

Abstract
Toxoplasma gondii, like all apicomplexan parasites, uses Ca2+ signaling pathways to activate gliding motility to power tissue dissemination and host cell invasion and egress. A group of “plant-like” Ca2+-dependent protein kinases (CDPKs) transduces cytosolic Ca2+ flux into enzymatic activity, but how they function is poorly understood. To investigate how Ca2+ signaling activates egress through CDPKs, we performed a forward genetic screen to isolate gain-of-function mutants from an egress-deficient cdpk3 knockout strain. We recovered mutants that regained the ability to egress from host cells that harbored mutations in the gene Suppressor of Ca2+-dependent Egress 1 (SCE1). Global phosphoproteomic analysis showed that SCE1 deletion restored many Δcdpk3-dependent phosphorylation events to near wild-type levels. We also show that CDPK3-dependent SCE1 phosphorylation is required to relieve its suppressive activity to potentiate egress. In summary, our work has uncovered a novel component and suppressor of Ca2+-dependent cell egress during Toxoplasma lytic growth.
Keywords: molecular genetics, parasite, proteomics, signaling, Toxoplasma gondii, Ca2+ signalling, apicomplexa, forward genetic screen, host cell egress
Introduction
The phylum Apicomplexa comprises a large group of obligate intracellular parasites, many of which have medical and agricultural significance. Plasmodium spp. are the causative agents of malaria, whereas Cryptosporidium spp. cause severe diarrhea. Toxoplasma gondii is the most ubiquitous of all apicomplexan parasites, chronically infecting >30% of the human population. Acute Toxoplasma infection can cause congenital birth defects, epilepsy, hydrocephalus, and stillbirth. Chronic infection is a leading cause of progressive blindness in some countries and can lead to neurological disease in immunocompromised patients (1–3).
To survive in the host, Toxoplasma, like all apicomplexan parasites, must actively invade host cells to initiate replication. After several rounds of division, unknown signals trigger tachyzoites to actively exit (egress from) host cells, allowing for subsequent rounds of invasion and intracellular growth. Thus, invasion and egress are critical for the establishment and persistence of infection and therefore represent potential therapeutic targets to treat infection.
Like all apicomplexan species, Toxoplasma initiates motility, invasion, and egress via an intracellular signaling cascade that activates exocytosis of adhesins from the microneme organelles and stimulates the glideosome, an actomyosin-based motor, required to drive motility. Ca2+, cGMP, and phospholipid signaling pathways all play important and interconnected roles in activating microneme release and parasite motility (4–6). Critical to transducing intracellular Ca2+ flux is a group of “plant-like” Ca2+-dependent protein kinases (CDPKs)2 (7). CDPKs are direct fusions between a kinase and calmodulin-like domains, an architecture that is not found in metazoans. CDPKs are localized to several intracellular cellular compartments where they presumably act on a defined set of substrates, altering their activity upon phosphorylation. CDPK1, a cytosolically localized ortholog, activates exocytosis of micronemes, releasing adhesins onto the tachyzoite surface, thus initiating gliding motility (8). CDPK3, on the other hand, localizes to the inner side of the parasite plasma membrane and potentiates rapid, Ca2+ ionophore-induced egress (9–11). In related Plasmodium species, Plasmodium falciparum CDPK1 has been implicated in parasite development and host cell invasion in blood stages (12–14), CDPK5 is required for parasite egress from erythrocytes (15), and Plasmodium berghei CDPK3 is necessary for motility during sexual stage development (16, 17). Interestingly, CDPK orthologs also play important roles in other cellular processes, such as parasite division, carbon metabolism, and stage differentiation (18–21).
Little is understood about how CDPKs activate motility. Recent efforts have utilized techniques such as substrate capture to identify the direct targets of CDPKs (22), and proximity ligation to identify putative interacting partners, the latter indicating that CDPK3 may directly phosphorylate components of the glideosome to regulate motility (23). Quantitative global phosphoproteomics have identified phosphoproteins that may be regulated by CDPK3 during egress, revealing a diversity of signaling pathways (24). In this study, we utilize a forward genetic screen to reveal novel components of Ca2+-dependent egress in Toxoplasma. By screening for gain-of-function mutants in a cdpk3-deficient background, we identify suppressor of Ca2+-dependent egress 1 (SCE1), which appears to act as a suppressor during egress. Further, we show that the functions of CDPK3 and SCE1 are interconnected and that phosphorylation of SCE1 by CDPK3 controls its suppressive activity during host cell egress.
Results
Identification of a suppressor of egress
To identify new factors involved in Toxoplasma Ca2+-dependent egress, we took advantage of previously described CDPK3 knockout (Δcdpk3) parasites (supplemental Table S1) (11), which show negligible egress following stimulation with the Ca2+ ionophore A23187. We devised a strategy to enrich and isolate parasites from the egress-deficient Δcdpk3 background that reverted to an egress-competent phenotype (hereafter referred to as Δcdpk3rev) through the acquisition of random mutations (Fig. 1A). Egress-deficient Δcdpk3 parasites were split into 10 independent populations and mutagenized using the chemical mutagen ethyl methane sulfonate (EMS) (hereafter referred to as Δcdpk3EMS.1–Δcdpk3EMS.10). Mutagenized populations were then treated with the Ca2+ ionophore A23187, which stimulates Ca2+-dependent egress in wild-type parasites, in medium supplemented with dextran sulfate to block reinvasion. The host cell monolayer was then washed to remove any parasites that had regained the capacity to egress, which were then transferred into a fresh culture flask for subsequent expansion.
Figure 1.

Forward genetic strategy for the enrichment of Δcdpk3rev mutants. A, outline of the strategy for the enrichment of Δcdpk3rev mutants from chemically mutagenized egress-deficient Δcdpk3. ν, host cell nucleus. B, egress of wild-type (RH:Δku80), Δcdpk3, Δcdpk3EMS.7.1, Δcdpk3EMS.7.2, Δcdpk3EMS.10.1, and Δcdpk3EMS.10,2 parasites stimulated with A23187 for 3 min. C, protein features of SCE1 (TGME49_269260), including 5× transmembrane (TM) domains, and point mutations seen in mutagenized parasites from population 7 and population 10. Error bars show mean ± S.D. *, adjusted p < 0.0001 using one-way ANOVA and Tukey test to correct for multiple comparisons. There is no statistical difference in egress capacity between any of the EMS-generated lines. The column data were derived from three independent biological replicates.
After eight rounds of enrichment, two populations (Δcdpk3EMS.7 and Δcdpk3EMS.10) showed a dramatic increase in sensitivity to stimulation of egress with A23187 compared with parental Δcdpk3 tachyzoites. From each population, we derived two clonal parasite strains, Δcdpk3EMS.7.1, Δcdpk3EMS.7.2, Δcdpk3EMS.10.1, and Δcdpk3EMS.10.2. The percentage of parasitophorous vacuoles showing egress of parasites after 2 min of A23187 stimulation was similar in all clones (∼60–70%, compared with ∼95% for the wild type (RH:Δku80), and ∼0.5% Δcdpk3), demonstrating that egress-competent Δcdpk3rev parasites had been recovered successfully from the egress-deficient Δcdpk3 population (Fig. 1B).
Whole-genome sequencing was performed for the mixed Δcdpk3EMS.7 and Δcdpk3EMS.10 populations, the clones Δcdpk3EMS.7.1, Δcdpk3EMS.7.2, Δcdpk3EMS.10.1, and Δcdpk3EMS.10,2, and the parental Δcdpk3 parasites. 880 unique mutations were identified in Δcdpk3EMS.7 and 858 in Δcdpk3EMS.10 compared with parental Δcdpk3. These mutations appeared fixed at every locus across all sequence reads, even in Δcdpk3EMS.7 and Δcdpk3EMS.10, suggesting that these lines were effectively clonal. Furthermore, this indicates that each population had likely arisen from an initial single Δcdpk3rev mutant and, thus, that the etiological mutation selected for by our strategy was rare. This is congruent with our finding that only 2 of the 10 mutagenized populations gave rise to Δcdpk3rev parasites.
To identify the etiological mutation for the rescue-of-egress phenotype in Δcdpk3rev parasites, we applied a stepwise ranking process with the following assumptions: the mutation must be non-synonymous; given the low rate of reversion across 10 populations and the identical egress levels in rescued populations, the same gene was mutated between populations; the mutated gene has different mutations in the different populations; the mutation is an SNP resulting in loss of protein function; and the mutated gene encodes a protein that localizes to the plasma membrane, like CDPK3. The full list of SNPs and the results from each of the ranking steps are presented in supplemental Table S2. Following this, independent mutations were identified in the coding sequence of the hypothetical protein TGME49_269260 in all EMS-mutagenized populations. TGME49_269260 is conserved in the closely related apicomplexan parasites Neospora caninum and Hammondia hammondi but contains no computationally predicted domains other than five C-terminal transmembrane domains (Fig. 1C). In Δcdpk3EMS.7, Δcdpk3EMS.7.1, and Δcdpk3EMS.7.2, the mutation in the TGME49_269260 gene resulted in the missense mutation H565R, whereas, in Δcdpk3EMS.10, Δcdpk3EMS.10.1 and Δcdpk3EMS.10,2, the mutation introduced a stop codon near the N terminus (Gln106STOP). We therefore hypothesized that mutation of the coding sequence of TGME49_269260 in both Δcdpk3EMS.7 and Δcdpk3EMS.10 parental populations resulted in an inactive gene product and, therefore, that this gene likely encoded a factor that inhibited Ca2+-dependent egress in Δcdpk3 parasites. The protein product of TGME49_269260 was therefore named suppressor of Ca2+ egress 1 (SCE1).
SCE1 is a suppressor of CDPK3-dependent egress
To begin investigation of the role of SCE1 in Toxoplasma, the endogenous locus was tagged with a 5×Ty epitope tag in RH:Δku80 parasites. By IFA, SCE1-Ty localized to the parasite periphery, including the residual body (a pocket of plasma membrane left behind during tachyzoite cell division), suggesting that SCE1 is associated with the plasma membrane (Fig. 2Ai). This localization is similar to that seen previously for CDPK3 (11). SCE1 is predicted to be ∼75 kDa, but, by reducing Western blot, this protein runs at approximately double the size (Fig. 2Aii).
Figure 2.

Characterization of the role of SCE1 in Toxoplasma egress. Ai, localization of SCE1-Ty and staining with α-Ty and DAPI merged with a phase-contrast image. Aii, reducing Western blot of SCE1-Ty, staining with α-Ty antibody, and α-GAP45 as loading control. B, egress of wild-type, Δsce1, Δcdpk3, and Δcdpk3Δsce1 parasites stimulated with A23187 for 3 min. Error bars show mean ± S.D. ns, not significant. All other comparisons, adjusted p = < 0.0001 using one-way ANOVA. Ci, localization of ectopically expressed SCE1wt in Δsce1 and staining with α-HA and α-GAP45. Cii, egress of Δsce1, Δcdpk3, Δcdpk3Δsce1, Δcdpk3EMS7.2, and Δcdpk3EMS10.2 with ectopically expressed SCE1wt-HA, stimulated with A23187 for 3 min. Error bars show mean ± S.D. All pairwise comparisons not labeled, p < 0.0001 using one-way ANOVA and Tukey test to correct for multiple comparisons. The column data were derived from three independent biological replicates.
To investigate the role of SCE1 in parasite egress, we genetically deleted sce1 in both RH:Δku80 and Δcdpk3 lines (giving rise to the lines Δsce1 and Δcdpk3Δsce1, respectively) (supplemental Fig. S1 and Table S1). We then tested whether the loss of SCE1 in Δcdpk3 parasites recapitulated the Δcdpk3rev phenotype. Following stimulation with A23187 for 3 min, parasites egressed from ∼65% of Δcdpk3Δsce1 parasitophorous vacuoles, similar to what was seen in EMS-mutagenized strains (Fig. 2B). Δsce1 parasites showed no significant difference in egress compared with wild-type parasites under these conditions.
To validate the role of loss of SCE1 activity in the EMS-induced Δcdpk3rev phenotype, a complementation construct was designed containing the sce1 cDNA sequence fused to a 3×HA epitope tag. This was expressed ectopically after integration at the uprt locus in Δsce1 (Δsce1:SCE1wt), Δcdpk3 (Δcdpk3:SCE1wt), Δcdpk3Δsce1 (Δcdpk3Δsce1:SCE1wt), Δcdpk3EMS.7.1 (Δcdpk3EMS.7.1:SCE1wt), and Δcdpk3EMS.10.1 (Δcdpk3EMS.10.1:SCE1wt) (supplemental Table S1). Ectopically expressed SCE1wt-HA localized to the plasma membrane and residual body, as observed for native SCE1 upon endogenous tagging (Fig. 2Ci). Some additional staining of an intracellular body was also observed, which may be a result of ectopic SCE1wt-HA overexpression.
We then tested the effect of complementation on egress capacity. Δsce1:SCE1wt and Δcdpk3:SCE1wt showed no significant difference in A23187 sensitivity relative to their parental strains, demonstrating that overexpression of SCE1wt did not modify egress competence (Fig. 2Cii). However, complementation with SCE1wt-HA restored the egress-deficient phenotype in Δcdpk3Δsce1, Δcdpk3EMS.7.1, and Δcdpk3EMS.10.1. Together, these data strongly suggest that loss of SCE1 activity in Δcdpk3rev confers the rescue-of-egress phenotype.
SCE1 does not significantly affect sensitivity to egress stimuli
We then wanted to determine what role SCE1 plays during egress. We previously hypothesized that CDPK3 acts to amplify egress capacity upon A23187 stimulation (11). We therefore reasoned that SCE1 may act in the opposite manner to dampen Ca2+ or cGMP signaling and the sensitivity of parasites to agonists of these pathways.
To determine whether SCE1 controls the rate of response to egress stimulation, we measured host cell exit at 2.5, 5, and 15 min after A23187 stimulation (Fig. 3A). At 2.5 min, egress capacity mimicked the results above (Fig. 2); however, by 5 min, Δcdpk3Δsce1 showed egress equivalent to wild-type parasites (Fig. 3A). At 15 min, Δcdpk3 showed some egress capacity, and Δcdpk3Δsce1 and wild-type egress remained equivalent. We observed no significant difference in egress capacity at any time point between Δsce1 and the wild type (Fig. 3A). We also tested whether loss of SCE1 altered the sensitivity of parasites to a range of concentrations of A23187. Here, although SCE1 knockout parasites (Δsce1 and Δcdpk3Δsce1) showed a trend toward increased sensitivity to low concentrations of A23187, this did not reach statistical significance, as analyzed by two-way ANOVA (Fig. 3B).
Figure 3.

Sensitivity of SCE1 mutants to egress agonists. Shown is egress of wild-type, Δsce1, Δcdpk3, and Δcdpk3Δsce1 parasites stimulated with either A23187 or BIPPO. A, egress assay measured at 2.5, 5, and 15 min. There was no difference in egress capacity between the wild type and Δsce1 at any time point. B, egress at 5 min over a concentration gradient of A23187. The only statistically significant differences observed are by comparison between all strains and with Δcdpk3 for the two highest concentrations of A23187. C, egress in response to BIPPO, measured at 2.5, 5, and 15 min. There was no statistically significant difference between the wild type and Δsce1 at any time point. D, egress in response to BIPPO at 5 min over a concentration gradient. The only statistically significant differences observed are by comparison between all strains and Δcdpk3 for the two highest concentrations of A23187. Error bars show mean ± S.D. All statistical tests were performed using two-way ANOVA and Tukey correction for multiple comparisons. The data were derived from three independent biological replicates.
We then investigated whether SCE1 plays a role in egress regulated by cGMP signaling. We previously described a potent Apicomplexa-specific PDE inhibitor, BIPPO, which stimulates egress through PKG (42). Using this inhibitor, we and others have also demonstrated a link between cGMP and Ca2+ signaling (21, 25, 26). To explore the rate of parasite egress following cGMP signaling, we monitored the egress of Δcdpk3, Δsce1, and Δcdpk3Δsce1 parasites at 2.5, 5, and 15 min after BIPPO treatment (Fig. 3C). Δcdpk3Δsce1 tachyzoites again showed significantly more egress than parental Δcdpk3 parasites, demonstrating that SCE1 also acts as a suppressor in cGMP-stimulated egress through a CDPK3-dependent mechanism (Fig. 3C). Although Δsce1 tachyzoites appeared to egress slightly higher than wild-type parasites, this was not statistically significant, as assessed by two-way ANOVA. Interestingly, using BIPPO, we found that, unlike A23187 treatment, there was no difference between wild-type and Δcdpk3Δsce1 at 2.5 min. We also examined the sensitivity of parasites to a range of BIPPO concentrations. We again observed a non-statistically significant trend (as analyzed by two-way ANOVA) toward increased egress of Δsce1 and Δcdpk3Δsce1 parasites at low BIPPO concentrations relative to wild-type parasites (Fig. 3D).
SCE1 and CDPK3 regulate phosphorylation signaling during egress, as demonstrated by global quantitative proteomics
CDPK3 influences the phosphorylation of hundreds of proteins, either as direct substrates or downstream of CDPK3 activity (24). We hypothesized that loss of SCE1 allows for CDPK3-independent egress in Δcdpk3Δsce1 by either up-regulating an alternative egress signaling pathway or by alleviating a cellular event that occurs downstream of CDPK3 that is required for subsequent signaling steps to occur. Although SCE1 is not predicted to be a kinase, we hypothesized that it may indirectly control egress by regulating the phosphorylation state of important Ca2+ signaling proteins that act downstream of CDPK3. Investigating this may therefore also identify the relationships between CDPK3 and other Ca2+-signaling proteins.
To answer this question, we utilized SILAC-based global quantitative phosphoproteomics (Fig. 4A). Wild-type, Δcdpk3Δsce1, and Δcdpk3 parasites were grown in “light” (L), “medium” (M), and “heavy” (H) SILAC labeling medium, respectively. It was assumed that all signaling processes relevant to regulating host cell egress occur within Toxoplasma, so HFF cell debris was removed by differential centrifugation, and human peptides were excluded from analysis (Fig. 4B). A total of 7483 unique peptides were identified, of which 5775 (77.2%) were phosphorylated. 5235 (70.0%) of the total peptides mapped to Toxoplasma proteins, of which 4423 (84.5%) were phosphorylated. This suggests that contaminating human peptides were largely dephosphorylated. Median-centered SILAC ratios of all quantified Toxoplasma sites showed a normal Gaussian distribution, indicating that the presence of host cell proteins, which were mostly unlabeled, did not significantly skew the expected distributions of SILAC ratios (Fig. 4C).
Figure 4.

Phosphoproteomic investigation of CDPK3 and SCE1 egress. A, overview of the strategy for phosphoproteomic investigation of CDPK3-regulated egress. Wild-type, Δcdpk3Δsce1, and Δcdpk3 parasites were grown in light, medium, or heavy SILAC labeling medium, respectively. After labeling, parasites were treated with A23187 for 1.5 min to trigger egress-signaling events, parasites were harvested by needle passage, and host cell debris was removed by differential centrifugation. SILAC-labeled samples were mixed 1:1:1 before tryptic digestion. Peptides were fractionated by strong cation exchange (SCX) chromatography, phosphopeptides were enriched on TiO2 beads, and the resulting samples were analyzed by MS/MS. B, the number of high-confidence Toxoplasma and human proteins, peptides, and phosphopeptides identified following MaxQuant analysis. C, histograms of median-centered log2 SILAC ratios, Δcdpk3Δsce1/wild type (M/L), Δcdpk3/wild type (H/L), and Δcdpk3/Δcdpk3Δsce1 (H/M). D, the number of high-confidence phosphosites that are significantly up- or down-regulated in Δcdpk3 and Δcdpk3Δsce1 (relative to wild-type parasites) and restored to near wild-type levels in Δcdpk3Δsce1 (relative to Δcdpk3).
We then filtered SILAC data for phosphopeptides showing evidence of regulation by CDPK3 or SCE1, according to the log2-converted SILAC ratios M/L (Δcdpk3Δsce1/wild type), H/L (Δcdpk3/wild type), and H/M (Δcdpk3/Δcdpk3Δsce1) (Fig. 4D). Only phosphopeptides with a minimum phosphosite localization score of 0.75 were considered (supplemental Table S3, tab 1). These were first determined to be misregulated (significantly up- or down-regulated relative to the wild type) in Δcdpk3 (supplemental Table S3, tab 2). Phosphopeptides were then filtered for those whose phosphorylation state was restored in Δcdpk3Δsce1 to a wild type-like phosphorylation state (e.g. down-regulated in H/L, up-regulated in H/M) (supplemental Table S3, tab 3). Following these criteria, a total of 308 phosphopeptides were misregulated in Δcdpk3 parasites relative to the wild type. 182 phosphopeptides that were misregulated in Δcdpk3 showed rescue in Δcdpk3Δsce1, representing 59% of all Δcdpk3 misregulated phosphopeptides. Compellingly, this represented 84% of all Δcdpk3Δsce1 misregulated phosphopeptides (214 phosphopeptides). Therefore, rescue of egress following knockout of SCE1 in Δcdpk3 is associated with the restoration of phosphorylation to a significant proportion of phosphorylation sites to near wild-type levels. This suggests that SCE1 does indeed indirectly regulate the phosphorylation of a large proportion of proteins downstream of CDPK3 activity during egress or that loss of this protein activates an alternative pathway that converges at common substrates for activation of egress.
Phosphopeptides regulated by CDPK3 and SCE1 are predicted to be involved in cell signaling
It has been shown previously that loss of CDPK3 influences the phosphorylation of diverse sets of proteins (24). We therefore aimed to determine whether particular sets of proteins or particular subcellular compartments were disproportionately influenced by CDPK3 and SCE1 activity. Filtered phosphopeptides described above were further grouped as follows: “Δcdpk3-misregulated,” all that fit the minimum criteria of being up- or down-regulated in Δcdpk3 relative to the wild type; “Δsce1-restored,” all that showed rescue to near wild-type levels in Δcdpk3Δsce1 relative to Δcdpk3; “Δsce1-unrestored,” all that showed no rescue in Δcdpk3Δsce1; and “Δcdpk3-independent,” all that showed no significant change in Δcdpk3 (supplemental Table S3, tabs 2–5, respectively). GO terms were annotated for all proteins where phosphopeptides were detected from within these groups. GO term enrichment analysis was performed, comparing representation of GO terms within each phosphoprotein group against a reference set of the whole Toxoplasma annotated proteome.
Several GO process terms were significantly enriched among proteins containing Δcdpk3-misregulated phosphosites compared with a reference set of the whole Toxoplasma annotated proteome (Fig. 5A). These included terms that are consistent with involvement in regulation of signaling processes, such as cell signaling processes (“signal transduction,” “cell communication,” and “intracellular signal transduction”), turnover of cyclic nucleotides (“phosphoric diester hydrolase activity”), and signaling through phospholipases (“phospholipase activity”). Interestingly, a number of GO process terms associated with nuclear transport were enriched in this dataset (“nuclear transport” and “nucleocytoplasmic transport”). Nuclear processes such as transcription are unlikely to play a direct role in the up-regulation of egress, which normally acts very rapidly (1–2 min) but may be altered as a downstream consequence of changes to egress efficiency or Ca2+ signaling following constitutive knockout of CDPK3.
Figure 5.

GO term enrichment of phosphopeptide datasets. Shown is an analysis of GO term enrichment in phosphoprotein datasets. Phosphopeptide data “test” sets were compared with a “reference” set comprising the complete Toxoplasma genome. A, all phosphosites showing up- or down-regulation in Δcdpk3. B, all Δcdpk3-misregulated phosphosites restored in Δcdpk3Δsce1. C, all Δcdpk3-misregulated phosphosites not restored in Δcdpk3Δsce1. D, all phosphosites showing no change in Δcdpk3. The color coding indicates the ontology source of GO terms: green, biological process; yellow, molecular function; purple, cellular component.
Many of these same GO terms were enriched for proteins containing Δsce1-restored phosphosites (Fig. 5B). However, certain terms were lacking, most noticeably those having to do with sphingolipid metabolism (“glycosphingolipid metabolic process” and “sphingolipid metabolic process”), organic substance transport, phosphoric diester hydrolase activity, sphingomyelin phosphodiesterase activity, and phospholipase activity. Instead, several of these terms are enriched in the Δsce1-unrestored group (Fig. 5C). This suggests that, although phosphorylation of proteins contributing to these processes may change in Δcdpk3 parasites, this is unlikely to directly relate to CDPK3- or SCE1-dependent egress signaling. This group of proteins was therefore not investigated further.
By contrast, proteins in the Δcdpk3-independent group showed no significant enrichment in GO terms relating to cell signaling (Fig. 5D). A slight enrichment in terms relating to protein and metabolite binding (“binding,” “nucleic acid binding,” “nucleotide binding,” “heterocyclic compound binding,” and “organic cyclic compound binding”) was seen, however. We also note an underrepresentation of membrane-associated proteins (“membrane,” “intrinsic component of membrane,” “integral component of membrane,” and “membrane part”); however, this may be a consequence of the general difficulty of solubilizing membrane proteins in proteomics.
We next interrogated our hits for phosphopeptides belonging to proteins that are known or predicted to be involved in apicomplexan motility and invasion signaling pathways. One phosphosite on each of the PDEs TGME49_202540 and TGME49_280410 appeared to be misregulated in Δcdpk3 and restored in Δcdpk3Δsce1, indicating that these may be crucial CDPK3- and SCE1-regulated sites in egress signaling (supplemental Table S4). In addition, the abundance of two phosphosites on the cAMP-dependent protein kinase (PKA) regulatory subunit shows regulation by CDPK3 and SCE1, indicating that such cyclic nucleotide signaling pathways may play a role in egress. Furthermore, the abundance of a single phosphopeptide of phospholipase C (PLC) was dependent on CDPK3 and SCE1. PLC has been implicated in regulating Ca2+ flux in Toxoplasma (6, 27). One phosphosite of the signaling kinase casein kinase 1 showed regulation by CDPK3 and SCE1. Casein kinase has been implicated in the phosphorylation of adhesin tails in P. falciparum (28). Interestingly, the uncharacterized Ca2+-dependent kinase CDPK2A showed several phosphosites regulated by CDPK3 that were independent of SCE1. CDPK2A has been implicated previously in egress, making it a very strong candidate going forward (24). Several hypothetical proteins also showed a high abundance of phosphosites regulated by CDPK3 and SCE1, such as TGME49_279100. For further discussion of identified CDPK3- and SCE1-dependent phosphopeptides see supplemental information. Together, these data highlight several cellular processes wherein phosphorylation is regulated by CDPK3 and indicate that SCE1 plays an indirect antagonistic role in regulating several of these, particularly those pertaining to cell signaling. This supports the hypothesis that CDPK3 and SCE1 regulate a common signaling pathway during Ca2+-stimulated egress.
Phosphorylation of SCE1 regulates Toxoplasma egress
During our interrogation of SILAC data, we noted that two phosphosites of SCE1 (Ser-641 and Ser-645) were consistently less abundant in Δcdpk3 (supplemental Table S4). Data from previous phosphoproteomic investigations of Toxoplasma egress also show lower levels of SCE1 phosphorylation in CDPK3-deficient parasites (24). Furthermore, recent experiments using the substrate capture technique suggested that SCE1 is likely to be a direct substrate of CDPK33 (supplemental Table S5 and Fig. 6A).
Figure 6.

Phosphorylation controls the activity of SCE1. A, schematic of SCE1 and position of identified phosphorylation sites. TM, transmembrane. B, localization of SCE1M1, SCE1M2, SCE1N1, and SCE1N2 and staining with α-HA and α-GAP45. C, Western blot of complemented lines and staining with α-HA and α-GAP45 as a loading control. D, SCE1-phosphomutant complementation egress assay, stimulated with A23187 for 3 min. Δsce1 (i), Δcdpk3 (ii), and Δcdpk3Δsce1 (iii) backgrounds were complemented with SCE1wt and two sets of phosphomimetic (SCE1M1, SCE1M2) or phospho-null (SCE1N1, SCE1N2) mutants. Error bars show mean ± S.D. *, p < 0.05; **, p < 0.001. All statistical tests were performed using two-way ANOVA and Tukey correction for multiple comparisons. The data were derived from three independent biological replicates.
To test the importance of phosphorylation in regulating SCE1 activity, we designed a set of complementation constructs expressing cDNA copies of SCE1 containing substitution mutations within detected phosphosites. Mutations were either phosphomimetic (Ser → Asp, SCE1M) or phospho-null (Ser → Ala, SCE1N constructs) (Fig. 6A). Constructs included one of two sets of mutations. Set 1 (i.e. M1 and N1) contained all mutations at phosphosites that were detected in a least one substrate capture experiment (supplemental Table S5). Set 2 (i.e. M2 and N2) contained only mutations at sites detected in more than one substrate capture experiment. Both sets 1 and 2 included mutations in Ser-641 and Ser-645 that were less abundant in global analyses (24) (supplemental Table S4).
To investigate the role of phosphorylation of SCE1 during egress, phosphosite mutants (and SCE1wt) were introduced ectopically into Δsce1, Δcdpk3, and Δcdpk3Δsce1 parasites (supplemental Table S1). Expression was confirmed by IFA, showing identical localization to SCE1wt (described above) (Fig. 6B). Therefore, phosphomutants did not affect the membrane localization of SCE1. Western blotting showed that the expression levels of the ectopic protein were comparable across all lines (Fig. 6C).
To assess the role of SCE1 phosphorylation during CDPK3-dependent egress, we assayed the response of phosphosite mutants to stimulation with 2 μm A23187 for 3 min. Expression of the phosphomimetic mutants in Δsce1 (parasite strains Δsce1:SCE1M1 and Δsce1:SCE1M2) did not affect A23187-stimulated egress relative to Δsce1:SCE1wt (Fig. 6Di). However, expression of phospho-null mutants (parasite strains Δsce1:SCE1N1 and Δsce1:SCE1N2) significantly inhibited Δsce1 parasite egress efficiency, with the percentage of parasitophorous vacuoles from which parasites egressed decreasing to ∼35% and 25% for Δsce1:SCE1N1 and Δsce1:SCE1N2, respectively. This suggests that phosphorylation of SCE1 is required for efficient egress even in the presence of functional CDPK3. Expression of wild-type or phosphomutant SCE1 in the Δcdpk3 background did not result in any changes in egress capacity (Fig. 6Dii). This suggests that mimicking phosphorylation of SCE1 alone cannot overcome loss of CDPK3 during egress.
Introduction of phosphomimetic SCE1 mutants into Δcdpk3Δsce1 (parasite strains Δcdpk3Δsce1:SCE1M1 and Δcdpk3Δsce1:SCE1M2) resulted in intermediate levels of egress (∼50%) (Fig. 6Diii). This suggests that mimicking constitutive phosphorylation of SCE1 partially replicates SCE1 knockout in Δcdpk3 parasites, in turn suggesting that suppression of egress by SCE1 is relieved by phosphorylation. In agreement with this, expression of phospho-null SCE1 mutants (parasite strains Δcdpk3Δsce1:SCE1N1 and Δcdpk3Δsce1:SCE1N2) completely suppressed Δcdpk3Δsce1 egress, with the strains having egress rates identical to that of Δcdpk3Δsce1:SCE1wt.
Overall, our data strongly suggests that phosphorylation of SCE1 is very important in relieving its suppressive activity during Ca2+-stimulated egress. It is interesting to note that, within all genetic backgrounds (Δsce1, Δcdpk3, and Δcdpk3Δsce1), expression of either phosphomimetic SCE1 (SCE1M1 and SCE1M2) or either phospho-null mutant (SCE1N1 and SCE1N2) yielded essentially the same egress phenotype. This demonstrates that the key phosphorylation sites that regulate SCE1 activity are captured in both groups of mutants (Ser-8, Ser-73, Ser-225, Ser-641, and Ser-645).
SCE1 does not affect cytosolic Ca2+ dynamics during egress
Our results suggest that SCE1 may regulate generic egress signaling events. We therefore wondered whether SCE1 may act as a Ca2+ transporter. Indeed, previous reports suggest that CDPK3 influences the resting cytosolic Ca2+ concentration ([Ca2+]cyt) (24) and, thus, that loss of SCE1 may restore this imbalance to promote egress. Although SCE1 appears to have no primary amino acid homology to Ca2+ transporters, the online structural homology prediction software iTasser suggests that SCE1 has greatest structural homology to transient receptor potential cation subfamily v (TRPV)-like channels (29). TRPV channels are typically found on the plasma membrane that typically transport cations such as Ca2+, Mg2+, and Na+ in response to a range of extracellular stimuli (30). To investigate the possibility that SCE1 could be a Ca2+ transporter or channel, we first measured [Ca2+]cyt in extracellular parasites suspended either in a Ca2+-free buffer or in buffers containing a range of free Ca2+ concentrations (Fig. 7A). In Ca2+-free buffer, we found that the [Ca2+]cyt of wild-type, Δcdpk3, Δsce1, and Δcdpk3Δsce1 parasites were all ∼40 nm. This value is similar to [Ca2+]cyt values obtained previously for extracellular Toxoplasma parasites in Ca2+-free solutions (31, 32). Upon exposing parasites to increasing extracellular Ca2+ concentrations, all strains were able to tightly regulate their [Ca2+]cyt, maintaining it below 140 nm up to extracellular Ca2+ concentrations ([Ca2+]o) of 1 mm (the highest concentration tested, Fig. 7A). No significant differences in [Ca2+]cyt were observed between any of the lines at any of the [Ca2+]o tested (p ≤ 0.4, one-way ANOVA). Our data suggest that neither SCE1 or CDPK3 are required to maintain a low [Ca2+]cyt in extracellular tachyzoites under the conditions of our experiments.
Figure 7.

SCE1 does not control resting concentration or amplitude of cytosolic Ca2+. A, the [Ca2+]cyt in extracellular WT parasites (gray), Δcdpk3 parasites (green), Δse1 parasites (blue), and Δcdpk3/Δse1 parasites (red) in the presence of varying [Ca2+]o values. Data are averaged from three to four independent experiments (performed on different days) for each parasite line. B, relative levels of cytosolic Ca2+ in intracellular parasites as measured by GCaMP6/mCherry ratio as described previously (25). C and D, the rate of rise in GCaMP6/mCherry fluorescence (C) and peak fluorescence level upon stimulation of egress with either A23187, BIPPO or saponin (D). Error bars show mean ± S.D.
It should be noted that a previous study reported that extracellular Δcdpk3 knockout parasites suspended in a Ca2+-free buffer had an elevated basal Ca2+ level compared with wild-type parasites, although these measurements were not calibrated (24). In the presence of [Ca2+]o, the values we obtained for [Ca2+]cyt were slightly higher in Δcdpk3 compared with the wild type. It is not clear why these differences are seen between our studies.
So far, we have only detected a phenotypic difference upon deletion of SE1 during egress, when tachyzoites are still intracellular. It therefore might be the case that SCE1 only functions when tachyzoites are intracellular. We therefore tested for differences in the levels of [Ca2+]cyt at this stage, when tachyzoites are egress-competent. To do this, we introduced a plasmid expressing the genetically encoded Ca2+ biosensor GCaMP6 into the uprt locus of wild-type, Δcdpk3, Δsce1, and Δcdpk3Δsce1 parasites (25). We observed negligible differences in resting [Ca2+]cyt between strains (Fig. 7B). Egress was then stimulated using A23187 or BIPPO or by semipermeabilization of host cells with saponin, and the subsequent rate of Ca2+ increase (Fig. 7C) and peak Ca2+ concentration (Fig. 7D) were monitored. We saw no significant difference in these parameters between any of the lines for any of these conditions. Overall, these data suggest that SCE1 does not have a role in maintaining [Ca2+]cyt while extracellular or intracellular and plays no role in Ca2+ levels during stimulated egress.
Discussion
Plant-like Ca2+-dependent protein kinases are important signal transducers that regulate motility, invasion, and host cell egress in apicomplexan parasites. We have shown previously that CDPK3 in Toxoplasma is critical for host cell egress (11), but how this kinase (or indeed any other CDPK) regulates egress remains unexplained. To uncover novel signaling proteins that participate in CDPK3-regulated egress, we designed a forward genetic screen to identify mutants that circumvent their dependence on CDPK3 during A23187-stimulated egress. Here we identified a novel regulator of Toxoplasma egress, the transmembrane protein SCE1. We demonstrated that SCE1 is a suppressor of Ca2+-dependent Toxoplasma host cell egress whose suppressive activity is relieved by phosphorylation. Furthermore, we demonstrated that SCE1 acts, through unknown mechanisms, to suppress the phosphorylation of an array of proteins predicted to be involved in CDPK3-dependent signal transduction pathways during egress.
Combining these findings with functional analysis of phosphorylation sites has allowed us to put together a model of CDPK3-dependent egress (Fig. 8). Previous work suggested that CDPK3 acts in an amplification loop to promote egress (30) (Fig. 8A, thicker arrows), and we propose here that CDPK3 also directly phosphorylates SCE1, relieving the suppressive activity of this protein, allowing for maximal stimulated host cell egress (Fig. 8A). The loss of CDPK3 results in the loss of phosphorylation of SCE1 and inhibition of egress. Under this condition, CDPK3/SCE1-independent egress still takes place, which is offset by the suppressive activity of SCE1 (Fig. 8B). Upon additional deletion of SCE1, the alternative egress pathway becomes more prominent, promoting greater but not full egress capacity (Fig. 8C). Prevention of SCE1 phosphorylation (by making phospho-null mutations) prevents relief of egress suppression even in the presence of CDPK3, thus dampening egress compared with the wild type (Fig. 8D). The suppressive activity of SCE1 can be somewhat overcome when phosphorylation events are mimicked with aspartic acids, resulting in increased egress compared with wild-type SCE1 (Fig. 8E).
Figure 8.
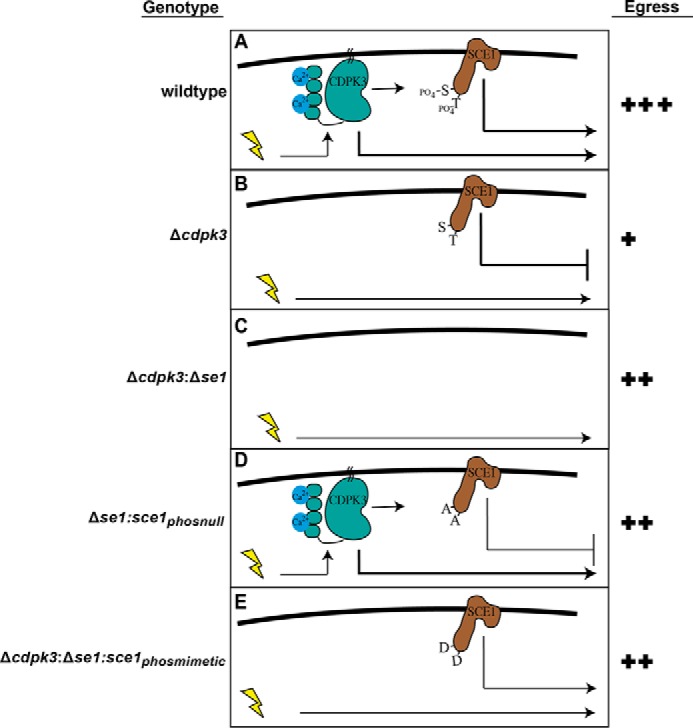
A hypothetical model of CDPK3- and SCE1-dependent egress. The yellow lightning bolt represents stimulation with A23187 or BIPPO. Arrows represent downstream events, including direct and indirect phosphorylation events, where the thickness of the line represents the hypothetical strength of the signal. A, model of CDPK3/SCE1-dependent egress in wild-type parasites. B, egress upon loss of CDPK3. C, egress upon deletion of both CDPK3 and SCE1. D, suppression of egress upon complementation of with the SCE1 phospho-null mutant. E, egress upon complementation of an SCE1 phosphomimetic mutant.
Interestingly, loss of SCE1 alone did not appear to result in premature egress of parasites, demonstrating that exogenous stimuli are still required to trigger egress in its absence. Our data demonstrate that egress signaling in Toxoplasma requires both activation of positive regulators and relief of suppression by negative regulators. Therefore, SCE1 knockout may, in fact, increase the speed and sensitivity of wild-type egress to exogenous stimuli. However, in our experiments, wild-type and Δsce1 egress are indistinguishable because wild-type egress is already so efficient in vitro, occurring just 1–2 min following A23187 or BIPPO treatment. In the future, it would be interesting to study the importance of SCE1 for the ability of Toxoplasma to infect cell types that are typically infected in vivo, such as macrophages. Such cells are more prone to immune pressure; thus, parasites within them may have to rapidly initiate egress earlier to escape targeting by the immune system.
SCE1 appears to localize to the tachyzoite plasma membrane, thus potentially co-localizing with CDPK3 and cGMP signaling proteins like PKG (5, 11). Our data presented here support a role for the plasma membrane as a key signaling hub for regulation of Ca2+ signaling and egress in Toxoplasma, with CDPK3 and SCE1 as key participants. We hypothesized that SCE1 may be a Ca2+ transporter or channel not easily identifiable by primary sequence homology; however, our data suggest that SCE1 does not significantly affect cytosolic Ca2+ levels before or during egress. Alternatively, SCE1 could suppress the activity of other signaling proteins or ion channels by blocking or sequestering them at the plasma membrane. In this model, phosphorylation of SCE1 could relieve its suppressive function, releasing sequestered proteins and thus promoting downstream signaling events. We attempted to identify interaction partners of SCE1 by using quantitative proteomics to identify proteins that differentially associate with SCE1 phospho-null versus phosphomimetic mutants, but these studies did not yield conclusive results (data not shown).
SCE1 is conserved in the closely related parasites N. caninum and H. hammondi but has no obvious homologs in other apicomplexan species. It is interesting to note that this group of closely related parasites are the only members of Apicomplexa to have the capacity to egress the host cell at any stage during intracellular growth. A checkpoint on egress, such as SCE1, may thus be essential to inhibit premature activation in this clade of apicomplexan parasites. By contrast, other apicomplexans utilize a range of asexual replication strategies (e.g. schizogony, endopolyogeny, and merogeny) wherein daughter cells are only formed after multiple rounds of nuclear division are completed. This places a developmental constraint on egress, meaning that signaling checkpoints may be largely unrequired.
Quantitative phosphoproteomics showed that a large portion of phosphosites misregulated in Δcdpk3 parasites were rescued to a wild type-like state in Δcdpk3Δsce1, supporting the notion that CDPK3 and SCE1 antagonistically regulate egress signaling events. These data also identified potentially important regulatory phosphosites on several proteins implicated in Ca2+-dependent signaling and egress. One phosphosite of the important Ca2+ flux regulator PLC (Ser-88) was observed to be down-regulated in Δcdpk3 parasites and rescued in Δcdpk3Δsce1 parasites, and it will be interesting to test the role of this phosphorylation site in Ca2+ signaling now that PLC mutants are available (6). Phosphosites on two PDEs and the regulatory subunit of PKA were observed to be down-regulated in Δcdpk3 parasites and rescued in Δcdpk3Δsce1 parasites, supporting a larger role for cyclic nucleotide kinases in egress. It is unknown whether these PDEs have cGMP hydrolysis activity or whether they are targeted by the known cGMP PDE inhibitors zaprinast and BIPPO. These data suggest that knocking out SCE1 may enhance egress by altering the levels of cyclic nucleotides in Toxoplasma, thereby increasing the activity of cyclic nucleotide-dependent kinases involved in egress, like PKG.
This study has shown that forward genetic screens using non-lethal knockout mutants with tightly defined phenotypes (such as Δcdpk3) can be a powerful means of unraveling signaling pathways in Toxoplasma and potentially other apicomplexan parasites. Moving forward, techniques such as this will be particularly useful to identify proteins such as SCE1 that display no phenotype when knocked out individually and are thus invisible by standard reverse genetics. As our appreciation for the complexity of pathogen cell signaling systems grows, these tools will become ever more useful, particularly when facilitated by whole-cell techniques such as quantitative phosphoproteomics and high-throughput genetic manipulation systems such as CRISPR/Cas9 (33, 34).
Experimental procedures
Parasite culture and transfection
Toxoplasma parasites were grown in human foreskin fibroblasts (HFFs) in D1 medium (DMEM supplemented with 1% FCS (Invitrogen) and 1% v/v Glutamax (Invitrogen)) at 37 °C with 10% CO2. Before parasite infection, HFFs were grown to confluency and maintained in DMEM supplemented with 10% Cosmic calf serum (Thermo Scientific). Transfection of parasites with DNA was performed by electroporation using a Bio-Rad Gene Pulser II module with conditions as described previously (35). Positive selection of recombinant parasites was performed using mycophenolic and xanthine, chloramphenicol, or phleomycin as described previously (35–37) or with 5′-fluo-2′-deoxyuridine-negative selection for integration at the uracil phosphoribosyl transferase locus (38).
DNA cloning
All sequence models were based on the Toxoplasma ME49 genome from ToxoDB.org (39). Oligonucleotide primers and synthesized gene fragments (gBlocks) were ordered from Integrated DNA Technologies. Assembly of DNA constructs was performed using the Gibson assembly cloning kit (New England Biolabs), according to the instructions of the manufacturer. Details regarding the construction of specific plasmids are presented in the supplemental information.
Forward genetic screen
Ten populations of Δcdpk3 parasites were mutagenized with EMS as described previously (40). After HFF lysis, ∼80,000 parasites from each population were added to fresh HFFs and allowed to invade for 2 h. Excess parasites were washed away three times with PBS. The medium was replaced with D1 supplemented with 25 mg/ml dextran sulfate (Sigma-Aldrich), and cultures were grown for 28 h. Flasks were washed twice with PBS, egress was stimulated for 2 min (4 μm A23187, 25 mg/ml dextran sulfate in DMEM-HEPES, 37 °C), and then cells were quenched with ice-cold PBS. Egressed parasites were washed off twice with ice-cold PBS; supernatant were removed and centrifuged to pellet parasites. Pellets were resuspended and added to fresh HFFs. These steps were repeated until an egress assay showed rescue of the Δcdpk3 egress-deficient phenotype. Clonal parasite lines were derived from rescue-of-egress populations using limited dilution. Details regarding sequencing of Δcdpk3rev mutants and egress assays are included in the supplemental information.
Cellular assays
Immunofluorescence assays (IFAs) were performed on 4% of formaldehyde-fixed samples using standard procedures. Images were taken with a DeltaVision Elite microscope (GE Healthcare) using a CoolSnap2 charged coupled device (CCD) detector and SoftWoRx software (GE Healthcare). Images were then processed and assembled using ImageJ, Adobe Photoshop, and Adobe Illustrator software. Extended methods are included in the supplemental information.
Phosphoproteomics of egress sample preparation
RH:Δku80, Δcdpk3, and Δsce1:Δcdpk3 parasites were grown on confluent HFFs in SILAC labeling medium for 1 week (DMEM without Arg or Lys (Gibco) with 1% dialyzed FCS, 1% Glutamax, 1% penicillin/streptomycin (Gibco) and supplemented with either (light) l-[U-12C6,14N4]arginine and l-[1H4]lysine, (medium) l-[U-13C6,14N4]arginine and l-[2H4]lysine, or (heavy) l-[U-13C6,15N4]arginine and l-[U-13C6,15N2]lysine (Cambridge Isotope Labs)). After complete HFF lysis, 7.5 × 107 labeled parasites of each strain were added to HFFs in 2 × 175 cm2 culture flasks each and allowed to invade in SILAC labeling medium for 2 h. Excess parasites were washed away with 3× PBS, the SILAC medium was replaced, and parasites were grown for a further 26 h. Egress stimulation medium was then added (2 μm A23187 in 5 ml of DMEM without Arg and Lys). After 1.5 min, egress was quenched by addition of 20 ml of ice-cold PBS, and the flasks were placed in an ice water bath for 1 min. Host cells and parasites were released from the surface of the culture flasks by cell scraping. Excess parasites and host cells were washed from the flasks using 5 ml of ice-cold PBS, and scraped cells and PBS wash were added to a 30-ml syringe and passed through a 27-gauge needle. Host cell debris was pelleted by centrifugation (600 × g, 2 min, 0 °C), the supernatant was transferred to a fresh centrifuge tube, and parasites were pelleted (1200 × g, 10 min, 0 °C). Parasite pellets were lysed immediately. More detailed methods for phosphoproteomic analysis are included in the supplemental information.
Ca2+ measurements with extracellular tachyzoites using Fura-2
Measurements of [Ca2+]cyt were performed with extracellular tachyzoites that had recently emerged from their host cells or that were harvested from their host cells by passage through a 26-gauge needle. The parasites were passed through a 3-μm filter to remove host cell material and then centrifuged (1500 × g for 10–20 min at room temperature). The supernatant medium was discarded, and the parasites were resuspended and then washed once (12,000 × g for 1 min) in a saline solution consisting of 125 mm NaCl, 5 mm KCl, 25 mm HEPES, 20 mm glucose, and 1 mm MgCl2 (pH 7.10). The parasites were resuspended in 2 ml of the saline solution and loaded with Fura-2 dye by incubation for 30 min at 37 °C in the presence of 6 μm Fura-2/AM and 0.04% pluronic acid F-127. The parasites were then washed twice (12,000 × g for 1 min) in saline solution to remove extracellular dye.
Fluorescence measurements were performed on Fura-2-loaded parasites (5–10 × 107) suspended either in Ca2+-free saline solution (identical to the saline solution described above but containing 1 mm EGTA) or in Ca2+-free saline solution to which sufficient CaCl2 was added to achieve a final free Ca2+ concentration of 1 μm, 100 μm, or 1 mm. The measurements were performed at 37 °C using a PerkinElmer Life Sciences LS 50B fluorescence spectrometer with a dual excitation Fast Filter accessory using excitation wavelengths of 340 nm and 380 nm and an emission wavelength of 515 nm. The measurements of [Ca2+]cyt were calibrated in situ (41) using a Kd of 120 nm for the Fura-2·Ca2+ complex.
For each external Ca2+ concentration, the [Ca2+]cyt value was determined from fluorescence measurements made within 40 min of the start of fluorescence recording (and within ∼60 min of having loaded the dye into the parasites). The order in which different [Ca2+]o values were tested varied between experiments.
Ca2+ measurements in intracellular tachyzoites and during egress
Intracellular Ca2+ measurements were undertaken by introducing GCaMP6 into the different parasite lines as described previously (25). This plasmid co-expresses mCherry to allow for internal normalization of the signal. Briefly, plasmids were integrated into the uprt locus by selecting parasites on fluorodeoxyribose. Relative Ca2+ levels were then measured on resting intracellular tachyzoites as well as on those stimulated to egress using either A23187, BIPPO, or saponin. Fluorescence intensities were then calculated using ImageJ, and ratios were calculated in an Excel spreadsheets and plotted using Prism software.
Author contributions
J.M.M., R.J.S., A.D.U., D.L., J.S., N.E.S., A.T.P., A.M.L., L.J.F., and C.J.T. designed the experiments and analyzed the data. J.M.M. and C.J.T. managed the project and wrote the manuscript. J.M.M. performed the forward genetic screen and, together with J.S. and A.T.P., undertook the next-generation sequencing and SNP detection. J.M.M. created parasite lines and, together with R.J.S., performed the phenotypic analysis of mutants. J.M.M., together with N.E.S. and L.J.F., performed the global quantitative phosphoproteomics. A.D.U. created phosphomutant lines and, together with J.M.M., performed phenotyping. Intracellular Ca2+ measurements using Fura-2 were performed by D.L. and A.M.L. Intracellular Ca2+ measurements by GCaMP were performed by R.J.S.
Supplementary Material
Acknowledgments
We thank Sebastian Lourido (Whitehead Institute) for sharing the unpublished CDPK3 thiophosphorylation data. We also thank Kelly Rogers and Lachlan Whitehead for help with live-cell imaging and analysis of the resulting data and the curators and administrators of ToxoDB.org, without which much of this project would not have been possible.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental text, Figure S1, and Tables S1–S5.
S. Lourido, Whitehead Institute, personal communication.
- CDPK
- Ca2+-dependent protein kinase
- EMS
- ethyl methane sulfonate
- IFA
- immunofluorescence assay
- ANOVA
- analysis of variance
- HFF
- human foreskin fibroblast
- PLC
- phospholipase C
- PDE
- phosphodiesterase
- BIPPO
- 5-benzyl-3-isopropyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one
- PKG
- protein kinase G
- SILAC
- stable isotope labeling with amino acids in cell culture
- GO
- gene ontology.
References
- 1. Tenter A. M., Heckeroth A. R., and Weiss L. M. (2000) Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30, 1217–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montoya J. G., and Liesenfeld O. (2004) Toxoplasmosis. Lancet 363, 1965–1976 [DOI] [PubMed] [Google Scholar]
- 3. Jones J., Lopez A., and Wilson M. (2003) Congenital toxoplasmosis. Am. Fam. Physician 67, 2131–2138 [PubMed] [Google Scholar]
- 4. Carruthers V. B., Giddings O. K., and Sibley L. D. (1999) Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell Microbiol. 1, 225–235 [DOI] [PubMed] [Google Scholar]
- 5. Donald R. G., Allocco J., Singh S. B., Nare B., Salowe S. P., Wiltsie J., and Liberator P. A. (2002) Toxoplasma gondii cyclic GMP-dependent kinase: chemotherapeutic targeting of an essential parasite protein kinase. Eukaryot. Cell 1, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bullen H. E., Jia Y., Yamaryo-Botté Y., Bisio H., Zhang O., Jemelin N. K., Marq J.-B., Carruthers V., Botté C. Y., and Soldati-Favre D. (2016) Phosphatidic acid-mediated signaling regulates microneme secretion in Toxoplasma. Cell Host Microbe 19, 349–360 [DOI] [PubMed] [Google Scholar]
- 7. Billker O., Lourido S., and Sibley L. D. (2009) Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe 5, 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lourido S., Shuman J., Zhang C., Shokat K. M., Hui R., and Sibley L. D. (2010) Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 465, 359–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garrison E., Treeck M., Ehret E., Butz H., Garbuz T., Oswald B. P., Settles M., Boothroyd J., and Arrizabalaga G. (2012) A forward genetic screen reveals that calcium-dependent protein kinase 3 regulates egress in Toxoplasma. PLoS Pathog. 8, e1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lourido S., Tang K., and Sibley L. D. (2012) Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J. 31, 4524–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCoy J. M., Whitehead L., van Dooren G. G., and Tonkin C. J. (2012) TgCDPK3 regulates calcium-dependent egress of Toxoplasma gondii from host cells. PLoS Pathog. 8, e1003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Azevedo M. F., Sanders P. R., Krejany E., Nie C. Q., Fu P., Bach L. A., Wunderlich G., Crabb B. S., and Gilson P. R. (2013) Inhibition of Plasmodium falciparum CDPK1 by conditional expression of its J-domain demonstrates a key role in schizont development. Biochem. J. 452, 433–441 [DOI] [PubMed] [Google Scholar]
- 13. Green J. L., Rees-Channer R. R., Howell S. A., Martin S. R., Knuepfer E., Taylor H. M., Grainger M., and Holder A. A. (2008) The motor complex of Plasmodium falciparum: phosphorylation by a calcium-dependent protein kinase. J. Biol. Chem. 283, 30980–30989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato N., Sakata T., Breton G., Le Roch K. G., Nagle A., Andersen C., Bursulaya B., Henson K., Johnson J., Kumar K. A., Marr F., Mason D., McNamara C., Plouffe D., Ramachandran V., et al. (2008) Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat. Chem. Biol. 4, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dvorin J. D., Martyn D. C., Patel S. D., Grimley J. S., Collins C. R., Hopp C. S., Bright A. T., Westenberger S., Winzeler E., Blackman M. J., Baker D. A., Wandless T. J., and Duraisingh M. T. (2010) A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science 328, 910–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siden-Kiamos I., Ecker A., Nybäck S., Louis C., Sinden R. E., and Billker O. (2006) Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Mol. Microbiol. 60, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishino T., Orito Y., Chinzei Y., and Yuda M. (2006) A calcium-dependent protein kinase regulates Plasmodium ookinete access to the midgut epithelial cell. Mol. Microbiol. 59, 1175–1184 [DOI] [PubMed] [Google Scholar]
- 18. Uboldi A. D., McCoy J. M., Blume M., Gerlic M., Ferguson D. J., Dagley L. F., Beahan C. T., Stapleton D. I., Gooley P. R., Bacic A., Masters S. L., Webb A. I., McConville M. J., and Tonkin C. J. (2015) Regulation of starch stores by a Ca2+-dependent protein kinase is essential for viable cyst development in Toxoplasma gondii. Cell Host Microbe 18, 670–681 [DOI] [PubMed] [Google Scholar]
- 19. Morlon-Guyot J., Berry L., Chen C.-T., Gubbels M.-J., Lebrun M., and Daher W. (2014) The Toxoplasma gondii calcium-dependent protein kinase 7 is involved in early steps of parasite division and is crucial for parasite survival. Cell Microbiol. 16, 95–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brochet M., Collins M. O., Smith T. K., Thompson E., Sebastian S., Volkmann K., Schwach F., Chappell L., Gomes A. R., Berriman M., Rayner J. C., Baker D. A., Choudhary J., and Billker O. (2014) Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol. 12, e1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Billker O., Dechamps S., Tewari R., Wenig G., Franke-Fayard B., and Brinkmann V. (2004) Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell 117, 503–514 [DOI] [PubMed] [Google Scholar]
- 22. Lourido S., Jeschke G. R., Turk B. E., and Sibley L. D. (2013) Exploiting the unique ATP-binding pocket of Toxoplasma calcium-dependent protein kinase 1 to identify its substrates. ACS Chem. Biol. 8, 1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaji R. Y., Johnson D. E., Treeck M., Wang M., Hudmon A., and Arrizabalaga G. (2015) Phosphorylation of a myosin motor by TgCDPK3 facilitates rapid initiation of motility during Toxoplasma gondii egress. PLoS Pathog. 11, e1005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Treeck M., Sanders J. L., Gaji R. Y., LaFavers K. A., Child M. A., Arrizabalaga G., Elias J. E., and Boothroyd J. C. (2014) The calcium-dependent protein kinase 3 of Toxoplasma influences basal calcium levels and functions beyond egress as revealed by quantitative phosphoproteome analysis. PLoS Pathog. 10, e1004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stewart R. J., Whitehead L., Nijagal B., Sleebs B. E., Lessene G., McConville M. J., Rogers K. L., and Tonkin C. J. (2016) Analysis of Ca2+ mediated signalling regulating Toxoplasma infectivity reveals complex relationships between key molecules. Cell Microbiol. 19, e12685. [DOI] [PubMed] [Google Scholar]
- 26. Sidik S. M., Hortua Triana M. A., Paul A. S., El Bakkouri M., Hackett C. G., Tran F., Westwood N. J., Hui R., Zuercher W. J., Duraisingh M. T., Moreno S. N., and Lourido S. (2016) Using a genetically encoded sensor to identify inhibitors of Toxoplasma gondii Ca2+ signaling. J. Biol. Chem. 291, 9566–9580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lovett J. L., Marchesini N., Moreno S. N., and Sibley L. D. (2002) Toxoplasma gondii microneme secretion involves intracellular Ca2+ release from inositol 1,4,5-triphosphate (IP3))/ryanodine-sensitive stores. J. Biol. Chem. 277, 25870–25876 [DOI] [PubMed] [Google Scholar]
- 28. Tham W. H., Lim N. T., Weiss G. E., Lopaticki S., Ansell B. R., Bird M., Lucet I., Dorin-Semblat D., Doerig C., Gilson P. R., Crabb B. S., and Cowman A. F. (2015) Plasmodium falciparum adhesins play an essential role in signalling and activation of invasion into human erythrocytes. PLoS Pathog. 11, e1005343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J.-L., Huang S.-Y., Li T.-T., Chen K., Ning H.-R., and Zhu X.-Q. (2016) Evaluation of the basic functions of six calcium-dependent protein kinases in Toxoplasma gondii using CRISPR-Cas9 system. Parasitol. Res. 115, 697–702 [DOI] [PubMed] [Google Scholar]
- 30. Ramsey I. S., Delling M., and Clapham D. E. (2006) An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647 [DOI] [PubMed] [Google Scholar]
- 31. Moreno S. N., and Zhong L. (1996) Acidocalcisomes in Toxoplasma gondii tachyzoites. Biochem. J. 313, 655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Borges-Pereira L., Budu A., McKnight C. A., Moore C. A., Vella S. A., Hortua Triana M. A., Liu J., Garcia C. R., Pace D. A., and Moreno S. N. (2015) Calcium signaling throughout the Toxoplasma gondii lytic cycle: a study using genetically encoded calcium indicators. J. Biol. Chem. 290, 26914–26926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sidik S. M., Hackett C. G., Tran F., Westwood N. J., and Lourido S. (2014) Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS ONE 9, e100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen B., Brown K. M., Lee T. D., and Sibley L. D. (2014) Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/Cas9. mBio 5, e01114–01114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soldati D., and Boothroyd J. C. (1993) Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science 260, 349–352 [DOI] [PubMed] [Google Scholar]
- 36. Asano T., Hakata M., Nakamura H., Aoki N., Komatsu S., Ichikawa H., Hirochika H., and Ohsugi R. (2011) Functional characterisation of OsCPK21, a calcium-dependent protein kinase that confers salt tolerance in rice. Plant Mol. Biol. 75, 179–191 [DOI] [PubMed] [Google Scholar]
- 37. Soldati D., Kim K., Kampmeier J., Dubremetz J. F., and Boothroyd J. C. (1995) Complementation of a Toxoplasma gondii ROP1 knock-out mutant using phleomycin selection. Mol. Biochem. Parasitol. 74, 87–97 [DOI] [PubMed] [Google Scholar]
- 38. Donald R. G., and Roos D. S. (1995) Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc. Nat. Acad. Sci. U.S.A. 92, 5749–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gajria B., Bahl A., Brestelli J., Dommer J., Fischer S., Gao X., Heiges M., Iodice J., Kissinger J. C., Mackey A. J., Pinney D. F., Roos D. S., Stoeckert C. J. Jr., Wang H., and Brunk B. P. (2008) ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 36, D553–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coleman B. I., and Gubbels M.-J. (2012) A genetic screen to isolate Toxoplasma gondii host-cell egress mutants. J. Vis. Exp. e3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grynkiewicz G., Poenie M., and Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 42. Howard B. L., Harvey K. L., Stewart R. J., Azevedo M. F., Crabb B. S., Jennings I. G., Sanders P. R., Manallack D. T., Thompson P. E., Tonkin C. J., and Gilson P. R. (2015) Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem. Biol. 10, 1145–1154 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.