

Abstract
NRF2 (nuclear factor erythroid 2-related factor 2) is a key transcriptional activator that mediates the inducible expression of antioxidant genes. NRF2 is normally ubiquitinated by KEAP1 (Kelch-like ECH-associated protein 1) and subsequently degraded by proteasomes. Inactivation of KEAP1 by oxidative stress or electrophilic chemicals allows NRF2 to activate transcription through binding to antioxidant response elements (AREs) and recruiting histone acetyltransferase CBP (CREB-binding protein). Whereas KEAP1-dependent regulation is a major determinant of NRF2 activity, NRF2-mediated transcriptional activation varies from context to context, suggesting that other intracellular signaling cascades may impact NRF2 function. To identify a signaling pathway that modifies NRF2 activity, we immunoprecipitated endogenous NRF2 and its interacting proteins from mouse liver and identified glucocorticoid receptor (GR) as a novel NRF2-binding partner. We found that glucocorticoids, dexamethasone and betamethasone, antagonize diethyl maleate-induced activation of NRF2 target genes in a GR-dependent manner. Dexamethasone treatment enhanced GR recruitment to AREs without affecting chromatin binding of NRF2, resulting in the inhibition of CBP recruitment and histone acetylation at AREs. This repressive effect was canceled by the addition of histone deacetylase inhibitors. Thus, GR signaling decreases NRF2 transcriptional activation through reducing the NRF2-dependent histone acetylation. Consistent with these observations, GR signaling blocked NRF2-mediated cytoprotection from oxidative stress. This study suggests that an impaired antioxidant response by NRF2 and a resulting decrease in cellular antioxidant capacity account for the side effects of glucocorticoids, providing a novel viewpoint for the pathogenesis of hypercorticosteroidism.
Keywords: gene transcription, glucocorticoid receptor, histone acetylation, Nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), oxidative stress, electrophile
Introduction
The liver is the most important organ for the detoxification of xenobiotics and endogenous reactive compounds. Many genes encoding detoxifying enzymes, antioxidant proteins, and drug transporters are abundantly expressed in hepatocytes, where they maintain redox homeostasis and prevent cellular damage. Whereas some genes encoding detoxifying enzymes are constitutively expressed, others are inducibly expressed following exposure to these chemicals. NRF2 is a master transcription activator that regulates the inducible expression of cytoprotective genes in response to xenobiotic electrophiles and reactive oxygen species by directly binding to specific DNA sequences called antioxidant response elements (AREs)4 (1). To date, protective roles of NRF2 in the liver have been demonstrated in numerous studies (2–8).
Under normal conditions, NRF2 activity is tightly regulated by KEAP1, a substrate adaptor protein for CULLIN3-based ubiquitin E3 ligases that constantly ubiquitinates NRF2 to target it for proteasomal degradation. KEAP1 is inactivated when cells are exposed to electrophiles and/or reactive oxygen species, resulting in NRF2 stabilization and induction of its target genes. Although the KEAP1-NRF2 system mainly contributes to cytoprotection and the antioxidant response, recent studies have highlighted cross-talk between NRF2 and various other cellular signaling pathways, expanding our understanding of the wide range of biological processes to which NRF2 contributes. For example, the NRF2 and NOTCH pathways engage in reciprocal transcriptional regulation during liver development and regeneration (9, 10), NRF2 cooperates with PI3K-AKT signaling to drive the metabolic reprogramming of proliferating cells (11–13), and NRF2 disturbs the proinflammatory NF-κB pathway to exert a potent anti-inflammatory function (14). However, studies that describe direct interactions between transcription factors and NRF2 are limited.
NRF2 possesses six functional domains: Neh1 for DNA binding and dimerization; Neh2 and Neh6 for stability control; and Neh3, Neh4, and Neh5 for transcriptional activation (15). Several transcription cofactors have been found to interact directly with NRF2. Small Maf proteins bind NRF2 within the Neh1 domain, thus conferring DNA binding ability on NRF2 (16, 17). KEAP1 and βTrCP bind NRF2 within the Neh2 and Neh6 domains, respectively, and ubiquitinate NRF2 to mark it for proteasomal degradation (18, 19). CBP, BRG1, and MED16 bind within the Neh4/Neh5 domains (20–22), and CHD6 binds within the Neh3 domain (23), supporting the NRF2-dependent transcriptional activation.
In this study, we aimed to explore cellular signaling pathways that exhibit cross-talk with NRF2 and modify its cytoprotective activity. We immunoprecipitated endogenous NRF2 and its interacting proteins from livers of hepatocyte-specific Keap1-deficient mice, in which NRF2 is constitutively stabilized and accumulates in the nucleus (24). Among various proteins identified by mass spectrometry, glucocorticoid receptor (GR) was reproducibly obtained, suggesting that glucocorticoid (GC) modulates NRF2 activity through GR-NRF2 interaction. We found that GR is recruited to AREs in response to GC and inhibits NRF2 activity. This inhibition was canceled by treatment with the histone deacetylase (HDAC) inhibitors, trichostatin A (TSA) and valproic acid (VA), suggesting that GR antagonizes NRF2 activity by reducing the NRF2-dependent histone acetylation surrounding the AREs. In our study, activation of GR signaling indeed sensitized cells to oxidative stress, suggesting that an impaired antioxidant response by NRF2 and a subsequent decrease in antioxidant capacity underlie pathological conditions caused by excessive activation of GR signaling, such as Cushing's syndrome.
Results
NRF2 interacts with GR
To clarify which intracellular signaling pathways influence the transcriptional activity of NRF2, we used an anti-NRF2 antibody to isolate NRF2-interacting proteins from liver of Keap1-deficient mice in which NRF2 is constitutively stabilized and localized in the nucleus. The immunoprecipitated proteins were analyzed through liquid chromatography-tandem mass spectrometry (LC-MS/MS). In addition to previously identified NRF2 interactors, such as CBP, MED16, and other subunits of the Mediator complex, we identified GR as a new NRF2-binding partner (Fig. 1A). Immunoblotting analysis using an anti-GR antibody confirmed the interaction between NRF2 and GR (Fig. 1B). To determine which NRF2 domains are responsible for this interaction, we conducted a GST-pulldown assay using GST-fusion proteins of various NRF2 mutants (Fig. 1C). Nuclear lysates from 293T cells transiently overexpressing FLAG-tagged GR were incubated with the GST-fusion NRF2 mutants (Fig. 1D). An anti-FLAG antibody detected the clear and specific association between GR and the Neh4/Neh5 domains of NRF2 (Fig. 1E). Thus, GR interacts with Neh4/Neh5, which are the transactivation domains of NRF2.
Figure 1.
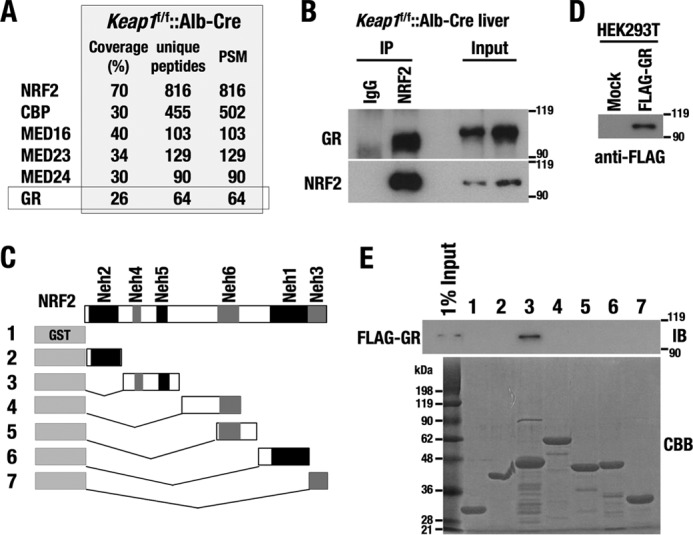
GR interacts with the transactivation domains of NRF2. A, list of NRF2-interacting proteins identified by LC-MS/MS from Keap1f/f::albumin-Cre mouse livers. PSM, total peptide spectrum matches. B, immunoblotting analysis of GR in the NRF2 complex. C, constructs of GST-fusion proteins of NRF2 mutants. 1, GST; 2, GST-Neh2(1–84); 3, GST-Neh4/5(85–210); 4, GST-Neh6u(211–407); 5, GST-Neh6d(319–408); 6, GST-Neh1(408–554); 7, GST-Neh3(555–598). D, FLAG-GR overexpressed in 293T cells, which was used for the interaction study with GST-fusion NRF2 mutants. E, GST-NRF2 pulldown assay with FLAG-GR. FLAG-GR was purified from FLAG-GR-expressing 293T cells with an anti-FLAG antibody and incubated with GST-NRF2 mutants. FLAG-GR was detected using an anti-FLAG antibody (top). A protein staining of GST-NRF2 mutants is shown (bottom). CBB, Coomassie Brilliant Blue.
GR signaling inhibits induction of NRF2 target genes in response to electrophiles
The association between GR and NRF2 led us to expect that GR signaling affects the transcriptional activity of NRF2. We examined the effect of GR on the inducible expression of NRF2 target genes using a mouse hepatoma cell line, Hepa1c1c7. Diethyl maleate (DEM)-induced expression of three representative NRF2 target genes, NAD(P)H-quinone oxidoreductase 1 (Nqo1), glucose-6-phosphate dehydrogenase X-linked (G6pdx), and glutamate-cysteine ligase modifier subunit (Gclm), were all decreased when cells were additionally treated with dexamethasone (Dex) and betamethasone (Bet) (Fig. 2A). To verify that the repression of the NRF2 target genes by Dex and Bet was GR-dependent, we knocked down GR and examined whether the repressive effect of Dex and Bet were canceled. Two different siRNAs against GR efficiently reduced the protein levels of GR (Fig. 2B). As we expected, GR knockdown canceled the inhibitory effect of Dex and Bet on Nqo1, G6pdx, and Gclm (Fig. 2C). Thus, both Dex and Bet repress NRF2-dependent transcriptional activation through GR, which indicates that GR signaling inhibits NRF2 activity.
Figure 2.

GR represses inducible expression of NRF2 target genes in response to DEM. A, relative expression levels of NRF2 target genes in Hepa1c1c7 cells treated with DEM, DEM + Dex, and DEM + Bet. B, protein levels of GR in Hepa1c1c7 cells with or without GR knockdown. C, relative expression levels of NRF2 target genes in Hepa1c1c7 cells with or without GR knockdown. Cells were treated with DEM, DEM + Dex, and DEM + Bet. -Fold changes were calculated against vehicle-treated samples, and the average and S.D. (error bars) of the -fold changes from three independent experiments are shown (A and C). Student's t test (A) and one-way ANOVA and Tukey's post hoc test (C) were performed as statistical analyses. *, p < 0.05; **, p < 0.01; ns, nonsignificant.
NRF2-mediated antioxidant response is attenuated in liver under hyperglucocorticoidism
To examine whether the repressive effect of GR signaling on NRF2-mediated transcriptional activation could be observed in vivo, we administered 2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im), a potent NRF2-inducing chemical, to mice with or without Dex treatment. -Fold inductions of NRF2 target genes by CDDO-Im were reduced when the mice were pretreated with Dex (Fig. 3A), indicating that Dex limits the inducible expression of NRF2 target genes in response to electrophiles.
Figure 3.

Dex treatment inhibits expression of NRF2 target genes in vivo. A, relative expression levels of NRF2 target genes in response to CDDO-Im with or without Dex treatment. The average and S.D. (error bars) are shown. Average expression levels of vehicle-treated animals are set as 1. B, relative expression levels of NRF2 target genes in livers of Ptenf/f::Keap1f/f::Alb-Cre mice with or without Dex treatment. The average and S.D. are shown. Average expression levels of vehicle-treated animals are set as 1. One-way ANOVA and Tukey's post hoc test were performed. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, nonsignificant.
We also examined whether constitutively activated NRF2 was repressed by Dex. To this end, we exploited hepatocyte-specific Pten:Keap1 double-deficient mice (Ptenf/f::Keap1f/f:: Alb-Cre mice), in which NRF2 markedly accumulates due to simultaneous inactivation of KEAP1-CUL3-dependent and βTrCP-CUL1-dependent degradation pathways (11, 12). The mice received a single injection of Dex. Our results showed that all NRF2 target genes were repressed by Dex treatment (Fig. 3B). These results confirm that GR signaling inhibits NRF2-mediated transcriptional activation in vivo.
GR signaling does not affect nuclear localization and DNA binding of NRF2
To understand the mechanism underlying the GR-mediated inhibition of NRF2-dependent transcriptional activation, we first examined the nuclear accumulation of NRF2 in the presence of Dex. DEM-induced NRF2 accumulation in the nucleus did not change following additional treatment with Dex in Hepa1c1c7 cells (Fig. 4, A and B). Similarly, CDDO-Im-induced nuclear accumulation of NRF2 in liver was comparable between the mice with and without Dex treatment (Fig. 4C).
Figure 4.
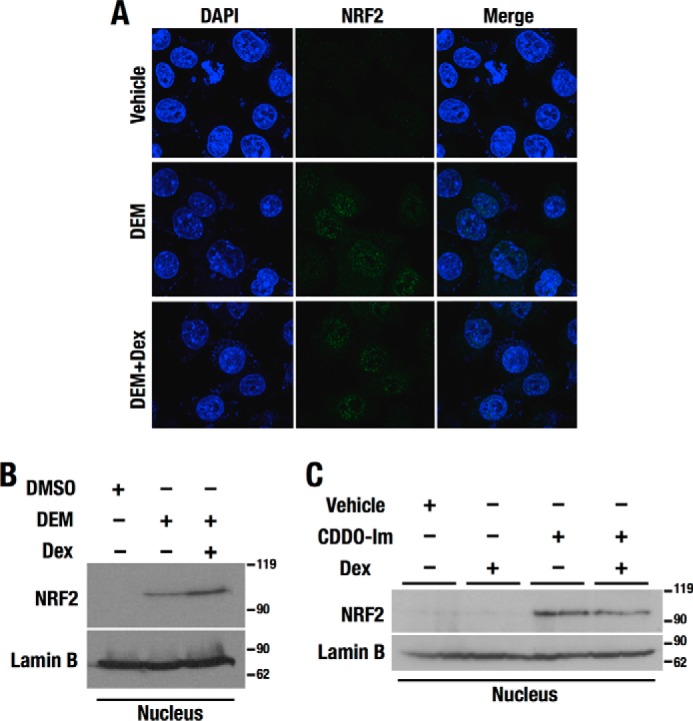
Nuclear localization of NRF2 is not affected by Dex. A, confocal microscopy analysis showing subcellular localization of NRF2 in Hepa1c1c7 cells treated with DEM and DEM + Dex. The nuclear staining by DAPI is shown in blue, and the endogenous NRF2 localization detected by anti-NRF2 antibody is shown in green. B and C, immunoblotting analysis of NRF2. Nuclear extracts of Hepa1c1c7 cells (B) and mouse livers (C) were analyzed. Lamin B was detected as a nuclear loading control.
We next examined NRF2 recruitment to AREs in NRF2 target gene loci in Hepa1c1c7 cells. Functional AREs are present in the promoter region of Nqo1, the second intron of G6pdx, and the upstream enhancer of Gclm (Fig. 5A). As expected from the finding that Dex treatment had no effects on the nuclear localization of NRF2 (Fig. 4, A and B), the ChIP assay showed that DEM-induced recruitment of NRF2 to the AREs was comparable between vehicle- and Dex-treated cells (Fig. 5B), indicating that Dex treatment does not influence the binding of NRF2 to AREs.
Figure 5.
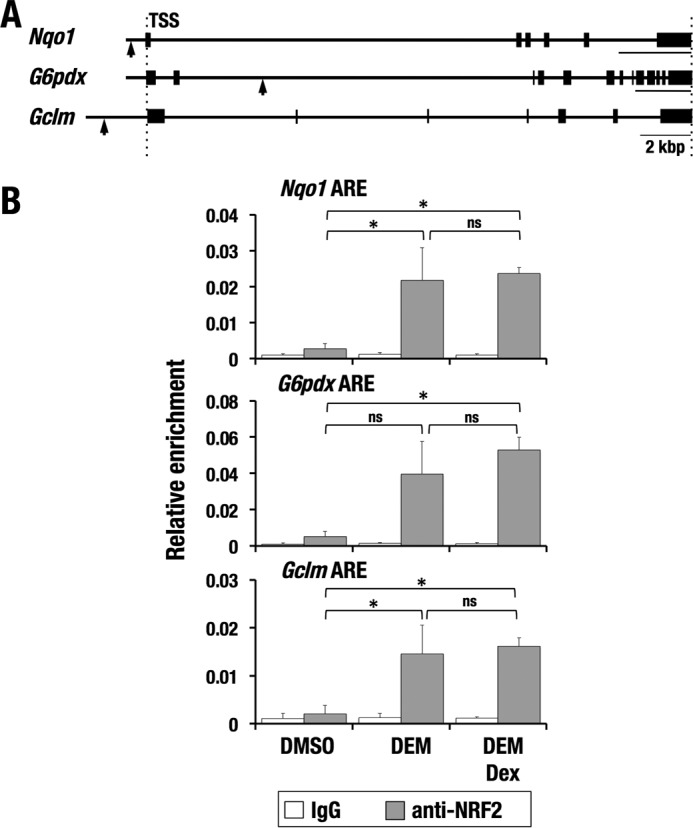
NRF2 recruitment to AREs is not affected by Dex. A, mouse gene loci of representative NRF2 target genes. Arrows, positions of AREs. B, ChIP assay with an anti-NRF2 antibody using Hepa1c1c7 cells treated with DEM and DEM + Dex. The average and S.D. (error bars) from triplicate reactions are shown. One-way ANOVA and Tukey's post hoc test were performed. *, p < 0.05; ns, nonsignificant.
GR inhibits NRF2-dependent transcriptional activation through AREs
One of the possible mechanisms for transcriptional repression by GR is through its direct binding to DNA at consensus sites termed glucocorticoid response elements (GREs). However, considering the interaction between GR and NRF2, we suspected that GR inhibits NRF2-dependent transcriptional activation through tethering to NRF2, which is referred to as transrepression. To test this hypothesis, we next examined whether AREs were sufficient for the inhibitory effect of GR signaling on NRF2 transcriptional activity. To this end, we conducted a reporter assay using a luciferase reporter gene driven by rabbit β-globin minimal promoter connected with triplicated AREs (3× ARE-LUC) (Fig. 6A). NRF2 overexpression resulted in increased reporter activity, and this increase was further enhanced by simultaneous treatment with DEM. The elevated reporter activity was dramatically repressed by Dex only when GR was overexpressed, which supports the notion that Dex represses NRF2 activity in a GR-dependent manner. Thus, AREs are likely to be sufficient to support GR-mediated inhibition of NRF2 transcriptional activity. This result suggests that interaction of GR and NRF2 is responsible for the repressive effect of GR.
Figure 6.
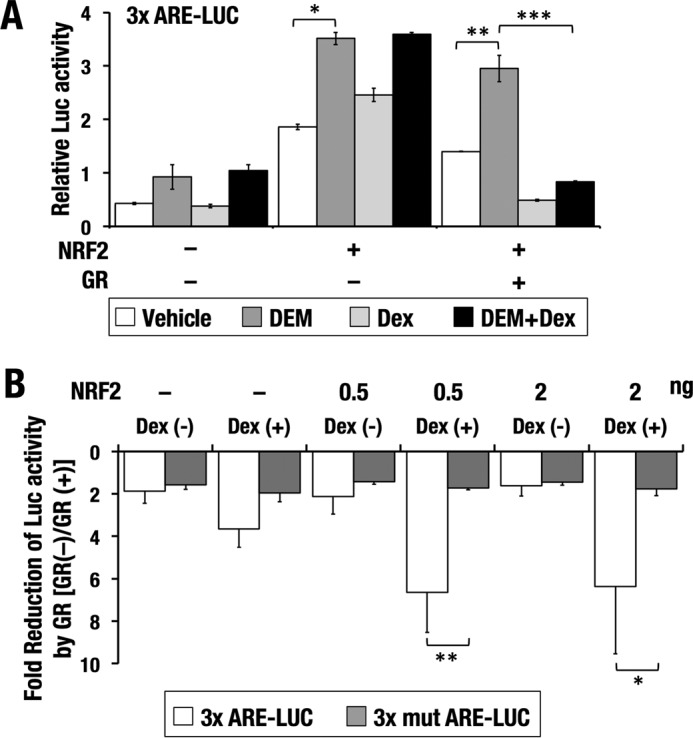
GR represses NRF2-dependent transcription through ARE. A, 3× ARE-LUC was introduced into 293T cells with or without NRF2 and GR expression vectors. Cells were treated with DEM and/or Dex for 18 h before luciferase activities were measured. The average and S.D. from three independent experiments are shown. One-way ANOVA and Tukey's post hoc test were performed. B, 3× ARE-LUC or 3× mut ARE-LUC was introduced into 293T cells with or without NRF2 and GR expression vectors. Cells were treated with or without Dex for 18 h before luciferase activities were measured. -Fold reduction of luciferase activity by GR was calculated. The average and S.D. (error bars) from three independent experiments are shown. Student's t test was performed. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We further verified whether the repressive effect of GR signaling on the reporter activity was specifically mediated through NRF2 binding to AREs. We compared the repressive activity of GR for 3× ARE-LUC and for 3× mut ARE-LUC, which harbored triplicated mutant AREs (Fig. 6B). Without Dex, the -fold reduction of luciferase activity was almost comparable between the two reporter genes irrespective of the NRF2 expression, suggesting that overexpressed GR elicited a nonspecific modest inhibitory effect on both reporter genes. In the presence of Dex, GR-mediated reduction was obvious for 3× ARE-LUC but not for 3× mut ARE-LUC. When NRF2 was additionally expressed, the GR-mediated reduction was further accentuated only in 3× ARE-LUC without any changes in 3× mut ARE-LUC. These results indicate that GR signaling represses NRF2-mediated transcriptional activation through AREs.
Dex treatment increases GR recruitment to AREs
GR has been shown to attenuate transcriptional activation without contacting DNA but by directly interacting with other transcription factors. Considering the interaction between GR and NRF2, we examined the recruitment of endogenous GR to AREs in the NRF2 target gene loci by a ChIP assay. Because available anti-GR antibodies worked for human GR but not for mouse GR in the ChIP assay, we used a human hepatoma cell line, HepG2. AREs of NQO1, G6PD, and GCLM were examined (Fig. 7A).
Figure 7.
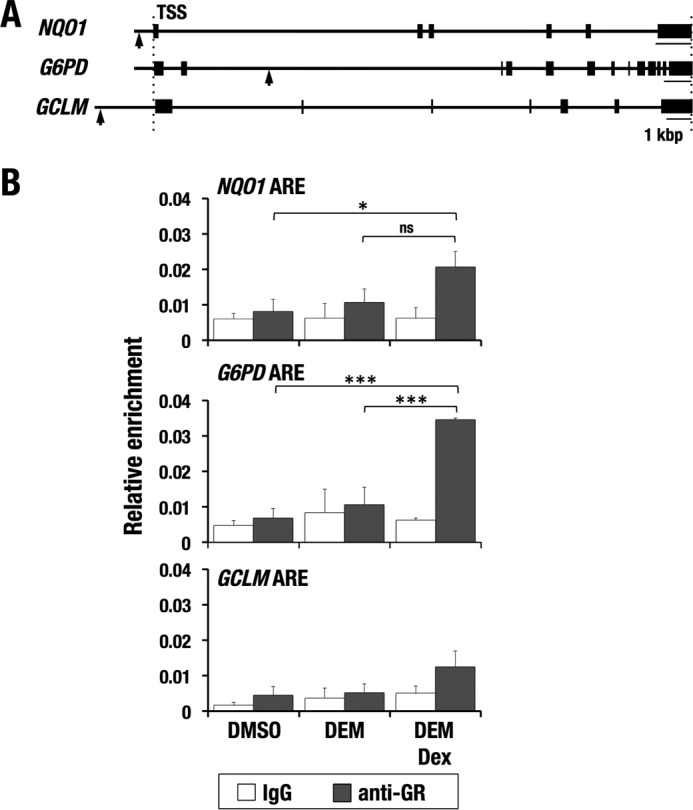
GR binds to AREs in Dex and DEM co-treated cells. A, human gene loci of representative NRF2 target genes (NQO1, GCLM, and G6PD). Arrows, positions of AREs. B, ChIP assay with anti-GR antibodies using HepG2 cells treated with DEM and DEM + Dex. The average and S.D. (error bars) from triplicate reactions are shown. One-way ANOVA and Tukey's post hoc test were performed. *, p < 0.05; ***, p < 0.001; ns, nonsignificant.
Although the GR recruitment to GCLM ARE did not reach statistical significance, the AREs of NQO1 and G6PD were significantly enriched by anti-GR antibody in DEM and Dex-co-treated cells (Fig. 7B), suggesting that GR interacts with NRF2 on the chromatin in response to Dex.
GR signaling reduces DEM-induced CBP recruitment and H3K27 acetylation
A previous study demonstrated that CBP interacts with NRF2 through its Neh4 and Neh5 domains to activate transcription (20). Because we found that GR also interacts with Neh4 and Neh5 domains of NRF2 (see Fig. 1, C–E) and is recruited to AREs during the antioxidant response (see Fig. 7B), we suspected that GR inhibited NRF2-dependent CBP recruitment to AREs. The CBP recruitment levels to the AREs at the Nqo1, G6pdx, and Gclm loci in DEM-Dex double-treated Hepa1c1c7 cells were consistently lower than those in DEM single-treated cells, although the results of three independent experiments did not reach statistical significance for G6pdx or Gclm (Fig. 8A and supplemental Figs. S1 and S2). Similar tendencies were observed for H3K27 acetylation levels in DEM-Dex double-treated cells compared with those of DEM single-treated cells (Fig. 8B and supplemental Fig. S3).
Figure 8.
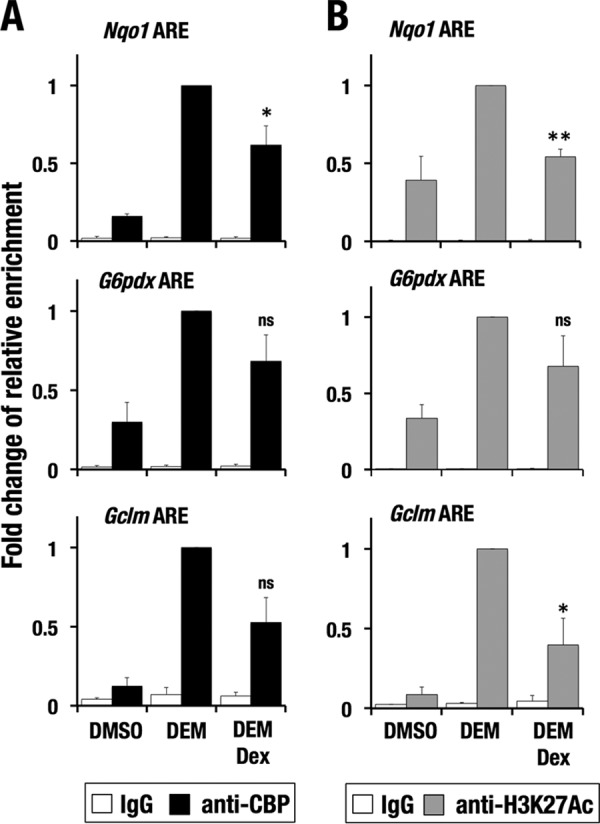
Dex treatment reduces CBP recruitment and H3K27 acetylation at AREs. A and B, ChIP assay with anti-CBP (A) and anti-H3K27Ac (B) antibodies using Hepa1c1c7 cells treated with DEM and DEM + Dex. -Fold changes of relative enrichments for DEM + Dex-treated and vehicle-treated cells against DEM-treated cells were calculated. The average and S.D. (error bars) of the -fold changes from triplicate reactions are shown. Confidence interval estimation was conducted for DEM + Dex-treated samples. *, α < 0.05; **, α < 0.01; ns, nonsignificant.
Previous reports described that active enhancers and promoters are marked by acetylation of H3K27 and H3K9, respectively (25, 26). We examined an effect of GR signaling on the acetylation status of H3K9. In contrast to H3K27 acetylation, Dex treatment did not induce apparent decrease in the H3K9 acetylation (Fig. 9). Moreover, DEM treatment did not increase the H3K9 acetylation at Gclm ARE. These results imply that the GR signaling affects the enhancer histone acetylation but not the promoter histone acetylation, although the molecular mechanisms underlying this selectivity are currently unknown. Thus, GR signaling reduces the CBP recruitment and H3K27 acetylation at NRF2 target gene loci.
Figure 9.
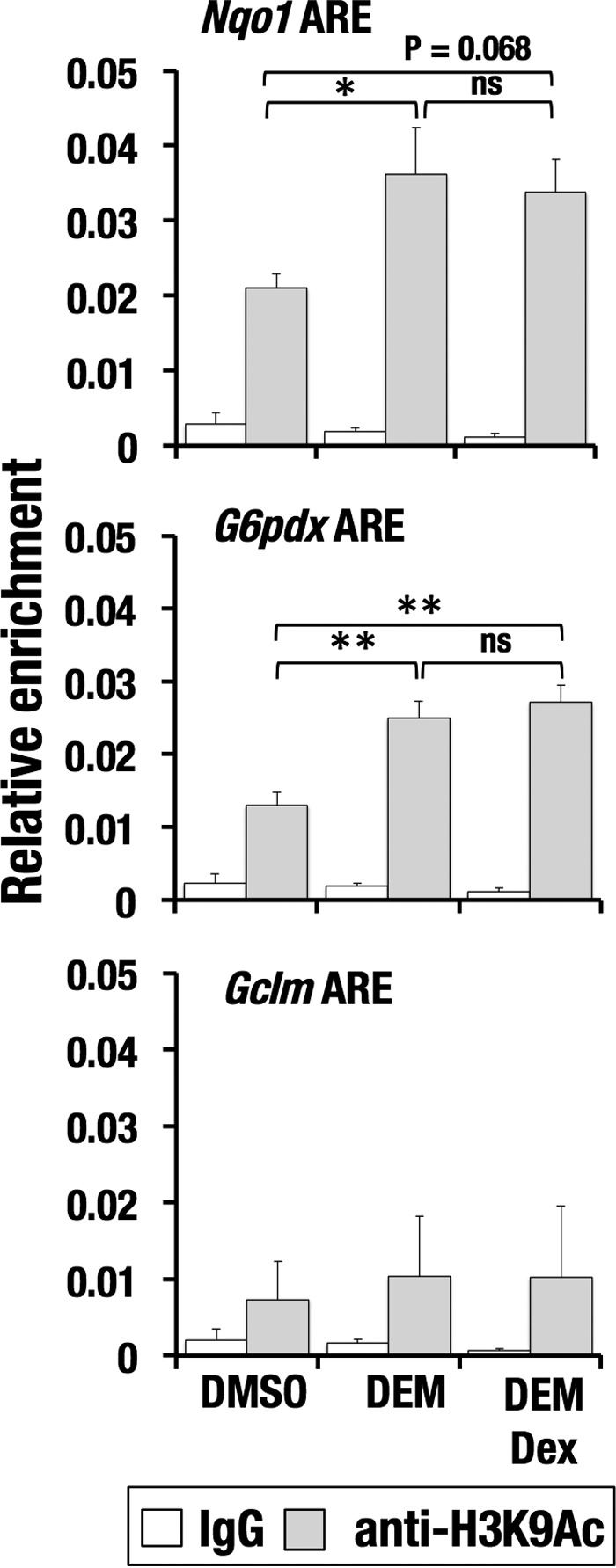
Dex treatment does not alter H3K9 acetylation at AREs. ChIP assay with anti-H3K9Ac antibody using Hepa1c1c7 cells treated with DEM and DEM + Dex. The average and S.D. (error bars) of the relative enrichments from triplicate reactions are shown. One-way ANOVA and Tukey's post hoc test were performed. *, p < 0.05; **, p < 0.01; ns, nonsignificant.
To investigate whether the status of histone acetylation affects the GR-dependent down-regulation of NRF2 target genes, we inhibited HDAC activity by applying two HDAC inhibitors, TSA and VA. The addition of TSA or VA alleviated the Dex-mediated inhibition of NRF2 transcriptional activation (Fig. 10, A and C), which was not accompanied by an increase in nuclear accumulation of NRF2 protein (Fig. 10, B and D). Statistical analysis of three independent results did not give significance to TSA-mediated derepression of Gclm (Fig. 10A, bottom) or VA-mediated derepression of G6pdx (Fig. 9C, middle), but both HDAC inhibitors reproducibly canceled the Dex-dependent repression of Gclm and G6pdx in each of the three independent experiments (supplemental Figs. S4 and S5). Thus, these results suggest that GR signaling inhibits histone acetylation and results in suppression of the NRF2-mediated antioxidant response.
Figure 10.
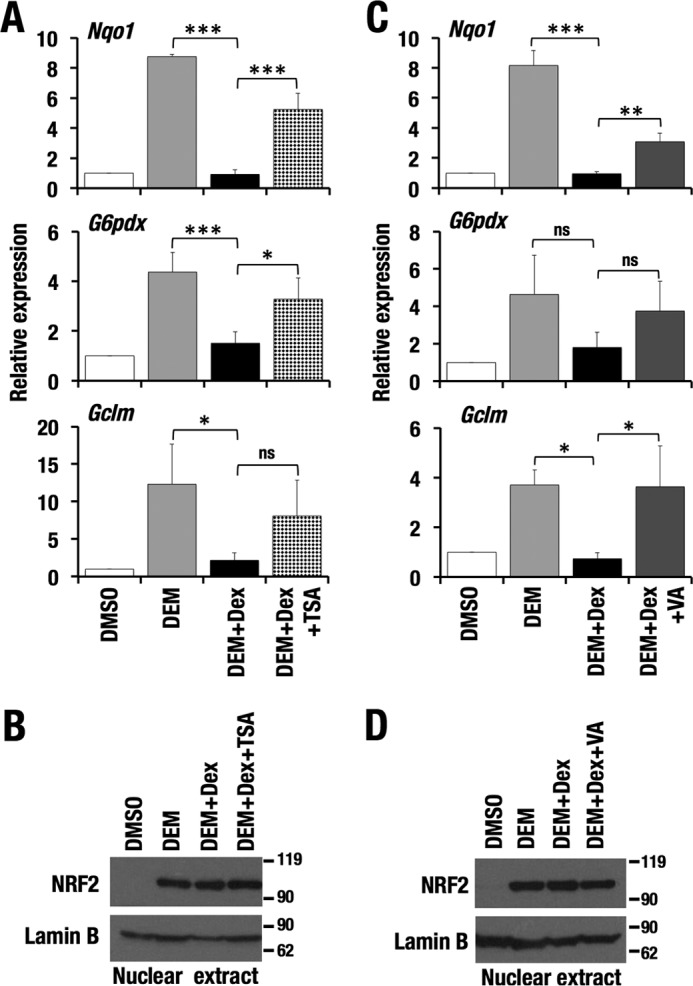
Dex-mediated suppression of NRF2 activity is canceled by HDAC inhibition. A and C, relative expression levels of NRF2 target genes in Hepa1c1c7 cells treated with DEM and DEM + Dex in combination with TSA (A) and VA (C). -Fold changes were calculated against vehicle-treated samples, and the average and S.D. (error bars) of the -fold changes from three independent experiments are shown. One-way ANOVA and Tukey's post hoc test were performed. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B and D, NRF2 abundance in nuclear extracts of Hepa1c1c7 cells. Lamin B was detected as a loading control.
NRF2-mediated cytoprotection is blocked by GR signaling
We finally examined the significance of GR signaling with respect to NRF2-dependent cytoprotection. Treatment of Hepa1c1c7 cells with DEM conferred resistance to menadione-induced oxidative stress. Co-treatment with DEM and Dex/Bet made the cells as susceptible to oxidative stress as vehicle- or Dex/Bet-treated cells (Fig. 11, A and B), indicating that GR signaling antagonizes the NRF2-directed antioxidant response.
Figure 11.
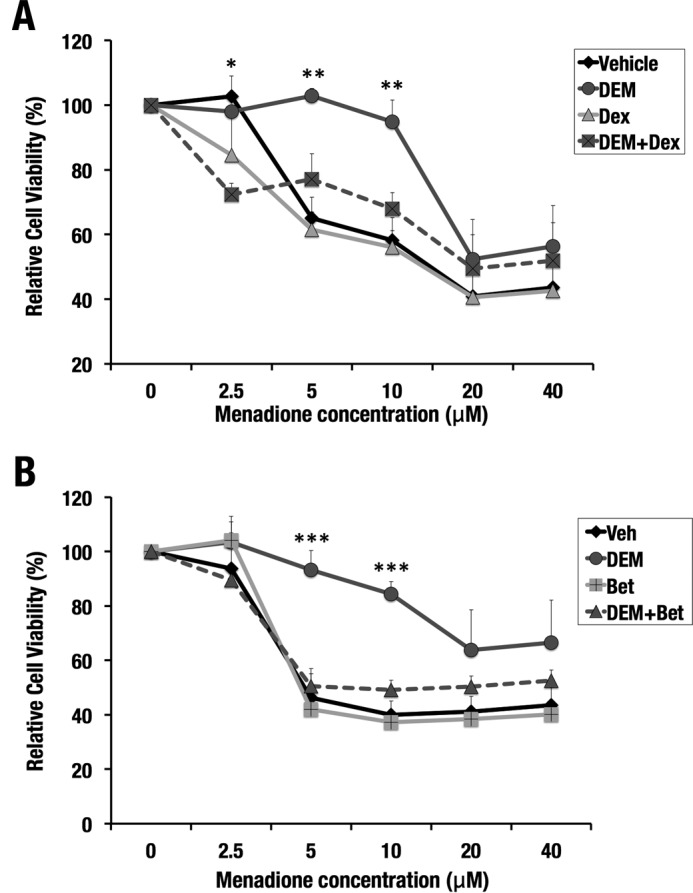
Dex or Bet treatment antagonizes DEM-induced cytoprotection against oxidative stress. Hepa1c1c7 cells were pretreated with DEM and/or Dex (A) and DEM and/or Bet (B) for 10 h and exposed to increasing concentrations of menadione. DMSO was used as vehicle. Cell viability was analyzed after 24 h. The number of cells without menadione was set as 100%. *, p < 0.05; **, p < 0.01; ***, p < 0.001 for the comparison of DEM-treated cells with DEM + Dex-co-treated (A) and DEM + Bet-co-treated (B) cells. One-way ANOVA and Tukey's post hoc test were performed. Error bars, S.D.
Discussion
We found a functional interaction between GR signaling and the KEAP1-NRF2 pathway based on the interaction between GR and NRF2. GR signaling represses NRF2-dependent transcriptional activation and abates the NRF2-mediated cytoprotection from oxidative stress. In the antioxidant response, NRF2 is stabilized and binds to AREs, resulting in the recruitment of CBP, enhancement of the histone acetylation, and transcriptional activation of its target genes. Dex treatment during the antioxidant response does not affect NRF2 recruitment to AREs but induces GR recruitment to AREs accompanied by CBP release, histone deacetylation, and decreased expression of the target genes. The inhibitory effect of Dex was abolished by the treatment with HDAC inhibitors, suggesting that GR signaling suppresses the NRF2-dependent antioxidant response through histone deacetylation. Thus, this work has revealed that NRF2 activity is under the control of GR-mediated transrepression through epigenetic regulation.
A previous study demonstrated that GR antagonizes the electrophile-induced expression of Gsta2 in a rat liver cell line, H4IIE cells, and suggested that this inhibitory effect of GR depends on a GRE in the 5′-region of Gsta2 based on the result of a reporter assay (27). The study also showed that Dex treatment reduces the basal expression levels of three other NRF2 target genes and implied that the decrease was mediated by the GR binding to GREs that are found in the 5′-regions of these genes. Although we consider that the GRE-mediated repression would be operative for some NRF2 target genes, our result strongly suggests that GR signaling generally inhibits the NRF2 target genes through the GR-mediated transrepression. We observed in the reporter assay that GR suppresses the NRF2-mediated transcriptional activation in an ARE-dependent manner. We also observed the GR recruitment to AREs and the CBP release from AREs in response to Dex during the antioxidant response. These results support the notion that GR interacts with NRF2 and reduces the expression of its target genes irrespective of whether GREs are present or not.
The transrepression activity of GR has been well studied, particularly in relation to immune-regulating transcription factors, such as NF-κB and AP-1 (28). GR, when bound to GCs, regulates transcription via GREs and also inhibits gene expression via transrepression, which is based on protein-protein interactions. In transrepression, GR attenuates gene expression by directly interacting with other transcription factors without contacting DNA. Two mechanisms have been described for transrepression by GR: tethering and squelching. In the former, the repressed transcription factors remain bound to the DNA, and in the latter, they are sequestered from the DNA. According to our results, inhibition of NRF2 activity by GR falls into the former category. Because GR has been shown to interact with HDAC2 (29), we surmise that GR recruits HDAC to AREs by associating with NRF2 resulting in the decreased transcriptional activity of NRF2 as a consequence of histone deacetylation. Because CBP and GR share Neh4/Neh5 domains for binding NRF2, GR is expected to put CBP away from NRF2 and thus facilitates deacetylation, whose precise mechanism needs to be further analyzed.
Although GR signaling makes a critical contribution to the maintenance of systemic energy homeostasis and stress response in cooperation with the sympathetic nervous system, excessive activation of GR signaling by increased GC causes many undesirable effects, such as obesity, insulin resistance, and steatosis. Especially in liver, excessive GR signaling has been implicated in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (30, 31). These similar liver disorders tend to be exacerbated in Nrf2-deficient mice (3–8), which is consistent with our observation that GR signaling antagonizes the NRF2 activity. Considering the high dose of Dex that was used in this study, the condition where NRF2-dependent cytoprotection is inhibited by GR signaling is likely to correspond to the pathological hyperglucocorticoidism status.
Circulating levels of GCs are controlled by the hypothalamic-pituitary-adrenal axis, whereas their tissue levels are controlled by enzymes that inactivate and regenerate GCs within cells (32, 33). Mice deficient in 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1), which regenerates GC and amplifies local GC action, are protected from hepatic steatosis induced by excessive GC administered in drinking water, suggesting that appropriate regulation of GC turnover in tissues is critical for the prevention of NAFLD and non-alcoholic steatohepatitis. Indeed, 11β-HSD1 inhibitors have been shown to effectively improve metabolic-syndrome parameters in rodents (34), and in a clinical trial, liver fat was modestly but significantly decreased in NAFLD patients after treatment with an 11β-HSD1 inhibitor (35). A previous report demonstrated that GC suppresses NRF2 activity, which is consistent with our results, and that the suppression is reversed by 11β-HSD1 inhibition (36). 11β-HSD1 inhibitors, by reducing the amount of GC available in tissue, may provide cellular environments that are favorable for the NRF2-driven cytoprotective response. Limited NRF2 activity may be one of the causes for the deleterious effects of excessive GC. We proposed that impairment of the NRF2-mediated defense mechanism against oxidative stress underlies the hypercorticosteroidism and its related metabolic disorders.
Experimental procedures
Mice
Keap1f/f mice and Ptenf/f mice were described previously (24, 37). Ptenf/f mice were a kind gift from Dr. Akira Suzuki (Kyushu University). The albumin-Cre transgenic mouse was purchased from the Jackson Laboratory (Bar Harbor, ME, USA) (38). Keap1f/f::albumin-Cre mice were obtained by mating Keap1f/f and albumin-Cre mice and were sacrificed for liver protein preparation. All mice were provided water and rodent chow ad libitum, maintained under specific-pathogen-free conditions, and treated according to the regulations of the Standards for Human Care and Use of Laboratory Animals of Tohoku University and Guidelines for Proper Conduct of Animal Experiments of the Ministry of Education, Culture, Sports, Science, and Technology of Japan. All animal experiments were approved by the Tohoku University Committee for Laboratory Animal Research.
Chemicals
DEM and DMSO were purchased from Wako Pure Chemicals (Osaka, Japan). Dex, Bet, menadione, TSA, VA, dimethyl pimelimidate dihydrochloride, and Complete (protease inhibitor mixture) were purchased from Sigma-Aldrich. CDDO-Im was obtained from Mochida Pharmaceuticals Co., Ltd. (Tokyo, Japan). Dithiobismaleimidoethane (DTME), dithiobis(succinimidyl propionate) (DSP), and ethylene glycol bis(succinimidyl succinate) were purchased from Thermo Fisher Scientific (Waltham, MA).
Plasmids
pGEX4T-1 mNRF2 mutant vectors expressing GST and GST-NRF2 mutants were used for recombinant protein production (22). For a reporter assay and protein expression in HEK293T cells, pRBGP2 (3× ARE-LUC), pRBGP4 (3× mut ARE-LUC), p3xFLAG-NRF2, pcDNA3-FLAG-hGRα, and pRL-LUC (internal control) were used (39, 40).
Cell culture
HEK293T and Hepa1c1c7 cells were maintained in high-glucose DMEM, and HepG2 cells were maintained in low glucose DMEM (Wako Pure Chemicals, Osaka, Japan) supplemented with 10% (v/v) fetal bovine serum (Sigma-Aldrich) and penicillin/streptomycin (Thermo Fisher Scientific) under 5.0% (v/v) CO2 at 37 °C.
Identification of NRF2-interacting proteins in mouse liver
We followed a protocol described previously (22). Briefly, Keap1f/f::albumin-Cre mouse livers were homogenized in 0.1× PBS containing 0.5 mm DTME and 0.5 mm DSP and incubated at 4 °C for 2 h followed by incubation in quenching buffer (20 mm Tris-HCl (pH 7.5), 5 mm cysteine) at 4 °C for 20 min. After washing with PBS, the sample was resuspended in lysis buffer (20 mm HEPES (pH 7.6), 20% (v/v) glycerol, 10 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA) and put on ice for 10 min. After centrifugation at 600 × g at 4 °C for 10 min, the pellet was sonicated in radioimmune precipitation assay buffer briefly and centrifuged at 10,000 × g at 4 °C for 10 min. The supernatant was subjected to anti-NRF2 affinity purification. An anti-NRF2 antibody (D1Z9C-XP, Cell Signaling Technology (Danvers, MA)) was cross-linked to Dynabeads anti-rabbit IgG (Thermo Fisher Scientific) with dimethyl pimelimidate dihydrochloride. The NRF2 complex was eluted from the beads by incubating at 37 °C for 20 min in elution buffer (50 mm Tris-HCl (pH 8.0), 0.2 m NaCl, 2% (w/v) SDS, 50 mm DTT). The eluate was subjected to gel-based LC-MS/MS analysis.
Gel-based LC-MS/MS analysis and protein sequence database searches
The detailed protocol was described previously (41, 42). Briefly, after SDS-PAGE using a 5–20% (w/v) polyacrylamide gradient gel (Oriental Instruments, Sagamihara, Japan) and Coomassie Brilliant Blue staining (43), each lane in the gel was divided into 17 sections. The resulting gel blocks were treated with DTT and acrylamide for reduction and alkylation of the sulfhydryl groups. After overnight tryptic digestion, the resulting peptides in each gel block were extracted, and one-half of each sample was subjected to LC-MS/MS using an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). The data acquisition of every sample was performed for 60 min after a 50-min LC gradient was started, where MS1 scans from m/z 321 to 1600 were carried out in the orbitrap with the resolution set at 60,000 with a lockmass at m/z 445.120025, followed by top-15 MS2 acquisition by collision-induced dissociation in the ion trap in the normal resolution mode. The settings for the MS2 scans were as follows: minimal signal intensity required = 500; AGC target = 5,000; and maximum ion injection time = 50 ms (44). The raw data files derived from samples in the same SDS-PAGE lane were converted together into a single MASCOT generic format file and were used for the database search by MASCOT (version 2.5.1; Matrix Science) against the mouse proteins in Swissprot (January 2016) and a custom database including contaminant proteins. The peptide expectation value cut-off was set at 0.05. Protein N-terminal acetylation (+42.0106), methionine oxidation (+15.9949), propionamidated cysteine (+71.0371), propionamidated DSP at lysine (+159.0354), and propionamidated DTME at cysteine (+246.0674) were considered as possible variable modifications. The false discovery rates were automatically adjusted to 1% by MASCOT Percolator in every search.
GST pulldown assay
The protocol for the GST-pulldown assay using NRF2 deletion mutant molecules was described previously (22). Briefly, the GST-fusion proteins of various NRF2 mutants were expressed in the E. coli strain Rosetta (DE3), and soluble lysates were prepared in PBS-T (PBS supplemented with 0.1% (v/v) Tween 20) by sonication. Glutathione-Sepharose-immobilized GST and GST-NRF2 mutants were incubated with nuclear extracts of 293T cells transiently expressing FLAG-GR and washed extensively with PBS-T. Proteins retained on beads were eluted in Laemmli sample buffer at 94 °C. Eluates were resolved by 6% (w/v) SDS-PAGE and analyzed by an immunoblotting assay for the presence of GR using an anti-FLAG antibody.
Nuclear protein preparation from cell lines
For nuclear extracts containing FLAG-GR, pcDNA3-FLAG-hGRα was transiently introduced into HEK293T cells. After 24 h, cells were lysed in Dignam lysis buffer A (20 mm HEPES (pH 7.6), 20% (v/v) glycerol, 10 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA) supplemented with 1 mm DTT, 0.1% (v/v) Triton X-100, 1 mm PMSF, and 1× Complete (Sigma-Aldrich). After centrifugation at 2,500 rpm for 5 min at 4 °C, the pellet was resuspended in Dignam extraction buffer B (20 mm HEPES (pH 7.6), 20% (v/v) glycerol, 400 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA) supplemented with 1 mm DTT, 1 mm PMSF, and 1× Complete and incubated for 30 min. The sample was centrifuged at 15,000 rpm for 10 min at 4 °C, and the supernatant was collected as a nuclear fraction containing FLAG-GR for the GST-pull-down assay. To extract nuclear proteins, including endogenous NRF2, Hepa1c1c7 cells treated with DMSO or 100 μm DEM and/or 100 nm Dex, 200 nm TSA, and 500 μm VA for 4 h were lysed with Dignam lysis buffer A containing 1 mm DTT, 0.1% (v/v) Triton X-100, 1 mm PMSF, 10 μm MG132, and 1× Complete and centrifuged. The pellet was lysed in 2× Laemmli buffer followed by boiling at 95 °C for 5 min. The samples were used for immunoblotting analysis.
RNA purification and quantitative RT-PCR
Total RNA samples were prepared using ISOGEN (Wako Pure Chemicals, Osaka, Japan) from Hepa1c1c7 cells treated with DMSO or 100 μm DEM and/or 100 nm Dex, 100 nm Bet, 300 nm TSA, and 500 μm VA for 16 h. cDNAs were synthesized from 0.5 μg of total RNA using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan). Real-time PCR was performed for each sample using a 7300 real-time PCR system (Applied Biosystems, Foster City, CA) in three independent experiments. Expression levels of hypoxanthine-guanine phosphoribosyltransferase (except for Fig. 3B) and rRNA were used as internal controls for normalization. Total RNA prepared from mouse liver was similarly processed for analysis. Primers used in the quantitative RT-PCR are listed in Table 1.
Table 1.
Sequences of primers used in quantitative RT-PCR
| Name | Sequence (5′–3′) |
|---|---|
| mNqo1 forward | AGC TGG AAG CTG CAG ACC TG |
| mNqo1 reverse | CCT TTC AGA ATG GCT GGC A |
| mNqo1 probe | FAM-ATT TCA GTT CCC ATT GCA GTG GTT TGG G-TAMRA |
| mG6pdx forward | GTC CAG AAT CTC ATG GTG CTG A |
| mG6pdx reverse | GCA ATG TTG TCT CGA TTC CAG A |
| mGclm forward | TGA CTC ACA ATG ACC CGA AA |
| mGclm reverse | GAT GCT TTC TTG AAG AGC TTC CT |
| mGclc forward | ATC TGC AAA GGC GGC AAC |
| mGclc reverse | ACT CCT CTG CAG CTG GCT C |
| mGclc probe | FAM-ACG GGT GCA GCA AGG CCC A-TAMRA |
| mTxnrd1 forward | AGA AAG TGC TGG TCT TGG ATT TTG |
| mTxnrd1 reverse | ACA CGT TCC TCC GAG ACC C |
| mTxnrd1 probe | FAM-TCT GGT CCC AAG AGG AGT CGG TGT G-TAMRA |
| mPgd forward | ATG CCA GGA GGG AAC AAA G |
| mPgd reverse | GTT CTC CGG TTC CCA CTT TT |
| mHPRT forward | CTG GTG AAA AGG ACC TCT CG |
| mHPRT reverse | TGA AGT ACT CAT TAT AGT CAA GGG |
| mHPRT probe | FAM-ATCCAACAAAGTCTGGCCTGTATCCAAC-TAMRA |
| mrRNA forward | CGG CTA CCA CAT CCA AGG AA |
| mrRNA reverse | GCT GGA ATT ACC GCG GCT |
| mrRNA probe | FAM-TGC TGG CAC CAG ACT TGC CCT C-TAMRA |
siRNA transfection
Hepa1c1c7 cells were transfected with 50 nm siRNA against GR using RNAiMAX (Invitrogen) according to the manufacturer's protocol. After 56 h of transfection, cells were treated with DMSO or 100 μm DEM and/or 100 nm Dex or 100 nm Bet. After additional culture for 16 h, total RNA samples were prepared. Predesigned siRNAs were purchased from Sigma-Aldrich (SASI_Mm01_00037535 and SASI_Mm01_00037536).
Administration of Dex and CDDO-Im to mice
As a model of pharmacological induction of NRF2, C57BL/6J male mice at an age of 5–7 weeks were used. The mice were i.p. administered PBS or Dex (10 mg/kg body weight) once a day for 3 days. On the fourth day, PBS, Dex (10 mg/kg of body weight), and/or CDDO-Im (30 μmol/kg of body weight) were intraperitoneally injected. 6 h later, mice were sacrificed for analysis. As a model of genetic induction of NRF2, Keap1f/f::Ptenf/f::albumin-Cre mice at 10 days after birth were intraperitoneally administered PBS or Dex (5 mg/kg of body weight, freshly diluted in PBS). 10 or 24 h later, mice were sacrificed for analysis. Livers were collected for RNA purification and nuclear protein preparation.
Nuclear protein preparation from mouse livers
Livers were homogenized in Dignam lysis buffer A supplemented with 1 mm DTT, 0.1% (v/v) Triton X-100, 1 mm PMSF, 10 μm MG132, and 1× Complete. The homogenates were kept on ice for 5 min, followed by centrifugation at 2,500 rpm for 5 min at 4 °C. The pellet was lysed in 2× Laemmli buffer, followed by boiling at 100 °C for 10 min. The samples were used for immunoblotting analysis.
Luciferase reporter assay
pRBGP2 (3× ARE-LUC) or pRBGP4 (3× mut ARE-LUC) was introduced into 293T cells along with NRF2 and GR expression vectors using Lipofectamine 3000 (Invitrogen). After 30 h of transfection, 100 μm DEM and/or 100 nm Dex were added to the medium. DMSO was added as vehicle. After additional culture for 18 h, luciferase activity was measured using a Dual Reporter Assay System (Promega, Madison, WI) and a luminometer (Berthold Japan, Tokyo, Japan). The averages and S.D. values were calculated from three independent experiments.
Immunocytochemistry analysis
Immunocytochemistry was performed based on the standard protocol. Briefly, Hepa1c1c7 cells treated with DMSO or 100 μm DEM and/or 100 nm Dex were fixed using fresh 4% (w/v) paraformaldehyde (Wako, Japan) for 15 min and permeabilized with 0.5% (v/v) Triton X-100 (Nacalai Tesque, Kyoto, Japan) for 10 min. The anti-NRF2 antibody (44) and Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody (A11034, Life Technologies) were used to examine subcellular localization of NRF2 at a dilution of 1:100 and 1:250, respectively. After the antibody reactions, cells were washed with PBS and refixed with 4% (w/v) paraformaldehyde followed by washing in PBS. Nuclei were imaged with DAPI (Dojindo, Kumamoto, Japan). Finally, coverslips were mounted with Vectashield (Vector Laboratories). Images were taken by using TCS SP8 confocal microscopy (Leica).
Immunoblotting analysis
Immunoblotting analysis was performed as described previously (45). Samples of mouse liver immunoprecipitated with an anti-NRF2 antibody (D1Z9C-XP, Cell Signaling Technology) were analyzed by immunoblotting analysis with anti-GR (sc-8992, Santa Cruz Biotechnology, Inc., Dallas, TX) and anti-NRF2 antibodies (46). Nuclear proteins prepared from Hepa1c1c7 cells, and mouse livers were analyzed by immunoblotting analysis with anti-NRF2 (46) and anti-Lamin B antibodies (sc-6217, Santa Cruz Biotechnology).
Chromatin immunoprecipitations
ChIP assays were performed in Hepa1c1c7 using anti-NRF2 antibody (D1Z9C-XP, Cell Signaling Technology), anti-H3K27Ac antibody (MABI0309, MAB Institute, Inc., Sapporo, Japan), anti-H3K9Ac antibody (catalog no. 04-1003, Merck Millipore), and anti-CBP antibody (sc-369X, Santa Cruz Biotechnology, Dallas, TX) and in HepG2 cells using anti-GR antibody (sc-1003X, Santa Cruz Biotechnology).
For ChIP assays with Hepa1c1c7 cells using anti-NRF2, anti-H3K9Ac, and anti-H3K27Ac antibodies, the cells were treated with DMSO, 100 μm DEM, and/or 100 nm Dex for 4 h, fixed with 1% (w/v) formaldehyde for 10 min, and lysed in cell lysis buffer (5 mm PIPES, pH 8.0, 85 mm KCl, 0.5% (v/v) Nonidet P-40) supplemented with 1× Complete and 1 mm PMSF. After centrifugation, the nuclear pellet was resuspended in the nuclear lysis buffer (50 mm Tris-HCl, pH 8.0, 10 mm EDTA, 1% (w/v) SDS) supplemented with 1× Complete and 1 mm PMSF and sonicated for DNA shearing. The chromatin solution was incubated overnight with anti-NRF2, anti-H3K9Ac, and anti-H3K27Ac antibodies at 4 °C. The former two antibodies were prebound to Dynabeads anti-rabbit IgG (Thermo Fisher Scientific), and the last antibody was prebound to Dynabeads anti-mouse IgG (Thermo Fisher Scientific).
For ChIP assays with Hepa1c1c7 cells using anti-CBP antibody, the cells were treated with DMSO, 100 μm DEM, and/or 100 nm Dex for 4 h; cross-linked with 1.5 mm ethylene glycol bis(succinimidyl succinate) for 20 min followed by 1% (w/v) formaldehyde for 10 min; and lysed in the cell lysis buffer. After centrifugation, the nuclear pellet was resuspended in the NUC buffer (15 mm HEPES, pH 7.5, 60 mm KCl, 15 mm NaCl, 0.32 mm sucrose, 3 mm CaCl2) and digested with micrococcal nuclease (New England Biolabs, Ipswich, MA) for DNA shearing. After double dilution with the sonication buffer (90 mm Hepes, pH 7.9, 220 mm NaCl, 10 mm EDTA, 1% (v/v) Nonidet P-40, 0.2% (w/v) sodium deoxycholate, 0.2% (w/v) SDS), the sample was briefly sonicated. The chromatin solution was incubated overnight with anti-CBP antibody bound to Dynabeads anti-rabbit IgG at 4 °C.
For ChIP assays with HepG2 cells using anti-GR antibody, the cells were treated with DMSO, 100 μm DEM, and/or 100 nm Dex for 2 h. The following procedure was the same as for the ChIP assay using anti-CBP antibody.
The precipitated DNA was analyzed by quantitative PCR using the primer sets listed in Table 2.
Table 2.
Sequences of primers used in ChIP-PCR assay
| Name | Sequence (5′–3′) |
|---|---|
| mNqo1 ARE forward | GCA CGA ATT CAT TTC ACA CGA GG |
| mNqo1 ARE reverse | GCT CAA ATT TTG CCG ACT CAC TG |
| mG6pdx ARE forward | CTC CTT CTT ACA CCC TGG CA |
| mG6pdx ARE reverse | TGA GCT GAC TTC TCC CTT GA |
| mGclm ARE forward | CGA GAC AAA AGA GCA GAC TC |
| mGclm ARE reverse | GTA ATC TAC ATT TCC TTT GGC TG |
| hNQO1 ARE forward | CAT GTC TCC CCA GGA CTC TC |
| hNQO1 ARE reverse | TTT TAG CCT TGG CAC GAA AT |
| hG6PD ARE forward | CTT TGG GGG AGT GCC AAC AT |
| hG6PD ARE reverse | ATC ACA AGG GCC ATG GGC TT |
| hGCLM ARE forward | GGA GAG CTG ATT CCA AAC TG |
| hGCLM ARE reverse | GAG TAA CGG TTA CGA AGC AC |
Cell viability test
Hepa1c1c7 cells (1 × 103) were seeded in 96-well plates, cultured for 48 h, and pretreated with DEM (100 μm) and/or Dex (100 nm) or Bet (100 nm) for 10 h before treatment with menadione. Cell viability was assessed 24 h after the menadione treatment using Cell Counting Kit-8 (Nacalai Tesque) according to the manufacturer's instructions.
Statistical analysis
Student's t test was used for comparison of two samples. One-way ANOVA and Tukey's post hoc test were used for comparison of three and more than three samples. p < 0.05 was considered to be statistically significant. The confidence interval was calculated for the evaluation of -fold changes. α < 0.05 was considered to be statistically significant.
Author contributions
M. M. A. and K. O. conducted the experiments, analyzed the data, and wrote early drafts of the paper. L. T. T. N., N. O., H. K., S. M., and H. Shima conducted the experiments and analyzed the data. K. I. contributed to the data analysis. H. Sekine and H. M. supervised the research, analyzed the data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Kyoko Ochiai for advice on protein purification and the Biomedical Research Core of the Tohoku University Graduate School of Medicine for technical support.
This work was supported by JSPS KAKENHI Grants 15H04692 (to H. M.) and 16K15228 (to H. M.), the Uehara Memorial Foundation (to H. M.), the Mitsubishi Foundation (to H. M.), the Naito Foundation (to H. M.), the Gonryo Medical Foundation (to H. S.), the Core Research for Evolutional Science and Technology from the AMED (to K. I.), and the Princess Takamatsu Cancer Research Fund 15-24728 (to H. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5.
- ARE
- antioxidant response element
- GR
- glucocorticoid receptor
- GC
- glucocorticoid
- HDAC
- histone deacetylase
- TSA
- trichostatin A
- VA
- valproic acid
- DEM
- diethyl maleate
- Dex
- dexamethasone
- Bet
- betamethasone
- CDDO-Im
- 2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide
- GRE
- glucocorticoid response element
- H3K9 and H3K27
- histone H3 Lys-9 and Lys-27, respectively
- H3K9Ac and H3K27Ac
- acetylated H3K9 and H3K27, respectively
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- NAFLD
- non-alcoholic fatty liver disease
- 11-HSD1
- 11β-hydroxysteroid dehydrogenase 1
- DTME
- dithiobismaleimidoethane
- DSP
- dithiobis(succinimidyl propionate)
- ANOVA
- analysis of variance
- FAM
- 6-carboxyfluorescein
- TAMRA
- tetramethylrhodamine.
References
- 1. Motohashi H., and Yamamoto M. (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549–557 [DOI] [PubMed] [Google Scholar]
- 2. Enomoto A., Itoh K., Nagayoshi E., Haruta J., Kimura T., O'Connor T., Harada T., and Yamamoto M. (2001) High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 59, 169–177 [DOI] [PubMed] [Google Scholar]
- 3. Sugimoto H., Okada K., Shoda J., Warabi E., Ishige K., Ueda T., Taguchi K., Yanagawa T., Nakahara A., Hyodo I., Ishii T., and Yamamoto M. (2010) Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G283–G294 [DOI] [PubMed] [Google Scholar]
- 4. Chowdhry S., Nazmy M. H., Meakin P. J., Dinkova-Kostova A. T., Walsh S. V., Tsujita T., Dillon J. F., Ashford M. L., and Hayes J. D. (2010) Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 48, 357–371 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y. K., Yeager R. L., Tanaka Y., and Klaassen C. D. (2010) Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 245, 326–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y. K., Wu K. C., and Klaassen C. D. (2013) Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS One 8, e59122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bae S. H., Sung S. H., Oh S. Y., Lim J. M., Lee S. K., Park Y. N., Lee H. E., Kang D., and Rhee S. G. (2013) Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 17, 73–84 [DOI] [PubMed] [Google Scholar]
- 8. Goto M., Kitamura H., Alam M. M., Ota N., Haseba T., Akimoto T., Shimizu A., Takano-Yamamoto T., Yamamoto M., and Motohashi H. (2015) Alcohol dehydrogenase 3 contributes to the protection of liver from nonalcoholic steatohepatitis. Genes Cells 20, 464–480 [DOI] [PubMed] [Google Scholar]
- 9. Wakabayashi N., Shin S., Slocum S. L., Agoston E. S., Wakabayashi J., Kwak M. K., Misra V., Biswal S., Yamamoto M., and Kensler T. W. (2010) Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci. Signal. 3, ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wakabayashi N., Skoko J. J., Chartoumpekis D. V., Kimura S., Slocum S. L., Noda K., Palliyaguru D. L., Fujimuro M., Boley P. A., Tanaka Y., Shigemura N., Biswal S., Yamamoto M., and Kensler T. W. (2014) Notch-Nrf2 axis: regulation of Nrf2 gene expression and cytoprotection by notch signaling. Mol. Cell. Biol. 34, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., and Motohashi H. (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 [DOI] [PubMed] [Google Scholar]
- 12. Taguchi K., Hirano I., Itoh T., Tanaka M., Miyajima A., Suzuki A., Motohashi H., and Yamamoto M. (2014) Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol. Cell. Biol. 34, 900–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shirasaki K., Taguchi K., Unno M., Motohashi H., and Yamamoto M. (2014) NF-E2-related factor 2 promotes compensatory liver hypertrophy after portal vein branch ligation in mice. Hepatology 59, 2371–2382 [DOI] [PubMed] [Google Scholar]
- 14. Kobayashi E. H., Suzuki T., Funayama R., Nagashima T., Hayashi M., Sekine H., Tanaka N., Moriguchi T., Motohashi H., Nakayama K., and Yamamoto M. (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J. D., and Yamamoto M. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Motohashi H., Katsuoka F., Engel J. D., and Yamamoto M. (2004) Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. U.S.A. 101, 6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katsuoka F., Motohashi H., Ishii T., Aburatani H., Engel J. D., and Yamamoto M. (2005) Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 25, 8044–8051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katoh Y., Iida K., Kang M. I., Kobayashi A., Mizukami M., Tong K. I., McMahon M., Hayes J. D., Itoh K., and Yamamoto M. (2005) Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 433, 342–350 [DOI] [PubMed] [Google Scholar]
- 19. Chowdhry S., Zhang Y., McMahon M., Sutherland C., Cuadrado A., and Hayes J. D. (2013) Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32, 3765–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katoh Y., Itoh K., Yoshida E., Miyagishi M., Fukamizu A., and Yamamoto M. (2001) Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 6, 857–868 [DOI] [PubMed] [Google Scholar]
- 21. Zhang J., Hosoya T., Maruyama A., Nishikawa K., Maher J. M., Ohta T., Motohashi H., Fukamizu A., Shibahara S., Itoh K., and Yamamoto M. (2007) Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem. J. 404, 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sekine H., Okazaki K., Ota N., Shima H., Katoh Y., Suzuki N., Igarashi K., Ito M., Motohashi H., and Yamamoto M. (2015) The Mediator subunit MED16 transduces NRF2-activating signals into antioxidant gene expression. Mol. Cell. Biol. 36, 407–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nioi P., Nguyen T., Sherratt P. J., and Pickett C. B. (2005) The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 25, 10895–10906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okawa H., Motohashi H., Kobayashi A., Aburatani H., Kensler T. W., and Yamamoto M. (2006) Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 339, 79–88 [DOI] [PubMed] [Google Scholar]
- 25. Shlyueva D., Stampfel G., and Stark A. (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286 [DOI] [PubMed] [Google Scholar]
- 26. Karmodiya K., Krebs A. R., Oulad-Abdelghani M., Kimura H., and Tora L. (2012) H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 13, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ki S. H., Cho I. J., Choi D. W., and Kim S. G. (2005) Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPb TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell. Biol. 25, 4150–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ratman D., Vanden Berghe W., Dejager L., Libert C., Tavernier J., Beck I. M., and De Bosscher K. (2013) How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol. Cell. Endocrinol. 380, 41–54 [DOI] [PubMed] [Google Scholar]
- 29. Ito K., Barnes P. J., and Adcock I. M. (2000) Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 20, 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rockall A. G., Sohaib S. A., Evans D., Kaltsas G., Isidori A. M., Monson J. P., Besser G. M., Grossman A. B., and Reznek R. H. (2003) Hepatic steatosis in Cushing's syndrome: a radiological assessment using computed tomography. Eur. J. Endocrinol. 149, 543–548 [DOI] [PubMed] [Google Scholar]
- 31. D'souza A. M., Beaudry J. L., Szigiato A. A., Trumble S. J., Snook L. A., Bonen A., Giacca A., and Riddell M. C. (2012) Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G850–G863 [DOI] [PubMed] [Google Scholar]
- 32. Mueller K. M., Themanns M., Friedbichler K., Kornfeld J. W., Esterbauer H., Tuckermann J. P., and Moriggl R. (2012) Hepatic growth hormone and glucocorticoid receptor signaling in body growth, steatosis and metabolic liver cancer development. Mol. Cell. Endocrinol. 361, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woods C. P., Corrigan M., Gathercole L., Taylor A., Hughes B., Gaoatswe G., Manolopoulos K., Hogan A. E., O'Connell J., Stewart P. M., Tomlinson J. W., O'Shea D., and Sherlock M. (2015) Tissue specific regulation of glucocorticoids in severe obesity and the response to significant weight loss following bariatric surgery (BARICORT). J. Clin. Endocrinol. Metab. 100, 1434–1444 [DOI] [PubMed] [Google Scholar]
- 34. Prasad Sakamuri S. S., Sukapaka M., Prathipati V. K., Nemani H., Putcha U. K., Pothana S., Koppala S. R., Ponday L. R., Acharya V., Veetill G. N., and Ayyalasomayajula V. (2012) Carbenoxolone treatment ameliorated metabolic syndrome in WNIN/Ob obese rats, but induced severe fat loss and glucose intolerance in lean rats. PLoS One 7, e50216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stefan N., Ramsauer M., Jordan P., Nowotny B., Kantartzis K., Machann J., Hwang J. H., Nowotny P., Kahl S., Harreiter J., Hornemann S., Sanyal A. J., Stewart P. M., Pfeiffer A. F., Kautzky-Willer A., et al. (2014) Inhibition of 11β-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2, 406–416 [DOI] [PubMed] [Google Scholar]
- 36. Kratschmar D. V., Calabrese D., Walsh J., Lister A., Birk J., Appenzeller-Herzog C., Moulin P., Goldring C. E., and Odermatt A. (2012) Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11β-HSD1-mediated glucocorticoid activation in hepatic cells. PLoS One 7, e36774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horie Y., Suzuki A., Kataoka E., Sasaki T., Hamada K., Sasaki J., Mizuno K., Hasegawa G., Kishimoto H., Iizuka M., Naito M., Enomoto K., Watanabe S., Mak T. W., and Nakano T. (2004) Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 113, 1774–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Postic C., and Magnuson M. A. (2000) DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26, 149–150 [DOI] [PubMed] [Google Scholar]
- 39. Igarashi K., Kataoka K., Itoh K., Hayashi N., Nishizawa M., and Yamamoto M. (1994) Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 367, 568–572 [DOI] [PubMed] [Google Scholar]
- 40. Sekine H., Mimura J., Yamamoto M., and Fujii-Kuriyama Y. (2006) Unique and overlapping transcriptional roles of arylhydrocarbon receptor nuclear translocator (Arnt) and Arnt2 in xenobiotic and hypoxic responses. J. Biol. Chem. 281, 37507–37516 [DOI] [PubMed] [Google Scholar]
- 41. Ando R., Shima H., Tamahara T., Sato Y., Watanabe-Matsui M., Kato H., Sax N., Motohashi H., Taguchi K., Yamamoto M., Nio M., Maeda T., Ochiai K., Muto A., and Igarashi K. (2016) The transcription factor Bach2 is phosphorylated at multiple sites in murine B cells but a single site prevents its nuclear localization. J. Biol. Chem. 291, 1826–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tanaka H., Muto A., Shima H., Katoh Y., Sax N., Tajima S., Brydun A., Ikura T., Yoshizawa N., Masai H., Hoshikawa Y., Noda T., Nio M., Ochiai K., and Igarashi K. (2016) Epigenetic regulation of the Blimp-1 gene (Prdm1) in B cells involves Bach2 and histone deacetylase 3. J. Biol. Chem. 291, 6316–6330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lawrence A. M., and Besir H. U. (2009) Staining of proteins in gels with Coomassie G-250 without organic solvent and acetic acid. J. Vis. Exp. 10.3791/1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalli A., Smith G. T., Sweredoski M. J., and Hess S. (2013) Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: focus on LTQ-Orbitrap mass analyzers. J. Proteome. Res. 12, 3071–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sekine H., Mimura J., Oshima M., Okawa H., Kanno J., Igarashi K., Gonzalez F. J., Ikuta T., Kawajiri K., and Fujii-Kuriyama Y. (2009) Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 29, 6391–6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maruyama A., Tsukamoto S., Nishikawa K., Yoshida A., Harada N., Motojima K., Ishii T., Nakane A., Yamamoto M., and Itoh K. (2008) Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch. Biochem. Biophys. 477, 139–145 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.