

Abstract
Bacterial phytochromes are dimeric light-regulated histidine kinases that convert red light into signaling events. Light absorption by the N-terminal photosensory core module (PCM) causes the proteins to switch between two spectrally distinct forms, Pr and Pfr, thus resulting in a conformational change that modulates the C-terminal histidine kinase region. To provide further insights into structural details of photoactivation, we investigated the full-length Agp1 bacteriophytochrome from the soil bacterium Agrobacterium fabrum using a combined spectroscopic and modeling approach. We generated seven mutants suitable for spin labeling to enable application of pulsed EPR techniques. The distances between attached spin labels were measured using pulsed electron-electron double resonance spectroscopy to probe the arrangement of the subunits within the dimer. We found very good agreement of experimental and calculated distances for the histidine-kinase region when both subunits are in a parallel orientation. However, experimental distance distributions surprisingly showed only limited agreement with either parallel- or antiparallel-arranged dimer structures when spin labels were placed into the PCM region. This observation indicates that the arrangements of the PCM subunits in the full-length protein dimer in solution differ significantly from that in the PCM crystals. The pulsed electron-electron double resonance data presented here revealed either no or only minor changes of distance distributions upon Pr-to-Pfr photoconversion.
Keywords: conformational change, dimerization, electron paramagnetic resonance (EPR), histidine kinase, photoreceptor, spectroscopy, PELDOR, SDSL, biliprotein, phytochrome
Introduction
Phytochromes are red-light-sensitive photoreceptors of plants, bacteria, and fungi (1). They comprise a conserved N-terminal chromophore-binding photosensory core module (PCM)5 and (most frequently) either a C-terminal regulatory histidine kinase or (less frequently) a histidine-kinase-like region. Typical phytochromes have two spectrally distinct and thermally stable forms, named Pr (red-absorbing form) and Pfr (far-red-absorbing form), that depend on the isomeric structure of the covalently bound bilin chromophore. In typical phytochromes, Pr is the dark state, whereas so-called bathy phytochromes have a Pfr ground state (2). Photoconversion, initialized by Z-to-E or E-to-Z isomerization around a double bond of the bilin chromophore results in protein conformational changes (3–5). In bacterial and fungal phytochromes with C-terminal histidine kinases, these conformational changes modulate the kinase activity. Despite a wealth of structural data on the N-terminal chromophore module (6–11) and the histidine kinases (12, 13), important structural details of this mechanism are so far unknown. According to electron microscopy studies, both subunits of the Deinococcus phytochrome homodimer are arranged in a parallel manner (14), in line with two kinase mechanisms, in which either the His substrate in one subunit is phosphorylated by the other (in trans) or phosphorylation takes place within the same subunit (in cis) (15). Strikingly, however, the subunits of the chromophore modules in PCM crystal structures are sometimes arranged in a parallel (6, 11, 16) and sometimes in an antiparallel fashion (7, 16). Therefore, an antiparallel arrangement, although less likely, was also considered for the full-length protein in solution.
Insights into the conformational changes that occur within the PCM of bacterial phytochromes during photoconversion have been gained by recent studies. Temperature-scan crystallography on the PCM of Pseudomonas aeruginosa phytochrome (a bathy phytochrome) showed that in the early steps of Pfr-to-Pr conversion, amino acids in the PXXDIP motif of the GAF domain are displaced (17). Crystal structures for normal and bathy phytochromes in their Pr and Pfr forms, respectively, revealed differences in the folding of the tip of the “tongue,” which is a part of the PHY domain that folds back to the GAF domain to close the chromophore pocket. A recent publication on the structure of the dark state and the illuminated state of the PCM of Deinococcus phytochrome confirms that during photoconversion the tongue refolds, as expected from the comparison of crystal structures (11, 18). Non-determined light-induced protein conformational changes within the PCM are also obvious from size-exclusion experiments on the PCM of cyanobacterial phytochrome Cph1 and of Agp1 (3, 4) and from ultracentrifugation studies with PCM of Cph1 (19), which indicate a switch from weak to strong subunit interaction upon photoconversion to the Pfr form. Intramolecular signal transduction from the sensory PCM to the regulatory output domain must also be associated with structural changes within the protein, which are expected to correspond to changes in distances between the monomers in both subunits. However, to date no experimental method was successful in providing such information on the full-length protein. Obtaining high-quality crystals is the major obstacle, which for X-ray crystallography on large protein complexes needs to be to overcome. The typical size of 85–120 kDa of full-length phytochrome also precludes taking advantage of high-resolution NMR spectroscopy, which can be applied to study the structure of proteins up to sizes of ∼50 kDa. In contrast, no size restriction exists for pulsed electron-electron double resonance (PELDOR; also called DEER for double electron-electron resonance) that can provide structural information on proteins in solution by mapping distances on a nanometer scale between two paramagnetic sites within a protein (20–22).
In the present study, we applied site-directed spin labeling (23, 24) in combination with PELDOR spectroscopy to investigate the structure and structural rearrangements of the 162-kDa full-length Agp1 phytochrome dimers of the soil bacterium Agrobacterium fabrum C58 (formerly Agrobacterium tumefaciens C58) in its dark Pr and red-light illuminated Pfr state, in analogy to recent studies on the Cph1 phytochrome from Synechocystis sp. PCC6803 (25). By site-directed spin labeling, a cysteine residue is introduced at a desired position by site-directed mutagenesis. Subsequently, a paramagnetic sulfhydryl-specific nitroxide probe is covalently attached to the free sulfhydryl to form the R1 side chain. By measuring the pairwise interaction between the magnetic moments of unpaired electron spins localized on the nitroxides, PELDOR reveals distances in a range between ∼1.5 and 8 nm (21, 26). A number of Agp1 mutants allowing introduction of spin labels at various positions within a monomer have been generated and exploited to gain structural information on the dimeric protein complex. Mapping of the PELDOR distances onto the PCM crystal structures (16), as well as on a structural model of the Agp1 histidine kinase that was predicted based on the Thermotoga maritima homolog (PDB code 2C2A) (12), corroborated the parallel orientation of the subunits in the full-length phytochrome. How well a PCM crystal structure will agree with the N-terminal sensory region of a full-length protein in vivo is still an open question. Currently, it is expected that the arrangement of PCM subunits in crystal structures reflects the situation of the full-length proteins in solution, as long as both subunits are arranged in parallel (11). We show here that this is not the case. Our solution data for the PCM distances clearly deviate from the respective distances in the PCM crystal structure. The solution distances between different subunits match well with structural models derived from a crystal structure of the inactive state.
Results and discussion
Phytochromes assemble as homodimers; thus, a mutation of one single cysteine in the monomeric protein introduces two spin labels into the dimeric structure. For site-directed mutagenesis, residues were selected based on the low-resolution PCM crystal structure of Agp1 (27) and a structural model based on the histidine kinase of the T. maritima homolog (12). For PELDOR measurements, distances between two unpaired electron spins are preferably in a range between 1.5 and 8 nm (21, 26). Thus, only such positions were selected for introduction of spin labels that resulted in distances that were expected to be within this range and that are accessible to spin label. For the structural investigation of Agp1, spin-label positions in various regions of the protein were chosen. We report here results for the positions (i) 122, which is in the link between the PAS and GAF domains; (ii) 362 in a β-sheet region of the PHY domain; (iii) 517 in the helix connecting the PHY domain with the histidine kinase; (iv) 528, the phosphorylation site of the histidine kinase; and (v) three more positions in the histidine kinase: 535, 554, and 603, where the last one is located in the ATP-binding domain of the histidine kinase. All these amino acids, as well as the position of the native cysteine (Cys-20), are indicated in the structural models shown in Fig. 1. A compilation of all experimental and computed interspin distances is given in Tables 1 and 2.
Figure 1.
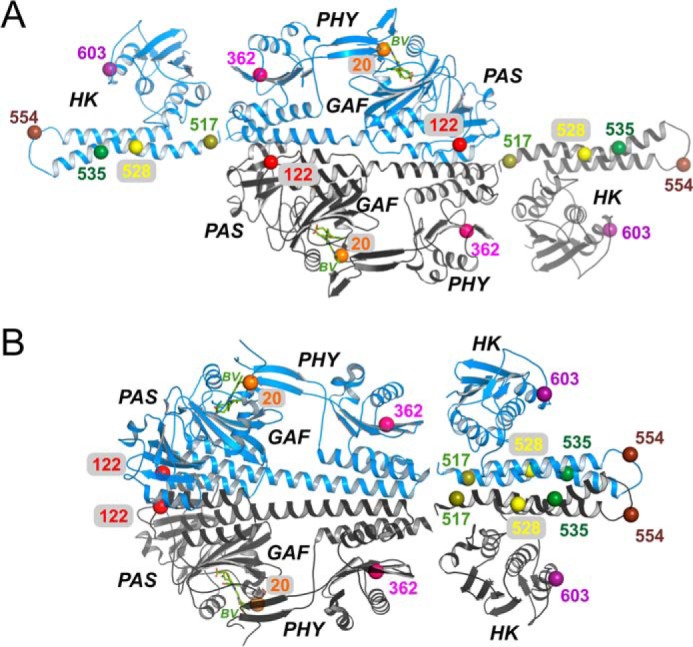
Locations of spin-label sites in the Agp1 phytochrome. A, crystal structure of the Agp1-PCM in antiparallel arrangement (PDB code 5HSQ) and two monomers of a homology model for the Agp1 His kinase (template from a T. maritima His-kinase (12)) placed manually close to the C termini of each PCM module. B, crystal structure of the Agp1-PCM in parallel arrangement (PDB code 5I5L) and a homology model for the Agp1 His-kinase dimer (12) placed manually next to the C termini of the PCM modules. The Cα atoms of the residues mutated to cysteines, as well as of the functional Cys-20, are shown as pairwise colored spheres and labeled according to their position in Agp1.
Table 1.
Experimental and computed PELDOR interspin distances for the labels in the PCM region
Parallel and antiparallel structures of PCM fragments (16). Half-widths at the half-maxima are given in parentheses.
| Computed distance |
||||
|---|---|---|---|---|
| PCM spin-label positions | Intramonomer, parallel/antiparallel | Intermonomer parallel PCM | Intermonomer antiparallel PCM | Experimental distance |
| nm | nm | nm | nm | |
| C20R1-C20R1 | 8.1 (0.1) | 6.1 (0.1); 6.4 (0.1) | ||
| S122R1-S122R1 | 1.2 (0.1); 1.9 (0.1) | 7.1 (0.1); 6.6 (0.1)a | 2.4 (0.5) | |
| S122R1-C20R1 | 5.1 (0.1); 4.6 (0.1)a/4.8 (0.1) | 5.5 (0.1); 4.8 (0.1) | 5.1 (0.1) | 4.7 (0.3) |
| A362R1-A362R1 | 4.9 (0.1) | 7.7 (0.1) | 4.7 (0.4) | |
| A362R1-C20R1 | 3.9 (0.1)/2.6 (0.1) | 7.3 (0.1) | 6.5 (0.1) | 3.4 (0.6) |
a The two different numbers stand for two maxima in the distance distribution.
Table 2.
Experimental and computed PELDOR interspin distances for the labels in the His-kinase region
Parallel and antiparallel structures of a homology model of the Agp1 histidine kinase that was predicted based on a T. maritima histidine kinase (12). Half-widths at the half-maxima are given in parentheses.
| His-kinase spin-label positions | Computed distance (parallel subunits) | Experimental distance |
|---|---|---|
| nm | nm | |
| K517R1-K517R1 | 2.3 (0.4) | 2.7 (0.3) |
| H528R1-H528R1 | 2.7 (0.3) | 3.1 (0.4) |
| R535R1-R535R1 | 2.7 (0.3) | 2.7 (0.4) |
| K554R1-K554R1 | 2.3 (0.4) | 3.2 (0.1) |
| R603R1-R603R1 (inactive) | 6.0 (0.7) | 2.6 (0.4) |
| R603R1-R603R1 (active) | 3.1 (0.15) | 2.6 (0.4) |
Native cysteines in Agp1 phytochrome
Three native cysteines, Cys-20, Cys-279, and Cys-295, all of them are potential binding sites for MTSL, can be found in the sequence of wild-type Agp1 phytochrome. Cys-279 and Cys-295 are deeply buried in the protein and not easily accessible for various labels (28). Nevertheless, they were successfully replaced by other amino acids without affecting the spectral properties or the enzymatic function of phytochrome. However, Cys-20 is necessary for covalent binding of the biliverdin (BV) cofactor. Efficient binding of BV, which was synthesized in the host cell, prevents the availability of Cys-20 to MTSL during the spin-labeling procedure. Depending on the sample, we have observed varying efficiency of chromophore binding with typical values of 65–80% for the mutants presented here. Consequently, unoccupied Cys-20 could be accessible for other reagents (28). Therefore, both the introduced cysteine and also to some extent Cys-20 were available for covalent linkage of cysteine-specific MTSL in each of the samples.
PCM region
We have attempted to investigate a larger number of mutants, in which the mutated residues are located in the PCM (photosensory core module or PAS-GAF-PHY) region of Agp1. However, labeling turned out to be inefficient for most of these mutants, even though the crystal-structure-based Multiscale Modeling of Macromolecules (MMM) computation predicted high accessibility for these positions. For the mutants that could be efficiently labeled, data analyses and interpretations turned out to be much more challenging than for the mutants located in the histidine kinase region. Below, the PELDOR data acquired for S122R1 and A362R1 are presented (Figs. 2 and 3). Residue 122 is located in the linker region between the PAS and GAF domains. Depending on the expression batch, a position in addition to position S122C was spin-labeled to a certain degree. As a consequence, the experimentally obtained distance distributions comprised two main components with maxima at 2.4 and 4.7 nm (Fig. 2B). The additional spin-label position was identified as Cys-20, the chromophore-binding site. The shorter component in the distance distributions was assigned to the S122R1-S122R1 distance and the longer was assigned to S122R1-C20R1. This assignment is based on the comparison of experimental and MMM-computed distance distributions. Two recently published PCM-Agp1 crystal structures, one with parallel and one with antiparallel arrangement of the subunits (16), have been used for the MMM computations.
Figure 2.
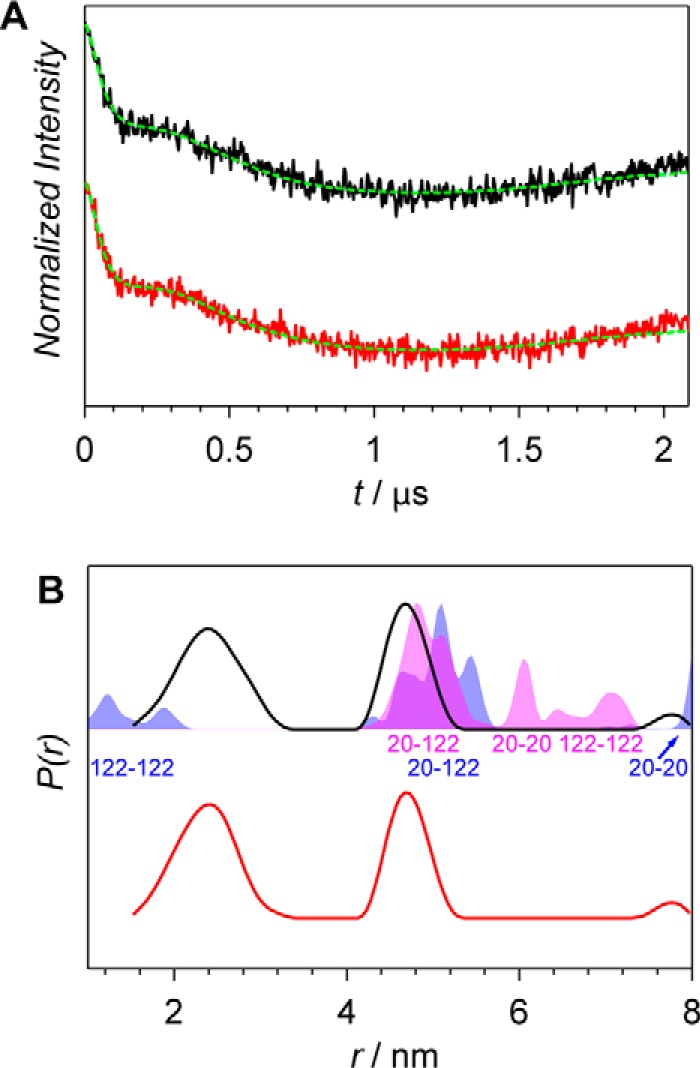
X-band PELDOR data of the Agp1 homodimer spin-labeled at position 122. A, PELDOR time-domain data after background correction of the S122R1 mutant of the Agp1 homodimer in the Pr and Pfr forms measured at 60 K (black and red lines, respectively). Fits obtained by Tikhonov regularization are shown as dashed green lines. B, experimental distance distributions of the Pr form (solid black line) and the Pfr form (solid red line) compared with the distance distribution predicted with MMM (shaded areas). MMM simulations are based on PCM-parallel (blue) and PCM-antiparallel (magenta) structures (16), and positions of amino acid residues contributing to the distributions are indicated below the peaks.
Figure 3.
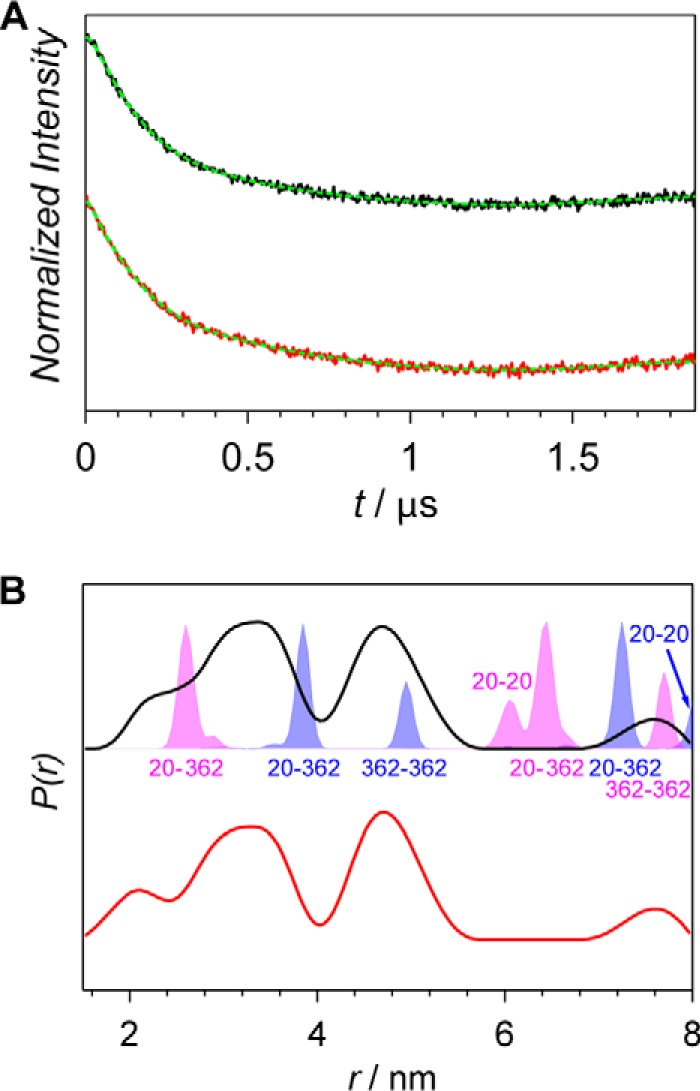
X-band PELDOR data of the Agp1 homodimer spin-labeled at position 362. A, PELDOR time-domain data after background correction of the A362R1 mutant of the Agp1 homodimer in the Pr and the Pfr form measured at 60 K (black and red lines, respectively). Fits obtained by Tikhonov regularization are shown as dashed green lines. B, experimental distance distributions of the Pr form (solid black line) and the Pfr form (solid red line) compared with the distance distribution predicted with MMM (shaded areas). MMM simulations are based on PCM-parallel (blue) and PCM-antiparallel (magenta) structures (16), and positions of amino acid residues contributing to the distributions are indicated either below or above the peaks.
For the parallel arrangement (Fig. 1B), a distance distribution with a mean distance of 1.6 nm was calculated for S122R1-S122R1 (Fig. 2B, blue-shaded area). A long distance of 8.1 nm was predicted for C20R1-C20R1. The distance distribution resulting from intramonomer interactions as well as intermonomer interactions, with a mean distance of 5.1 nm was computed for S122R1-C20R1. The overlap of computed and experimental distance distributions is fairly good in the region between 4.2 and 5.2 nm. In the short-distance range, the calculated distances are much shorter than the experimental ones, resulting in an only very small overlap of distributions.
Distances predicted based on an antiparallel dimer of PCM-Agp1 (Fig. 1A) are in a range between 4 and 8 nm and do not overlap with the region of shorter distances of the experimental distance distribution (Fig. 2B, magenta-shaded areas). The predicted S122R1-S122R1 distance distribution has two components with maxima at 6.6 and 7.1 nm. The C20R1-S122R1 distance is shorter and centered at 5.1 nm, whereas the C20R1-C20R1 distance is located in-between with its main maximum at 6.1 nm.
Overall, from the two MMM predictions, much better agreement with experimental data is found for the parallel than for the anti-parallel dimer of PCM-Agp1. Therefore, the experimental distance distribution with maxima at 2.4 and 4.7 nm were assigned to distances S122R1-S122R1 and C20R1-S122R1, respectively. The C20R1-C20R1 distance is not observed in the experimental distance distribution, mainly because of its length beyond the PELDOR range but also because of relatively small probability of finding both Cys-20 residues labeled in the dimer. This assumption is further corroborated by the fact that no contribution from the C20R1-C20R1 distance was found for proteins with spin labels in the histidine kinase (see “Histidine kinase region”). Importantly, one has to keep in mind that MMM assumes complete spin labeling of all cysteines in a protein, which is, however, not the case for proteins with the majority of Cys-20 bound to the BV chromophore. No changes in the distance distributions were observed for the sample before (Pr form) and after (Pfr form) red-light illumination (Fig. 2B).
An interaction with the spin label bound to Cys-20 is also expected to contribute to the PELDOR data obtained for A362R1 (Fig. 3) that is localized in the PHY domain. The obtained distance distributions (Fig. 3B) are dominated by two contributions with maxima at 3.4 and 4.7 nm. These were assigned to distances C20R1-A362R1 and A362R1-A362R1, respectively (16). The predicted distance distribution for the parallel dimer of PCM-Agp1 comprises three main contributions: A362R1-A362R1 at ∼5.0 nm, C20R1-A362R1 at ∼3.9 nm, and 7.3 nm for intra- and interchain distances, respectively, and C20R1-C20R1 at 8.1 nm (Fig. 3B, blue-shaded areas, and Tables 1 and 2). The calculated distance distributions are very narrow and overlap only partially with peaks of the experimental distance data. Similarly, narrow distance distributions are predicted for the antiparallel dimer of PCM-Agp1 (magenta-shaded areas in Fig. 3B), which overlap with the experimental distance distributions even less. For the latter structure, the A362R1-A362R1 distance of 7.7 nm is the longest of the predicted distances. The C20R1-C20R1 distance is 6.1 nm, and two C20R1-A362R1 distances are 2.6 and 6.4 nm for intra- and interchain distances, respectively (see also Tables 1 and 2). Overall, the relatively small overlap of experimental distance distributions with predicted ones makes an unambiguous assignment rather difficult. Nevertheless, the basic pattern of the experimental data is much better reproduced by the MMM prediction for the parallel dimer, thus supporting the parallel arrangement of monomers in the full-length protein. Consequently, a distance distribution with a maximum at 4.7 nm was assigned to the interspin distance between both A362R1 labels in the dimer and the distance distribution with a maximum at 3.4 nm to the C20R1-A362R1 distance. The experimental data suggest that at position 362 both subunits are slightly closer together in the full-length protein in solution as compared with the parallel PCM crystal structure. Therefore, the subunit arrangement in the crystal again does not reflect the arrangement in the full-length protein. A very small shift from 2.3 nm (Pr form) to 2.1 nm (Pfr form) in the shoulder of the contribution assigned to the C20R1-A362R1 distance is observed upon red-light illumination with all the other contributions remaining unchanged (Fig. 3B).
Histidine kinase region
PELDOR data of four spin-labeled proteins with the mutated residues being located on one of the helices from the dimerization and histidine phosphotransfer domain of the histidine kinase are shown in Fig. 4. K517R1 is localized in the region between the PHY domain and the histidine kinase, whereas the others (H528R1, R535R1, and K554R1) are in the histidine kinase itself (Fig. 1C) with His-528 being the phosphorylation site of the histidine kinase. Narrow distance distributions were obtained for each of the mutants (Fig. 4B, black and red lines). The experimental distance distributions overlap significantly with the distance distributions predicted for the structural model of the histidine kinase based on the T. maritima homolog (12) (Fig. 4B, shaded areas) for all the mutants but K554R1. For the latter, the predicted distance is significantly shorter than the measured one. Because Lys-554 is located in a loop, it is most likely that this loop in the Agp1 protein is folded differently in solution than in the model structure that was used for theoretical analysis. No contributions from couplings to C20R1 are observed for any of the four proteins (which is expected because of the very large separations between these positions) and also not from the C20R1-C20R1 interaction itself (thus supporting our assignments for PAS/GAF/PHY region) (Fig. 4B). The good agreement between experimental and MMM-predicted distance distributions supports the parallel alignment of the monomers in the full-length Agp1 phytochrome. In the antiparallel arrangement, the estimated distances range from 9 nm for Lys-517 to almost 20 nm for Lys-554 (see also Fig. 1A). Such distances are beyond the PELDOR range. For all mutants, signals of dark-adapted and red-light-illuminated samples are nearly identical (Fig. 4B, black and red lines, respectively). The very small differences are most probably due to the slightly different signal-to-noise ratios obtained in the respective measurements and are not necessarily due to conformational changes of the protein upon photoconversion.
Figure 4.
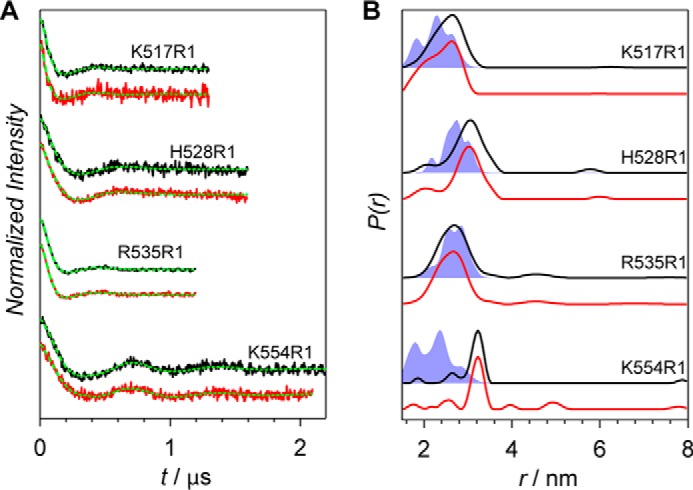
X-band PELDOR data of the Agp1 homodimer spin-labeled at positions 517, 528, 535, and 554. A, PELDOR time-domain data of the K554R1, R535R1, H528R1, and K517R1 mutants of the Agp1 phytochrome homodimer at 60 K. Experimental data after background correction of the Pr and Pfr forms are shown in black and red, respectively. Fits obtained by Tikhonov regularization are shown in green. B, experimental distance distributions of the Pr form (black lines) and the Pfr form (red lines) are compared with MMM predictions (shaded blue areas).
Subunit arrangement
Despite the two crystal structures available for the PCM of Agp1 (16) differ in their subunit arrangements, it is difficult to decide whether the spin label data for positions 122 and 362 reflect a parallel or an anti-parallel arrangement. Complex multimodal distance distributions were predicted for both positions in the parallel-aligned as well as in the antiparallel-aligned structures (Figs. 2B and 3B). The predicted distributions comprise relatively narrow components that suggest tight locations of the labels in the crystal structures. Differently, experimental distance distributions exhibit broad components, indicating a rather unconstrained environment of the spin labels attached to both positions in the full-length protein in solution. The only limited agreement between experimental and MMM-predicted distance distributions makes an unambiguous assignment of the components difficult. Nevertheless, the basic pattern of the distance contributions agrees better with the PCM structure arranged in parallel. This assignment is supported by the PELDOR results for the proteins with spin-label positions located within the histidine kinase unit. All interspin distances between residues located on the helices of the dimerization and histidine phosphotransfer domain of that region agree well with the parallel-arranged structure of the histidine kinase as derived based on the T. maritima homolog (PDB code 2C2A) (12). Consequently, only the parallel arrangement of the PCM region of Agp1 is likely in the full-length Agp1 phytochrome. However, the subunit arrangement in the PCM region of the full-length dimer in solution differs clearly from the subunit arrangement in the crystal structure (Tab. 1).
ATP-binding domain of the histidine kinase
None of the so-far discussed mutations is localized in the ATP-binding domain of the Agp1 phytochrome. No clear light-triggered structural rearrangements have been observed for any of those mutants. In the study on the structural basis of the histidine kinase autophosphorylation by Dago et al. (29), a structural model of the active kinase conformations was derived from the crystal structure of the histidine kinase module of T. maritima sensory kinase HK853 (12). The obtained active conformation of the histidine kinase differed from the inactive one in particular such that the ATP-binding domain has rotated by 90° around the four-helix-bundle axis of the kinase and positioned on top of the functional histidine, thus enabling autophosphorylation. Upon the rotational movement of the ATP-binding domains, the interdimer distances between the corresponding amino acids of the domains change significantly. Following this idea, we have investigated position Arg-603 (homologous to position Asn-338 in the histidine kinase of T. maritima) in the ATP-binding domain, for which the 603–603 distance changes from 6 nm in the inactive form to 3 nm in the active form of the histidine kinase (Fig. 5B, blue- and magenta-shaded areas, respectively). The experimental distance distribution of the dark-adapted sample with its maximum at 2.7 nm overlaps very well with the predicted distance distribution for the model structure of the active conformation of the histidine kinase. This observation is in line with a high phosphorylation activity observed for Agp1 in the dark-adapted Pr form. There was no overlap of the experimental and calculated distance distributions when the homology model based on the crystal structure of the histidine kinase of T. maritima (12) was used that corresponds to the inactive form. Within the histidine kinase module, the orientation of the ATP-binding domain in the dark-adapted form of Agp1 does not agree with the one that is found in the structure of T. maritima, which was assigned to the inactive form of the histidine kinase, but agrees well with the structure of the active form of the histidine kinase proposed in Ref. 29.
Figure 5.
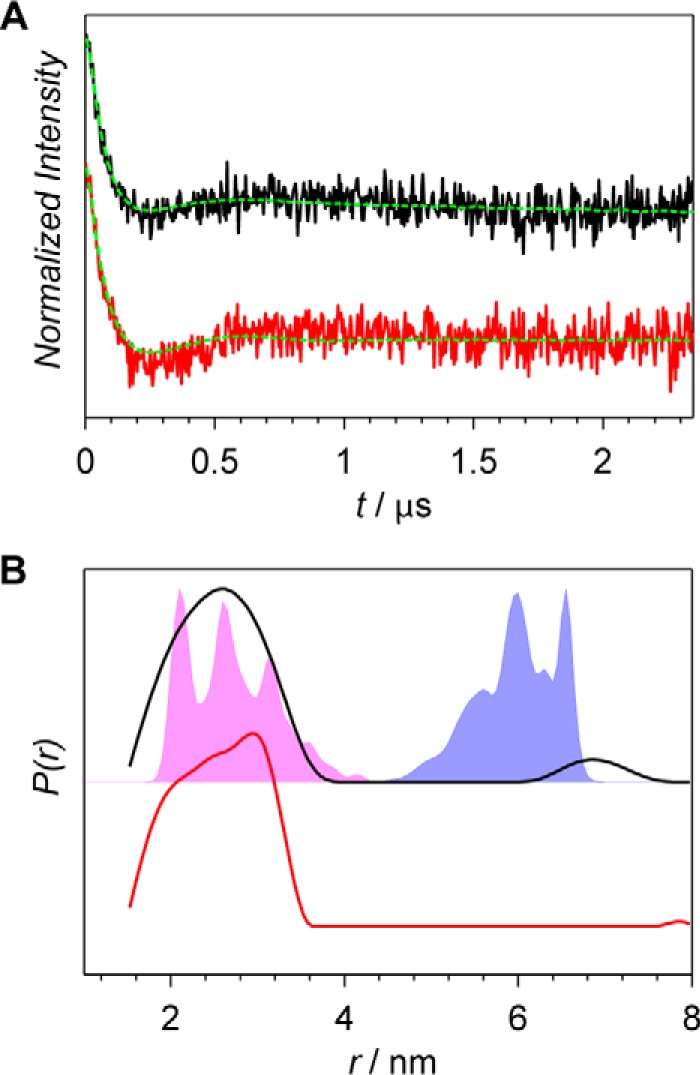
X-band PELDOR data of the Agp1 homodimer spin-labeled at position 603. A, PELDOR time-domain data after background correction of the R603R1 mutant of the Agp1 homodimer in the Pr form (black line) and the Pfr form (red line) measured at 60 K. Fits obtained by Tikhonov regularization are shown as dashed green lines. B, experimental distance distributions of the Pr form (black line) and the Pfr form (red line) are compared with the distance distribution predicted with MMM (shaded areas). MMM simulations are based on the inactive (blue) and the active (magenta) form of the histidine kinase. The structural models of the inactive and active forms of the Agp1 histidine kinase was based on the T. maritima homolog and the structure from Ref. 29, respectively.
Conformational changes upon illumination
Based on limited proteolysis and other methods, it is expected that Agp1 undergoes conformational changes during photoconversion (4, 28, 30). Such changes are thought to be responsible for spectral changes, modulation of enzyme activity, and differences in the interaction with other proteins. It was suggested that such changes would also result in distance changes at various positions of the protein (3, 31). However, the PELDOR data presented here did not reveal any substantial distance changes upon Pr-to-Pfr conversion. Hence, it is likely that these are not large enough to affect the subunit distances in a significant manner. Detection of distance changes might be hampered by an unfavorable fraction of dimers fulfilling all requirements for the structural rearrangements to take place: red-light irradiation of Agp1 typically results in formation of only ∼80% Pfr. Taking into account that ∼80% of all protein is assembled with BV, only 64% of all molecules could undergo photoconversion to Pfr. Therefore, at best 40% of the protein is expected to be in a Pfr/Pfr conformation (both halves of the dimer complex in Pfr form) after irradiation. Despite this rather intricate challenging situation, distance changes should, however, still be detectable.
We have also carried out PELDOR measurements at Q-band microwave frequency using smaller sample-tube diameters typically chosen at the higher microwave frequency. The obtained distance distributions for illuminated and non-illuminated samples were nearly identical to those from X-band PELDOR (data not shown).
For the PCM of Deinococcus bacteriophytochrome, based on Pr/Pfr crystal structures and SAXS measurements, an increase of up to 15 Å of the distances between both subunits in the region of the PHY domain upon Pr-to-Pfr photoconversion has been reported (11). The K517R1 mutant examined in our study is located in the region of the long helix connecting the PHY domain and the histidine kinase. We observed no Pr/Pfr differences at these positions, which supports that the distance change in that region is negligible. We therefore consider that the PAS-GAF-PHY protein segment alone behaves differently as compared with the full-length protein, in which the histidine kinase forms an additional dimerization site that might keep the PHY domains together. In a recent SAXS analysis on full-length Deinococcus phytochrome, such distance changes were indeed not observed (32).
An activation mechanism predicted for the histidine kinase based on computer molecular simulations (29) assumes that the ATP-binding sites rotate by 90° around the four-helix-bundle axis of the kinase. According to this mechanism, the distances between respective residues in both subunits change significantly like for the position of Arg-603 of Agp1, a homolog of Asn-338 in T. maritima, where the inter-residue distance would change from 6 nm in the inactive form to 3 nm in the active form of the histidine kinase. This substantial rearrangement could not be confirmed by our PELDOR experiments (Fig. 5B) because for both dark-adapted and red-light-illuminated samples only the short distance centered at 2.7 nm was detected. Thus, our measurements do not confirm the major conformational changes of the quaternary structure predicted by recent SAXS studies (11) and MAGMA simulations (29). However, our data have shown that the dark-adapted structure of the histidine kinase region in full-length Agp1 in the region of the ATP-binding domain does not correspond to the histidine-kinase crystal structure of T. maritima but corresponds to an activated form as presented in Ref. 29.
In summary, sensory proteins such as Agp1 phytochrome of A. fabrum must relay structural signals from the sensory site over large distances to regulatory output domains. Our results clearly show that the dimer of the full-length Agp1 phytochrome in the Pr form adopts parallel conformation. However, the weak agreement between experimental distance distributions and those obtained based on the parallel-aligned crystal structure of the PCM fragment suggests considerable differences between the solution structure of the full-length protein and the crystal structure of the PCM fragment. Additional information on the orientation of the ATP-binding domains relative to the four helices of the histidine kinase has been obtained that agrees well with the activated form of histidine kinase (29). The PELDOR studies presented here did not reveal any significant changes in the interspin distances upon Pr-to-Pfr photoconversion. We propose that the intramolecular signal transduction is not realized by a change of distance between both subunits or by distance changes that are too small to be detected by PELDOR.
Experimental procedures
Site-directed mutagenesis
All mutants used in this study were generated based on the expression vector pNM3c, which encodes for Agrobacterium heme oxygenase and full-length Agp1 with a C-terminal hexahistidine tag for Ni2+-affinity purification of the protein (4). Heme oxygenases catalyze the conversion of heme to BV, the chromophore of Agp1. As a result, the Escherichia coli cells produce holo-Agp1. Agp1 has three native cysteines: Cys-20 for covalent attachment of the BV chromophore to the apoprotein to generate holoprotein, and most bacterial phytochromes contain the conserved Cys-279 and Cys-295 (Agp1 numbering) of yet unknown function. Cys-279 and Cys-295 were replaced by site-directed mutagenesis to serines using the QuikChange site-directed mutagenesis approach (Stratagene, La Jolla, CA) with two complementary primers (Sigma) with desired mutations in the center. The resulting plasmid was used to generate various cysteine mutants for PELDOR experiments. All primers were bought from Sigma. The primer sequences for each mutant (sense strand) were as follows: C295A, ACGTGAAGCCGCTGATTTTGCAGCGCA; C279S, GGCTTGATTGCCAGCCACCACGCGACACCGC; S122C, GGAGTGGGCGAATGTGCTGAAAAGCTGATG; A362C, CGCGGATGGCGCATGTCTTGTCCTGGGCGACG; K517C, GTTGCAGCGCACCAATTGCGAGCTGGAGGCTTTTTC; H528C, CCTATTCCGTTTCGTGCGACCTGCGC; R535CCCTGCGCGCGCCGTTCTGCCATATTGTCG; K554C, CGATGCCCTGGACGAGTGCTCCCTGCATTATCTGC; and R603C, GAGCGAAGTTCGCTGCTCTCTTTCACACGCGG. After mutagenesis, the plasmids were checked for correctness by sequencing at GATC (Konstanz, Germany).
Protein expression and purification
E. coli XL1-blue cells with the desired expression vector were grown in 4 liters of lysogeny broth medium with 0.3 μm final concentration of ampicillin and tetracycline at 37 °C until an A600 of 0.6 was reached. Specific protein expression was induced by adding isopropyl-β-d-1-thiogalactopyranoside to a final concentration of 50 μm. The bacterial cultures were shaken for 48 h in darkness at 18 °C; the A600 reached ∼2.3. All subsequent steps were carried out under green safelight or in darkness at 4 °C. The cells were harvested and centrifuged at 5000 × g for 10 min, and the pellet was suspended in 20 ml of basic buffer (300 mm NaCl, 50 mm Tris·HCl, 5 mm EDTA, pH 7.8) with freshly added 2 mm (final concentration) DTT. The cells were lysed with a French pressure cell (Aminco) with two passages at 1380 bars and clarified by centrifugation at 20,000 × g for 30 min. The soluble proteins in the supernatant were precipitated with 50% ammonium sulfate, and the pellet was dissolved in dilute imidazole buffer (300 mm NaCl, 50 mm Tris·HCl, 10 mm imidazole, pH 7.8). The sample was centrifuged again, and the supernatant was loaded onto a 3 cm × 5 cm Ni2+-affinity chromatography column (Qiagen), which was equilibrated with dilute imidazole buffer. Concentrated imidazole buffer (300 mm NaCl, 50 mm Tris·HCl, 250 mm imidazole, pH 7.8) was used to elute the protein from the column. The eluted protein fractions were combined, and protein was precipitated with 50% ammonium sulfate. The resulting pellet was suspended in basic buffer and again centrifuged. Subsequently, the protein was used for characterization and spin labeling. Protein concentrations were estimated from the respective absorption at 280 nm (33).
Characterization of mutants
Absorption spectra of all samples 250 were recorded with a JASCO V-550 photometer. The scan speed was usually 1000 nm min−1. The spectra were measured between 900 and 250 nm at room temperature. Throughout this work, a 780-nm far-red light source with 100 μmol m−2 s−1 intensity and a 655-nm red light source with 20 μmol m−2 s−1 intensity light emitting diodes were used for photoconversion. For autophosphorylation, an earlier protocol (3, 33) was adopted. Phosphorylation experiments with the holoproteins were performed under blue-green safelight (λmax = 505 nm) or in darkness. The protein concentration was 12 μm. Sample volumes of 5 μl were irradiated with either red or with far-red light. The irradiation time was always 3 min, and the temperature during irradiation was 25 °C. Directly after irradiation, 15 μl of phosphorylation buffer (final concentrations were 25 mm Tris·HCl, 5 mm MgCl2, 4 mm β-mercaptoethanol, 0.2 mm EDTA, pH 7.8, 50 mm KCl, 5% ethylene glycol, 0.45 μm (50 MBq/ml) [γ-32P]ATP, pH 7.8) was added to each sample. After mixing, the samples were immediately incubated in a thermal block that was preset to 25 °C for 30 min. After this time, the phosphorylation reaction was stopped by adding 10 μl of loading buffer (30% glycerol, 6% SDS, 300 mm DTT, 0.01% bromphenol blue, 240 mm Tris·HCl, pH 6.7) to each reaction mixture. Then 10 μl of each sample were loaded to SDS-PAGE gels (10% acrylamide in separating gel). After electrophoresis, the protein was transferred onto a PVDF membrane (Millipore) with a Trans-Blot semi-dry blot apparatus (Bio-Rad). The dried membrane was exposed to a phosphorimaging plate (Fuji) for ∼5 min, followed by quantification using the fluorescent image analyzer FLA 2000 (Fuji) and integrated analysis software. The protein on the PVDF membrane was stained with Simply Blue SafeStain (Invitrogen).
Spin labeling
Spin labeling of Agp1 samples was performed at 4 °C in degassed labeling buffer (300 mm NaCl, 50 mm Tris·HCl, pH 7.8). The sample was first reduced by adding a 3-fold molar excess of DTT followed by incubation at room temperature for 40 min. Reduced Agp1 was precipitated with 50% ammonium sulfate to remove the excess of DTT from the sample. The resulting pellet was washed with labeling buffer and dissolved in the same buffer containing a 10-fold molar excess of MTSL (Toronto Research Chemicals, North York, Canada). The sample was immediately irradiated with far-red light for 5 min and incubated at 4 °C overnight in darkness. The spin-labeled protein was precipitated, and the supernatant containing unbound spin label was discarded. The resulting pellet was washed with labeling buffer, dissolved in 1 ml of the same buffer, and passed through a PD-10 desalting column (GE Healthcare) to further remove unbound spin label from the samples. The samples were concentrated to a final concentration of 100–150 μm using Amicon ultrafiltration tubes with a pore size corresponding to a nominal molecular weight limit of 30 kDa (Merck-Millipore) and 20% (v/v) glycerol was added. Concentrated samples were loaded into 3.8-mm (outer diameter) synthetic-quartz EPR tubes (Qsil GmbH, Langewiesen, Germany) and quickly frozen in liquid nitrogen, either directly or after 1 min red-light irradiation. Control UV-visible measurements were routinely performed to confirm that the samples (before freezing) were in the Pr state (unirradiated samples) or converted to ∼80% Pfr (irradiated samples at photoequilibrium).
PELDOR measurements
Dead-time-free four-pulse PELDOR experiments (34) were carried out at X-band frequencies (9.5–9.6 GHz) with a Bruker Elexsys E680 spectrometer equipped with a PELDOR unit. Microwave pulses were amplified by using a 1-kW TWT amplifier. A Bruker dielectric resonator ER4118X-MD-5-W1 was used that was immersed in a continuous-flow helium gas-flow cryostat (CF-935; Oxford Instruments). The temperature during the measurements was 60 K and was stabilized to ±0.1 K by an Oxford Instruments ITC-503 temperature controller. In all measurements, the pump frequency was set to the center of the resonator dip, as well as to the maximum of the nitroxide EPR spectrum. The observer frequency was 60 MHz higher. All measurements were performed with observer pulse lengths of 16 and 32 ns for π/2 and π pulses, respectively, and a pump pulse length of 24 ns. Proton modulation was suppressed by adding eight traces acquired at different τ1 values starting from τ1 = 300 ns with Δτ = 16 ns. Depending on the interspin distances, τ2 values from 1400 to 4000 ns were used. The total measurement time for each sample was 12–16 h. Experimental data were analyzed with a home-written program employing a model-free iterative Tikhonov regularization in combination with a non-negative constraint. Structure-based PELDOR time traces were additionally calculated using the open-source package Multiscale Modeling of Macromolecules (31). MMM uses the rotamer-library approach to model spin-label conformations at desired positions in a model structure. The computed rotamer distributions are then used to calculate distance distributions between pairs of sites that are automatically transformed into dipolar-evolution time traces of the PELDOR experiment (31). The structural models that were used as input into MMM were the crystal structure of Agp1-PCM with parallel subunits arrangement (PDB code 5I5L), the crystal structure of Agp1-PCM with antiparallel subunit arrangement (PDB code 5HSQ), and homology models of the Agp1 histidine kinase modules in the inactive and active states that were based on the crystal structure of the histidine kinase module of the T. maritima sensory kinase HK853 (PDB code 2C2A) and a computational model of the active conformation of the same protein (available as supplementary information from reference (29)), respectively.
Author contributions
S. K., S. W., I. N., and T. L. wrote the manuscript; S. K., W. L., T. L., and S. W. coordinated experiments; S. K., I. N., A. R., J. F., and E. R. performed experiments; S. K. analyzed the data; S. N., N. K., P. S., and T. L. contributed structure data; and all authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Stephan Rein for implementing a PELDOR analysis program and for fruitful discussions.
This work was supported by Deutsche Forschungsgemeinschaft Grants WE 2376/3 -1 and LA 799/11-1 and by the Max Planck Gesellschaft. This work was also supported in part by Deutsche Forschungsgemeinschaft Grants SFB740-B6 and SFB1078-B6 (to P. S.), by Deutsche Forschungsgemeinschaft Cluster of Excellence “Unifying Concepts in Catalysis” Research Field Grant D3/E3-1 (to P. S.), by Struktur- und Innovationsfond für die Forschung (to S. W. and S. K.), and by Deutsche Forschungsgemeinschaft Grant INST 39/928-1 FUGG (to S. W. and S. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- PCM
- photosensory core module
- PELDOR
- pulsed electron-electron double resonance
- PDB
- Protein Data Bank
- BV
- biliverdin
- SAXS
- small-angle X-ray scattering
- MTSL
- 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate
- MMM
- Multiscale Modeling of Macromolecules
- GAF
- cGMP phosphodiesterase/adenylcyclase/FhlA
- PAS
- Per-Arnt-Sim.
References
- 1. Lamparter T. (2004) Evolution of cyanobacterial and plant phytochromes. FEBS Lett. 573, 1–5 [DOI] [PubMed] [Google Scholar]
- 2. Rottwinkel G., Oberpichler I., and Lamparter T. (2010) Bathy phytochromes in Rhizobial soil bacteria. J. Bacteriol. 192, 5124–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Esteban B., Carrascal M., Abian J., and Lamparter T. (2005) Light-induced conformational changes of cyanobacterial phytochrome Cph1 probed by limited proteolysis and autophosphorylation. Biochemistry 44, 450–461 [DOI] [PubMed] [Google Scholar]
- 4. Noack S., Michael N., Rosen R., and Lamparter T. (2007) Protein conformational changes of Agrobacterium phytochrome Agp1 during chromophore assembly and photoconversion. Biochemistry 46, 4164–4176 [DOI] [PubMed] [Google Scholar]
- 5. Song P. S. (1999) Inter-domain signal transmission within the phytochromes. J. Biochem. Mol. Biol. 32, 215–225 [Google Scholar]
- 6. Yang X., Kuk J., and Moffat K. (2008) Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc. Natl. Acad. Sci. U.S.A. 105, 14715–14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Essen L. O., Mailliet J., and Hughes J. (2008) The structure of a complete phytochrome sensory module in the Pr ground state. Proc. Natl. Acad. Sci. U.S.A. 105, 14709–14714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wagner J. R., Brunzelle J. S., Forest K. T., and Vierstra R. D. (2005) A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331 [DOI] [PubMed] [Google Scholar]
- 9. Li H., Zhang J., and Vierstra R. D. (2010) Quaternary organization of a phytochrome dimer as revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U.S.A. 107, 10872–10877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang X., Kuk J., and Moffat K. (2009) Conformational differences between the Pfr and Pr states in Pseudomonas aeruginosa bacteriophytochrome. Proc. Natl. Acad. Sci. U.S.A. 106, 15639–15644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takala H., Björling A., Berntsson O., Lehtivuori H., Niebling S., Hoernke M., Kosheleva I., Henning R., Menzel A., Ihalainen J. A., and Westenhoff S. (2014) Signal amplification and transduction in phytochrome photosensors. Nature 509, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marina A., Waldburger C. D., and Hendrickson W. A. (2005) Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. EMBO J. 24, 4247–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamada S., Akiyama S., Sugimoto H., Kumita H., Ito K., Fujisawa T., Nakamura H., and Shiro Y. (2006) The signaling pathway in histidine kinase and the response regulator complex revealed by X-ray crystallography and solution scattering. J. Mol. Biol. 362, 123–139 [DOI] [PubMed] [Google Scholar]
- 14. Burgie E. S., Wang T., Bussell A. N., Walker J. M., Li H., and Vierstra R. D. (2014) Crystallographic and electron microscopic analyses of a bacterial phytochrome reveal local and global rearrangements during photoconversion. J. Biol. Chem. 289, 24573–24587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casino P., Rubio V., and Marina A. (2009) Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139, 325–336 [DOI] [PubMed] [Google Scholar]
- 16. Nagano S., Scheerer P., Zubow K., Michael N., Inomata K., Lamparter T., and Krauss N. (2016) The crystal structures of the N-terminal photosensory core module of Agrobacterium phytochrome Agp1 as parallel and anti-parallel dimers. J. Biol. Chem. 291, 20674–20691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang X., Ren Z., Kuk J., and Moffat K. (2011) Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Nature 479, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burgie E. S., Zhang J., and Vierstra R. D. (2016) Crystal structure of Deinococcus phytochrome in the photoactivated state reveals a cascade of structural rearrangements during photoconversion. Structure 24, 448–457 [DOI] [PubMed] [Google Scholar]
- 19. Strauss H. M., Schmieder P., and Hughes J. (2005) Light-dependent dimerisation in the N-terminal sensory module of cyanobacterial phytochrome 1. FEBS Lett. 579, 3970–3974 [DOI] [PubMed] [Google Scholar]
- 20. Jeschke G. (2012) DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446 [DOI] [PubMed] [Google Scholar]
- 21. Schiemann O., and Prisner T. F. (2007) Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q. Rev. Biophys. 40, 1–53 [DOI] [PubMed] [Google Scholar]
- 22. Borbat P., and Freed J. (2013) Pulse dipolar rlectron spin resonance: distance measurements. In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences (Timmel C. R., and Harmer J. R., eds) pp. 1–82, Springer; Berlin Heidelberg [Google Scholar]
- 23. Hubbell W. L., Gross A., Langen R., and Lietzow M. A. (1998) Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8, 649–656 [DOI] [PubMed] [Google Scholar]
- 24. Hubbell W. L., Cafiso D. S., and Altenbach C. (2000) Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7, 735–739 [DOI] [PubMed] [Google Scholar]
- 25. Heyes D. J., Khara B., Sakuma M., Hardman S. J., O'Cualain R., Rigby S. E., and Scrutton N. S. (2012) Ultrafast red light activation of Synechocystis phytochrome Cph1 triggers major structural change to form the Pfr signalling-competent state. PLoS One 7, e52418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeschke G., and Polyhach Y. (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 9, 1895–1910 [DOI] [PubMed] [Google Scholar]
- 27. Scheerer P., Michael N., Park J. H., Nagano S., Choe H.-W., Inomata K., Borucki B., Krauss N., and Lamparter T. (2010) Light-induced conformational changes of the chromophore and the protein in phytochromes: bacterial phytochromes as model systems. ChemPhysChem 11, 1090–1105 [DOI] [PubMed] [Google Scholar]
- 28. Borucki B., and Lamparter T. (2009) A polarity probe for monitoring light-induced structural changes at the entrance of the chromophore pocket in a bacterial phytochrome. J. Biol. Chem. 284, 26005–26016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dago A. E., Schug A., Procaccini A., Hoch J. A., Weigt M., and Szurmant H. (2012) Structural basis of histidine kinase autophosphorylation deduced by integrating genomics, molecular dynamics, and mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 109, E1733–E1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noack S., and Lamparter T. (2007) Light modulation of histidine-kinase activity in bacterial phytochromes monitored by size exclusion chromatography, crosslinking, and limited proteolysis. Methods Enzymol. 423, 203–221 [DOI] [PubMed] [Google Scholar]
- 31. Polyhach Y., Bordignon E., and Jeschke G. (2011) Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys. 13, 2356–2366 [DOI] [PubMed] [Google Scholar]
- 32. Björling A., Berntsson O., Lehtivuori H., Takala H., Hughes A. J., Panman M., Hoernke M., Niebling S., Henry L., Henning R., Kosheleva I., Chukharev V., Tkachenko N. V., Menzel A., Newby G., et al. (2016) Structural photoactivation of a full-length bacterial phytochrome. Sci. Adv. 2, e1600920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lamparter T., Michael N., Mittmann F., and Esteban B. (2002) Phytochrome from Agrobacterium tumefaciens has unusual spectral properties and reveals an N-terminal chromophore attachment site. Proc. Natl. Acad. Sci. U.S.A. 99, 11628–11633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pannier M., Veit S., Godt A., Jeschke G., and Spiess H. W. (2000) Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson. 142, 331–340 [DOI] [PubMed] [Google Scholar]