

Abstract
β-1,2-Glucan is an extracellular cyclic or linear polysaccharide from Gram-negative bacteria, with important roles in infection and symbiosis. Despite β-1,2-glucan's importance in bacterial persistence and pathogenesis, only a few reports exist on enzymes acting on both cyclic and linear β-1,2-glucan. To this end, we purified an endo-β-1,2-glucanase to homogeneity from cell extracts of the environmental species Chitinophaga arvensicola, and an endo-β-1,2-glucanase candidate gene (Cpin_6279) was cloned from the related species Chitinophaga pinensis. The Cpin_6279 protein specifically hydrolyzed linear β-1,2-glucan with polymerization degrees of ≥5 and a cyclic counterpart, indicating that Cpin_6279 is an endo-β-1,2-glucananase. Stereochemical analysis demonstrated that the Cpin_6279-catalyzed reaction proceeds via an inverting mechanism. Cpin_6279 exhibited no significant sequence similarity with known glycoside hydrolases (GHs), and thus the enzyme defines a novel GH family, GH144. The crystal structures of the ligand-free and complex forms of Cpin_6279 with glucose (Glc) and sophorotriose (Glc-β-1,2-Glc-β-1,2-Glc) determined up to 1.7 Å revealed that it has a large cavity appropriate for polysaccharide degradation and adopts an (α/α)6-fold slightly similar to that of GH family 15 and 8 enzymes. Mutational analysis indicated that some of the highly conserved acidic residues in the active site are important for catalysis, and the Cpin_6279 active-site architecture provided insights into the substrate recognition by the enzyme. The biochemical characterization and crystal structure of this novel GH may enable discovery of other β-1,2-glucanases and represent a critical advance toward elucidating structure-function relationships of GH enzymes.
Keywords: enzyme structure; glycosidase; Gram-negative bacteria; oligosaccharide; polysaccharide; β-1,2-glucan; Chitinophaga pinensis; endo-β-1,2-glucanase; novel glycoside hydrolase family; sophorooligosaccharide
Introduction
Glycoside hydrolase (GH)2 is a general term for enzymes that catalyze hydrolytic breakdown of the glycosidic linkages of glycosides, glycans, and glycoconjugates. GHs are distributed in almost all organisms and play important roles not only in various biological phenomena but also industrial processes. These enzymes are categorized into GH families based on their amino acid sequences in the Carbohydrate-Active enZyme (CAZymes) database, forming numerous classes in the database (1–5). The number of GH families has been increasing and currently ranges from GH1 to more than GH140. However, whereas GHs acting on ubiquitously available sugars have been well characterized, there have been few studies on GHs acting on sugars that are difficult to obtain.
β-1,2-Glucan is a polysaccharide difficult to obtain abundantly unlike other glucose (Glc) homopolymers such as cellulose (β-1,4-glucan) and laminarin (β-1,3/1,6-glucan). In nature, this polysaccharide is mainly found as a cyclic β-1,2-glucan (also referred to as cyclosophoraose) produced by certain Gram-negative bacteria such as Agrobacterium, Rhizobium, Shinorhizobium, and Brucella species with degrees of polymerization (DP) of 17–25 (6–10). Cyclic β-1,2-glucan plays an important role in infection of or symbiosis with animals or plants and acts as a modulator of intracellular osmotic pressure after it has accumulated in the periplasmic compartment (9–12). Linear β-1,2-glucan is also known as an extracellular or periplasmic glucan produced by certain bacteria. Some species of Acetobacter and Xanthomonas produce linear β-1,2-glucan with DP of 6–42 and 8–20, respectively (13, 14). Escherichia coli and Pseudomonas syringae produce linear β-1,2-glucan consisting of 5–13 Glc residues that branch at the β-1,6-glucosidic bonds (15–17). In contrast to studies on the physiological role of cyclic β-1,2-glucan in bacteria, there have been few studies on β-1,2-glucan-related enzymes.
Recently, we cloned and characterized two β-1,2-glucan-related enzymes from Listeria innocua in the same gene cluster. The first identified enzyme is 1,2-β-oligoglucan phosphorylase (LiSOGP, EC 2.4.1.333) belonging to GH94, which phosphorolyzes β-1,2-glucooligosaccharides (sophorooligosaccharides, Sopns) with DP ≥3 from the non-reducing end to release α-glucose 1-phosphate (18). The second one is a GH3 β-glucosidase (LiBGL, EC 3.2.1.21) that preferentially hydrolyzes sophorose (Glc-β-1,2-Glc) (19). Although the characteristics of LiSOGP and LiBGL suggest that the two enzymes degrade Sopns cooperatively, these enzymes are both exo-type enzymes.
The endo-β-1,2-glucanase activities (EC 3.2.1.71) were found in some fungi and a bacterium (20–22). In fungi, an endo-β-1,2-glucanase activity, which degrades cyclic β-1,2-glucan to release sophorose as a main product, was found in a culture filtrate of Acremonium sp. 15 and purified to homogeneity (21). Bacterial endo-β-1,2-glucanase activity has been reported only in Chitinophaga arvensicola (formerly known as Cytophaga arvensicola). The partially purified enzyme degrades cyclic β-1,2-glucan to produce Sopns (22). These endo-β-1,2-glucanase activities were remarkably induced in the presence of β-1,2-glucan. However, the gene encoding this enzyme has not been identified.
Recently, we established a method of a large-scale enzymatic synthesis of linear β-1,2-glucan from inexpensive sugars, which makes it easier to study β-1,2-glucan-related enzymes and proteins (23). From now on, we denote linear β-1,2-glucan simply as “β-1,2-glucan” unless otherwise noted. In this report, we propose a new GH family of bacterial endo-β-1,2-glucanases on the basis of the structural and functional characteristics of the C. arvensicola and Chitinophaga pinensis enzymes. Furthermore, we determined the crystal structures of the C. pinensis enzyme in a ligand-free form and a complex form with Sop3 and Glc.
Results
Purification of endo-β-1,2-glucanase
In a previous study, bacterial endo-β-1,2-glucanase activity was induced with cyclic β-1,2-glucan and was detected only in cell extracts of C. arvensicola (22). We first cultured C. pinensis, which is closely related to C. arvensicola, with Glc or β-1,2-glucan as a sole carbon source in addition to C. arvensicola because the genome sequence of C. pinensis is completely known (24). After cultivation, the β-1,2-glucan-hydrolyzing activities in both culture filtrates and cell extracts were measured. β-1,2-Glucan-hydrolyzing activity was detected in cell extracts of both C. pinensis and C. arvensicola (Fig. 1, A and B). However, purification of endo-β-1,2-glucanase from C. pinensis was unsuccessful. Therefore, we selected C. arvensicola as a source of endo-β-1,2-glucanase for purification. The endo-β-1,2-glucanase activity from C. arvensicola was investigated using a β-glucosidase inhibitor, gluconic acid δ-lactone (GDL), by TLC analysis. GDL only inhibited β-glucosidase activity, i.e. it did not inhibit endo-β-1,2-glucanase activity (Fig. 1C). A 43-kDa protein was purified to homogeneity from a cell extract of C. arvensicola in fraction 9 (Fig. 2A). The protein showed endo-β-1,2-glucanase activity but did not show β-glucosidase activity even in the absence of GDL (Fig. 2B).
Figure 1.

TLC analysis of β-1,2-glucan-hydrolyzing activity of C. arvensicola and C. pinensis. A, C. pinensis (culture filtrates). B, C. arvensicola (culture filtrates and cell extracts). Culture filtrates and cell extracts (30 μl) were mixed with the same volume of 1% (w/v) β-1,2-glucan (average DP 64) and then incubated at 25 °C. An aliquot of each mixture was spotted onto a TLC plate. Lane M contains markers (1 μl of 0.2% each sugar). C, TLC analysis of β-glucosidase inhibition by GDL. A reaction mixture (20 μl) containing 12.5% (v/v) cell extract of C. arvensicola, 0.5% (w/v) β-1,2-glucan, 100 mm MOPS-NaOH buffer (pH 6.5), and 20–50 mm GDL was incubated at 30 °C for 2 days, and then each reaction mixture was spotted onto a TLC plate. Lanes M1 and M2 contain markers (M1, 1 μl of 0.2% each sugar; M2, 1 μl of 20 mm GDL). The asterisk indicates the origin on the TLC plate.
Figure 2.

Purification of endo-β-1,2-glucanase from a C. arvensicola cell extract. A, fractionated proteins were subjected to SDS-PAGE, followed by silver staining (bottom), and then the enzymatic activity was measured (top). Lane M contains protein standard markers. Endo-β-1,2-glucanase activity was evaluated as the ratio of β-1,2-glucan-hydrolyzing activity to β-glucosidase activity. The enzymatic reaction was conducted in a mixture (50 μl) containing 80% (v/v) enzyme fraction, 0.2% (w/v) β-1,2-glucan (average DP 64), and 50 mm MOPS-NaOH buffer (pH 6.5) at 30 °C for 16 h. An aliquot of the reaction mixture (40 μl) was mixed with 160 μl of a 1% (w/v) PAHBAH-HCl solution and then heated at 100 °C for 5 min, followed by measurement of absorbance at 405 nm. The arrow indicates the protein band subjected to N-terminal amino acid sequence analysis. B, TLC analysis of endo-β-1,2-glucanase in fraction 9. The enzymatic reaction was carried out in a mixture (10 μl) containing 85% (v/v) fraction 9 concentrated using Amicon Ultra 30,000 molecular weight cutoff (Millipore), 0.2% (w/v) β-1,2-glucan (average DP 64), and 50 mm MOPS-NaOH buffer (pH 6.5) at 30 °C for 16 h, and then an aliquot of the mixture was spotted onto a TLC plate. Lane M contains markers (1 μl of 0.2% (w/v) each sugar). The asterisk indicates the origin on the TLC plate. Minus and plus indicate whether fraction 9 was added to the reaction mixture or not.
Sequence analysis
The 43-kDa protein was subjected to in-gel digestion to obtain its internal peptides. Based on a BLAST search limited to C. pinensis using the sequences constructed as described under “Experimental procedures,” five sequences matched the same hypothetical protein (locus tag; Cpin_6279) (Table 1). C. pinensis did not have paralogs of Cpin_6279. Cpin_6279 showed no sequence similarity to known GHs, although this protein is annotated as a glycoamylase (PF10091) in the Pfam database (25), implying that Cpin_6279 belongs to a novel GH family (described below).
Table 1.
Constructed internal sequences of endo-β-1,2-glucanase
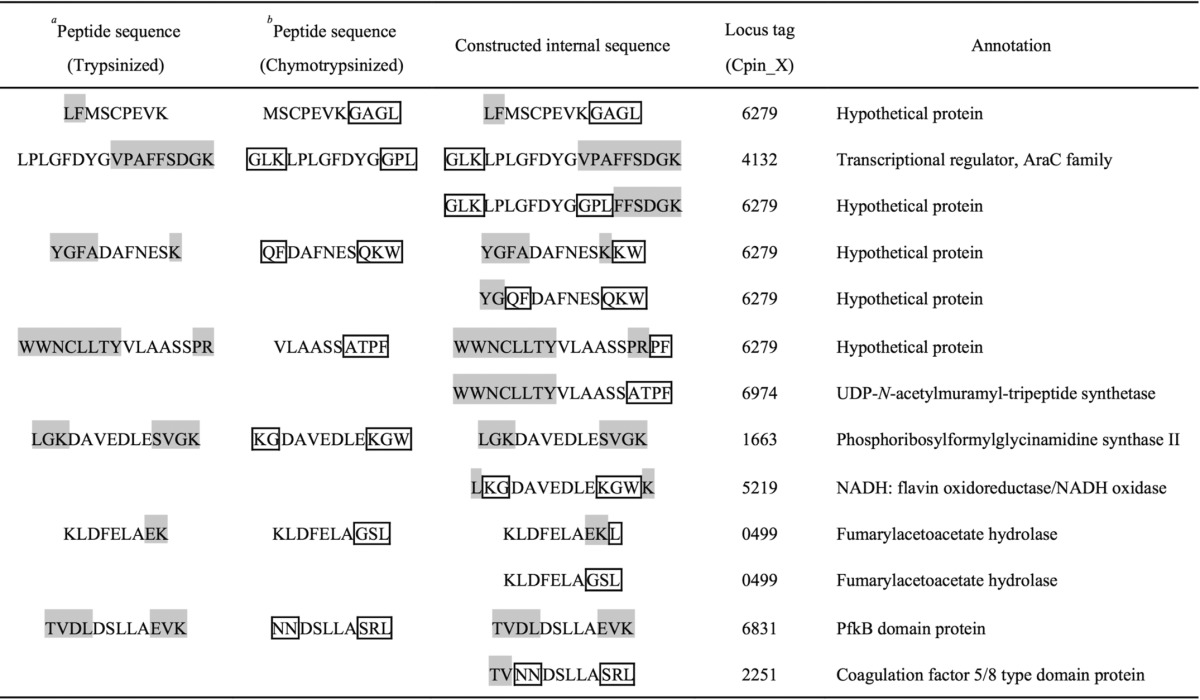
a Gray highlighted letters indicate amino acid residues inherent to trypsinized peptides.
b Boxed letters indicate amino acid residues inherent to chymotrypsinized peptides.
The protein sequence of Cpin_6279 available in the RefSeq database (accession number; WP_044220121) consists of 457 amino acids. The N-terminal sequence of the 43-kDa protein purified from C. arvensicola (QVARATLAFDRT) corresponds to the sequence starting from the 32nd amino acid of Cpin_6279, whose N-terminal 18 amino acids were predicted to be a signal sequence by the SignalP 4.1 Server (26). This suggests that the predicted signal sequence plus 13 amino acids had been cleaved during the inner membrane translocation and protein purification. Indeed, the β-1,2-glucan-degrading activity of C. pinensis was not detected in culture filtrates (Fig. 1B). Thus, Cpin_6279 may be localized in the periplasmic space. The β-glucosidase activity in cell extracts seems to be derived from a GH3 LiBGL homolog (Cpin_1816), which contains a signal peptide (19). Cpin_6279 and Cpin_1816 seem to be involved in degradation of β-1,2-glucan in C. pinensis.
Identification of β-1,2-glucanase and its enzymatic properties
To determine whether the Cpin_6279 protein is an endo-β-1,2-glucanase, we expressed and purified the recombinant C-terminally His6-tagged Cpin_6279 (Cpin_6279rC). The purified protein migrated as a single band of 45 kDa on SDS-PAGE (Fig. 3A), nearly corresponding to the theoretical molecular mass (51,215 Da) of Cpin_6279rC. Cpin_6279rC obviously catalyzed hydrolysis of β-1,2-glucan to produce Sopns with DP of 2–5 (Fig. 3B).
Figure 3.

SDS-PAGE analysis of Cpin_6279rC (A) and TLC analysis of β-1,2-glucan-degrading activity of Cpin_6279rC (B). A, SDS-PAGE of the purified Cpin_6279rC. Lane M contains protein standard markers. B, lane M contains markers (1 μl of 0.2% each sugar). The enzymatic reaction was conducted in a reaction mixture (50 μl) containing various concentrations of Cpin_6279rC, 0.2% (w/v) β-1,2-glucan (average DP 64), and 50 mm MOPS-NaOH buffer (pH 6.5) at 30 °C for 10 min. The asterisk indicates the origin on the TLC plate.
Cpin_6279rC exhibited high activity at 50 °C and pH 4.5–6.5. This enzyme was stable up to 50 °C on incubation for 1 h at pH 6.5 and in the range of pH 3.5–10 at 30 °C for 1 h (Fig. 4). Among the various tested polysaccharides (see “Experimental procedures”), Cpin_6279rC showed high hydrolytic activity only toward β-1,2-glucan (36.6 units/mg), exhibiting only <0.2% relative activity toward other polysaccharides, demonstrating that it is an endo-β-1,2-glucanase highly specific for β-1,2-glucan.
Figure 4.

pH (A) and temperature (B) profiles of Cpin_6279rC. The stability (dashed line) and optimum (solid line) are shown. A, buffers used for incubation and enzymatic reaction were sodium citrate (pH 3.0–5.5, closed circles), MES-NaOH (pH 5.5–6.5, open circles), MOPS-NaOH (pH 6.5–7.5, closed triangles), 2-hydroxy-3-[4-(2-hydroxyethyl)-1-piperazinyl]propanesulfonic acid-NaOH (pH 7.5–8.5, open triangles), and glycine-NaOH (pH 8.6–10, closed rhombi). The highest activity was defined as 100% (stability, pH 6.5; and optimum, pH 6.0). B, highest activity was defined as 100% (50 °C). As for the enzymatic reaction at 60 and 70 °C, the data from 0 to 1.5 min were used to calculate the initial velocity.
Action patterns of Cpin_6279 on β-1,2-glucan and sophorooligosaccharides
The action patterns on linear and cyclic β-1,2-glucans were analyzed by TLC. Both β-1,2-glucans were broken down into Sop2 to Sop5 at the early stage of the reaction (Fig. 5, A and B). These results confirmed that Cpin_6279 is an endo-type GH. Finally, both β-1,2-glucans were completely broken down into Sop2 to Sop5 in 60 min in contrast to the case of the partially purified endo-β-1,2-glucanase from C. arvensicola, which releases Sop3 to Sop5 as final reaction products (20).
Figure 5.

TLC analysis of the action patterns of Cpin_6279rC on β-1,2-glucan and Sopns. β-1,2-Glucan (average DP 64) (A), cyclic β-1,2-glucan (DP 17–24) (B), Sop5 (C), Sop6 (D), and Sop7 (E) were used as substrates. Lane M contains markers (0.2% each sugar). Asterisks represent the origins of the TLC plates.
The action patterns on Sopns were also analyzed by TLC. All tested Sopns were decomposed into oligosaccharides with DP of 2–5 (Fig. 5, C–E). The hydrolytic velocities of Cpin_6279rC for Sop6 and Sop7 were higher than that for Sop5. Notably, Sop7 is predominantly cleaved into Sop3 and Sop4, suggesting that the subsites of the enzyme extend from −3 to +4 or −4 to +3. Sop3 and Sop4 were not degraded (data not shown). Based on HPLC analysis, the specific activities as to Sop5, Sop6, and Sop7 were 3.0, 15, and 19 units/mg, respectively, and the ratios of the reaction products for Sop6 and Sop7 were (Sop2 + Sop4):(Sop3 + Sop3) = 4:3 and (Sop2 + Sop5):(Sop3 + Sop4) = 1:2, respectively (Fig. 6). This result is consistent with the action pattern obtained on TLC analysis.
Figure 6.

HPLC analysis of the action patterns of Cpin_6279rC on Sopns. A, reference chromatogram of 2 mm Sop2–5. B–D, monitoring of the reaction products derived from Sop5 (B), Sop6 (C), and Sop7 (D). The solid line represents the sample before incubation with Cpin_6279rC. The dotted and dashed lines represent the samples after incubation with Cpin_6279rC for 5 and 10 min, respectively.
Subsite characterization of Cpin_6279
To determine the detailed action pattern of Cpin_6279, we examined the incorporation of 18O atoms into the newly produced reducing ends using electrospray ionization (ESI)-MS after enzymatic hydrolysis of Sop7 in the presence or absence of 18O-labeled water (Fig. 7). The observed hydrolytic patterns are schematically shown in Fig. 7A. When Sop7 is broken down into Sop3 and Sop4 in the presence of H218O, patterns I and II are conceivable. In the case of pattern I, the ion signals of [Sop3 + Na]+ and [Sop4 + Na (18O)]+ will be detected. As shown in Fig. 7, D and E, the ion signals corresponding to [Sop3 + Na]+ and [Sop4 + Na]+ were detected in the absence of H218O. In the presence of H218O, the ion signals corresponding to [Sop3 + Na]+ and [Sop4 + Na (18O)]+ were detected in ratios of 76 and 68%, respectively. The ratio of oligosaccharide incorporating 18O was calculated from the ratio of the peak area of each 18O-incorporating oligosaccharide to the peak area of the total amount of the corresponding oligosaccharide produced. These results demonstrate that Cpin_6279 mainly binds Sop7 at subsites −4 to +3 as shown in pattern I and suggest that recognition at subsite +4 is weaker than that at subsite −4. We also investigated the hydrolytic pattern when Sop7 was decomposed into Sop2 and Sop5 (Fig. 7, F and G). In the absence of H218O, the ion signals corresponding to [Sop2 + Na]+ and [Sop5 + Na]+ were detected. In the presence of H218O, the ion signals corresponding to [Sop2 + Na]+ and [Sop5 + Na (18O)]+ were detected as major peaks. In contrast, the intensity of [Sop2 + (18O)]+ was very weak compared with that of [Sop2 + Na]+, and the signal of [Sop5 + Na (18O)]+ showed a sufficiently high ratio of 78%, indicating that Cpin_6279 can also bind to subsites −5 to +2, as shown in pattern III but not −2 to +5. Therefore, subsite −3 is essential for binding.
Figure 7.

Mass spectra of the reaction products from Sop7 hydrolyzed by Cpin_6279rC in 18O-labeled water. A, hydrolysis patterns of Cpin_6279rC as to Sop7 observed in D–G. Each reducing end is indicated by a slanting line. The numbers above schematic diagrams of oligosaccharides represent subsites. The numbers in parentheses indicate minor subsites. B and C, reference spectra of Sop3 and Sop4. D–G, enlarged views of mass spectra of Sop3 (D), Sop4 (E), Sop2 (F), and Sop5 (G) produced by enzymatic hydrolysis of Sop7. Solid and dashed lines show the spectra of the reaction products in the presence and absence of 18O-labeled water, respectively. The values above the peaks represent the molecular weights of the reaction products when using H218O. The asterisks denote the peaks derived from natural 13C-containing oligosaccharides. Schematic diagrams in parentheses represent the minor products.
Kinetic analysis
The kinetic parameters of Cpin_6279rC were determined using β-1,2-glucan and Sop5 as substrates (Table 2). The kcat and Km values were sufficiently high and low compared with those of other GHs. The higher kcat value and the lower Km value for β-1,2-glucan than those for Sop5 imply the preference of more than six subsites on the enzyme, consistent with the HPLC analysis where the specific activities increased in proportion to DP.
Table 2.
Kinetic parameters of Cpin_6279rC
| Substrate | kcat | Km | Ki | kcat/Km |
|---|---|---|---|---|
| s−1 | mm | mm | s−1 mm−1 | |
| β-1,2-Glucan | 49 ± 4 | 0.068 ± 0.014 (0.71 ± 0.15)a | 720 ± 110 (69 ± 10)a | |
| Sop5 | 4.7 ± 1.3 | 1.5 ± 0.5 | 1.9 ± 0.8 | 3.2 ± 0.3 |
a Values in parentheses represent the values when the substrate concentration used is expressed as (mg ml−1).
Stereochemistry of hydrolysis catalyzed by Cpin_6279
To determine the stereochemical course of the Cpin_6279-catalyzed hydrolysis, we performed 1H NMR using β-1,2-glucan as a substrate (Fig. 8). Standard spectra (Sop2–5 and β-1,2-glucan with average DP 25) are shown in Fig. 8B. A reference spectrum (t = 0 min) was recorded prior to the addition of the enzyme to the reaction mixture. Because the signals of the anomeric axial protons of liberated Sopns (H1β), which appeared around δ 4.7, overlapped with those of water and anomeric axial protons at internal glucosides, we compared the signals of the anomeric equatorial protons of liberated Sopns (H1α, δ 5.4) with the signals of the C2 protons of the non-reducing end glucoside in the liberated Sopns (H2NR, δ 3.35) regardless of the anomeric configuration. After the addition of the enzyme, the H1α signals increased along with the H2NR signals (t = 4 and 27 min) (Fig. 8, C and D). As the reaction proceeded (t = 55 and 105 min and 24 h), the ratio of the H1α signals to the internal standard decreased, which is attributed to the mutarotation of anomers. The ratio of the H2NR signals to the internal standard gradually increased at the late stage of the reaction (Fig. 8, C and D), which seems to be due to slower hydrolysis of Sop5. This finding indicates that Cpin_6279 is an inverting GH.
Figure 8.

1H NMR spectra and polarimetric analysis of the Cpin_6279rC-catalyzed reaction. A, schematic representation of hydrolysis catalyzed by Cpin_6279rC. −1 and +1 denote subsites. H1α and H1β represent anomeric equatorial and axial protons at the reducing end of the reaction product, respectively. H2NR represents the C2 proton at the non-reducing end of the reaction product. B, reference spectra of Sop2–5 and β-1,2-glucan (average DP 25). C, 1H NMR time course analysis of the reaction products. H1α and H2NR represent the resonance signals corresponding to A. D, time course of the ratio of peak area to internal standard. The ratios of the peak areas of H1α and H2NR to the internal standard are represented by closed triangles and closed circles, respectively. E, time course of observed optical rotation during β-1,2-glucan-hydrolysis by Cpin_6279rC. The arrow indicates the time of the addition of a droplet of an ammonia solution (6 min). β-1,2-Glucan (average DP25) was used as a substrate.
We also carried out polarimetric analysis for easier identification of the reaction mechanism (Fig. 8E). The observed optical rotation increased to a positive value on addition of Cpin_6279rC, quickly decreased after addition of an ammonia solution, and then reached equilibrium. This indicates that optical rotation derived from the reducing end α-anomer in β-1,2-glucan is greater than that from the β-anomer, as is the case with Glc released from substrates by inverting exo-type β-1,3-glucanases (27, 28). Thus, polarimetric analysis can be used for determination of the stereochemical course of β-1,2-glucan hydrolysis.
Crystal structure of Cpin_6279
We first determined the ligand-free structure of Cpin_6279rC at 1.8 Å resolution (Table 3). Although these data were collected for a crystal soaked in a solution supplemented with Sop3 (a main reaction product) as a cryoprotectant, the sugar ligand was not bound to the protein, probably due to crystal packing hindrance by the C-terminal His6 tag (described below). The complex structure of N-terminally His6-tagged Cpin_6279 (Cpin_6279rN) with Sop3 and Glc was next determined at 1.7 Å resolution by a co-crystallization method (Fig. 9A). Although Glc was not added to the crystallization sample at any preparation step, weak electron density of a Glc monosaccharide, which seems to be derived from Sop3, was observed in the binding groove (Fig. 9, B and C). These ligand-free (Cpin_6279rC) and Sop3-Glc complex (Cpin_6279rN) crystals contained two molecules per asymmetric unit and belonged to space groups P31 and C2, respectively. The primary structure of the ligand-free form (Cpin_6279rC) consists of 449 residues, and the amino acid region in the determined structure extends from 24 to 449 in both chains. The C-terminal His6 tags on both chains (residues 444–449) were visible in the electron density map. The primary structure of the complex form (Cpin_6279rN) consists of 461 residues, and the amino acid region in the determined structure extends from 43 to 460 (corresponding to 23–440 in Cpin_6279rC) in both chains. The root mean square deviations (r.m.s.d.) for the Cα atoms between all pairs of four molecules (two chains in the two structures) are within 0.1 Å. Hereafter, we focus on chain A unless otherwise noted. The overall structure exhibits an (α/α)6-barrel (Fig. 9A). A surface model depicts a cleft structure suitable for accommodating a large substrate (Fig. 9B). A structural homology search using the DALI server revealed that the structure of Cpin_6279 is very similar to those of homologous hypothetical proteins BF9343_0330 from Bacteroides fragilis (PDB code 3EU8), BACCAC_03554 from Bacteroides caccae (PDB code 4QT9), and BACUNI_03963 from Bacteroides uniformis (PDB code 4GL3) determined by means of structural genomics. The r.m.s.d. values for Cα atoms are less than 1.3 Å, and the Z-scores are more than 63, reflecting their high sequence identity of ∼67% with Cpin_6279. The structures of these homologous proteins also show the large cleft observed in Cpin_6279. In addition, the structure of Cpin_6279 exhibits modest similarity (r.m.s.d. values of 3.5–4.0 Å and Z-scores of < 20) but low sequence identity (<12%) with GH15 glucoamylase, cellobiose 2-epimerase, GH8 chitosanase, N-acetylglucosamine 2-epimerase, GH8 endo-β-1,4-glucanase, and GH126 α-amylase.
Table 3.
Crystallographic statistics
| Data set | Ligand-free (Cpin_6279rC) | Sop3-Glc complex (Cpin_6279rN) |
|---|---|---|
| Data collection | ||
| PDB entry | 5GZH | 5GZK |
| Beamline | AR-NW12A | AR-NW12A |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Space group | P31 | C2 |
| Unit cell parameters (Å, °) | a = b = 91.0, c = 124.1 | a = 126.1, b = 117.4, c = 75.6, β = 92.6 |
| Resolution (Å)a | 50.00–1.80 (1.83–1.80) | 50.00–1.70 (1.73–1.70) |
| Total reflections | 562,221 | 450,422 |
| Unique reflectionsa | 105,566 (5,394) | 120,399 (6,010) |
| Completeness (%)a | 99.0 (100.0) | 99.9 (100.0) |
| Redundancya | 5.3 (5.6) | 3.7 (3.6) |
| Mean I/σ(I)a | 21.3 (2.3) | 13.1 (2.0) |
| Rmerge (%)a | 9.8 (68.2) | 11.0 (68.0) |
| CC1/2 (%)a | (82.3) | (69.6) |
| Refinement | ||
| Resolution (Å) | 48.7–1.80 | 49.5–1.70 |
| No. of reflections | 100,115 | 114,356 |
| Rwork/Rfree (%) | 16.8/20.4 | 16.6/19.9 |
| Root mean square deviation from ideal values | ||
| Bond lengths (Å) | 0.020 | 0.025 |
| Bond angles (°) | 1.852 | 2.158 |
| Average B-factors (Å2) | ||
| Protein (chain A/B) | 27.9/27.9 | 17.9/18.4 |
| Ligand (Sop3/Glc) | 28.9/43.1 | |
| Water | 35.5 | 27.1 |
| Trehalose | 41.1 | |
| Iodide ion | 66.1 | |
| Phosphate ion | 29.5 | |
| Potassium ion | 44.7 | |
| Chloride ion | 43.5 | |
| Tetraethylene glycol | 34.4 | |
| Di(hydroxyethyl)ether | 35.5 | |
| Pentaethylene glycol | 37.1 | |
| Ramachandran plot (%) | ||
| Favored | 97.2 | 97.2 |
| Allowed | 2.6 | 2.5 |
| Outlier | 0.2 | 0.2 |
a Values in parentheses represent the highest resolution shell.
Figure 9.

Crystal structure of Cpin_6279rN. The bound ligands are shown as green sticks. A, schematic representation of the overall structure of Cpin_6279rN complexed with Sop3 and Glc. The structure is color-ramped from the N terminus (blue) to the C terminus (red). B, surface representation of the Cpin_6279rN structure. The highly conserved catalytic residue candidates (Glu-54, Asp-135, Asp-139, Glu-142, Glu-211, and Asp-400) are colored red. C, active-site architecture. Amino acid side chains engaged in the ligand recognition are represented by gray sticks (ligand-free) and green sticks (Sop3-Glc complex). The candidate catalytic residues are given in red. The water molecules are represented as red spheres. Hydrogen bonds are represented by yellow dashed lines. The σA-weighted mFo − DFc omit electron density map of the ligands is shown as a blue mesh (contoured at 2.5 σ).
Active-site architecture
In the complex structure, Sop3 and Glc were observed at the center of the barrel (Fig. 9C). All Glc residues adopt a standard 4C1 conformation, and the C6 hydroxyl group is in a gauche-gauche orientation. The hydroxyl groups of Sop3 form hydrogen bonds with the NE2 atom of His-193, the ND2 atom of Asn-258, the ND2 atom of Asn-210, and six water molecules, of which four are held by the OD2 atom of Asp-135, the OD2 atom of Asp-139, the OE2 atom of Glu-142, and the NE1 atom of Trp-269. The carboxylic group of Glu-211 forms bifurcated hydrogen bonds with the O3 and O4 hydroxyl groups of the central Glc unit of Sop3. The O3 and O4 hydroxyl groups of the Glc form bifurcated hydrogen bonds with the ϵ-amino group of Arg-55. O4 and O6 of Glc form direct and water-mediated hydrogen bonds with Glu-54. In addition, O1 of Glc is hydrogen bonded to a water molecule. In the ligand-free form structure, the side chain of Glu-142 slightly turns downward and that of Arg-55 turns to a slightly distal position compared with the complex structure (Fig. 9C). The displacements in the ligand-free form structure are partly due to the crystal packing of the P31 space group crystal, in which the active site is completely blocked with the C-terminal His6 tag from a symmetry mate molecule. In the binding pocket, six acidic amino acid residues (Glu-54, Asp-135, Asp-139, Glu-142, Glu-211, and Asp-400) are highly conserved in Cpin_6279 homologs (Figs. 9B and 10), indicating that these residues are candidates for the catalytic residues. Only Asp-400 does not form any hydrogen bonds with the Sop3 or Glc molecule. The aromatic rings of Phe-131, Trp-192, Phe-204, Trp-269, and Tyr-330 are involved in hydrophobic interactions.
Figure 10.

Sequence alignment of Cpin_6279 and its homologs. Multiple sequence alignment was performed with Clustal Omega (55). Identical amino acids are highlighted in red, and conservative residues are boxed. Helices (α), strands (β), and 310 helices (η) are denoted above the alignment. Letters in parentheses correspond to the secondary structural elements shown in Fig. 9A. The residues corresponding to highly conserved acidic amino acids in the active site are indicated by solid stars. The GenBankTM accession numbers of the proteins used for the alignment are as follows: Cpin_6279 (ACU63684.1); BF9343_0330 (CAH06109.1); BACCAC_03554 (EDM19439.1); BACUNI_03963 (EDO52350.1); BT_3566 (AAO78672.1); Bovatus_03682 (ALJ48288.1); Fjoh_3523 (ABQ06537.1); and Dfer_4452 (ACT95653.1). The figure was prepared with ESPript (56).
Mutational analysis
To predict the catalytic residues, we carried out mutational analysis by replacing the highly conserved acidic amino acid residues in the active site with isosteric residues (Asn or Gln) (Table 4). Mutations at Asp-139, Glu-142, and Glu-211, which are located around the non-reducing end of Sop3 (Fig. 9C), reduced the specific activity to <0.2% compared with the wild type. As for E54Q and D135N, the specific activity also decreased, but it was not as critical as the above three mutants. D400N was less effective on the specific activity, consistent with the structural observation that Asp-400 does not participate in any ligand recognition (Fig. 9C).
Table 4.
Specific activity of the wild type and mutant Cpin_6279rC
| Enzyme | Specific activitya | Relative activity |
|---|---|---|
| units/mg | % | |
| Wild type | 26 ± 3 | 100 |
| E54Q | 0.32 ± 0.23 | 1.2 |
| D135N | 0.21 ± 0.01 | 0.81 |
| D139N | 0.045 ± 0.043 | 0.17 |
| E142Q | 0.024 ± 0.017 | 0.092 |
| E211Q | 0.039 ± 0.029 | 0.15 |
| D400N | 5.3 ± 0.6 | 20 |
a The errors represent the standard deviation for triplicate measurements.
Discussion
Although several studies on bacterial and fungal endo-β-1,2-glucanases were reported over 30 years ago (20–22), their genes have not been identified. In this study, we could purify a bacterial endo-β-1,2-glucanase and clone its gene from a Chitinophaga species by using enzymatically synthesized linear β-1,2-glucan. Biochemical and structural characterizations were carried out using the recombinant Cpin_6279 protein, revealing that the enzyme has at least seven subsites ranging from −4 to +3 in a long cleft of the (α/α)6 barrel molecule. Because Cpin_6279 showed no significant amino acid sequence similarity with any known enzymes, the enzyme and its homologs will be classified as a novel GH family, GH144.3 Among the uncharacterized proteins homologous to Cpin_6279, only the ligand-free crystal structures of three hypothetical proteins from Bacteroides have become available by means of structural genomics. These hypothetical proteins were incorrectly annotated as glucoamylases because they were classified into the glycoamylase family (PF10091) by Pfam. Cpin_6279 showed no glucoamylase activity (data not shown). Therefore, the structure of the complex with Sop3 is important evidence that shows the correct function of Cpin_6279.
Bioinformatic analysis of Cpin_6279 homologs showed that the homologous proteins are mainly distributed in Gram-negative bacteria and not in eukaryotes. A phylogenetic tree illustrates that the proteins in the new GH family are mainly from the Bacteroidetes and Proteobacteria and can be divided into distinct clades (Fig. 11). Notably, not only soil bacteria but also gut bacteria such as Bacteroides thetaiotaomicron, Bacteroides fragilis, and Bacteroides ovatus have a homologous protein.
Figure 11.

Phylogenetic tree of a novel GH family. Sequences for phylogenetic analysis were retrieved for each genus in the KEGG database and confined to one paralog per species. After multiple sequence alignment using MUSCLE had been performed (57), a phylogenetic tree was constructed using MEGA7 (58) based on the neighbor-joining method. Species harboring the gene of a Cpin_6279 homolog are categorized into phyla and surrounded by solid lines. The organism possessing the gene cloned in this study is shown with a black background and white letters. Cpin_6279 is denoted by a closed circle.
C. pinensis does not have a gene cluster for dissimilation of β-1,2-glucan, although the genomes of some other bacteria encode such gene clusters. Many members of the Bacteroidetes harboring Cpin_6279 homologs seem to use the SusC (TonB-dependent receptor)/SusD (carbohydrate-binding protein)-like system for the catabolism of β-1,2-glucan. These susCD-like genes are located around the Cpin_6279 homolog gene, which is a feature of the Bacteroidetes for degradation of complex polysaccharides known as polysaccharide utilization loci (29). Other bacteria appear to utilize ABC proteins or the other systems for dissimilation. In addition to these genes, GHs such as LiBGL homologs (GH3) and LiSOGP homologs (GH94) and LacI transcription factors tend to form gene clusters together with these uptake systems. Functional predictions suggest these proteins are involved in the synergetic decomposition of β-1,2-glucan.
Some cyclic β-1,2-glucan-producing bacteria such as Agrobacterium tumefaciens and Rhizobium meliloti possess a Cpin_6279 homolog. In a number of bacterial exopolysaccharide secretion systems, a GH or polysaccharide lyase is found in the operon of that system. These degrading enzymes are suggested to be important for efficient production of exopolysaccharides, clearing the residual exopolysaccharides in the periplasmic space and forming a scaffold for secretion of the polymers into the extracellular space (30–33). The cellular concentrations of cyclic β-1,2-glucan in Rhizobium and Agrobacterium species range from 5 to 20% of the whole-cell dry weight, and the concentration depends on the species, medium composition, and growth phase (7). Although almost all the Cpin_6279 homologs from root nodule bacteria do not form a gene cluster with cyclic β-1,2-glucan synthase and related proteins, they may play a role in clearance of the residual cyclic β-1,2-glucan in the periplasm.
The crystal structure of Cpin_6279 bound with Sop3 and Glc provided structural evidence for binding of Sopns. Considering the preferable binding of Sop7 to subsites −4 to +3 suggested by ESI-MS analysis (Fig. 7A), the Sop3 and Glc observed in the crystal structure may occupy subsites +1 to +3 and subsite −3, respectively (Fig. 9C). In the binding cleft, highly conserved Glu-54, Asp-135, Asp-139, Glu-142, Glu-211, and Asp-400 are located (Fig. 9B). These residues except for Asp-400 form direct or indirect hydrogen bonds with the hydroxyl groups of Sop3 and Glc (Fig. 9C). Mutation at these residues, especially at Asp-139, Glu-142, and Glu-211, obviously decreases the activity (Table 4), suggesting that these residues play an important role in catalysis. However, none of the three residues are in the direct hydrogen transfer distance range as to the O2 atoms of all the Glc units in Sop3 (Fig. 9C) or are proximal to space between Sop3 and Glc. Fig. 12 shows a comparison of the topological positions of the conserved acidic residues in Cpin_6279 with the catalytic residues in two representative inverting GHs (GH15 and GH8) having an (α/α)6-barrel fold. It has been known that the majority of inverting GHs with an (α/α)6-barrel, including GH15 (Fig. 10B), possess catalytic residues at loops between α5 and α6 (catalytic acid) and between α11 and α12 (catalytic base), whereas several families possess them at circularly permutated positions (34). GH8 (Fig. 10C) and GH48 have catalytic residues in loops of α1–α2 (catalytic acid) and α7–α8 (catalytic base), and GH9 has them in α11–α12 (catalytic acid) and α5–α6 (catalytic base). Glu-211 (α5–α6) of Cpin_6279 is topologically equivalent with the general acid residue in GH15 (Fig. 10A). However, Glu-211 does not appear to be involved in proton transfer to the glycosidic bond but anchors the central Glc unit of Sop3 (Fig. 9C). The positions of Asp-139 and Glu-142 (α3–α4) are unprecedented in known inverting GHs. These observations may imply a non-canonical reaction mechanism for the novel GH enzymes, as in the case of several exceptional inverting GHs such as GH6 Cel6A (35), GH45 PcCel45A (36), GH55 Lam55A (37), GH95 α-1,2-l-fucosidase (38), and GH124 Cel124A (39). Presumption of the positions of subsite −1 and a nucleophilic water is difficult from the complex structure in this study.
Figure 12.

Structural comparison with other GH family enzymes. The α-helices are colored cyan. Catalytic residues (GH15 and GH8) and strictly conserved amino acids in the active site of Cpin_6279 are colored magenta and shown as a stick model. A, overall structure of Cpin_6279rN. Glu-54, Asp-135, Asp-139, Glu-142, Glu-211, and Asp-400 are strictly conserved residues. Glu-54 is located between α1 and α2. Asp-135, Asp-139, and Glu-142 are located between α3 and α4. Glu-211 is located between α5 and α6. Asp-400 is located between α11 and α12. B, overall structure of GH15 glucoamylase (PDB code 1AGM). Glu-179 (catalytic acid) is located between α5 and α6. Glu-400 (catalytic base) is located between α11 and α12. C, overall structure of GH8 endoglucanase (PDB code 1KWF). Glu-95 (mutated to Gln-95, catalytic acid) is located between α1 and α2. Glu-278 (catalytic base) is located between α7 and α8.
C. pinensis is a Gram-negative bacterium that exhibits potential as to the discovery of novel CAZymes (24, 40). This bacterium possesses diverse carbohydrate-active enzymes including 191 GHs that can be classified into 56 different families. In addition, C. pinensis shows poor growth on cellulose and starch, which are the most abundant sugar resources on earth. Therefore, it is expected that novel GHs will be further found in this organism.
Studies on endo-β-1,2-glucanase did not greatly advance from 1987 until recently because it had been difficult to prepare large amounts of β-1,2-glucan. The identification, characterization and crystal structure of this enzyme will facilitate further discovery of β-1,2-glucan-related enzymes and help us better understand the structure-function relationships of GH enzymes. Moreover, our study will provide solid foundations for a wide range of research concerning β-1,2-glucan and Sopns.
Experimental procedures
Chemicals
β-1,2-Glucan (average DP 25) was prepared as described previously (23). β-1,2-Glucan (average DP 64) was prepared through the synthetic reaction of LiSOGP in the presence of Glc and α-glucose 1-phosphate (18). β-1,2-Glucan (average DP 25) was used for large-scale culture of Chitinophaga, stereochemical analysis, and mutational analysis. β-1,2-Glucan (average DP 64) was used for small-scale culture of Chitinophaga and measurement of the hydrolytic activity of enzymes. Cyclic β-1,2-glucan (DP 17–24) was kindly provided by Dr. M. Hisamatsu (6). Sop2, Sop3, Sop4, and Sop5 were produced by synthetic reaction of LiSOGP in the presence of Sop2 and α-glucose 1-phosphate (18) and enzymatic hydrolysis of β-1,2-glucan (average DP 25) catalyzed by Cpin_6279rC (endo-β-1,2-glucanase identified in this study). The chain length of the hydrolysates was increased by LiSOGP in the presence of α-glucose 1-phosphate. After ionic compounds had been removed with Amberlite MB4 (Organo, Tokyo, Japan), a sample was mixed with an aliquot of acetonitrile and then passed through a 0.45-μm regenerated cellulose membrane filter (Minisart RC4 Syringe Filter 17822; Sartorius, Goettingen, Germany). Then, the sample was loaded onto an Asahipak NH2P-90 20F column (particle size, 9 μm; inner diameter, 20 mm × 300 mm; Showa Denko, Tokyo, Japan). Sop6, Sop7, and Sop8 were fractionated by elution with 60% acetonitrile at the flow rate of 5.0–7.0 ml/min at 25 °C.
GDL was purchased from Wako Pure Chemical Industries (Osaka, Japan). Laminarin, carboxymethyl (CM)-cellulose, and p-nitrophenyl-β-d-glucopyranoside (pNP-β-Glc) were purchased from Sigma. CM-pachyman, CM-curdlan, lichenan, β-glucan from barley, xyloglucan (tamarind), glucomannan, arabinogalactan, arabinan, and polygalacturonate were purchased from Megazyme (Wicklow, Ireland). Pustulan was purchased from Calbiochem.
Strain and culture conditions
C. arvensicola (JCM2839) and C. pinensis (DSM2588) were purchased from the Japan Collection of Microorganisms (Ibaraki, Japan), and the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany), respectively.
C. arvensicola was pre-cultured in 5 ml of YP medium (1% polypeptone, 0.5% yeast extract, and 0.5% NaCl, adjusted to pH 7.2) at 30 °C for 24 h. C. pinensis was pre-cultured in 5 ml of CY medium (0.3% casitone, 0.14% CaCl2, 2H2O, and 0.1% yeast extract, adjusted to pH 7.2) at 25 °C for 48 h. For investigation of the β-1,2-glucan-hydrolyzing activity, these pre-cultured bacteria were inoculated into 5 ml of chemically defined medium (22) containing 0.5% β-1,2-glucan (average DP 64) or 0.5% Glc as a sole carbon source. C. arvensicola was grown at 30 °C for 24 h. C. pinensis was grown at 30 °C for 2 days (the Glc-containing medium) or 12 days (the β-1,2-glucan-containing medium). These bacteria were collected by centrifugation at 13,000 × g for 4 min, and the collected cells were suspended in 500 μl of 20 mm sodium phosphate (pH 7.0). The cells were then disrupted by sonication and centrifuged at 20,000 × g for 10 min to obtain cell extracts. The culture filtrates and cell extracts were concentrated and buffered with 20 mm MOPS-NaOH (pH 7.0) using Amicon Ultra 10,000 molecular weight cutoff (Millipore, Billerica, MA) to remove low molecular weight compounds such as Glc in the medium. For purification of the endo-β-1,2-glucanase, the pre-cultured C. arvensicola was inoculated into 1 liter of chemically defined medium containing 0.5% β-1,2-glucan (average DP 25) and then grown at 30 °C for 24 h.
Purification of endo-β-1,2-glucanase from C. arvensicola
C. arvensicola cultured in 1 liter of chemically defined medium was harvested by centrifugation at 16,000 × g for 15 min, and the harvested cells (3.8 g) were suspended in 20 ml of 5 mm MOPS-NaOH buffer (pH 7.5) (buffer A). The cells were disrupted by sonication and then centrifuged at 27,000 × g for 10 min to obtain a cell extract. The cell extract was applied onto an anion-exchange column (DEAE-Sepharose CL-6B (10 ml); GE Healthcare, Buckinghamshire, UK) pre-equilibrated with buffer A. After the column had been washed with buffer A, bound proteins were eluted with a 100-ml linear gradient of 0–500 mm NaCl in buffer A at a flow rate of 2.0 ml/min and collected in 2.5-ml aliquots. The fractions containing β-1,2-glucanase activity were pooled and dialyzed against 5 mm sodium phosphate buffer (pH 6.5) (buffer B). The solution was applied onto a hydroxyapatite column (Bio-Scale MiniCHT Ceramic Hydroxyapatite Cartridges (5 ml); Bio-Rad) pre-equilibrated with buffer B. After the column had been washed with buffer B, bound proteins were eluted with a 50-ml linear gradient of 5–500 mm sodium phosphate buffer (pH 6.5) at a flow rate of 2.0 ml/min and collected in 2.5-ml aliquots. The fractions containing β-1,2-glucanase activity were concentrated using Amicon Ultra 30,000 molecular weight cutoff (Millipore). The concentrated fraction was applied onto a Superdex 200 column (10/300 GL; GE Healthcare) pre-equilibrated with 50 mm sodium phosphate buffer (pH 6.5) containing 150 mm NaCl at a flow rate of 1.0 ml/min and collected in 0.5-ml aliquots. The fractions containing β-1,2-glucanase activity were applied onto a hydroxyapatite column again as described above except that the elution was carried out with a 30-ml linear gradient, and the eluate was collected in 1.0-ml aliquots. ÄKTA (GE Healthcare) was used for all fractionations. The fractionated proteins were subjected to SDS-PAGE, followed by staining with 2D-Silver Stain II (COSMO BIO Co., Ltd., Tokyo, Japan) or Coomassie Brilliant Blue R-250 (Nacalai Tesque, Kyoto, Japan). Precision Plus ProteinTM unstained standards (Bio-Rad) were used as protein markers.
Measurement of hydrolytic activity during purification
β-1,2-Glucan-hydrolyzing activity (the sum of the activities of β-1,2-glucanase and β-glucosidase) was determined by measuring the increase in reducing ends using the p-hydroxybenzoic acid hydrazide (PAHBAH) method (41). A reaction mixture (50 μl) containing 40% (v/v) enzyme fraction, 0.2% (w/v) β-1,2-glucan (average DP 64), and 50 mm MOPS-NaOH buffer (pH 6.5) was incubated at 30 °C for 24 h and then heated at 100 °C for 5 min to stop the reaction. An aliquot of the reaction mixture (20 μl) was mixed with 120 μl of a 1% (w/v) PAHBAH-HCl solution and heated at 100 °C for 5 min, followed by measurement of absorbance at 405 nm on a Spectramax 190 (Molecular Devices) with a 96-well microplate (EIA/RIA plate, 96-well half-area; Corning, NY). These instruments were used for all spectrophotometric measurements. During the purification from the cell extract of C. arvensicola, β-1,2-glucan-hydrolyzing activity was defined as the amount of released reducing ends (Glc equivalence) in ∼24 h based on a Glc standard curve.
Endo-β-1,2-glucanase activity was analyzed by TLC in the presence of GDL, a known β-glucosidase inhibitor (42). The enzymatic reaction was carried out in the reaction mixture described above in the presence of 50 mm GDL, and an aliquot of the reaction mixture was analyzed by TLC.
β-Glucosidase activity was determined by measuring the increase in pNP using pNP-β-Glc as a substrate. A reaction mixture (100 μl) containing 40% (v/v) enzyme fraction, 5 mm pNP-β-Glc, and 50 mm MOPS-NaOH buffer (pH 6.5) was incubated at 30 °C for 24 h, and an aliquot of the reaction mixture (20 μl) was mixed with 180 μl of 0.2 m Na2CO3. The increase in p-nitrophenol (pNP) was calculated from absorbance at 405 nm based on a pNP standard curve. β-Glucosidase activity was defined as the amount of released pNP in 24 h based on a pNP standard curve.
Sequence analysis
The protein band of the 43-kDa candidate for the target enzyme was transferred to a Sequi-BlotTM PVDF membrane (Bio-Rad) and then stained with Coomassie Brilliant Blue. The N-terminal sequence of the protein was analyzed by GenoStaff Co., Ltd. (Tokyo, Japan). Internal sequences of the protein were analyzed as described below. The protein detected on SDS-PAGE was routinely digested with trypsin (Promega, WI) or chymotrypsin (Promega). The obtained peptides were subjected to LC/MS/MS analysis as described in the previous report (43). The peptide sequences of the target enzyme were analyzed with an in-house MASCOT server as described in the previous report (43) except that the search parameters were as follows: taxonomy, other bacteria, and fragment mass tolerance ±0.6 Da. The peptide sequences were also analyzed by de novo sequencing using SPIDER 5 (44) on PEAKS Online 6 (Bioinformatics Solutions Inc., Waterloo, Canada) (45). From the trypsinized peptides, we selected ones that possessed more than six sequential amino acids with confidence above 80% with SPIDER 5. Then we searched for peptides from chymotrypsinized peptides that have more than five sequential common amino acids with the above selected trypsinized peptides. The internal sequences of the purified enzyme were constructed by combining these peptides. All amino acids that differ between these two peptides were arranged based on either trypsinized or chymotrypsinized peptides.
Cloning, expression, and purification
The gene encoding the Cpin_6279 protein (GenBankTM accession number ACU63684.1) was amplified by PCR (94 °C for 2 min and then 30 cycles of 94 °C for 15 s, 40 °C for 30 s, and 68 °C for 2 min) with a forward primer 5′-CATACCACATATGATGTCATGTGGAGGTTC-3′ (NdeI site underlined) and a reverse primer 5′-ATCAACTCGAGCTTCAGGTAAGGGCTCTG-3′ (XhoI site underlined) using TaKaRa ExTaqHS (Takara Bio, Shiga, Japan) and the genomic DNA as a template. The genomic DNA of C. pinensis was extracted from the cells with InstaGene Matrix (Bio-Rad). The amplified Cpin_6279 gene was inserted between the XhoI and NdeI sites of the pET-30a vector (Novagen, Madison, WI) to add a His6 tag to the target protein at the C terminus. For preparation of Cpin_6279rN, the Cpin_6279 gene was amplified by PCR (30 cycles of 94 °C for 15 s, 60 °C for 30 s, and 68 °C for 2 min) with a forward primer (as described above) and a reverse primer 5′-ATCAACTCGAGTCACTTCAGGTAAGGGCT-3′ (XhoI site underlined) using KOD Plus (TOYOBO, Osaka, Japan) and pET-30a inserted with Cpin_6279 as a template. The amplified gene was inserted to the corresponding site of the pET-28b vector (Novagen).
The constructed plasmid was transformed into E. coli BL21(DE3), and the transformant was cultured in 1 liter of Luria-Bertani medium containing 30 mg/liter kanamycin at 37 °C until the absorbance at 600 nm reached 0.6. Then, expression was induced with 0.1 mm isopropyl β-d-1-thiogalactopyranoside, and the transformant was further cultured at 20 °C for 24 h. The cells were centrifuged at 3,900 × g for 10 min and then suspended in 20 mm MOPS-NaOH buffer (pH 7.0) containing 500 mm NaCl (buffer C). The suspended cells were disrupted by sonication and centrifuged at 27,000 × g for 15 min to obtain a cell extract. The cell extract was applied onto a HisTrap FF crude column (5 ml; GE Healthcare) pre-equilibrated with buffer C containing 20 mm imidazole. After the column had been washed with the same buffer, the target protein was eluted with a linear gradient of 20–300 mm imidazole in buffer C. Amicon Ultra 30,000 molecular weight cutoff (Millipore) was used for the concentration of a portion of the fractionated protein and for exchanging the buffer in the protein solution to 20 mm MOPS-NaOH (pH 6.5).
As for the crystallization sample of Cpin_6279rC, ammonium sulfate was added to the peak fractions obtained on nickel affinity chromatography to 40% saturated concentration after the fractions had been collected and diluted to 0.5 mg/ml with 20 mm MOPS-NaOH (pH 6.5). The enzyme solution was applied onto a HiTrap Butyl HP column (5 ml; GE Healthcare) pre-equilibrated with 20 mm MOPS-NaOH (pH 6.5) containing 40% saturated ammonium sulfate. After the column had been washed with the same buffer, the target protein was eluted with a linear gradient of 40 to 0% saturated concentration of ammonium sulfate. Appropriate fractions were concentrated and buffered with 5 mm MOPS-NaOH (pH 6.5).
For preparation of Cpin_6279rN for crystallization, hydrophobic interaction chromatography was performed as described above except that 20 mm MOPS-NaOH (pH 7.0) and 30% saturated ammonium sulfate were used for purification instead of 20 mm MOPS-NaOH (pH 6.5) and 40% saturated ammonium sulfate, respectively. Then, Cpin_6279rN was applied onto a Superdex 200 pg 16/60 column (GE Healthcare) pre-equilibrated with 20 mm MOPS-NaOH (pH 7.0) containing 150 mm NaCl. Appropriate fractions were concentrated and buffered with 10 mm MOPS-NaOH (pH 7.0). Protein concentrations were determined by measurement of the absorbance at 280 nm, and calculation was based on the theoretical extinct coefficients of Cpin_6279rC and Cpin_6279rN (134,815 m−1·cm−1). The purity was checked by SDS-PAGE, and DynaMarker Protein MultiColor Stable (BioDynamics Laboratory Inc., Tokyo) was used as a protein marker.
Hydrolytic activity of the recombinant enzyme
The substrate specificity for various polysaccharides was determined by measuring the increase in reducing ends using PAHBAH. The enzymatic reaction was performed in a reaction mixture (200 μl) containing 4.0 μg/ml Cpin_6279rC, 0.2% (w/v) β-1,2-glucan (average DP 64), and 50 mm MES-NaOH buffer (pH 6.0) at 30 °C for 10 min. These conditions were defined as the standard conditions. To investigate the substrate specificity of Cpin_6279rC, the enzymatic reaction was performed under the standard conditions using an appropriate concentration of the enzyme, by substituting 0.2% β-1,2-glucan with 0.2% barley β-glucan, 0.0125% lichenan, 0.0125% laminarin, 0.006% arabinogalactan, 0.05% CM-curdlan, 0.025% CM-pachyman, 0.1% konjac glucomannan, 0.2% xyloglucan (tamarind), 0.1% polygalacturonate, 0.1% CM-cellulose, 0.05% pustulan, or 0.2% arabinan. Then, an aliquot of the reaction mixture (35 μl) was mixed with 105 μl of a 1% (w/v) PAHBAH-HCl solution and incubated at 100 °C for 5 min, followed by measurement of the absorbance at 405 nm. Because Sopns do not reduce alkaline copper or dinitrosalicylic acid (20, 22) and show low sensitivity toward PAHBAH, we used Sop4 as a standard for calculation of the enzymatic activity toward β-1,2-glucan. Sop4 is the main hydrolysate of β-1,2-glucan at the early stage of the reaction catalyzed by Cpin_6279rC. The enzymatic activity toward other polysaccharides was determined based on a Glc standard curve. One unit was defined as the amount of enzyme that released Sop4 or Glc equivalents/1 min under the standard conditions.
The action patterns for β-1,2-glucan and Sopns were analyzed by TLC. The enzymatic reaction was performed under the standard conditions with 0.2% each substrate. After the reaction had been stopped by heat treatment, an aliquot of the reaction mixture was analyzed by TLC.
Specific activity toward Sop5–7 was determined by HPLC analysis. The enzymatic reaction was performed in the presence of 10 mm each substrate at 30 °C for 5 and 10 min and then was stopped by heat treatment at 80 °C for over 3 min. The concentrations of the enzyme used for degradation of Sop5, Sop6, and Sop7 were 50, 10, and 5 μg/ml, respectively. The samples (25 μl) were injected onto a Shodex Asahipak NH2P-50 4E column (particle size, 5 μm; inner diameter 4.6 mm × 250 mm; Showa Denko K. K.) equilibrated with 70% (v/v) acetonitrile/water. Elution of the reaction products was performed with the same solution at the flow rate of 1 ml/min at 40 °C for 40 min by HPLC (Prominence; Shimadzu, Kyoto, Japan). A refractive index detector (RID-10A, Shimadzu) was used for detection of the eluates. The concentrations of the reaction products (Sop2–5) were determined using mixtures of Sop2–5 (0, 0.5, and 2 mm each) as standards. Specific activity was calculated from velocity of release of each reaction product (Sop2–3, Sop2–4, and Sop2–5 for Sop5, Sop6, and Sop7 substrates, respectively).
ESI-MS analysis
A substrate solution (20 μl) containing 5 mm MES-NaOH (pH 6.0) and 0.2% (w/v) Sop7 was mixed with 70 μl of 18O-labeled water (97%, Sigma) or unlabeled water as a control. The enzymatic reaction was conducted by adding 10 μl of 40 μg/ml Cpin_6279rC in 20 mm MOPS-NaOH (pH 6.5) to the substrate solution at 30 °C for 1 h. After the reaction, the reaction mixture was applied to Amicon Ultra 10,000 molecular weight cutoff (Millipore), and the flow-through solution was collected. An aliquot of the solution was dried in a centrifugal evaporator, dissolved in 50% (v/v) methanol/water, and then subjected to positive ESI-MS analysis using a JMS-T100LC AccuTOF spectrometer (JEOL, Tokyo, Japan). Reference spectra of Sop3 and Sop4 were measured with the same procedure as for the reaction solution.
pH and temperature profiles
To investigate the pH and temperature profiles, the hydrolytic activity of Cpin_6279rC was determined by measuring Glc after degradation of the β-1,2-glucan-hydrolysate produced by Cpin_6279rC into Glc by LiBGL for enhancement of sensitivity. The enzymatic reaction was carried out under the standard conditions, and an aliquot of the reaction mixture (20 μl) was incubated at 100 °C for 3 min to stop the reaction. The solution (20 μl) was mixed with 20 μl of 0.2 mg/ml LiBGL in 50 mm MES-NaOH (pH 6.0) and then incubated at 25 °C for 1 h. As for the effect of pH on Cpin_6279rC, 0.5 m MES-NaOH was used instead of 50 mm MES-NaOH (pH 6.0). The precipitate generated during heat treatment was removed by centrifugation. The supernatant (20 μl) was mixed with 140 μl of GOPOD Format kit (Megazyme) and then incubated at 40 °C for 20 min. The absorbance was measured at 510 nm, and the Glc concentration was determined based on a Glc standard curve.
The effect of pH on Cpin_6279rC activity was determined with 4.0 μg/ml Cpin_6279rC under the standard conditions by replacing 50 mm MES-NaOH buffer (pH 6.0) with 20 mm each buffer. The effect of temperature on Cpin_6279rC activity was determined with 2.0 μg/ml Cpin_6279rC, and the reaction was performed at each temperature for 7.5 min. pH and temperature stability were determined from the residual activity under the standard conditions after incubation of 0.4 mg/ml Cpin_6279rC in each buffer at 30 °C for 1 h and 0.04 mg/ml Cpin_6279rC in 20 mm MOPS-NaOH (pH 6.5) at each temperature for 1 h, respectively.
Kinetic analysis
To determine the kinetic parameters for β-1,2-glucan, the enzymatic reaction was performed in a 250-μl reaction mixture containing 3.0 μg/ml Cpin_6279rC and 0.019–0.39 mm β-1,2-glucan (average DP 64) under the standard conditions. The concentration of β-1,2-glucan was calculated based on its average DP. An aliquot of the reaction mixture (40 μl) was mixed with 120 μl of a 1% (w/v) PAHBAH-HCl solution, followed by spectrophotometric measurement and calculation of the enzymatic activity as described above. Kinetic parameters were determined by fitting experimental data to a Michaelis-Menten equation, v/[E]0 = kcat [S]/(Km + [S]), using GraFit version 7.0.3 (Erithacus Software Ltd., London, UK), where v is the initial velocity; [E]0 is the enzyme concentration; [S] is the substrate; kcat is the turnover number; and Km is the Michaelis constant.
Kinetic analysis for Sop5 was performed in a reaction mixture containing 10 μg/ml Cpin_6279rC and 0.3–10 mm Sop5. An aliquot of the reaction mixture (10 μl) was mixed with 10 μl of 200 mm sodium acetate buffer (pH 5.0) containing 1.0 mg/ml β-glucosidase from almonds (Oriental Yeast, Tokyo, Japan) and then incubated at 50 °C for 2 h to hydrolyze sophorose released by Cpin_6279rC. Because the β-glucosidase from almonds did not act on Sopns with DP ≥3,4 only sophorose was decomposed to Glc. An aliquot of the reaction mixture (15 μl) was mixed with 105 μl of GOPOD Format kit (Megazyme), and the Glc concentration was determined based on a Glc standard curve as described above. Sop5-hydrolyzing activity was calculated by halving the Glc concentration. Kinetic parameters were determined by fitting experimental data to a theoretical equation of substrate inhibition: v/[E]0 = kcat [S]/{(Km + [S]) (1 + [S]/Ki)} using GraFit version 7.0.3 (Erithacus), where Ki is the substrate inhibition constant, and the others are as given above.
Stereochemical analysis of reaction products
The anomeric configurations of the reaction products arising on enzymatic hydrolysis of β-1,2-glucan were determined by polarimetry and 1H NMR. Polarimetric analysis was performed with a JASCO P1010 polarimeter (JASCO Co., Tokyo, Japan). Prior to the addition of the enzyme, the control optical rotation was measured (0 min). The enzymatic reaction was conducted at room temperature in a reaction mixture (1.52 ml) containing 15.8 mm sodium phosphate (pH 6.0), 1.97% (w/v) β-1,2-glucan (average DP 25), and 0.64 mg/ml Cpin_6279rC, and the time course of the optical rotation was recorded (1.5–10 min, with 14 recordings). At 6 min, 100 μl of a 35% ammonia solution was added to the reaction mixture to accelerate mutarotation.
1H NMR analysis was performed with a Bruker Advance 600 spectrometer (Bruker Biospin, Rheinstetten, Germany) at 20 °C. The enzymatic reaction was conducted in a reaction mixture (600 μl) containing 10 mm sodium acetate (pH 5.0), 1.5% (w/v) β-1,2-glucan (average DP 25), 98.5% (v/v) D2O, and 0.2 mg/ml Cpin_6279rC. Before addition of the enzyme, a reference spectrum was recorded. After addition of the enzyme, the time course of the spectral change was recorded (3 min to 24 h, with 14 recordings). Sodium acetate was used as an internal standard for peak area calculation. Acetone was added after the reaction for use as an external standard for chemical shift adjustment. Reference spectra of Sop2–5 and β-1,2-glucan were obtained under the same conditions as for the reaction solution with a Bruker Advance 400 spectrometer. α-Anomers of the compounds were assigned based on the assignment of NMR spectra of β-1,2-glucan, Sop2, Sop3, and Sop4 in the previous study (18, 46).
TLC analysis
An aliquot of the reaction mixture (1 μl) after the reaction was stopped was spotted onto a TLC plate (7.5 × 5 cm, Kieselgel 60 F254; Merck, Darmstadt, Germany). The TLC plate was developed with a 75% (v/v) acetonitrile/water solution. The TLC plate was soaked in a 5% (w/v) sulfuric acid/ethanol solution and then heated in an oven until sugars were sufficiently detected on the plate.
Crystallography
Crystals of ligand-free Cpin_6279 were grown at 25 °C using the hanging drop vapor diffusion method in a 500-μl aliquot of a reservoir solution by mixing 1 μl of a 10 mg/ml Cpin_6279rC solution in 5 mm MOPS-NaOH (pH 6.5) with an equal volume of the reservoir solution containing 0.1 m ammonium iodide and 7% (w/v) PEG 3350. The crystals were soaked and cryoprotected with a solution containing 0.1 m ammonium iodide, 16% (w/v) PEG 3350, 25% (w/v) trehalose, and 5 mm Sop3. Despite the supplementation of Sop3, the crystal structure was obtained as a ligand-free form (see “Results”). Co-crystals of Cpin_6279 with Sop3 were produced at 25 °C using the sitting drop vapor diffusion method in a 70-μl aliquot of the reservoir solution by mixing 0.5 μl of a 28 mg/ml Cpin_6279rN solution containing 10 mm Sop3 in 9 mm MOPS-NaOH (pH 7.0) with an equal volume of the reservoir solution from the JCSG core I suite (Qiagen, Hilden, Germany), number 51 (0.2 m sodium chloride, 0.1 m sodium/potassium phosphate (pH 6.2), and 10% (w/v) PEG 8000). Crystals appropriate for X-ray diffraction experiments grew at least in 1 week. The crystals were soaked in the reservoir solution supplemented with 30% (v/v) PEG 400 for cryoprotection. The crystals were flash-cooled at 100 K in a stream of liquid nitrogen. X-ray diffraction data were collected using a charge-coupled device camera on beamline AR-NW12A at the Photon Factory of the High Energy Accelerator Research Organization (KEK, Japan). The data set was indexed, integrated, and scaled using HKL2000 (47). The initial phase was determined by the molecular replacement method using Molrep (48), and BF9343_0330 from B. fragilis NCTC9343 (PDB code 3EU8) was used as a search model. Automated model building was carried out using Buccaneer (49). Manual model building and refinement were carried out using Coot (50) and Refmac5 (51), respectively. The refined structures were validated using Molprobity (52) and Rampage (53). A model structure of β-Sop3 was built using JLigand (54). The structural figures were prepared using PyMOL (DeLano Scientific, Palo, Alto, CA).
Preparation and hydrolytic activity of mutant enzymes
The plasmids for expression of Cpin_6279r mutants were constructed using a PrimeSTAR mutagenesis basal kit (Takara Bio) with pET-30a-Cpin_6279 as a template. The primer pairs used for amplification of the mutant genes were as follows: 5′-GCAAGGCAACGTTATCATGTAGACGGT-3′ and 5′-ATAACGTTGCCTTGCCATACCACTAAC-3′ (E54Q); 5′-CAAAAAAACAATGGCGGAGATCTCGTA-3′ and 5′-GCCATTGTTTTTTTGTCCGAATGGCTT-3′ (D135N); 5′-GGCGGAAATCTCGTAGAGACCTCTTTT-3′ and 5′-TACGAGATTTCCGCCATTGTCTTTTTG-3′ (D139N); 5′-CTCGTACAGACCTCTTTTATGATACAA-3′ and 5′-AGAGGTCTGTACGAGATCTCCGCCATT-3′ (E142Q); 5′-TATAATCAATGCCTGATCATGTATATA-3′ and 5′-CAGGCATTGATTATAACCGGTCACGGG-3′ (E211Q); and 5′-GCAATAAATCAGGGACCTATCGTTGTG-3′ and 5′-TCCCTGATTTATTGCCAGGTACTGCTG-3′ (D400N). The underlining indicates the mutated nucleotides. The mutant enzymes were produced and purified basically in the same way as described for the wild type. The enzymatic reaction was carried out under the standard conditions using β-1,2-glucan (average DP 25) as a substrate instead of that of average DP 64.
Author contributions
M. N., H. T., H. N., and S. F. designed the experiments; K. A., M. N., H. M., S. K., T. Y., T. N., Y. T., and N. S. performed the experiments; K. A., M. N., A. M., H. T., T. A., and S. F. analyzed the data; and K. A. wrote the manuscript with help from the other authors.
Acknowledgments
We thank the staff of the Photon Factory for the X-ray data collection. We also thank Dr. M. Hisamatsu and Dr. N. Isono for providing the cyclic β-1,2-glucan and Dr. M. Kitaoka for helping with the substrate preparation.
This work was supported in part by a Noda Institute for Scientific Research Grant (2013 Young Investigator Research Grant). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5GZH and 5GZK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
B. Henrissat, personal communication.
M. Nakajima, unpublished data.
- GH
- glycoside hydrolase
- Cpin_6279rC
- recombinant C-terminal His6-tagged Cpin_6279 protein
- Cpin_6279rN
- recombinant N-terminal His6-tagged Cpin_6279 protein
- DP
- degree of polymerization
- GDL
- gluconic acid δ-lactone
- LiBGL
- β-glucosidase from L. innocua
- LiSOGP
- 1,2-β-oligoglucan phosphorylase from L. innocua
- Sopns
- sophorooligosaccharides (β-1,2-glucooligosaccharides)
- ESI
- electrospray ionization
- PDB
- Protein Data Bank
- pNP
- p-nitrophenol
- CM
- carboxymethyl
- PAHBAH
- p-hydroxybenzoic acid hydrazide.
References
- 1. Henrissat B. (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Henrissat B., and Bairoch A. (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 293, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Henrissat B., and Bairoch A. (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316, 695–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davies G., and Henrissat B. (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859 [DOI] [PubMed] [Google Scholar]
- 5. Henrissat B., and Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 [DOI] [PubMed] [Google Scholar]
- 6. Hisamatsu M. (1992) Cyclic (1→2)-β-d-glucans (cyclosophorans) produced by Agrobacterium and Rhizobium species. Carbohydr. Res. 231, 137–146 [DOI] [PubMed] [Google Scholar]
- 7. Breedveld M. W., and Miller K. J. (1994) Cyclic β-glucans of members of the family Rhizobiaceae. Microbiol. Rev. 58, 145–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hisamatsu M., Amemura A., Matsuo T., Matsuda H., and Harada T. (1982) Cyclic (1→2)-β-d-glucan and the octasaccharide repeating-unit of succinoglycan produced by Agrobacterium. Microbiology 128, 1873–1879 [Google Scholar]
- 9. Briones G., Iñón de Iannino N., Roset M., Vigliocco A., Paulo P. S., and Ugalde R. A. (2001) Brucella abortus cyclic β-1,2-glucan mutants have reduced virulence in mice and are defective in intracellular replication in HeLa cells. Infect. Immun. 69, 4528–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gay-Fraret J., Ardissone S., Kambara K., Broughton W. J., Deakin W. J., and Le Quéré A. (2012) Cyclic-β-glucans of Rhizobium (Sinorhizobium) sp. strain NGR234 are required for hypo-osmotic adaptation, motility, and efficient symbiosis with host plants. FEMS Microbiol. Lett. 333, 28–36 [DOI] [PubMed] [Google Scholar]
- 11. Rigano L. A., Payette C., Brouillard G., Marano M. R., Abramowicz L., Torres P. S., Yun M., Castagnaro A. P., Oirdi M. E., Dufour V., Malamud F., Dow J. M., Bouarab K., and Vojnov A. A. (2007) Bacterial cyclic β-1,2-glucan acts in systemic suppression of plant immune responses. Plant Cell 19, 2077–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arellano-Reynoso B., Lapaque N., Salcedo S., Briones G., Ciocchini A. E., Ugalde R., Moreno E., Moriyón I., and Gorvel J. P. (2005) Cyclic β-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nat. Immunol. 6, 618–625 [DOI] [PubMed] [Google Scholar]
- 13. Amemura A., Hashimoto T., Koizumi K., and Utamura T. (1985) Occurrence of extracellular (1→2)-β-d-glucans and (1→2)-β-d-gluco-oligosaccharides in Acetobacter. Microbiology 131, 301–307 [Google Scholar]
- 14. Amemura A., and Cabrera-Crespo J. (1986) Extracellular oligosaccharides and low-Mr polysaccharides containing (1,2)-β-d-glucosidic linkages from strains of Xanthomonas, Escherichia coli and Klebsiella pneumoniae. J. Gen. Microbiol. 132, 2443–2452 [DOI] [PubMed] [Google Scholar]
- 15. Bohin J. P. (2000) Osmoregulated periplasmic glucans in Proteobacteria. FEMS Microbiol. Lett. 186, 11–19 [DOI] [PubMed] [Google Scholar]
- 16. Talaga P., Fournet B., and Bohin J. P. (1994) Periplasmic glucans of Pseudomonas syringae pv. syringae. J. Bacteriol. 176, 6538–6544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller K. J., Kennedy E. P., and Reinhold V. N. (1986) Osmotic adaptation by Gram-negative bacteria: possible role for periplasmic oligosaccharides. Science 231, 48–51 [DOI] [PubMed] [Google Scholar]
- 18. Nakajima M., Toyoizumi H., Abe K., Nakai H., Taguchi H., and Kitaoka M. (2014) 1,2-β-Oligoglucan phosphorylase from Listeria innocua. PLoS ONE 9, e92353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakajima M., Yoshida R., Miyanaga A., Abe K., Takahashi Y., Sugimoto N., Toyoizumi H., Nakai H., Kitaoka M., and Taguchi H. (2016) Functional and structural analysis of a β-glucosidase involved in β-1,2-glucan metabolism in Listeria innocua. PLoS ONE 11, e0148870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reese E. T., Parrish F. W., and Mandels M. (1961) β-d-1,2-Glucanases in fungi. Can. J. Microbiol. 7, 309–317 [DOI] [PubMed] [Google Scholar]
- 21. Kitahata S., and Edagawa S. (1987) Cyclic 1,2-β-d-glucan-hydrolyzing enzymes from Acremonium sp.15. Purification and some properties of endo-1,2-β-d-glucanase and β-d-glucosidase. Agric. Biol. Chem. 51, 2701–2708 [Google Scholar]
- 22. Mendoza N. S., and Amemura A. (1983) (1,2)-β-d-Glucan-hydrolyzing enzymes in Cytophaga arvensicola: partial purification an some properties of endo-(1,2)-β-glucanase and β-d-glucosidase specific for (1,2)-and (1,3)-linkages. J. Ferment. Technol. 61, 473–481 [Google Scholar]
- 23. Abe K., Nakajima M., Kitaoka M., Toyoizumi H., Takahashi Y., Sugimoto N., Nakai H., and Taguchi H. (2015) Large-scale preparation of 1,2-β-glucan using 1,2-β-oligoglucan phosphorylase. J. Appl. Glycosci. 62, 47–52 [Google Scholar]
- 24. Glavina Del Rio T., Abt B., Spring S., Lapidus A., Nolan M., Tice H., Copeland A., Cheng J. F., Chen F., Bruce D., Goodwin L., Pitluck S., Ivanova N., Mavromatis K., Mikhailova N., et al. (2010) Complete genome sequence of Chitinophaga pinensis type strain (UQM 2034). Stand. Genomic Sci. 2, 87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., Potter S. C., Punta M., Qureshi M., Sangrador-Vegas A., Salazar G. A., Tate J., and Bateman A. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 27. Nelson T. E. (1970) The hydrolytic mechanism of an exo-β-(1→3)-d-glucanase. J. Biol. Chem. 245, 869–872 [PubMed] [Google Scholar]
- 28. Sova V. V., Shirokova N. I., Kusaykin M. I., Scobun A. S., Elyakova L. A., and Zvyagintseva T. N. (2003) β-1,3-Glucanase from unfertilized eggs of the sea urchin Strongylocentrotus intermedius. Comparison with β-1,3-glucanases of marine and terrestrial mollusks. Biochemistry 68, 529–533 [DOI] [PubMed] [Google Scholar]
- 29. Hemsworth G. R., Déjean G., Davies G. J., and Brumer H. (2016) Learning from microbial strategies for polysaccharide degradation. Biochem. Soc. Trans. 44, 94–108 [DOI] [PubMed] [Google Scholar]
- 30. Kawano S., Tajima K., Kono H., Erata T., Munekata M., and Takai M. (2002) Effects of endogenous endo-β-1,4-glucanase on cellulose biosynthesis in Acetobacter xylinum ATCC23769. J. Biosci. Bioeng. 94, 275–281 [DOI] [PubMed] [Google Scholar]
- 31. Köseoğlu V. K., Heiss C., Azadi P., Topchiy E., Güvener Z. T., Lehmann T. E., Miller K. W., and Gomelsky M. (2015) Listeria monocytogenes exopolysaccharide: origin, structure, biosynthetic machinery and c-di-GMP-dependent regulation. Mol. Microbiol. 96, 728–743 [DOI] [PubMed] [Google Scholar]
- 32. Bakkevig K., Sletta H., Gimmestad M., Aune R., Ertesvåg H., Degnes K., Christensen B. E., Ellingsen T. E., and Valla S. (2005) Role of the Pseudomonas fluorescens alginate lyase (AlgL) in clearing the periplasm of alginates not exported to the extracellular environment. J. Bacteriol. 187, 8375–8384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jain S., and Ohman D. E. (2005) Role of an alginate lyase for alginate transport in mucoid Pseudomonas aeruginosa. Infect. Immun. 73, 6429–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stam M. R., Blanc E., Coutinho P. M., and Henrissat B. (2005) Evolutionary and mechanistic relationships between glycosidases acting on α- and β-bonds. Carbohydr. Res. 340, 2728–2734 [DOI] [PubMed] [Google Scholar]
- 35. Koivula A., Ruohonen L., Wohlfahrt G., Reinikainen T., Teeri T. T., Piens K., Claeyssens M., Weber M., Vasella A., Becker D., Sinnott M. L., Zou J. Y., Kleywegt G. J., Szardenings M., Ståhlberg J., and Jones T. A. (2002) The active site of cellobiohydrolase Cel6A from Trichoderma reesei: the roles of aspartic acids D221 and D175. J. Am. Chem. Soc. 124, 10015–10024 [DOI] [PubMed] [Google Scholar]
- 36. Nakamura A., Ishida T., Kusaka K., Yamada T., Fushinobu S., Tanaka I., Kaneko S., Ohta K., Tanaka H., Inaka K., Higuchi Y., Niimura N., Samejima M., and Igarashi K. (2015) “Newton's cradle” proton relay with amide-imidic acid tautomerization in inverting cellulase visualized by neutron crystallography. Sci. Adv. 1, e1500263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bianchetti C. M., Takasuka T. E., Deutsch S., Udell H. S., Yik E. J., Bergeman L. F., and Fox B. G. (2015) Active site and laminarin binding in glycoside hydrolase family 55. J. Biol. Chem. 290, 11819–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J., Zheng M., Zhang C., and Xu D. (2013) “Amide resonance” in the catalysis of 1,2-α-l-fucosidase from Bifidobacterium bifidum. J. Phys. Chem. B 117, 10080–10092 [DOI] [PubMed] [Google Scholar]
- 39. Brás J. L., Cartmell A., Carvalho A. L., Verzé G., Bayer E. A., Vazana Y., Correia M. A., Prates J. A., Ratnaparkhe S., Boraston A. B., Romão M. J., Fontes C. M., and Gilbert H. J. (2011) Structural insights into a unique cellulase fold and mechanism of cellulose hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 5237–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McKee L. S., and Brumer H. (2015) Growth of Chitinophaga pinensis on plant cell wall glycans and characterisation of a glycoside hydrolase family 27 β-l-arabinopyranosidase implicated in arabinogalactan utilisation. PLoS ONE 10, e0139932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lever M. (1972) A new reaction for colorimetric determination of carbohydrates. Anal. Biochem. 47, 273–279 [DOI] [PubMed] [Google Scholar]
- 42. Reese E. T., Parrish F. W., and Ettlinger M. (1971) Nojirimycin and d-glucono-1,5-lactone as inhibitors of carbohydrases. Carbohydr. Res. 18, 381–388 [Google Scholar]
- 43. Nakajima M., Yamashita T., Takahashi M., Nakano Y., and Takeda T. (2012) Identification, cloning, and characterization of β-glucosidase from Ustilago esculenta. Appl. Microbiol. Biotechnol. 93, 1989–1998 [DOI] [PubMed] [Google Scholar]
- 44. Han Y., Ma B., and Zhang K. (2005) SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 3, 697–716 [DOI] [PubMed] [Google Scholar]
- 45. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., and Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 46. Roslund M. U., Tähtinen P., Niemitz M., and Sjöholm R. (2008) Complete assignments of the 1H and 13C chemical shifts and JH,H coupling constants in NMR spectra of d-glucopyranose and all d-glucopyranosyl-d-glucopyranosides. Carbohydr. Res. 343, 101–112 [DOI] [PubMed] [Google Scholar]
- 47. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 48. Vagin A., and Teplyakov A. (1997) MOLREP: an Automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 49. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 50. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lovell S. C., Davis I. W., Arendall W. B. 3rd., de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., and Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 54. Lebedev A. A., Young P., Isupov M. N., Moroz O. V., Vagin A. A., and Murshudov G. N. (2012) JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D Biol. Crystallogr. 68, 431–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McWilliam H., Li W., Uludag M., Squizzato S., Park Y. M., Buso N., Cowley A. P., and Lopez R. (2013) Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 41, W597–W600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gouet P., Robert X., and Courcelle E. (2003) ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31, 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kumar S., Stecher G., and Tamura K. (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]