

Abstract
Xylose fermentation is a rare trait that is immensely important to the cellulosic biofuel industry, and Candida tenuis is one of the few yeasts that has been reported with this trait. Here we report the isolation of two strains representing a candidate sister species to C. tenuis. Integrated analysis of genome sequence and physiology suggested the genetic basis of a number of traits, including variation between the novel species and C. tenuis in lactose metabolism due to the loss of genes encoding lactose permease and β-galactosidase in the former. Surprisingly, physiological characterization revealed that neither the type strain of C. tenuis nor this novel species fermented xylose in traditional assays. We reexamined three xylose-fermenting strains previously identified as C. tenuis and found that these strains belong to the genus Scheffersomyces and are not C. tenuis. We propose Yamadazyma laniorum f.a. sp. nov. to accommodate our new strains and designate its type strain as yHMH7 (=CBS 14780 = NRRL Y-63967T). Furthermore, we propose the transfer of Candida tenuis to the genus Yamadazyma as Yamadazyma tenuis comb. nov. This approach provides a roadmap for how integrated genome sequence and physiological analysis can yield insight into the mechanisms that generate yeast biodiversity.
Keywords: Yamadazyma, Candida tenuis, Scheffersomyces, novel species, genome sequence, xylose fermentation
Here we report the discovery, genome sequence, physiological characterization and taxonomic description of a new species of yeast that is sister to a species previously believed to ferment xylose.
INTRODUCTION
Bioethanol derived from lignocellulosic feedstocks has considerable environmental advantages when compared to starch-derived bioethanol (U.S. DOE 2006, 2015; Parreiras et al. 2014; Sato et al. 2016). Pretreatment of lignocellulosic feedstocks for ethanol production usually produces D-xylose as a major constituent, along with D-glucose. While a handful of yeast species can natively ferment xylose, including members of the genera Spathaspora and Scheffersomyces (Nguyen et al. 2006; Jeffries et al. 2007; Wohlbach et al. 2011; Hittinger et al. 2015; Riley et al. 2016), Saccharomyces cerevisiae is incapable of fermenting this pentose (Batt et al. 1986; Parreiras et al. 2014). Recombinant strains of S. cerevisiae can be engineered to ferment xylose by adding genes expressing three yeast enzymes: a xylose reductase (Xyl1), a xylitol dehydrogenase (Xyl2) and a xylulokinase (Xyl3) (Karhumaa et al. 2007; Sato et al. 2014; Konishi et al. 2015). These enzymes convert xylose to xylulose 5-phosphate, which enters the pentose phosphate pathway and is metabolized into ethanol.
Candida tenuis (Diddens and Lodder 1942) is a member of the Yamadazyma clade (Kurtzman 2011; Lachance et al. 2011) and has been extensively studied, in part due to its apparent ability to ferment the pentose sugar D-xylose (Toivola, Yarrow and Van Den Bosch 1984; Wohlbach et al. 2011). The putative xylose reductase of C. tenuis has been characterized structurally (Kavanagh et al. 2002, 2003) and functionally (Neuhauser et al. 1997, 1998; Nidetzky, Klimacek and Mayr 2001). This enzyme has even been altered into a NADH-specific aldose reductase (Petschacher and Nidetzky 2008) to begin to overcome the redox challenges inherent to the three-enzyme pathway used by yeasts to consume xylose (Jin, Laplaza and Jeffries 2004). In addition, the genome of the type strain of C. tenuis has been sequenced, and many of its genes have been implicated in xylose utilization using comparative genomic approaches, including several genes that significantly improved the utilization of xylose when engineered into S. cerevisiae (Wohlbach et al. 2011).
The genus Yamadazyma was described by Billon-Grand (1989) to accommodate 16 species that form hat-shaped ascospores, produce pseudohyphae and have coenzyme Q-9 as the main ubiquinone. In addition, they share the traits of fermenting sugars and requiring an external source of vitamins for growth (Kurtzman 2011). However, a phylogenetic analysis of the gene encoding the D1/D2 LSU rRNA revealed the polyphyletic nature of Yamadazyma (Kurtzman and Robnett 1998). For this reason, the genus was generally not accepted as originally proposed. Subsequent phylogenetic work by Kurtzman and Suzuki (2010) on species of Pichia that produce CoQ-9 resulted in the transfer of many species to the newly described genera Babjeviella, Meyerozyma, Millerozyma and Priceomyces. As a result, the revised genus Yamadazyma was well supported as a monophyletic clade of 17 species that included 11 asexual species that remain assigned to Candida. In the most recent edition of The Yeasts: A Taxonomic Study (Kurtzman, Fell, and Boekhout 2011), 6 sexual species and 23 asexual species, belonging to the genus Candida, were placed in the Yamadazyma clade with Yamadazyma philogaea as the type species (Kurtzman 2011). Since then, additional asexual species of this clade have been identified (Groenewald, Robert and Smith 2011), and three have already been transferred to Yamadazyma as novel combinations (Nagatsuka et al. 2016). Additionally, many novel species of Yamadazyma have been recently described from various substrates and environments (Ciafardini et al. 2013; Kaewwichian et al. 2013; Junyapate, Jindamorakot and Limtong 2014; Jindamorakot et al. 2015; Lopes et al. 2015; Wang et al. 2015; Burgaud et al., 2016; Khunnamwong and Limtong 2016; Nagatsuka et al. 2016). To date (February 2017), YeastIP (Weiss et al. 2013) lists 59 members of this clade, 18 species of the genus Yamadazyma and 41 asexual species still assigned to the genus Candida, making it one of the largest genera tentatively assigned to the family Debaryomycetaceae.
During an ongoing study of yeast biodiversity, we isolated two strains representing a novel xylose-assimilating species from two Wisconsin ecosystems. Analyses of their ITS and D1/D2 sequences suggest that these strains belong to the Yamadazyma clade and represent a species sister to C. tenuis. Sporulation was not observed for either isolate, and they are described here as the species Yamadazyma laniorum f.a. sp. nov. In addition, we identify three erroneously classified strains of C. tenuis as cryptic strains of Scheffersomyces and conclude that C. tenuis does not ferment the sugar xylose. Finally, we report a draft genome of sequence of Y. laniorum and correlate several physiological traits with genome content.
METHODS
Yeast isolation, identification and characterization
Strain yHMH7 was obtained from the bark of Acer saccharum (sugar maple) collected on the 23 May 2015 from the Baird Creek Nature Preserve (+44.500476, –87.923857), a 34.5-acre parcel of land containing areas of old growth forest within the city of Green Bay, Wisconsin, USA. Strain yHMH613 was obtained from the bark of A. saccharum collected on the 8 October 2016 from Wyalusing State Park, Wisconsin (+42.993546, –91.104548). Both strains were isolated at 30°C. Enrichment and isolation protocols were followed as previously described (Sylvester et al. 2015), except isolation was done using a 0.8% glucose enrichment medium.
Species identification was accomplished by sequencing the rDNA locus encoding the internal transcribed spacer (ITS) – 5.8S region and the D1/D2 variable domains of the large subunit of rRNA gene, using the primers ITS1 and ITS4 (McCullough et al. 1998) and NL1 and NL4 (Kurtzman and Robnett 1998), respectively. The methods of DNA extraction, amplification and sequencing were followed as described previously (Sylvester et al. 2015). All ITS and D1/D2 sequences generated in this study have been deposited in GenBank under the following accession numbers: KY588337–KY588341 (ITS) and KY588136–KY588140 (D1/D2).
Physiological characterization of strains yHMH7 and yHMH613 was conducted using standard methods described by Kurtzman et al. (2011). Carbon and nitrogen assimilation tests were performed using liquid media, and growth was observed for up to 4 weeks. Carbon fermentation was tested in a YP base media with 2% sugar, and Durham tubes were used to visualize carbon dioxide production. Growth at various temperatures (4°C, 10°C, 22°C, 30°C and 37°C) was assessed by streaking cells onto yeast extract peptone glucose (YPD) agar plates and incubating them for ∼2 weeks. Formation of true (septate) hyphae and pseudohyphae was investigated using the Dalmau plate method on both YPD and 5% malt extract agar plates. Ascospore formation was investigated by growing the two strains on 5% malt extract agar and YPD agar at 22°C for 2 months with periodic examination under the microscope. The two strains were also mixed and tested for mating and subsequent ascospore formation as described above.
Phylogenetic placement of the novel species was inferred using concatenated DNA sequences encoding the ITS and D1/D2 regions of the LSU rRNA (Groenewald, Robert and Smith 2011), after removing the oligonucleotide sequences used during PCR. Sequences for species in the Yamadazyma clade were obtained using the online tool YeastIP (Weiss et al. 2013) and aligned using MAFFT v.7.273 (Katoh and Standley 2014) with the G-INS-I algorithm; all ambiguous sequence positions were removed. Maximum-likelihood (ML) phylogenetic inference was performed using the program RAxML v.8.2.4 (Stamatakis 2014) with a partitioned multilocus (ITS and D1/D2) matrix under a general time reversible model with a gamma distribution of site rate variation (GTRGAMMA). ML support was estimated by rapid bootstrap analysis with 500 replicates.
Genome sequencing, assembly, annotation and phylogenetics
Genomic DNA (gDNA) was isolated from strain yHMH7. gDNA was sonicated and ligated to Illumina sequencing adaptors as previously described (Hittinger et al. 2010). The paired-end library was sequenced on an Illumina HiSeq 2500 instrument, conducting a Rapid 2 × 250 run.
To generate whole genome assemblies, paired-end Illumina reads were used as input to a meta-assembler pipeline iWGS (Zhou et al. 2016). Briefly, this pipeline performed quality-based read trimming, followed by k-mer length optimization, and used a range of state-of-the-art assemblers to generate several genome sequence assemblies. The quality of the assemblies was assessed using QUAST v3.1 (Gurevich et al. 2013), and the best assembly for the newly described species was chosen based on N50 statistics. This genome was assembled with SPAdes (Bankevich et al. 2012) to a total of 92 scaffolds (>1000 bp), with a total length of 10.9 Mb, and a N50 value of 800 Kb. Raw sequencing data were deposited in GenBank under Bioproject ID PRJNA374875.
Phylogenomic placement of the newly described species was assessed using an approach previously described in Lopes et al. (2016). Briefly, BUSCO v2 (Simão et al. 2015) was used to identify 534 conserved single-copy orthologs in the best genome assembly of the newly described species and 32 published genome assemblies of species closely related to the families Debaryomycetaceae and Metschnikowiaceae, as well as in three outgroup Saccharomycetaceae species (Hittinger et al. 2015; Shen et al.2016). Then, ML phylogenies were generated for each conserved gene, as well as for a superalignment of all genes concatenated together (346 412 sites). Finally, individual gene trees were used to calculate internode certainty scores for each branch of the ML tree, as proposed by Salichos and Rokas (2013) and implemented in RAxML 8.2.4 (Stamatakis 2014).
Assigning genes to enzyme functions
Predicted proteins were identified and assigned a function using BLAST homology methods as previously described (Riley et al.2016). TBLASTX and BLASTN searches were made using query sequences from the characterized pathways in model organisms (e.g. Saccharomyces cerevisiae) versus the genome assemblies, using an e-value threshold of 10−10 to assign copy number of each gene. This e-value threshold was relaxed to 10−1 for the fast-evolving mating-type idiomorphs. The genome was annotated using the MAKER pipeline (Holt and Yandell 2011), which identified 5744 predicted genes. Genome synteny comparisons were performed using the program MUMmer version 3.0 (Kurtz et al., 2004). Alignments were made using the PROmer script.
RESULTS AND DISCUSSION
Species delineation, classification and ecology
Phylogenetic analyses of ITS and D1/D2 sequences placed strains yHMH7 and yHMH613 as members of the Yamadazyma clade, forming a sister pair with Candida tenuis (Fig. 1). Strain yHMH7 differs by twelve substitutions and eight indels in the ITS region and four substitutions and one indel in the D1/D2 region, which we interpret as evidence that it represents a species distinct from C. tenuis. Strain yHMH613 differs by twelve substitutions and eight indels in the ITS region and five substitutions and one indel in the D1/D2 region. Despite one substitution between strains yHMH7 and yHMH613, the two strains formed a highly supported sister group with C. tenuis. The monophyly of the Yamadazyma clade was well supported when both ITS and D1/D2 regions were analyzed.
Figure 1.

ML phylogenetic inference indicating novel species phylogenetic placement among other species of the Yamadazyma clade. The monophyly of Yamadazyma is supported based on a partitioned ITS and D1/D2 alignment matrix. The alignment contained 1005 positions. Bootstrap values of 50 and greater are shown. Strain numbers are followed by GenBank accession numbers for the ITS and D1/D2 regions, and all represent type strains for the species, except for yHMH613.
Strains were tested for sporulation individually and in pairs, and neither strain produced ascospores. We examined the mating-type locus of Yamadazyma laniorum (strain yHMH7T) by BLASTing the two mating type idiomorphs of C. albicans (Hull and Johnson 1999) to the genome of Y. laniorum. We were able to identify only genes corresponding to a MATα idiomorph, which is syntenic with C. tenuis, Spathaspora passalidarum and Suhomyces tanzawaensis, suggesting a similar heterothallic mating-type locus organization (Riley et al. 2016). We searched for genes implicated in mating in C. albicans (Johnson 2003) and found that homologs of STE2, STE6, STE7, STE12, STE20, FUS3, KEX2 and KSS1 (names of Saccharomyces cerevisiae homologs given) were all present and lacked obvious inactivating mutations in Y. laniorum. Since attempted mating of the two strains failed, strain yHMH613 may also be MATα. Alternatively, the species may mate in specialized conditions or may be asexual for genetic reasons yet to be discovered. Regardless of whether the species is sexual or not, its phylogenetic placement within the Yamadazyma clade is well supported. Thus, the epithet Y. laniorum f.a. sp. nov. is proposed to accommodate the strains yHMH7 and yHMH613. The addition of f.a. or forma asexualis is added to indicate that the sexual state is unknown (Lachance 2012).
Both strains were isolated from the bark of the common Wisconsin tree Acer saccharum (sugar maple), possibly suggesting an association with this substrate. However, the presence of Y. laniorum on the bark of A. saccharum may be due to movement by an unknown vector. In fact, C. tenuis has been repeatedly isolated from tree-invading beetles and other insects (Kurtzman et al. 2011). Given the close relationship between C. tenuis and Y. laniorum, they may occupy a similar ecological niche. Several species in the Yamadazyma clade have been isolated from plant matter (Nakase et al. 2008; Groenewald, Robert and Smith 2011; Kaewwichian et al. 2013; Junyapate, Jindamorakot and Limtong 2014; Jindamorakot et al. 2015; Lopes et al. 2015), and many show associations with insects (Kurtzman et al. 2011). Thus, it appears that these are common habitats for many Yamadazyma species. A notable exception is the phylogenetic clade of C. atmosphaerica, C. spencermartinsiae, C. atlantica, C. oceani, C. taylorii and Y. barbieri, which has been isolated from various marine ecosystems and deep-sea hydrothermal vents (Burgaud et al. 2011, 2016). Considering the low internode support within the Yamadazyma clade and the diverse set of defined phenotypes and ecologies, it is likely the Yamadazyma clade will be revised in the near future, perhaps with genome-scale phylogenies. For this reason, we defer the transfer of the many affiliated Candida species to Yamadazyma. We note that Y. laniorum and C. tenuis are both closely related to the type species of the genus, Y. philogaea (Fig. 1).
Phylogenomic analyses
To understand the broader relationship of Y. laniorum with related species and other yeast genera, we took a phylogenomic approach. We compared the genome of the novel species to 32 publicly available yeast species genomes (Hittinger et al.2015; Shen et al.2016). We identified 534 conserved, single-copy orthologous genes for ML phylogenetic analyses (Fig. 2). We also calculated internode certainty (IC) support values of each branch to test the robustness of our ML approach. Unfortunately, only one genome in the Yamadazyma clade, C. tenuis, has been published to date (Wohlbach et al. 2011), so our analyses were unable to further resolve the relationships among species within the Yamadazyma clade. We note that several genome-scale analyses have now found C. tenuis to be an outgroup to the Debaryomycetaceae and Metschnikowiaceae species with sequenced genomes, suggesting the tentative assignment of the genus Yamadazyma to the family Debaryomycetaceae may be premature (Riley et al.2016; Shen et al.2016). Regardless, our analyses showed that Y. laniorum lies firmly within the Yamadazyma clade, is a member of the CUG-Ser clade that uses an alternative genetic code and is strongly supported as a distinct phylogenetic species from C. tenuis. The related taxonomic groups, Candida/Lodderomyces, Spathaspora and Metschnikowia, were resolved to their accepted taxonomy (Lachance, Hurtado and Hsiang 2016; Riley et al.2016). Deep phylogenetic relationships between some taxa remained difficult to resolve, even with genome-level analyses and increased taxon sampling (Shen et al. 2016). These nodes may require improved datasets or analytical techniques, or they may be due to true polytomies, reticulation or incomplete lineage sorting.
Figure 2.

Phylogenetic placement of Y. laniorum obtained from a concatenated alignment of 534 conserved, single-copy orthologous genes from the newly described species, 32 closely related Debaryomycetaceae or Metschnikowiaceae species with publicly available genome sequence data and Saccharomycetaceae species as the outgroup. Values above branches indicate their IC support, obtained based on 534 individual gene trees. IC values close to 1 indicate the absence of conflict between genes for a given branch among the gene trees, IC values close to zero indicate equal support for a given branch and the second most-frequent alternative, and IC values close to –1 indicate lower support for a given branch as compared to its second most-frequent alternative.
While the ITS and D1/D2 sequences showed limited sequence divergence between Y. laniorum and C. tenuis, the genome sequences revealed substantial sequence divergence between the species. When compared to the well-defined species pair C. albicans and C. dubliniensis, Y. laniorum and C. tenuis are nearly three times more divergent at the genome sequence level, further supporting their classification as separate species (Fig. 2). As expected, the genomes of Y. laniorum and C. tenuis are largely syntenic, supporting their close relationship (Fig. 3A, Fig. S1a, Supporting Information). Nonetheless, we observed numerous translocations and inversions between the two species. When we compared the Y. laniorum genome to more distantly related yeast species, synteny quickly dissipated (see Fig. S1b–d, Supporting Information).
Figure 3.

Synteny analysis between C. tenuis and Y. laniorum (A). Scaffolds are labeled on the X and Y axis, and a * indicates the reverse complement of the scaffold is used in the alignment. The zoomed-in region (100 Kb) shows the GALactose cluster and flanking regions (B). Numbers indicate genes of interest, (1) GAL1, (2) GAL10, (3) GAL7, (4) ORF-Y, (5) ORF-X, (6) K1F, (7) the gene encoding a putative lactose permease. Colored regions indicate regions of homology, while black colored ORFs indicate non-homologous intervening genes.
Correlation of metabolic traits and genome content
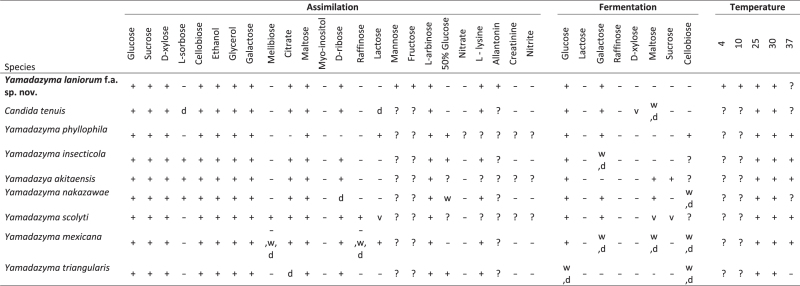
In the era of genomics, the traditional physiological traits scored for species diagnosis (Kurtzman et al. 2011) now afford an exciting opportunity to understand the genetic mechanisms that generate yeast biodiversity (Hittinger et al.2015). In addition to characterizing Y. laniorum physiology using traditional assays (Table 1), we sought to determine how some of these traits are genetically encoded in its genome. Thus, we compared the genome content and metabolic capabilities of Y. laniorum to related species (Fig. 4). Generally, genome content and metabolic capabilities were congruent: if the required genes were present in the genome, the species could utilize the substrate; otherwise, the species could not.
Table 1.
Physiological characteristics of Y. laniorum that differentiates it from closely related species.
|
Figure 4.

Metabolic traits and their underlying genes for the sugars galactose, D-xylose, L-sorbose, maltose, raffinose, sucrose, melibiose, lactose and cellobiose and for the nitrogen source allantoin. Numbered boxes indicate the copy number of each gene determined by BLAST analyses. Yellow branches correspond to members of Debaryomycetaceae and blue to members of Saccharomycetaceae.
For the substrates galactose, xylose, maltose, cellobiose, lactose and allantoin, we found that the gene content of Y. laniorum is consistent with its observed growth phenotype (Fig. 4). In S. cerevisiae, galactose metabolism is carried out by three enzymes encoded by the genes GAL1 (galactokinase), GAL7 (galactose-1-phosphate uridyl-transferase) and GAL10 (UDP-glucose-4-epimerase), which convert D-galactose to glucose-1-phosphate, which then enters glycolysis after conversion by the phosphoglucomutases encoded by PGM1 and PGM2 (Johnston 1987). We find that Y. laniorum has one copy of each gene encoding the above enzymes, which is consistent with its ability to both assimilate and ferment the sugar. Consistent with its growth on xylose, we find that Y. laniorum has one copy of the XYL1 and XYL3 genes and two copies of the XYL2 gene, which is the same as C. tenuis (Fig. 4). Maltose is a disaccharide composed of two glucose moieties linked via an α-1,4 bond, which is cleaved by α-glucosidases, which are encoded by MAL12 and paralogs in S. cerevisiae (Charron, Dubin and Michels 1986). Consistent with its positive growth on maltose, we find that Y. laniorum has two copies of genes encoding α-glucosidases (Fig. 4). Cellobiose is another disaccharide composed of two glucose moieties, but it is connected via a β-1,4 bond. Yamadazyma laniorum has three copies of genes encoding β-glucosidases (BGL), which are required to cleave cellobiose and enable its assimilation, which is consistent with its observed positive growth on cellobiose (Fig. 4).
Yamadazyma laniorum is unable to utilize the sugar lactose, a β-1,4 disaccharide of glucose and galactose, while C. tenuis is able to do so. In the yeast Kluyveromyces lactis, LAC4 encodes a β-galactosidase (Sheetz and Dickson 1981) and LAC12 encodes a lactose permease (Chang and Dickson 1988); both are required to metabolize the sugar. We searched the genomes of Y. laniorum and C. tenuis for LAC4 and LAC12 homologs and found that Y. laniorum possesses neither a homolog of LAC4 nor LAC12, while C. tenuis possesses a clear homolog of each. The putative C. tenuis LAC4 homolog is located on the same scaffold as the GAL cluster, but nearly 400 kb upstream of GAL7. Interestingly, the putative C. tenuis LAC12 homolog is located only ∼30 kb upstream of GAL7 (Fig. 3), a novel linkage pattern not previously noted in other yeasts. Thus, the difference in lactose metabolism between the two species is defined at the genetic level by the loss of the genes encoding both the β-galactosidase (LAC4) and lactose permease (LAC12) in Y. laniorum. The loss of these two genes in Y. laniorum therefore occurred sometime after it and C. tenuis diverged, perhaps signifying a recent ecological shift in Y. laniorum (Hittinger, Rokas and Carroll 2004). We note that the ability to utilize lactose is variable across the genus Yamadazyma (Kurtzman et al. 2011), suggesting that there may have been several independent loses of lactose metabolism across the genus.
In S. cerevisiae, the genes involved in allantoin utilization are organized into a large cluster, while in C. albicans they are not clustered (Wong and Wolfe 2005; Naseeb and Delneri 2012). Based on the relatively close phylogenetic relationship to C. albicans, we expected these genes to be unclustered in Y. laniorum. As expected, we identified unclustered homologs of all enzymatic genes required for allantoin degradation, consistent with the observed phenotype of Y. laniorum.
The gene content of Y. laniorum did not correlate with the observed growth phenotype on the sugars L-sorbose, meliobiose and sucrose. One of the diagnostic characteristics that separates Y. laniorum from C. tenuis is its inability to utilize the sugar L-sorbose. In the yeast C. albicans, the gene SOU1, encoding a sorbose reductase, is required for the assimilation of L-sorbose (Greenberg et al. 2005). While Y. laniorum appears to have three apparent homologs of the SOU1 gene, it is unable to grow on the sugar. This discordance between genome content and phenotype was also observed in the species Sp. passalidarum and Su. tanzawaensis (Riley et al. 2016), suggesting another mechanism beyond simple gene loss confers the observed phenotype. Possibilities include changes in L-sorbose transportation, variations in enzyme kinetics, changes in gene regulation or an undetermined pathway of sugar utilization. In S. cerevisiae, SUC2 encodes an invertase, which hydrolyzes the sugar sucrose, a β-1,2 disaccharide of glucose and fructose (Carlson, Osmond and Botstein 1981). We found that, although Y. laniorum is able to utilize the sugar sucrose, it does not have any identifiable homolog of the SUC2 gene. The ability to utilize sucrose in the absence of an identifiable SUC2 homolog is a widespread but unexplained feature in CUG-Ser clade of yeasts (Riley et al.2016). Conversely, the ability to utilize the sugar melibiose is infrequent among CUG-Ser yeasts. Melibiose is formed by an α-1,6 linkage between galactose and glucose, which is broken down into its component saccharides by an α-galactosidase, such as the enzyme encoded by MEL1 (Sumner-Smith et al.1985). Surprisingly, we found that, although both Y. laniorum and C. tenuis have identifiable homologs of the MEL1 gene, they were incapable of growth on the sugar. These results suggest that the MEL1 homologs may perform a different function or may be insufficiently induced to promote melibiose assimilation. We conclude that many yeast metabolic traits can be successfully predicted from genome sequence, while other traits remain a challenge.
GALactose cluster analysis
Galactose catabolism is a well-characterized pathway in S. cerevisiae (Johnston 1987), S. kudriavzevii (Hittinger et al. 2010), S. uvarum (Kuang et al.2016) and C. albicans (Dalal et al.2016). The ability to utilize galactose is highly variable across yeast species and seems to be driven mainly by gene loss from an ancestor that could consume the sugar (Hittinger, Rokas and Carroll 2004; Slot and Rokas 2010; Riley et al. 2016). In the subphylum Saccharomycotina, the GAL genes are physically linked in a gene cluster. In S. cereviseae and related species, the cluster is comprised of three genes: GAL7, GAL10, and GAL1; GAL7 is downstream of GAL10 and they have the same gene orientation, while GAL1 is upstream of GAL10 and they are divergently transcribed. In C. albicans, the GAL cluster is nearly identical, but there are two intervening genes, ORF-Y and ORF-X, between GAL7 and GAL10. ORF-Y encodes a dTDP-glucose 4,6-dehydratase-like enzyme, and ORF-X encodes one of the subunits of the transport protein particle complex of the cis-Golgi. We expected both Y. laniorum and C. tenuis to share the GAL cluster configuration of C. albicans. Instead, we found a novel arrangement of the GAL cluster in these species: the orientations of GAL1 and GAL10 are the same as C. albicans; however, GAL7 and GAL10 have opposite gene orientations and are convergently transcribed, which places the three GAL genes in an uninterrupted cluster (Fig. 3B). In addition, the gene K1F, which is immediately downstream of GAL1 in C. albicans, is around 40 kb downstream of GAL1 in Y. laniorum and around 20 kb upstream of GAL1 in C. tenuis.
The flanking region of the GAL cluster in Y. laniorum and C. tenuis is nearly syntenic for about 300 kb, but we observed an inversion involving the GAL genes between C. tenuis and Y. laniorum (Fig. 3B). The boundaries of the inversion are ORF-X and a gene encoding a hypothetical protein (C. tenuis accession: CANTEDRAFT_115058). In C. tenuis, the gene encoding the hypothetical protein appears to have been truncated and likely inactivated, perhaps by the inversion itself, suggesting that the inversion occurred in the lineage leading to C. tenuis, rather than Y. laniorum. Inversions within this genomic region could be one mechanism by which allelic variants in this gene cluster are maintained across evolutionary timescales because inversions suppress recombination. Altogether, these findings offer a more complex and complete view of the evolution of the GAL gene cluster and suggest that multiple inversions have affected this region.
Xylose reductase (XR) analysis and identification of cryptic strains of Scheffersomyces
The purported ability of C. tenuis to ferment xylose was first demonstrated in 1984 in a screen for xylose-fermenting yeasts (Toivola, Yarrow and Van Den Bosch 1984). Three strains of C. tenuis were scored as xylose fermenters (CBS 4113, CBS 4285 and CBS 4435), while the type strain, CBS 615T, produced no gas in Durham tubes and produced very little ethanol from xylose. Candida tenuis CBS 615T was also recently tested for xylose fermentation using gas chromatography, and only minimal ethanol production was observed aerobically, while no ethanol was produced anaerobically (Wohlbach et al. 2011). To confirm the xylose fermentation phenotypes of CBS 615, 4113, 4285 and 4345, we performed fermentation tests in 2% xylose using Durham tubes. As predicted, CBS 615 did not produce gas, while CBS 4113, 4285 and 4435 partially filled the Durham tubes with gas (see Table S1, Supporting Information), confirming xylose fermentation.
When we compared the xylose reductase (Xyl1) amino acid (AA) sequences of CBS 615 and CBS 4435 to other yeast species, we were surprised to discover that the Xyl1 of CBS 4435 was most similar to the Xyl1 of Scheffersomyces shehatae (see Fig. S2, Supporting Information). To more closely analyze the relationships of the Xyl1 of Y. laniorum, C. tenuis and other related yeasts, we inferred their Xyl1 phylogeny (Fig. 5). These analyses conclusively placed CBS 4435 in the Scheffersomyces clade, suggesting that CBS 4435 is a cryptic strain of Scheffersomyces. While the genes encoding the Xyl1 proteins of CBS 4113 and 4285 have not been sequenced, their phenotypic similarity to CBS 4435 prompted us to determine the sequence of their ITS and D1/D2 regions (see Table S2, Supporting Information). All three strains were closely related, but not identical, to Sch. shehatae, Sch. insectosa and Sch. lignosus, indicating these xylose-fermenting strains were previously misidentified and are strains of Scheffersomyces, rather than C. tenuis.
Figure 5.

ML phylogenetic inference of the amino acid sequence of the XYL1 gene. ML phylogenetic inference was performed using the program RAxML under a general time reversible protein substitution model with GAMMA model of rate heterogeneity (PROTGAMMAGTR). ML support was estimated by rapid bootstrap analysis with 500 replicates. Sequences of interest are in bold font.
While the ITS and D1/D2 regions are often sufficient for identification at the species level, a multilocus approach has been used in the Scheffersomyces clade to delineate species more finely (Urbina and Blackwell 2012; Urbina et al.2013; Suh et al.2013; Ren et al.2014; Liu et al.2016). In particular, the sequence of XYL1 has been used to recognize previously cryptic species within the Scheffersomyces clade (Urbina and Blackwell 2012; Urbina et al.2013; Suh et al.2013; Ren et al.2014), and many more cryptic species likely remain in this and other xylose-fermenting clades (Urbina, Schuster and Blackwell 2013). The XYL1 gene typically differs among members of the genus Scheffersomyces by 5%–12% sequence divergence. The XYL1 gene of strain CBS 4435 differs from that of its closest described relative, Sch. shehatae, by 11% (36 substitutions and 1 gap). Thus, the strain CBS 4435 and possibly the strains CBS 4113 and CBS 4285 may eventually be recognized as one or more novel species. Given how closely related these strains are, we are reluctant to propose species names and diagnoses in the absence of genome sequence data, but we suggest their reevaluation in the near future.
Given that the only three putative strains of C. tenuis reported to ferment xylose (Toivola, Yarrow and Van Den Bosch 1984) are now identified as members of the genus Scheffersomyces, we conclude that C. tenuis should no longer be recognized as a D-xylose-fermenting yeast and that the genus Yamadazyma is likely not a clade of xylose-fermenting yeasts. Furthermore, these newly identified cryptic strains of Scheffersomyces refine the germplasm base of xylose-fermenting yeasts and substantially clarify the phylogenetic distribution of this rare metabolic trait.
Description of Yamadazyma laniorum Haase, Kominek, Langdon, Kurtzman, & Hittinger sp. nov.
Yamadazyma laniorum (lan.i.or.um gen. plural. n.) pertaining to butchers, in honor of the hardworking people of the historic meat packing industry of Green Bay, Wisconsin, USA, the location where it was first isolated from the bark of Acer saccharum.
After 3 days in liquid YPD at 30°C (Fig. 6), cells are ovoid to elongate (1.4–3 μm × 1.1–7 μm) and occur singularly or in pairs with the formation of pseudohyphae; budding is multilateral; sediment is formed. After 2 weeks on YPD agar, colonies are white, cream, butyrous, smooth and glistening; the margin is entire. Formation of true hyphae is not observed. Ascospore formation is not observed on either YPD or 5% YM after 2 months. Yamadazyma laniorum differs from Candida tenuis in the assimilation of L-sorbose and lactose and in the fermentation of maltose. Additional growth characteristics are reported in Table 1. The type strain yHMH7 (=CBS 14780 = NRRL Y-63967T) is cryopreserved as a glycerol stock at the University of Wisconsin-Madison, Madison, WI, USA. The species can be further diagnosed by its whole-genome shotgun sequencing project, which has been deposited at DDBJ/ENA/GenBank under the accession MVNT00000000. The version described in this paper is version MVNT01000000. The MycoBank no. is 819969.
Figure 6.
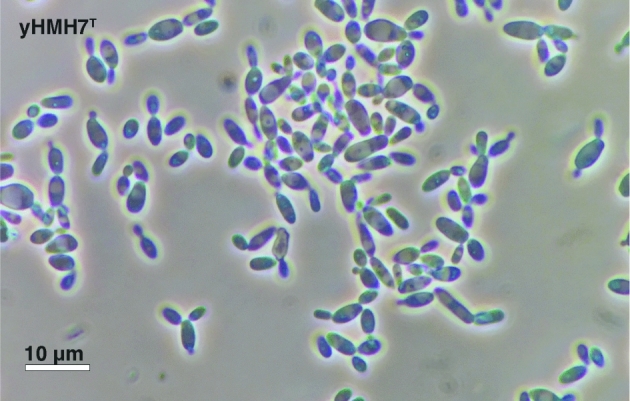
Budding cells of Y. laniorum f.a. sp. nov. after 3 day growth in YPD liquid media at room temperature (∼22°C).
Description of Yamadazyma tenuis (Diddens & Lodder) Haase, Kominek, Langdon, Kurtzman, & Hittinger comb. nov.
Basionym
Candida tenuis Diddens & Lodder, Die anaskosporogenen Hefen, II Hälfte: 488 (1942); MycoBank no. MB284782
Type strain
CBS 615T (=ATCC 10573T = CCRC 21748T = DBVPG 6159T = IFO 10315T = JCM 9827T = NRRL Y-1498T = VKM Y-70T)
MycoBank no.
819970
CONCLUSIONS
Yamadazyma laniorum has been placed within the genus Yamadazyma, along with its sister species, Y. tenuis. Its genome sequence also revealed the underlying genetic basis of multiple physiological traits. The inability of Y. laniorum to metabolize the sugar lactose is one of the diagnostic differences between Y. laniorum and Y. tenuis, a difference that is likely due to the loss of two genes encoding lactose permeases and β-galactosidases, respectively, in Y. laniorum. Analysis of the GALactose cluster in Y. laniorum led to the identification of a novel cluster configuration and an inversion between Y. laniorum and Y. tenuis. Finally, we identified three Scheffersomyces strains that were originally identified as strains of Y. tenuis and showed that Y. tenuis is not a xylose-fermenting species. These results clarify the phylogenetic distribution of xylose fermentation and more accurately and narrowly circumscribe the germplasm base that encodes this unusual trait. We conclude that genomics offers a powerful tool to describe new yeast species, while simultaneously illuminating the genetic mechanisms that generate yeast biodiversity and their diverse metabolisms.
Supplementary Material
Supplementary data are available at FEMSYR online.
Acknowledgments
We thank Jim McKeown for critical input on the correct Latin grammar of the novel species epithet, Thomas W. Jeffries for strain yHCT166 (=NRRL Y-27907) of Spathaspora passalidarum, Dana A. Opulente and EmilyClare Baker for preparing Illumina sequencing libraries, Lucigen Corporation (Middleton, WI) for use of their Covaris for gDNA sonication, and the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing Illumina and Sanger sequencing facilities and services.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSYR online.
FUNDING
This material is based upon work supported by the National Science Foundation under Grant No. DEB-1442148 to CTH and CPK, a National Science Foundation Graduate Research Fellowship under Grant No. DGE-1256259 to QKL, by USDA National Institute of Food and Agriculture Hatch Project 1003258 to CTH, and funded in part by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494 to Timothy J. Donohue). QKL was also supported by the Predoctoral Training Program in Genetics, funded by the National Institutes of Health (5 T32 GM007133-40). CTH is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Conflict of interest. None declared.
REFERENCES
- Bankevich A, Nurk S, Antipov D et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt CA, Caryallo S, Easson DD et al. Direct evidence for a xylose metabolic pathway in Saccharomyces cerevisiae. Biotechnol Bioeng 1986;28:549–53. [DOI] [PubMed] [Google Scholar]
- Billon-Grand G. A new ascosporogenous yeast genus: Yamadazyma gen. nov. Mycotaxon 1989;35:201–204. [Google Scholar]
- Burgaud G, Arzur D, Sampaio JP et al. Candida oceani sp. nov., a novel yeast isolated from a Mid-Atlantic Ridge hydrothermal vent (-2300 meters). Anton Leeuw 2011;100:75–82. [DOI] [PubMed] [Google Scholar]
- Burgaud G, Coton M, Jacques N et al. Yamadazyma barbieri f.a. sp. nov., an ascomycetous anamorphic yeast isolated from a Mid-Atlantic Ridge hydrothermal site (-2300 m) and marine coastal waters. Int J Syst Evol Micr 2016;66:3600–6. [DOI] [PubMed] [Google Scholar]
- Carlson M, Osmond BC, Botstein D. Mutants of yeast defective in sucrose utilization. Genetics 1981;98:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YD, Dickson RC. Primary structure of the lactose permease gene from the yeast Kluyveromyces lactis Presence of an unusual transcript structure. J Biol Chem 1988;263:16696–703. [PubMed] [Google Scholar]
- Charron MJ, Dubin RA, Michels CA. Structural and functional analysis of the MAL1 locus of Saccharomyces cerevisiae. Mol Cell Biol 1986;6:3891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciafardini G, Zullo BA, Antonielli L et al. Yamadazyma terventina sp. nov., a yeast species of the Yamadazyma clade from Italian olive oils. Int J Syst Evol Micr 2013;63:372–6. [DOI] [PubMed] [Google Scholar]
- Dalal CK, Zuleta IA, Mitchell KF et al. Transcriptional rewiring over evolutionary timescales changes quantitative and qualitative properties of gene expression. eLife 2016;5:e18981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diddens HA, Lodder J. Die Anaskosporogenen Hefen, Ii Hälfte. Amsterdam: North-Holland Publ. Co, 1942. [Google Scholar]
- Greenberg JR, Price NP, Oliver RP et al. Candida albicans SOU1 encodes a sorbose reductase required for L-sorbose utilization. Yeast 2005;22:957–69. [DOI] [PubMed] [Google Scholar]
- Groenewald M, Robert V, Smith MT. The value of the D1/D2 and internal transcribed spacers (ITS) domains for the identification of yeast species belonging to the genus Yamadazyma. Persoonia 2011;26:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N et al. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013;29:1072–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Carroll SB. Parallel inactivation of multiple GAL pathway genes and ecological diversification in yeasts. P Natl Acad Sci USA 2004;101:14144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Gonçalves P, Sampaio JP et al. Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature 2010;464:54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Bai F-Y et al. Genomics and the making of yeast biodiversity. Curr Opin Genet Dev 2015;35:100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 2011;12:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull CM, Johnson AD. Identification of a mating type-like locus in the asexual pathogenic yeast Candida albicans. Science 1999;285:1271–5. [DOI] [PubMed] [Google Scholar]
- Jeffries TW, Grigoriev IV, Grimwood J et al. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol 2007;25:319–26. [DOI] [PubMed] [Google Scholar]
- Jindamorakot S, Am-In S, Kaewwichian R et al. Yamadazyma insecticola f.a., sp. nov. and Yamadazyma epiphylla f.a., sp. nov., two novel yeast species. Int J Syst Evol Micr 2015;65:1290–6. [DOI] [PubMed] [Google Scholar]
- Jin YS, Laplaza JM, Jeffries TW. Saccharomyces cerevisiae engineered for xylose metabolism exhibits a respiratory response. Appl Environ Microb 2004;70:6816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. The biology of mating in Candida albicans. Nat Rev Microbiol 2003;1:106–16. [DOI] [PubMed] [Google Scholar]
- Johnston M. A model fungal gene regulatory mechanism: the GAL genes of Saccharomyces cerevisiae. Microbiol Rev 1987;51:458–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junyapate K, Jindamorakot S, Limtong S. Yamadazyma ubonensis f.a., sp. nov., a novel xylitol-producing yeast species isolated in Thailand. Anton Leeuw 2014;105:471–80. [DOI] [PubMed] [Google Scholar]
- Kaewwichian R, Yongmanitchai W, Kawasaki H et al. Yamadazyma siamensis sp. nov., Yamadazyma phyllophila sp. nov. and Yamadazyma paraphyllophila sp. nov., three novel yeast species isolated from phylloplane in Thailand and Taiwan. Anton Leeuw 2013;103:777–88. [DOI] [PubMed] [Google Scholar]
- Karhumaa K, Fromanger R, Hahn-Hägerdal B et al. High activity of xylose reductase and xylitol dehydrogenase improves xylose fermentation by recombinant Saccharomyces cerevisiae. Appl Microbiol Biot 2007;73:1039–46. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT: Iterative refinement and additional methods. Methods Mol Biol 2014;1079:131–46. [DOI] [PubMed] [Google Scholar]
- Kavanagh KL, Klimacek M, Nidetzky B et al. The structure of apo and holo forms of xylose reductase, a dimeric aldo-keto reductase from Candida tenuis. Biochemistry 2002;41:8785–95. [DOI] [PubMed] [Google Scholar]
- Kavanagh KL, Klimacek M, Nidetzky B et al. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochem J 2003;373:319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khunnamwong P, Limtong S. Yamadazyma endophytica f.a. sp. nov., an ascomycetous yeast species isolated from leaf tissue. Int J Syst Evol Micr 2016;66:2717–23. [DOI] [PubMed] [Google Scholar]
- Konishi J, Fukuda A, Mutaguchi K et al. Xylose fermentation by Saccharomyces cerevisiae using endogenous xylose-assimilating genes. Biotechnol Lett 2015;37:1623–30. [DOI] [PubMed] [Google Scholar]
- Kuang MC, Hutchins PD, Russell JD et al. Ongoing resolution of duplicate gene functions shapes the diversification of a metabolic network. eLife 2016;5:e19027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL et al. Versatile and open software for comparing large genomes. Genome Biol 2004;5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman CP. Yamadazyma Billon-Grand (1989). in: Kurtzman CP, Fell JW, Boekhout T (eds.). The Yeasts: A Taxonomic Study. Amsterdam: Elsevier Science, 2011, 919–29. [Google Scholar]
- Kurtzman CP, Fell JW, Boekhout T et al. Methods for isolation phenotypic characterization and maintenance of yeasts. In: Kurtzman CP, Fell JW, Boekhout T (eds.). The Yeasts: A Taxonomic Study. Amsterdam: Elsevier Science, 2011, 87–110. [Google Scholar]
- Kurtzman CP, Fell JW, Boekhout T. The Yeasts: A Taxonomic Study, 5th edn Amsterdam: Elsevier, 2011. [Google Scholar]
- Kurtzman CP, Robnett CJ. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Anton Leeuw 1998;73:331–71. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP, Suzuki M. Phylogenetic analysis of ascomycete yeasts that form coenzyme Q-9 and the proposal of the new genera Babjeviella, Meyerozyma, Millerozyma, Priceomyces, and Scheffersomyces. Mycoscience 2010;51:2–14. [Google Scholar]
- Lachance MA. In defense of yeast sexual life cycles: the forma asexualis – an informal proposal. Yeast Newsl 2012;61:24–5. [Google Scholar]
- Lachance MA, Boekhout T, Scorzetti G et al. Candida Berkhout (1923). in: Kurtzman CP, Fell JW, Boekhout T (eds.). The Yeasts: A Taxonomic Study. Amsterdam: Elsevier Science, 2011, 987–1278. [Google Scholar]
- Lachance MA, Hurtado E, Hsiang T. A stable phylogeny of the large-spored Metschnikowia clade. Yeast 2016;33:261–75. [DOI] [PubMed] [Google Scholar]
- Liu XJ, Cao WN, Ren YC et al. Taxonomy and physiological characterisation of Scheffersomyces titanus sp. nov., a new D-xylose-fermenting yeast species from China. Sci Rep 2016;6:32181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes MR, Ferreira MC, Carvalho TF et al. Yamadazyma riverae sp. nov., a yeast species isolated from plant materials. Int J Syst Evol Micr 2015;65:4469–73. [DOI] [PubMed] [Google Scholar]
- Lopes MR, Morais CG, Kominek J et al. Genomic analysis and D-xylose fermentation of three novel Spathaspora species: Spathaspora girioi sp. nov., Spathaspora hagerdaliae f. a., sp. nov. and Spathaspora gorwiae f. a., sp. nov. FEMS Yeast Res 2016;16:1–12. [DOI] [PubMed] [Google Scholar]
- Mccullough MJ, Clemons KV, Mccusker JH et al. Intergenic transcribed spacer PCR ribotyping for differentiation of Saccharomyces species and interspecific hybrids. J Clin Microbiol 1998;36:1035–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsuka Y, Ninomiya S, Kiyuna T et al. Yamadazyma kitorensis f.a., sp. nov. and Zygoascus biomembranicola f.a., sp. nov., novel yeasts from the stone chamber interior of the Kitora tumulus, and five novel combinations in Yamadazyma and Zygoascus for species of Candida. Int J Syst Evol Micr 2016;66:1692–704. [DOI] [PubMed] [Google Scholar]
- Nakase T, Jindamorakot S, Ninomiya S et al. Candida kanchanaburiensissp. nov., a new ascomycetous yeast species related to Pichia nakazawae isolated in Thailand. J Gen Appl Microbiol 2008;54:259–65. [DOI] [PubMed] [Google Scholar]
- Naseeb S, Delneri D. Impact of chromosomal inversions on the yeast DAL cluster. PLoS One 2012;7:e42022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhauser W, Haltrich D, Kulbe KD et al. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis. Isolation, characterization and biochemical properties of the enzyme. Biochem J 1997;326:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhauser W, Haltrich D, Kulbe KD et al. Noncovalent enzyme-substrate interactions in the catalytic mechanism of yeast aldose reductase. Biochemistry 1998;37:1116–23. [DOI] [PubMed] [Google Scholar]
- Nguyen NH, Suh SO, Marshall CJ et al. Morphological and ecological similarities: wood-boring beetles associated with novel xylose-fermenting yeasts, Spathaspora passalidarum gen. sp. nov. and Candida jeffriesii sp. nov. Mycol Res 2006;110:1232–41. [DOI] [PubMed] [Google Scholar]
- Nidetzky B, Klimacek M, Mayr P. Transient-state and steady-state kinetic studies of the mechanism of NADH-dependent aldehyde reduction catalyzed by xylose reductase from the yeast Candida tenuis. Biochemistry 2001;40:10371–81. [DOI] [PubMed] [Google Scholar]
- Parreiras LS, Breuer RJ, Avanasi Narasimhan R et al. Engineering and two-stage evolution of a lignocellulosic hydrolysate-tolerant Saccharomyces cerevisiae strain for anaerobic fermentation of xylose from AFEX pretreated corn stover. PLoS One 2014;9:e107499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petschacher B, Nidetzky B. Altering the coenzyme preference of xylose reductase to favor utilization of NADH enhances ethanol yield from xylose in a metabolically engineered strain of Saccharomyces cerevisiae. Microb Cell Fact 2008;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Chen L, Niu Q et al. Description of Scheffersomyces henanensis sp. nov., a new D-xylose-fermenting yeast species isolated from rotten wood. PLoS One 2014;9:e92315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R, Haridas S, Wolfe KH et al. Comparative genomics of biotechnologically important yeasts. P Natl Acad Sci USA 2016;113:9882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salichos L, Rokas A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 2013;497:327–31. [DOI] [PubMed] [Google Scholar]
- Sato TK, Liu T, Parreiras LS et al. Harnessing genetic diversity in Saccharomyces cerevisiae for fermentation of xylose in hydrolysates of alkaline hydrogen peroxide-pretreated biomass. Appl Environ Microb 2014;80:540–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TK, Tremaine M, Parreiras LS et al. Directed evolution reveals unexpected epistatic interactions that alter metabolic regulation and enable anaerobic xylose use by Saccharomyces cerevisiae. PLoS Genet 2016;12:e1006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz RM, Dickson RC. Lac4 is the structural gene for beta-galactosidase in Kluyveromyces lactis. Genetics 1981;98:729–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XX, Zhou X, Kominek J et al. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 (Bethesda) 2016;6:3927–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- Slot JC, Rokas A. Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. P Natl Acad Sci USA 2010;107:10136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SO, Houseknecht JL, Gujjari P et al. Scheffersomyces parashehatae f.a., sp. nov., Scheffersomyces xylosifermentans f.a., sp. nov., Candida broadrunensis sp. nov. and Candida manassasensis sp. nov., novel yeasts associated with wood-ingesting insects, and their ecological and biofuel implications. Int J Syst Evol Micr 2013;63:4330–9. [DOI] [PubMed] [Google Scholar]
- Sumner-Smith M, Bozzato RP, Skipper N et al. Analysis of the inducible MEL1 gene of Saccharomyces carlsbergensis and its secreted product, alpha-galactosidase (melibiase). Gene 1985;36:333–40. [DOI] [PubMed] [Google Scholar]
- Sylvester K, Wang QM, James B et al. Temperature and host preferences drive the diversification of Saccharomyces and other yeasts: a survey and the discovery of eight new yeast species. FEMS Yeast Res 2015;15:fov002. [DOI] [PubMed] [Google Scholar]
- Toivola A, Yarrow D, Van Den Bosch E. Alcoholic fermentation of D-xylose by yeasts. Appl Environ Microb 1984;47:1221–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina H, Blackwell M. Multilocus phylogenetic study of the Scheffersomyces yeast clade and characterization of the N-terminal region of xylose reductase gene. PLoS One 2012;7:e39128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina H, Frank R, Blackwell M. Scheffersomyces cryptocercus: a new xylose-fermenting yeast associated with the gut of wood roaches and new combinations in the Sugiyamaella yeast clade. Mycologia 2013;105:650–60. [DOI] [PubMed] [Google Scholar]
- Urbina H, Schuster J, Blackwell M. The gut of Guatemalan passalid beetles: a niche colonized by cellobiose- and xylose-fermenting yeasts. Fungal Ecol 2013;6:339–55. [Google Scholar]
- U.S. DOE Breaking the Biological Barriers to Cellulosic Ethanol: A Joint Research Agenda, DOE/SC-0095, 2006. www.genomicscience.energy.gov/biofuels/ (21 Feburary 2017, date last accessed).
- U.S. DOE Lignocellulosic Biomass for Advanced Biofuels and Bioproducts, DOE/SC-1070, 2015. http://genomicscience.energy.gov/biofuels/lignocellulose/ (21 Feburary 2017, date last accessed).
- Wang Y, Ren YC, Li Y et al. Molecular phylogeny and taxonomy of Yamadazyma dushanensis f.a., sp. nov., a cellobiose-fermenting yeast species from China. Curr Microbiol 2015;71:268–73. [DOI] [PubMed] [Google Scholar]
- Weiss S, Samson F, Navarro D et al. YeastIP: A database for identification and phylogeny of Saccharomycotina yeasts. FEMS Yeast Res 2013;13:117–25. [DOI] [PubMed] [Google Scholar]
- Wohlbach DJ, Kuo A, Sato TK et al. Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. P Natl Acad Sci USA 2011;108:13212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S, Wolfe KH. Birth of a metabolic gene cluster in yeast by adaptive gene relocation. Nat Genet 2005;37:777–82. [DOI] [PubMed] [Google Scholar]
- Zhou X, Peris D, Kominek J et al. In silico Whole Genome Sequencer & Analyzer (iWGS): a computational pipeline to guide the design and analysis of de novo genome sequencing studies. G3 (Bethesda) 2016;6:3655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at FEMSYR online.