol exists in two major pools: unesterified (free cholesterol), which accounts for 95% of cholesterol in the brain, and the esterified cholesterol, which constitutes 5% of total cholesterol. Free cholesterol resides in cell membranes and functions in cellular membrane fluidity, electrical transduction, structure, repair, and permeability 37. Conversely, esterified cholesterol is found intracellularly and is sequestered into lipid droplets. Esterified cholesterol is thought to function as cholesterol storage in the brain 38,39. HDL, which contains free cholesterol and esterified cholesterol, is produced by astrocytes and binds to apoE via lipidation by ABC transporters. It is subsequently transported to neurons via apoE for the purposes of membrane repair and cellular maintenance 40,41.
Multiple studies have confirmed the role free cholesterol plays in AD models. Schneider and colleagues demonstrated in neuronal cultures that reduced free cholesterol is associated with reduction in Aβ. Furthermore, when free cholesterol was increased, Aβ aggregation also increased 42. Concurrently, Ferrara and colleagues found that free cholesterol promotes Aβ-mediated neurotoxicity and is associated with a reduction of cell viability markers. Additionally, Abramov and colleagues used hippocampal neuronal cultures to demonstrate that increasing membrane cholesterol caused neuronal death when treated with Aβ 43.
In neurons, lipid rafts are membrane micro-domains that are enriched for free cholesterol and GM1 ganglioside and are thought to promote β-secretase cleavage of APP, producing the aggregation-prone Aβ42 44,45. Consistent with this long-held hypothesis, experiments by Djelti and colleagues observed significantly elevated free cholesterol when the mRNA of the metabolic enzyme CYp46a1, which regulates neuronal cholesterol content, was inhibited 46. When investigators isolated lipid rafts from mouse hippocampi on sucrose gradients, APP and Aβ peptide levels were increased and tau phosphorylation was observed. The increased cholesterol was accompanied by endosomal enlargement, and animals exhibited cognitive deficits and neuronal death.
Historically, most neuronal cholesterol literature has focused on free cholesterol due to its abundance. However, several studies have sought to manipulate neuronal cholesterol dynamics to investigate potential mechanisms in AD. For example, work by Kovacs and colleagues has shown that inhibition of acyl-coenzyme A:cholesterol acyltransferase (ACAT147), which catalyzes cholesterol ester formation, reduces Aβ generation via alteration of the intracellular distribution of cholesterol 36,48. For example, in mice containing the London and Swedish mutations, treatment with ACAT inhibitors reduced esterified cholesterol by 86%, and Aβ plaque level by 88%-99%, along with a slight improvement in spatial learning in mice 48. Similarly, Bryleva and colleagues eliminated ACAT1 in triple transgenic mice and saw a 60% reduction in human APP, and observed improved cognitive function in mice with cognitive deficits. In this work, lack of ACAT1 caused an increase in 24-S-OH and cholesterol 39. ACAT inhibition has also been shown to enhance autophagy and reduce tau in the triple transgenic AD mouse model 49. Taken together, this evidence suggests that the ratio of esterified cholesterol to free cholesterol is a primary determinant of AD neuropathology.
Another study investigated associations between neurotoxic Aβ fibrils, intracellular vesicular trafficking, and cholesterol homeostasis by labeling esterified cholesterol and free cholesterol in rat neuronal cell cultures 47. Neurotoxic Aβ fibril treatment increased free cholesterol, reduced esterified cholesterol, and increased vesicular exocytotic markers. These results suggest that Aβ fibrils alter cholesterol homeostasis through vesicular trafficking mechanisms and that free cholesterol may regulate vesicular trafficking that are involved in cellular cholesterol homeostasis. Another hypothesis postulates that Aβ fibrils bind membrane cholesterol and attenuate the conversion of free cholesterol to esterified cholesterol. Along this line, our group previously reported a marked increase in free cholesterol and ganglioside GM1 in Aβ-positive synaptosomes from AD cortex 50, another result that may reflect increased rafts in synaptic membrane, or the binding of cholesterol to synaptic Aβ.
In contrast, a study conducted by Hutter-Paier and colleagues showed that ACAT inhibition reduced esterified cholesterol levels by 89%, reduced accumulation of Aβ plaques by 88%-99%, reduced insoluble Aβ by 83%-96%, and reduced soluble Aβ by 34% in brain homogenates of transgenic mice expressing human APP 48. Furthermore, spatial learning and memory in mice improved and correlated with reduced Aβ levels. These results suggest that reduced esterified cholesterol through ACAT inhibition may reduce AD pathogenesis. Similarly, several studies demonstrate that ACAT1 deficiency in mice (ACAT-/-) results in reduced APP and Aβ levels 39,49. These studies imply that reduced esterified cholesterol levels prevent Aβ accumulation in AD. Overall, the literature concerning esterified cholesterol in AD is varied. Because cholesterol pool dynamics associated with AD pathogenesis remain unclear and difficult to measure due to fast turnover of cholesterol pools, more research is needed to investigate the potential mechanisms underlying cholesterol esterification. Furthermore, functional studies and imaging studies in total cholesterol versus esterified and free cholesterol turnover need to be conducted in human models.
In recent reports, cholesterol in AD has been found to be mediated by APOE. Arold and colleagues measured free cholesterol across apoE isoforms in synaptosomes from both human and targeted replacement mice expressing human apoE isoforms (apoETR). Consistent with the hypothesis that synaptic Aβ is cleared by apoE, apoE levels were increased in Aβ-positive synapses in normal samples compared to late AD samples, suggesting effective clearance in normal samples. Furthermore, the apoE2 isoform expressed higher levels of apoE and free cholesterol compared to apoE4 samples. These results suggest that the increased lipidation capacity of the apoE2 isoform is optimal for Aβ clearance compared to the apoE4 isoform 51. Additionally, Hu and colleagues demonstrate that apoETR mice expressing human apoE4 have reduced apoE-associated cholesterol and increased Aβ accumulation compared to mice expressing apoE3 and apoE2 isoforms 52. Adding important data from human subjects, Shinohara and colleagues show that the APOE2 allele is associated with lower incidence of cognitive decline during aging using retrospective clinical data 53. This was validated with experiments on aged apoE2TR mice that exhibited reduced synaptic loss and increased apoE levels when compared to either apoE3TR or apoE4TR mice. Additionally, free cholesterol was reduced among apoE2 mice, but not in apoE4 mice. Overall, despite the lack of consensus, there is strong experimental evidence suggesting APOE isoform mediates cholesterol dynamics contributing to pathological changes in AD.
Inflammation and the innate immune system in AD
ApoE and inflammation
A wide body of evidence suggests that aggregated proteins can initiate an immune response that can exacerbate neurodegenerative conditions such as AD. Injured neurons are thought to release distress signals, initiating a strong response from microglia, the immune cells of the CNS. This inflammatory response may itself injure neurons and synapses, but may ultimately protect circuits. Much work is needed to understand the early sequence of events that occur in vivo 54. ApoE, genetic variants of which contribute differentially to AD risk, is the predominant apolipoprotein present in the CNS. It is mainly produced by astrocytes and activated microglia in the brain 55. Recent lines of evidence make it clear that apoE is a key mediator of the immune response to stressors both in the brain and in the periphery 56,57.
Studying APOE effects in the periphery, Gale and colleagues evaluated the innate immune response to organ injury in sepsis using whole blood from healthy APOE3/3 and APOE3/4 humans. Healthy volunteers were given lipopolysaccharide (LPS) to induce an inflammatory response. The APOE3/4 volunteers exhibited higher temperatures, TNFα levels, and IL-6 levels compared to the APOE3/3 volunteers. This suggests that APOE genotype modulates immune response to TLR signaling during systemic stressors in the periphery 57. Similarly, Ringman and colleagues analyzed 77 plasma signaling proteins in a small cohort of ADAD subjects with PSEN1 or APP mutations (mean age, 33 - 42 years, mostly asymptomatic). Surprisingly, signaling proteins were not altered in mutation carriers, but APOE genotype affected the level of 17 proteins. They found that in persons with the e2/3 and e3/3 genotypes, apoE and superoxide dismutase (SOD1) levels were significantly higher than in e3/4 genotypes. Furthermore, IL-13, IL-3, IL-4, IL-5, and IL-12p40 levels were elevated in e3/4 compared to e3/3 and e2/3 genotypes. These results suggest that APOE genotype modulates inflammatory mediators in AD, with e4 carriers demonstrating elevated inflammatory mediators 58.
Focused on chronic neuroinflammation, Tai and colleagues reported results from transgenic mice expressing five familial AD mutations and human apoE isoforms (EFAD mice) 56. Results showed that impaired apoE4 function modulates Aβ-induced neuroinflammatory signaling; increases were observed in detrimental TLR4-p38a signaling, while the beneficial IL-4R-nuclear receptor pathway was suppressed. However, the differential modulation varied with progression of disease. In mixed glial cultures from such mice, oligomeric Ab-induced TNFα secretion was increased by apoE4 via the TLR4 pathway, suggesting TNFα and TLR4 as potential targets for APOE-modulated Aβ-induced neuroinflammation.
Furthermore, data from Rodriguez and colleagues support the hypothesis that APOE genotype affects Aβ-associated neuroinflammation. Investigators studied the impact of APOE genotype on Aβ-associated microglial activity in EFAD mouse models. E4FAD mice had increased levels of Aβ plaques compared to E3FAD and E2FAD mice. Moreoever, IL-1β levels and microglial reactivity were increased in E4FAD mice compared to E3FAD and E2FAD mice 59. Overall, results from these reports strongly suggest that APOE genotype influences Aβ-mediated microglial activation and neuroinflammatory processes in AD progression.
In a series of experiments in APP transgenic mouse models, Chakrabarty and colleagues studied interleukin-10 (IL-10) expression and its effects on apoE and Aβ 60. IL-10 is a cytokine that attenuates inflammatory cascades by inhibiting inflammatory cytokines. Investigators found that IL-10 expression exacerbated AD pathology by increasing plaque burden and reduced synaptic markers, and also produced cognitive decline as measured by fear tone conditioning tasks. Importantly, IL-10 also increased apoE expression and apoE colocalization with insoluble Aβ, and both IL-10 and apoE reduced phagocytosis of Aβ by microglia 60. Despite these studies, more work is needed to determine whether apoE isoforms differentially affect this Aβ-mediated inflammatory response in AD.
Microglia in AD
Microglia are the innate immune cells of the CNS and have been known to play an essential role in mediating and responding to inflammatory signals in the brain. Like the importance of cholesterol metabolism, the evidence showing that inflammatory mediators and innate immunity are critical mediators in AD has exploded in recent years 6. For example, it has been known for decades that inflammation plays a role in the pathogenesis of AD and early studies looking at the use of NSAIDs and AD have highlighted the inverse correlation between NSAID use and AD development 61. Furthermore, the innate immune system has been implicated in AD, for example complement pathway proteins were observed near plaques and tangles in 1989 and microglia have been shown to interact with and internalize senile plaques 62,63.
Recent genome-wide association studies have identified single nucleotide polymorphism (SNPs) in immune genes that confer an elevated risk in developing AD. Of particular interest to the field is the microglial-enriched triggering receptor expressed on myeloid cells 2 (TREM2), which has sharply highlighted the strength of the association between inflammatory pathways and innate immunity and AD 64,65. TREM2 is a cell surface receptor expressed in microglia that signals via its co-adaptor protein TYROBP, a tyrosine kinase protein. R47H, the most frequently studied TREM2 SNP, has been shown in some meta-analysis studies to increase the risk of developing AD to the same degree as possessing a single allele of APOE4, which has long been the leading risk factor in late-onset AD.6 Although the function of TREM2 is not fully defined, recent studies have shown that it plays a role in sustaining microglial phagocytosis of Aβ and neuronal debris, promoting microglia survivability, compacting Aβ-plaques, and lipid sensing via apolipoproteins E and J 66-68. Efforts in establishing the exact role TREM2 plays in AD have identified a connection between microglia and neuronal health. In one study, a set of key observations were made by Wang and colleagues, who examined TREM2-/- 5XFAD mice and demonstrated that the lack of TREM2 expression was associated with increased Aβ accumulation in the hippocampus and increased loss of layer-V cortical neurons compared to controls. TREM2 deficiency was shown to impair microglial colocalization with Aβ plaques and the mechanism proposed was that TREM2 acts as a sensor of anionic and zwitterionic lipids that accompany neuronal damage and Aβ accumulation 66. The discovery of the link between microglial-TREM2 and AD has highlighted the importance in looking at microglial phagocytosis of CNS substrates such as neuronal synapses and beta-amyloid.
Recent focus on microglial dysfunction across age-related neurological diseases like Parkinson-like diseases and AD have highlighted the heterogeneity in microglia morphology and function. In a study conducted by Bachstetter and colleagues, investigators assessed microglia in human brain tissue from dementia with Lewy body, hippocampal sclerosis, and AD patients using immunohistochemistry analysis of microglia/macrophage markers IBA1 and CD68. The authors first found that microglia exhibited varying morphology based on disease and classified them according to 5 forms: ramified, hypertrophic, dystrophic, rod-shaped, and amoeboid microglia. In agreement with previous literature, there was increased dystrophic/deteriorated microglia level in AD samples, implying hypoactivation of microglia in AD pathology 69. Of note, rod-like microglia have been highlighted to be a morphology associated with age and many neurological diseases, such as in vitro microglial aging, CNS injury, and a transgenic Tau-APP mouse models 70-72. Rod-like microglia are observed in close proximity with damaged and degenerating neurons have been implicated in synaptic stripping in the CNS. These studies emphasize microglial response to neurodegenerative diseases and have further highlighted their role in maintaining neuronal homeostasis.
A recent pioneering study assessed neuronal synapse health in AD using transgenic mouse models. Hong and colleagues utilized the J20 mouse model of AD to investigate the hypothesis that synaptic loss is mediated by the complement cascade and microglia early in AD. The initiating complement protein C1q level was elevated and localized to the synapse prior to plaque formation, and C1q was necessary for the downstream toxic effects of soluble Aβ oligomers on synapses. Inhibition of C1q, C3, and the C3 receptor, CR3 (CD11b-CD18/MAC-1), reduced the number of activated microglia and the degree of early synapse loss. Importantly, microglial phagocytosis of Aβ treated synaptic material was found to be in part via a CR3-dependent process. Overall, the findings of this study suggest that synaptic loss early in AD involves activation of the complement cascade and phagocytosis by microglia. Thus, these elements of innate immunity early in AD may represent novel therapeutic targets with microglia being a cell-target 73.
To begin to further understand microglia in disease and potentially develop therapies aimed at AD, new tools and technologies must be developed. One major challenge in studying microglia in vivo has been distinguishing them from their macrophage counterparts under disease or injury states. Mounting evidence highlights the importance of peripheral-derived macrophages and their role in CNS diseases and recent studies have suggested that macrophages may be TREM2- positive cells despite reports highlighting TREM2 being microglial-enriched in the CNS72,74. Recently, Bennet and colleagues developed novel rabbit monoclonal antibodies to detect Tmem119 intracellular and extracellular domains in mice. Investigators were able to utilize Tmem119 antibody in immunohistochemical identification of microglia and FACS isolation of microglia. They found that TMEM119 immunoreactivity remains constant, independent of microglia activation state and the TMEM119 antibody can be used to isolate and study either quiescent or activated microglia 75. However, in an SOD1 mutation model of ALS, TMEM119 mRNA levels decreased compared to WT controls. TMEM119 appears to be a superior method for identifying microglia versus other myeloid cells in the CNS and its use will advance the studying of microglia in transgenic models. Studies using in vitro models like mouse microglial cell lines have allowed for studying molecular pathways involved in phagocytosis or inflammation. A recent study looking at a mouse AD model deficient of T- and B- cells (Rag5xfAD) revealed increased beta-amyloid expression compared to immune-intact AD model 76. Renewed analysis of the gene expression data of Rag5xfAD mice revealed an increased expression of the phagocytosis regulator enzyme SHIP1 (INPP5D; Fig 2A). Inhibiting SHIP1 in BV2 microglia using a SHIP1-specific inhibitor increased phagocytosis of fibrillar Aβ greater than vehicle and similar to IgG-stimulated increased phagocytosis (Fig 2B). New tools have also been developed for studying human microglial lines using induced-pluripotent lines. In collaboration, we have begun to investigate how TREM2+ iPS-derived microglia-like cells (iMGLs) internalize human derived synaptosomes from patient cases (Figure 2D). Utilizing this platform, we will continue to study the role of microglia-mediated inflammation in AD models as well as their role in synaptic pruning and phagocytosis in health and disease.
Fig. 2.
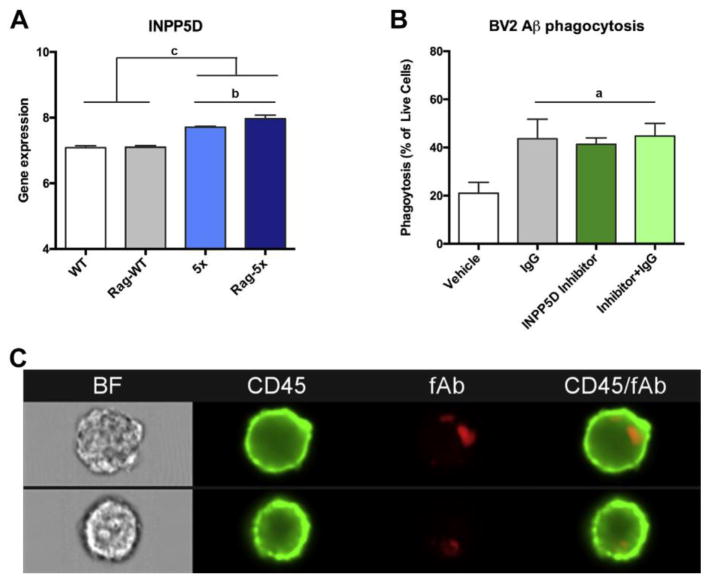
Modeling and targeting microglial genes and their functional pathways in neurode- generative diseases using in vitro models. Microglial molecular pathways implicated in AD can be modeled and their pathways interrogated using murine mouse models. (A) A recent study highlights that immune-deficient AD mice (Rag-5×) have elevated beta-amyloid compared with 5× littermates.75 Analysis of deposited whole-brain mRNA data reveal elevated expression of the phagocytosis-modulator INPP5D/SHIP1. (B) Bar graphs of murine BV2 microglia phagocytosis of fibrillary Ab on Amnis ImagestreamX flow cytometer. BV2 microglia preincubated with nonspecific mouse IgG, INNP5D inhibitor, or both increases microglia phagocytosis. (C) Representative images captured on Amnis ImagestreamX flow cytometer of BV2 microglia internalizing fibrillary beta-amyloid. Statistical analysis was determined using 1-way analysis of variance followed by Tukey's post hoc test. (A) n 5 4 per group, (B) n 5 2 to 4 per group; a P<.001, b P<.01, and c P<.05.
Astrocytes
Astrogliosis is a prominent feature of AD pathobiology. Synaptic pruning by astrocytes is not only important in brain development but may be aberrantly activated in neurodegenerative diseases including AD. Barres and colleagues have utilized murine synaptosome phagocytosis assays to identify important cell surface receptors that are necessary for synaptic pruning (MERTK) 77. A newly developed technology, Amnis Imagestream, couples traditional flow cytometry to imaging, and will greatly facilitate studies examining pruning in development as well as neurological diseases like AD. Astrocyte phagocytosis can be assessed by incubating astrocytes with human synaptosomes pre-labeled with pHrodoRed, which only emits fluorescence at low pH, which is present in intracellular acidified lysosomes. Therefore, fluorescent signal is indicative of bonafide phagocytosis. As shown in Figure 3A, traditional flow analysis can gate for phagocytic events within iPS-derived astrocytes that lack fluorescence in the absence of synaptosomes (left panel), but upon incubation with synaptosomes emit fluorescence (right panel). In Figure 3B, the imaging capability allows for the visualization of every cell and the potential to determine co-localization between the pHrodoRed-emitting synaptosomes and the MERTK receptor, which has been implicated in synaptic pruning. Quantification of co-localization indicates that essentially all cells that demonstrated phagocytic activity also expressed MERTK (Figure 2D). The synaptosome phagocytosis assay can be utilized to assess astrocyte function. In Figure 2C, the synaptosome phagocytosis is used to demonstrate that the ability of iPS-derived astrocytes to phagocytose synaptosomes is similar to fetal-derived astrocytes. Therefore, this assay can be used to investigate how APOE genotype influences astrocyte-mediated synaptic pruning during development or in response to neurotoxic insults like Aβ or inflammatory cytokines.
Fig. 3.
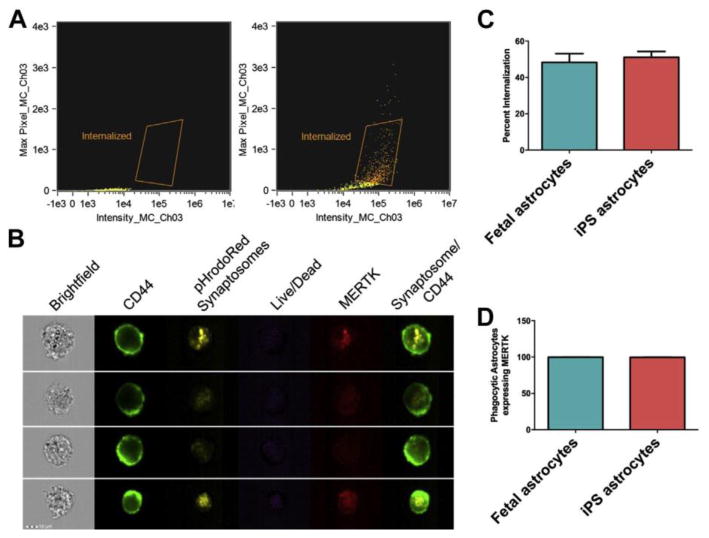
Amnis Imagestream can be used to examine astrocyte phagocytosis of human synap- tosomes (A–D). Synaptosomes were labeled with pHrodoRed (Lifetech) and incubated with iPS-derived astrocytes. Astrocytes are then labeled with CD44 and MERTK and cells that were CD44-positive were examined for the presence of pHrodoRed. (A) Flow cytometry analysis of astrocytes to gate for phagocytic events. (B) Representative images of iPS-derived astrocytes can evaluate viability, and can be used to examine colocalization between synaptosome- containing cellular compartments and MERTK. (C) iPS astrocytes can functionally phagocy- tose synaptosomes to the same extent as fetal astrocytes. (D) Essentially all iPS-derived astrocytes that demonstrate a phagocytic event express MERTK, which has been shown to be essential for astrocyte-mediated synaptic pruning.
Conclusion
Research into AD mechanisms has produced an immense and complex body of literature, reflecting the multifactorial nature of the disease. This short review is not meant to be comprehensive, but is intended to highlight some of recent advances and directions in which the basic science is moving. Given the lack of success in amyloid and other clinical trials to date, new directions offer the possibility for translation into the clinic for prevention and treatment of a costly and devastating disease.
Acknowledgments
AG16573 (W.W.P.) We are indebted to Jenn Atwood and the Immunology Institute Flow Core
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levy-Lahad E, Wijsman EM, Nemens E, et al. A familial Alzheimer's disease locus on chromosome 1. Science. 1995;269(5226):970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 2.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 3.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 5.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nature genetics. 2013;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO molecular medicine. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78(1):94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pooler AM, Polydoro M, Maury EA, et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer's disease. Acta neuropathologica communications. 2015;3:14. doi: 10.1186/s40478-015-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 11.Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. The American journal of pathology. 1999;155(3):853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bilousova T, Miller CA, Poon WW, et al. Synaptic Amyloid-beta Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. The American journal of pathology. 2016;186(1):185–198. doi: 10.1016/j.ajpath.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. The American journal of pathology. 2004;165(5):1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickett EK, Koffie RM, Wegmann S, et al. Non-Fibrillar Oligomeric Amyloid-beta within Synapses. Journal of Alzheimer's disease : JAD. 2016 doi: 10.3233/JAD-160007. [DOI] [PubMed] [Google Scholar]
- 15.Capetillo-Zarate E, Gracia L, Yu F, et al. High-resolution 3D reconstruction reveals intra-synaptic amyloid fibrils. The American journal of pathology. 2011;179(5):2551–2558. doi: 10.1016/j.ajpath.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8(348) doi: 10.1126/scitranslmed.aaf6667. 348ra396. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. 2011;96(4):529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Frontiers in cellular neuroscience. 2015;9:191. doi: 10.3389/fncel.2015.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esparza TJ, Zhao H, Cirrito JR, et al. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73(1):104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 21.de Calignon A, Polydoro M, Suarez-Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73(4):685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO reports. 2013;14(4):389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada K, Holth JK, Liao F, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211(3):387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sokolow S, Henkins KM, Bilousova T, et al. Presynaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease. Journal of neurochemistry. 2014 doi: 10.1111/jnc.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pooler AM, Noble W, Hanger DP. A role for tau at the synapse in Alzheimer's disease pathogenesis. Neuropharmacology. 2014;76(Pt A):1–8. doi: 10.1016/j.neuropharm.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann A, Spires-Jones T. Clearing the way for tau immunotherapy in Alzheimer's disease. Journal of neurochemistry. 2015;132(1):1–4. doi: 10.1111/jnc.12845. [DOI] [PubMed] [Google Scholar]
- 27.Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116–125. doi: 10.1016/j.ceb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiology of aging. 2014;35(8):1792–1800. doi: 10.1016/j.neurobiolaging.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohamed NV, Herrou T, Plouffe V, Piperno N, Leclerc N. Spreading of tau pathology in Alzheimer's disease by cell-to-cell transmission. The European journal of neuroscience. 2013;37(12):1939–1948. doi: 10.1111/ejn.12229. [DOI] [PubMed] [Google Scholar]
- 30.Polanco JC, Scicluna BJ, Hill AF, Gotz J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. The Journal of biological chemistry. 2016;291(24):12445–12466. doi: 10.1074/jbc.M115.709485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain : a journal of neurology. 2015;138(Pt 6):1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuyama K, Sun H, Usuki S, et al. A potential function for neuronal exosomes: sequestering intracerebral amyloid-beta peptide. FEBS Lett. 2015;589(1):84–88. doi: 10.1016/j.febslet.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 34.Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer's disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2014 doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato N, Morishita R. The roles of lipid and glucose metabolism in modulation of beta-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front Aging Neurosci. 2015;7:199. doi: 10.3389/fnagi.2015.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puglielli L, Konopka G, Pack-Chung E, et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nature cell biology. 2001;3(10):905–912. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- 37.Petrov AM, Kasimov MR, Zefirov AL. Brain Cholesterol Metabolism and Its Defects: Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. Acta Naturae. 2016;8(1):58–73. [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy SR, Chang CC, Dogbevia G, et al. Acat1 knockdown gene therapy decreases amyloid-beta in a mouse model of Alzheimer's disease. Mol Ther. 2013;21(8):1497–1506. doi: 10.1038/mt.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryleva EY, Rogers MA, Chang CC, et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3081–3086. doi: 10.1073/pnas.0913828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390(4):287–293. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mauch DH, Nagler K, Schumacher S, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 42.Schneider A, Schulz-Schaeffer W, Hartmann T, Schulz JB, Simons M. Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiology of disease. 2006;23(3):573–577. doi: 10.1016/j.nbd.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Abramov AY, Ionov M, Pavlov E, Duchen MR. Membrane cholesterol content plays a key role in the neurotoxicity of beta-amyloid: implications for Alzheimer's disease. Aging Cell. 2011;10(4):595–603. doi: 10.1111/j.1474-9726.2011.00685.x. [DOI] [PubMed] [Google Scholar]
- 44.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160(1):113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nature medicine. 1998;4(6):730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 46.Djelti F, Braudeau J, Hudry E, et al. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer's disease. Brain : a journal of neurology. 2015;138(Pt 8):2383–2398. doi: 10.1093/brain/awv166. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Peterson DA, Schubert D. Amyloid beta peptide alters intracellular vesicle trafficking and cholesterol homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):13266–13271. doi: 10.1073/pnas.95.22.13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hutter-Paier B, Huttunen HJ, Puglielli L, et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron. 2004;44(2):227–238. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 49.Shibuya Y, Niu Z, Bryleva EY, et al. Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer's disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiology of aging. 2015;36(7):2248–2259. doi: 10.1016/j.neurobiolaging.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gylys KH, Fein JA, Yang F, Miller CA, Cole GM. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer's disease cortex. Neurobiology of aging. 2007;28(1):8–17. doi: 10.1016/j.neurobiolaging.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 51.Arold S, Sullivan P, Bilousova T, et al. Apolipoprotein E level and cholesterol are associated with reduced synaptic amyloid beta in Alzheimer's disease and apoE TR mouse cortex. Acta neuropathologica. 2012;123(1):39–52. doi: 10.1007/s00401-011-0892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu J, Liu CC, Chen XF, Zhang YW, Xu H, Bu G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Molecular neurodegeneration. 2015;10:6. doi: 10.1186/s13024-015-0001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shinohara M, Kanekiyo T, Yang L, et al. APOE2 eases cognitive decline during Aging: Clinical and preclinical evaluations. Ann Neurol. 2016 doi: 10.1002/ana.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Czirr E, Wyss-Coray T. The immunology of neurodegeneration. J Clin Invest. 2012;122(4):1156–1163. doi: 10.1172/JCI58656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.LaDu MJ, Shah JA, Reardon CA, et al. Apolipoprotein E and apolipoprotein E receptors modulate A beta-induced glial neuroinflammatory responses. Neurochemistry international. 2001;39(5-6):427–434. doi: 10.1016/s0197-0186(01)00050-x. [DOI] [PubMed] [Google Scholar]
- 56.Tai LM, Ghura S, Koster KP, et al. APOE-modulated Abeta-induced neuroinflammation in Alzheimer's disease: current landscape, novel data, and future perspective. Journal of neurochemistry. 2015;133(4):465–488. doi: 10.1111/jnc.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gale SC, Gao L, Mikacenic C, et al. APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. 2014;134(1):127–134. doi: 10.1016/j.jaci.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ringman JM, Elashoff D, Geschwind DH, et al. Plasma signaling proteins in persons at genetic risk for Alzheimer disease: influence of APOE genotype. Archives of neurology. 2012;69(6):757–764. doi: 10.1001/archneurol.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J Neuroinflammation. 2014;11:111. doi: 10.1186/1742-2094-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakrabarty P, Li A, Ceballos-Diaz C, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85(3):519–533. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szekely CA, Town T, Zandi PP. NSAIDs for the chemoprevention of Alzheimer's disease. Subcell Biochem. 2007;42:229–248. doi: 10.1007/1-4020-5688-5_11. [DOI] [PubMed] [Google Scholar]
- 62.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neuroscience letters. 1989;107(1-3):341–346. doi: 10.1016/0304-3940(89)90843-4. [DOI] [PubMed] [Google Scholar]
- 63.Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta neuropathologica. 1989;77(6):569–575. doi: 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- 64.Benitez BA, Jin SC, Guerreiro R, et al. Missense variant in TREML2 protects against Alzheimer's disease. Neurobiology of aging. 2014;35(6):1510. doi: 10.1016/j.neurobiolaging.2013.12.010. e1519-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. The New England journal of medicine. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan P, Condello C, Keene CD, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016;90(4):724–739. doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melchior B, Garcia AE, Hsiung BK, et al. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro. 2010;2(3):e00037. doi: 10.1042/AN20100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bachstetter AD, Van Eldik LJ, Schmitt FA, et al. Disease-related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta neuropathologica communications. 2015;3:32. doi: 10.1186/s40478-015-0209-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caldeira C, Oliveira AF, Cunha C, et al. Microglia change from a reactive to an age-like phenotype with the time in culture. Frontiers in cellular neuroscience. 2014;8:152. doi: 10.3389/fncel.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan TF, Liang YX, Peng B, Lin B, So KF. Local proliferation is the main source of rod microglia after optic nerve transection. Scientific reports. 2015;5:10788. doi: 10.1038/srep10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen W, Abud EA, Yeung ST, et al. Increased tauopathy drives microglia-mediated clearance of beta-amyloid. Acta neuropathologica communications. 2016;4(1):63. doi: 10.1186/s40478-016-0336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsieh CL, Koike M, Spusta SC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. Journal of neurochemistry. 2009;109(4):1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(12):E1738–1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marsh SE, Abud EM, Lakatos A, et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(9):E1316–1325. doi: 10.1073/pnas.1525466113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chung WS, Allen NJ, Eroglu C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol. 2015;7(9):a020370. doi: 10.1101/cshperspect.a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]