

Summary
Impaired T helper type 1 (Th1) function is implicated in the susceptibility of patients with chronic obstructive pulmonary disease (COPD) to respiratory infections, which are common causes of acute exacerbations of COPD (AECOPD). To understand the underlying mechanisms, we assessed regulatory T (Treg) cells and the expression of an inhibitory T‐cell receptor, cytotoxic T‐lymphocyte‐associated antigen 4 (CTLA‐4). Cryopreserved peripheral blood mononuclear cells (PBMC) from patients with AECOPD (n = 17), patients with stable COPD (sCOPD; n = 24) and age‐matched healthy non‐smoking controls (n = 26) were cultured for 24 hr with brefeldin‐A or monensin to detect intracellular or surface CTLA‐4 (respectively) by flow cytometry. T cells in PBMC from AECOPD (n = 9), sCOPD (n = 14) and controls (n = 12) were stimulated with anti‐CD3 with and without anti‐CTLA‐4 blocking antibodies and cytokines were quantified by ELISA. Frequencies of circulating T cells expressing intracellular CTLA‐4 were higher in sCOPD (P = 0·01), whereas patients with AECOPD had more T cells expressing surface CTLA‐4 than healthy controls (P = 0·03). Increased frequencies of surface CTLA‐4+ CD4+ T cells and CTLA‐4+ Treg cells paralleled increases in plasma soluble tumour necrosis factor receptor‐1 levels (r = 0·32, P = 0·01 and r = 0·29, P = 0·02, respectively) in all subjects. Interferon‐γ responses to anti‐CD3 stimulation were inversely proportional to frequencies of CD4+ T cells expressing intracellular CTLA‐4 (r = −0·43, P = 0·01). Moreover, CTLA‐4 blockade increased the induction of interferon‐γ, tumour necrosis factor‐α and interleukin‐6 in PBMC stimulated with anti‐CD3. Overall, chronic inflammation may expand sub‐populations of T cells expressing CTLA‐4 in COPD patients and therefore impair T‐cell function. CTLA‐4 blockade may restore Th1 function in patients with COPD and so aid the clearance of bacterial pathogens responsible for AECOPD.
Keywords: acute exacerbations of chronic obstructive pulmonary disease, chronic obstructive pulmonary disease, cytotoxic T‐lymphocyte‐associated antigen 4, regulatory T cells, T cells
Abbreviations
- AECOPD
acute exacerbations of COPD
- APC
allophycocyanin
- COPD
chronic obstructive pulmonary disease
- CRP
C‐reactive protein
- CTLA‐4
cytotoxic T‐lymphocyte‐associated antigen‐4
- FCS
fetal calf serum
- GOLD
Global initiative for chronic obstructive lung disease
- HC
healthy controls
- IFN‐γ
interferon‐γ
- IL
interleukin
- NTHI
non‐typeable Haemophilus influenzae
- PBMC
peripheral blood mononuclear cells
- sCOPD
stable COPD
- sTNFR1
soluble tumour necrosis factor receptor‐1
- Th1
T helper type 1
- TNF‐α
tumour necrosis factor‐α
- Treg
regulatory T
- UWA
University of Western Australia
Introduction
Chronic obstructive pulmonary disease (COPD) is one of the top five causes of morbidity and mortality worldwide. Smoking is the most common cause of COPD, but genetic factors and environmental pollutions are also implicated.1 The disease is characterized by progressive limitation of airflow, with abnormal inflammatory responses to inhaled particles or gases.2 The chronic inflammation of the airways involves infiltration and activation of neutrophils, macrophages and T cells.3, 4, 5 Subsequent systemic inflammation contributes to co‐morbidities (e.g. cardiovascular disease and cachexia).6
Most patients are relatively stable with treatment (sCOPD), but some experience acute exacerbations of COPD (AECOPD), which increase mortality and morbidity.7 AECOPD is defined as ‘an event in the natural course of the disease characterized by a change in the patient's baseline dyspnoea, cough and/or sputum, and beyond normal day‐to‐day variations, that is acute in onset and may warrant a change in regular medication in a patient with underlying COPD’.2 Respiratory bacterial infections account for up to 50% of AECOPD events. Non‐typeable Haemophilus influenzae (NTHI), Streptococcus pneumoniae and Moraxella catarrhalis are the major bacterial pathogens isolated from patients with AECOPD.8 As NTHI oral vaccines do not reduce the frequency and severity of AECOPD,9 the capacity to mount a protective anti‐bacterial immune response may be limited in patients with COPD.
Despite its inflammatory aetiology, COPD is considered as an immune‐deficient state as the abundant activated T cells in the airways of COPD patients do not eradicate bacterial infections. Indeed, T helper type 1 (Th1) immune responses [e.g. production of interferon‐γ (IFN‐γ) and phagocytosis] are impaired in patients with COPD.10, 11, 12, 13 Exogenous IFN‐γ can enhance killing of NTHI by monocytes from patients with bronchiectasis,14 confirming the necessity for appropriate Th1 responses for clearance of bacterial infections. Here we address the regulators of T‐cell responses in patients with COPD and search for means to improve host production of IFN‐γ.
An important regulator of T‐cell function is cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4), which blocks the CD28‐mediated activation of T cells.15 CTLA‐4 is constitutively expressed by regulatory T (Treg) cells and activated effector T cells. It inhibits the proliferation of T cells and the production of pro‐inflammatory cytokines, and so prevents continuous T‐cell activation.15, 16 Polymorphisms in the gene encoding CTLA‐4 (e.g. rs231775 and rs5742909) and increased serum levels of the soluble form of CTLA‐4 have been associated with dysregulated T‐cell responses and increased susceptibility to COPD and chronic bronchitis.17, 18, 19, 20 Furthermore, increased levels of soluble CTLA‐4 in the serum of patients with COPD paralleled decreased lung function and increased C‐reactive protein (CRP).21 Frequencies of circulating CTLA‐4+ T cells and CTLA‐4+ Treg cells were also higher in patients with COPD than in controls.22, 23 Kalathil et al. reported that CTLA‐4 blockade in vitro increased the proliferation of CD4+ and CD8+ T cells and production of IFN‐γ by peripheral blood mononuclear cells (PBMC) from three patients with COPD.24
Here in a larger patient cohort, we address the possibility that chronic inflammation in patients with COPD may increase CTLA‐4 expression or proportions of Treg cells which constitutively express CTLA‐4, so limiting protective Th1‐cell responses (e.g. IFN‐γ production). Little is known about the role of CTLA‐4 in AECOPD in terms of levels of expression and anti‐bacterial function. Furthermore, most studies have only assessed intracellular expression as surface expression is complicated by the rapid endocytosis of CTLA‐4. Hence we have addressed the expression of intracellular and surface CTLA‐4 using novel assays and hypothesized that the expression of CTLA‐4 is elevated in AECOPD, which reduces antibacterial responses such as IFN‐γ production.
Methods
Study subjects and sample collection
Patients with AECOPD (n = 17; 7 current smokers and 10 ex‐smokers) were recruited on admission to the Emergency Department in Royal Perth Hospital in Western Australia. Patients with stable COPD (sCOPD; n = 24, all ex‐smokers) were recruited from a dedicated COPD clinic at Royal Perth Hospital. All AECOPD and sCOPD patients had a smoking history of > 15 pack‐years and ex‐smokers were defined as those who had ceased smoking > 1 year earlier. The diagnosis and severity of COPD was established by a respiratory physician according to the GOLD criteria (Stages 2–4).25 All patients with COPD had been treated with anticholinergic drugs, long‐acting beta agonists and inhaled corticosteroids for >3 months before participating in the study. Co‐morbidities included hypertension, osteoporosis and ischaemic heart disease. No patients were receiving systemic corticosteroids or had diabetes, neuromuscular, allergic or rheumatological disease. Age‐matched healthy non‐smoking controls with no clinical evidence of COPD and not taking any antibiotics or anti‐inflammatory medications were tested in parallel (HC; n = 26). This study was approved by the Royal Perth Hospital Human Research Ethics Committee (EC2012/23) and all participants gave informed consent.
Blood samples were collected in lithium heparin tubes, centrifuged at 1000 g for 10 min and plasma was stored in aliquots at −80°. PBMC were isolated by Ficoll‐Paque PLUS density gradient centrifugation (GE Healthcare, Uppsala, Sweden) and cryopreserved in 10% DMSO/fetal calf serum (FCS; Gibco by Invitrogen, Carlsbad, CA).
T‐cell subsets
The PBMC (1 × 106 cells/ml) were cultured at 37° in 5% CO2 for 24 hr in polypropylene tubes on a 5‐degree incline in 10% FCS/RPMI. BD GolgiPlug™ (Brefeldin‐A 1 μg/ml) or BD GolgiStop™ (monensin; 2 μm) plus CTLA‐4‐allophycocyanin (APC) antibodies (BD Biosciences, San Jose, CA) was added after 2 hr. Brefeldin‐A prevents secretion of proteins including CTLA‐4 and hence measures intracellular CTLA‐4. As surface expression of CTLA‐4 is transient due to rapid endocytosis, PBMC were cultured in the presence of CTLA‐4‐APC antibodies plus monensin (which prevents acidification and subsequent degradation of endocytosed CTLA‐4 antibody complexes) before staining for immunophenotypic markers. Cells were washed and stained with the BD Horizon™ Fixable Viability Stain 450, followed by surface‐staining with CD3‐APC‐H7, CD4‐V500, CD8‐Peridinin chlorophyll protein‐Cy5.5 and CD45RA‐phycoerythrin‐Cy7 antibodies. Intracytoplasmic staining was done using the BD Pharmingen™ Human Foxp3 buffer sets with Foxp3‐phycoerythrin and CTLA‐4‐APC antibodies (BD Biosciences). A total of 200 000 events were acquired using a BD FACSCanto II cytometer and analysed with flowjo v5·7·2 software (Tree Star, Ashland, OR). The expression of intracellular or surface CTLA‐4 was measured in Treg cells (CD25+ Foxp3+ CD4+ CD3+ lymphocytes), total CD4+ T cells (CD4+ CD3+ lymphocytes) or total CD8+ T cells (CD8+ CD3+ lymphocytes). Gating strategies are shown in the Supplementary material (Fig. S1).
CTLA‐4 blocking assay
The PBMC (2 × 105 cells/well at 1 × 106 cells/ml) from nine patients with AECOPD, 14 patients with sCOPD and 12 controls were stimulated with 10 ng/ml anti‐CD3 (Mabtech, Nacka Strand, Sweden), with or without 10 μg/ml anti‐CTLA‐4 antibody (clone BNI3; BD Biosciences) for 24 hr in 96‐well flat‐bottom plates. Culture supernatants were stored at −80°C. To measure T‐cell production of IFN‐γ or tumour necrosis factor‐α (TNF‐α), PBMC (1 × 106 cells/ml) from six participants (AECOPD, n = 2; sCOPD, n = 2; controls, n = 2) were cultured with 10 ng/ml anti‐CD3 for 24 hr, with or without 10 μg/ml anti‐CTLA‐4 antibody. Brefeldin‐A was added 2 hr after the start of culture. Cells were washed and stained with the BD Horizon™ Fixable Viability Stain 450, followed by surface‐staining with CD3‐V500, CD4‐PerCP and CD8‐APC‐H7 antibodies (BD Biosciences). Intracytoplasmic staining was performed using the BD Cytofix Cytoperm™ buffer sets, IFN‐γ‐BV421 and TNF‐α‐phycoerythrin antibodies (BD Biosciences). In all, 200 000 events were acquired using a BD FACSCanto II cytometer and analysed with flowjo v5·7·2 software (Tree Star).
Quantification of soluble biomarkers
Plasma levels of CRP and soluble TNF receptor‐1 (sTNFR1) were measured by ELISA (R&D Systems, Minneapolis, MN). Concentrations of IFN‐γ, interleukin‐6 (IL‐6), IL‐10 and TNF‐α (BD Biosciences) were measured in culture supernatants by ELISA with the diluent (10% FCS/PBS) as a negative control. A sample with known cytokine concentrations (QC) was assayed on each plate to assess inter‐plate variation (coefficient of variance was < 10%).
Data analysis
Non‐parametric Mann–Whitney U‐tests were used to compare data between groups. Wilcoxon matched‐pairs signed rank tests were used to compare data within groups (e.g. with and without CTLA‐4 blocking). Correlations were assessed using Spearman's rank correlation coefficients. All analyses were performed with graphpad prism 5·04 software (GraphdPad Software, La Jolla, CA). Statistically significant differences (P < 0·05) are indicated in the figures.
Results
Systemic inflammation in patients with AECOPD is higher than in sCOPD patients
Study demographics are presented in Table 1. Relative to healthy controls, AECOPD and sCOPD patients exhibited increased systemic inflammation as defined by higher levels of plasma CRP (P < 0·001 for both) and sTNFR1 (P = 0·007 and P < 0·001, respectively). Levels of CRP and sTNFR1 were also higher in patients with AECOPD than in sCOPD (P = 0·02 and P = 0·04, respectively; Table 1).
Table 1.
Plasma biomarkers of systemic inflammation are increased in patients with chronic obstructive pulmonary disease
| Healthy controls | Stable COPD | AECOPD | |
|---|---|---|---|
| N | 26 | 24 | 17 |
| Sex (male:female)a | 14:12 | 13:11 | 9:8 |
| Age in yearsb | 75 [53–90] | 71 [60–89] | 78 [53–92] |
| Plasma CRP (mg/l) | 0·66 [0·15–6·8] | 8·4 [0·15–73]c | 33 [2·2–157]c , e |
| Plasma sTNFR1 (ng/ml) | 0·52 [0·35–1·0] | 1·3 [0·79–2·48]d | 1·89 [1·13–6·41]c , e |
Abbreviations:AECOPD, acute exacerbation of COPD; COPD, chronic obstructive pulmonary disease; CRP, C‐reactive protein; sTNFR1, soluble tumour necrosis factor receptor‐1.
Data are presented as median [range].
Similar in all groups (Fisher's test, P > 0·05).
Similar in all groups (Mann–Whitney U‐tests, P > 0·05).
Higher than controls, P < 0·001.
Higher than controls, P < 0·01.
Higher than sCOPD, P < 0·05.
Patients with AECOPD exhibit higher frequencies of circulating Treg cells and CTLA‐4+ T cells
The frequency of Treg cells (CD4+ CD25+ Foxp3+) was higher in patients with AECOPD than in sCOPD (P = 0·01) or healthy controls (P = 0·001), but similar in patients with sCOPD and controls (Fig. 1a). Frequencies of total and memory (CD45RAneg) CD4+ and CD8+ T cells were similar in all groups (data not shown). The frequency of Treg cells expressing intracellular CTLA‐4 was higher in patients with sCOPD than in controls (P = 0·01; Fig. 1b) and correlated with plasma levels of sTNFR1 (r = 0·26, P = 0·04). The frequency of CD4+ T cells expressing intracellular CTLA‐4 was marginally higher in patients with sCOPD than controls (P = 0·06; Fig. 1c). Expression of intracellular CTLA‐4 was low in CD8+ T cells (< 5%) and did not differ between groups (Fig. 1d).
Figure 1.
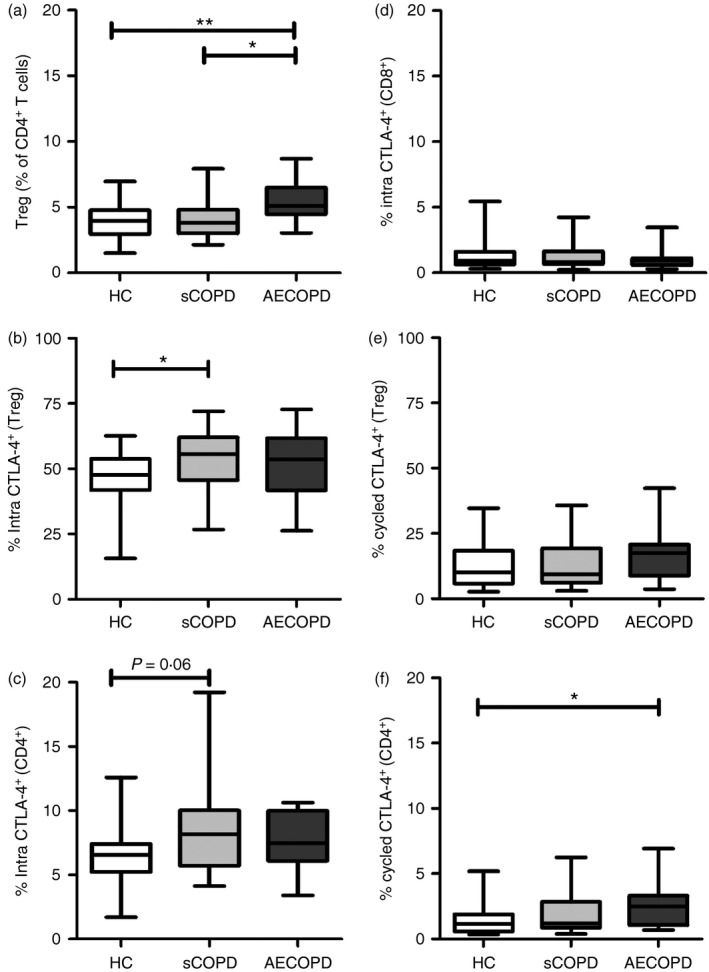
Proportion of regulatory T (Treg) cells and cytotoxic T lymphocyte‐associated antigen 4 (CTLA‐4)‐expressing T cells were increased in patients with chronic obstructive pulmonary disease (COPD). Circulating frequency of (a) Treg cells (as % of CD4+ T cells), (b) intracellular CTLA‐4+ Treg cells, (c) intracellular CTLA‐4+ CD4+ T cells, (d) surface CTLA‐4+ Treg cells, (e), surface CTLA‐4+ CD4+ T cells, and (f) surface CTLA‐4+ CD8+ T cells. **P < 0·01; *P < 0·05, Mann‐Whitney U‐test. The graphs show the interquartile range (box), median (middle line) and range (whiskers) for each group.
Interestingly, the frequency of surface CTLA‐4+ Treg cells or CD4+ T cells are significantly lower than the frequency of intracellular CTLA‐4+ Treg cells or CD4+ T cells, respectively (P < 0·0001; Fig. 1b versus 1e and Fig. 1c versus 1f, respectively). The median frequency of surface CTLA‐4+ Treg cells was higher in patients with AECOPD than in patients with sCOPD (P = 0·15) or controls (P = 0·17), but the difference was not significant (Fig. 1e). Patients with AECOPD had a higher frequency of CD4+ T cells with surface CTLA‐4+ than controls (P = 0·03), but there was no increase in patients with sCOPD (Fig. 1f). Levels of sTNFR1 correlated with the proportion of surface CTLA‐4+ Treg cells (r = 0·29, P = 0·02) and surface CTLA‐4+ CD4+ T cells (r = 0·32, P = 0·01). Few CD8+ T cells expressed surface CTLA‐4 (< 1%). The proportions of Treg cells, CD4+ T cells or CD8+ T cells expressing intracellular or surface CTLA‐4 did not differ between patients with AECOPD who were current smokers compared with those who were ex‐smokers (P = 0·67 to P = 1·00).
Levels of pro‐inflammatory cytokines induced by anti‐CD3 were increased by CTLA‐4 blockade in all subjects
Sufficient PBMC were available from nine patients with AECOPD, 14 patients with sCOPD and 12 healthy controls. PBMC were cultured with anti‐CD3 to induce a pan T‐cell response, with or without added anti‐CTLA‐4 antibody. Levels of IFN‐γ, TNF‐α, IL‐6 and IL‐10 in supernatants of PBMC cultured with anti‐CD3 only (no CTLA‐4 blockade) were similar in the three groups (Fig. 2a–d). Overall the production of IFN‐γ correlated inversely with the proportions of Treg cells (r = −0·46, P = 0·004) and intracellular CTLA‐4+ CD4+ T cells (r = −0·52, P < 0·001).
Figure 2.
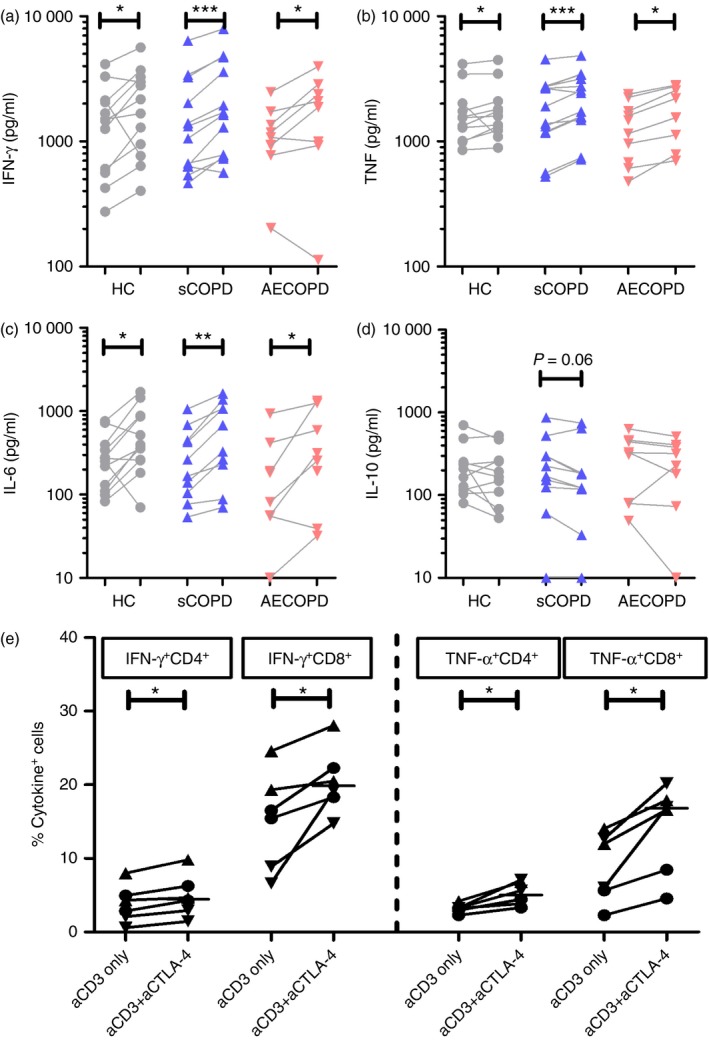
Cytotoxic T lymphocyte‐associated antigen 4 (CTLA‐4) blockade increased the production of interferon‐γ (IFN‐γ), tumour necrosis factor‐α (TNF‐α) and interleukin‐6 (IL‐6) by peripheral blood mononuclear cells (PBMC) from patients with an acute exacerbation of COPD (AECOPD) patients, patients with stable COPD (sCOPD) and controls. Levels of (a) IFN‐γ, (b) TNF‐α, (c) IL‐6 and (d) IL‐10 in cell culture supernatants of anti‐CD3 stimulated PBMC with (right) or without (left) the addition of anti‐CTLA‐4 blocking antibodies. (e) Frequencies of IFN‐γ‐ and TNF‐α‐producing CD4+ and CD8+ T cells with CTLA‐4 blockade (right) or without CTLA‐4 blockade (left) from six participants (two AECOPD [▼], two sCOPD [▲], two controls [●]). ***P < 0·001; **P < 0·01; *P < 0·05, Wilcoxon matched‐pairs signed rank tests. [Colour figure can be viewed at wileyonlinelibrary.com]
CTLA‐4 blockade increased the anti‐CD3‐induced IFN‐γ, TNF‐α and IL‐6 responses of PBMC from healthy controls, and patients with sCOPD and AECOPD (Fig. 2a–c), whereas IL‐10 levels were marginally reduced (P = 0·10 to P = 0·32; Fig. 2d). In a pilot study of two patients with AECOPD, two with sCOPD and two healthy controls analysed as a group, CTLA‐4 blockade increased the proportions of IFN‐γ + or TNF‐α + CD4+ and CD8+ T cells (Fig. 2e). Hence CTLA‐4 blockade may promote production of pro‐inflammatory cytokines while inhibiting the production of IL‐10.
Discussion
Acute exacerbations in patients with COPD are commonly associated with respiratory infections,8 reflecting impaired Th1 responses to bacteria.10, 11, 12, 13 For example; in vitro lymphoproliferative responses to NTHI protein P6 were lower in patients with AECOPD whose sputum was culture positive for NTHI than in patients with COPD with no exacerbations attributed to NTHI.12 However, the underlying mechanisms have not been elucidated. Here, we provide novel data to support a role for CTLA‐4 and Treg cells in limiting protective Th1 responses in patients with AECOPD. Expression of surface (not intracellular) CTLA‐4 in patients with AECOPD was increased and associated with impaired IFN‐γ responses. Furthermore, the blockade of CTLA‐4 improved production of Th1 cytokines.
Compared with controls, the frequencies of circulating CD4+ T cells and Treg cells expressing intracellular CTLA‐4 were higher in patients with sCOPD, whereas patients with AECOPD exhibited increased frequencies of circulating Treg cells and surface CTLA‐4+ CD4+ T cells, with no significant increase in surface CTLA‐4+ Treg cells. Our data extend previous studies, which have only shown increased circulating Treg cells in patients with AECOPD.26 Importantly, we have linked the increased proportions of CD4+ T cells and Treg cells expressing surface CTLA‐4 with increased levels of plasma sTNFR1. We suggest that chronic inflammation experienced by all patients with COPD may expand populations of intracellular CTLA‐4+ CD4+ T cells and CTLA‐4+ Treg cells. Episodes of AECOPD may then further activate these cells, promoting the cycling of CTLA‐4 between intracellular compartments and the cell surface, as shown previously in PHA‐activated PBMC.27 This is the first demonstration of the critical need to distinguish surface from intracellular CTLA‐4 in patients with AECOPD and patients with sCOPD.
Splice variants of the CTLA‐4 gene may result in the expression of the transmembrane isoform (Tm‐CTLA‐4) or soluble isoform which lacks the transmembrane domain (sCTLA‐4). The expression of both isoforms can be induced by activated CD4+ T cells and both are immunosuppressive. Hence, increased cycling of CTLA‐4 (e.g. during an AECOPD episode) could reflect increases in both transmembrane and soluble CTLA‐4 because the commercially available clone of anti‐CTLA‐4 antibody (BNI3) used in our study targets both CTLA‐4 isoforms. Future study should investigate the role of sCTLA‐4 versus Tm‐CTLA‐4 in AECOPD using an anti‐CTLA‐4 antibody clone that specifically targets the sCTLA‐4 isoform (e.g. those generated by Esposito et al.28).
The production of IFN‐γ was associated inversely with the proportions of circulating Treg cells and intracellular CTLA‐4+ CD4+ T cells. Furthermore, the production of IFN‐γ, TNF‐α and IL‐6 by PBMC and proportions of IFN‐γ‐ or TNF‐α‐producing CD4+ and CD8+ T cells from all subjects (including patients with AECOPD) were increased by CTLA‐4 blockade. Increased production of IFN‐γ and TNF‐α after CTLA‐4 blockade may reflect the increased proportions of IFN‐γ‐ or TNF‐α‐producing CD4+ and CD8+ T cells found after CTLA‐4 blockade, even though the expression of CTLA‐4 is much lower in CD8+ T cells compared with CD4+ T cells. As the regulation of T‐cell responses by CTLA‐4 may be in an intrinsic or extrinsic manner,16 CTLA‐4 blockade may enhance the activation of CTLA‐4+ T cells and neighbouring T cells, which may have low or no expression of CTLA‐4 (e.g. CD8+ T cells). Furthermore, improvement in CD4+ T‐cell responses upon CTLA‐4 blockade may also improve general Th1 responses (e.g. increased production in IFN‐γ), which may induce the activation of other Th1 cells such as CD8+ T cells. Hence, increased proportions of CTLA‐4‐expressing CD4+ T cells or Treg cells in patients with COPD may impair Th1 responses needed for clearance of infections.29, 30
Our data support the use of CTLA‐4 blockade to restore Th1 responses (e.g. IFN‐γ production) in patients with COPD, which in turn should aid clearance of bacterial and/or viral pathogens, so reducing the risk of AECOPD. The blockade of ‘immune checkpoints’ such as CTLA‐4 has been used to inhibit Treg cell function and promote effector T‐cell responses,31 showing promising clinical results in anti‐tumour treatment.32 Similarly in mice infected with Nippostrongylus brasiliensis, Listeria monocytogenes or Mycobacterium bovis, CTLA‐4 blockade enhanced the production of IFN‐γ and TNF‐α.33, 34, 35
Although we had no access to cells from the lung, the observed increase in proportions of Treg cells matches that seen in bronchoalveolar lavage and lymphoid follicles from patients with COPD.23, 36 As responses by PBMC may not reflect respiratory mucosal responses to NTHI, studies of CTLA‐4 expression by T‐cells from the lung and challenge with bacteria implicated in AECOPD episodes (e.g. NTHI) are warranted to establish CTLA‐4 blockade as a viable therapeutic strategy to improve the production of Th1 cytokines.
Disclosure
None of the authors has any potential financial conflict of interest related to this manuscript.
Supporting information
Figure S1. Gating strategies used to identify CTLA‐4+ T‐cell subsets. (a) Singlets were defined by forward scatter‐area (FCS‐A) and FCS‐Height. (b) Dead cells were then excluded and (c) lymphocytes were gated based on side scatter (SSC)‐A and FSC‐A. (d) CD4+ and (e) CD8+ T cells were identified by co‐expression of CD3 with CD4 or CD8, respectively. (f) Co‐expression of CTLA‐4 and Foxp3 was assessed in CD4+ T cells.
Acknowledgements
DT, PP and YM conceived the project. DT designed the experiments. DT, TT, AS, NO and MZ performed the experiments and analysed the data. All authors contributed to data interpretation. DT, PP, LK and YM prepared the manuscript and all authors provided critical review. DT was supported by a UWA‐MRF postdoctoral fellowship and the work was funded by research grants from the Ada Bartholomew Medical Research Trust Grant, Faculty of Medicine, Dentistry and Health Sciences, University of Western Australia (UWA) and from the Royal Perth Hospital Medical Research Foundation. The authors thank the staff at the Centre for Clinical Research in Emergency Medicine, Harry Perkins Institute of Medical Research and the COPD Linkage clinic, Royal Perth Hospital for patient recruitment, and the patients and controls who volunteered for this study. The authors also acknowledge the contributions of V. Seenarain, E. Dikstaal and A. Anderson who contributed to data collection as part of their UWA medical course.
References
- 1. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet 2007; 370:765–73. [DOI] [PubMed] [Google Scholar]
- 2. Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A et al Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–65. [DOI] [PubMed] [Google Scholar]
- 3. Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 2009; 360:2445–54. [DOI] [PubMed] [Google Scholar]
- 4. Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc 2007; 4:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Donnell R, Breen D, Wilson S, Djukanovic R. Inflammatory cells in the airways in COPD. Thorax 2006; 61:448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Res J 2009; 33:1165–85. [DOI] [PubMed] [Google Scholar]
- 7. MacIntyre N, Huang YC. Acute exacerbations and respiratory failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2008; 5:530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med 2002; 347:465–71. [DOI] [PubMed] [Google Scholar]
- 9. Teo E, House H, Lockhart K, Purchuri SN, Pushparajah J, Cripps AW et al Haemophilus influenzae oral vaccination for preventing acute exacerbations of chronic bronchitis and chronic obstructive pulmonary disease. Cochrane Database Systematic Rev 2014; 9:CD010010. [DOI] [PubMed] [Google Scholar]
- 10. Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis 2006; 194:1375–84. [DOI] [PubMed] [Google Scholar]
- 11. Taylor AE, Finney‐Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA et al Defective macrophage phagocytosis of bacteria in COPD. Eur Res J 2010; 35:1039–47. [DOI] [PubMed] [Google Scholar]
- 12. Abe Y, Murphy TF, Sethi S, Faden HS, Dmochowski J, Harabuchi Y et al Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 165:967–71. [DOI] [PubMed] [Google Scholar]
- 13. Knobloch J, Schild K, Jungck D, Urban K, Muller K, Schweda EK et al The T‐helper cell type 1 immune response to gram‐negative bacterial infections is impaired in COPD. Am J Respir Crit Care Med 2011; 183:204–14. [DOI] [PubMed] [Google Scholar]
- 14. King P, Ngui J, Oppedisano F, Robins‐Browne R, Holmes P, Holdsworth S. Effect of interferon‐γ and CD40 ligation on intracellular monocyte survival of nontypeable Haemophilus influenzae . APMIS 2008; 116:1043–9. [DOI] [PubMed] [Google Scholar]
- 15. Bour‐Jordan H, Esensten JH, Martinez‐Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T‐cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev 2011; 241:180–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell‐extrinsic regulator of T cell responses. Nat Rev Immunol 2011; 11:852–63. [DOI] [PubMed] [Google Scholar]
- 17. Simone R, Pesce G, Antola P, Rumbullaku M, Bagnasco M, Bizzaro N et al The soluble form of CTLA‐4 from serum of patients with autoimmune diseases regulates T‐cell responses. BioMed Res Int 2014; 2014:215763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fernandez‐Mestre M, Sanchez K, Balbas O, Gendzekhzadze K, Ogando V, Cabrera M et al Influence of CTLA‐4 gene polymorphism in autoimmune and infectious diseases. Hum Immunol 2009; 70:532–5. [DOI] [PubMed] [Google Scholar]
- 19. Zhu G, Agusti A, Gulsvik A, Bakke P, Coxson H, Lomas DA et al CTLA4 gene polymorphisms are associated with chronic bronchitis. Eur Respir J 2009; 34:598–604. [DOI] [PubMed] [Google Scholar]
- 20. Wood AM, Newby PR, Gough SC, Stockley RA. CTLA4 polymorphisms and COPD. Eur Respir J 2010; 35:457–8. [DOI] [PubMed] [Google Scholar]
- 21. Shen Y, Yang T, Wang T, Chen L, Wen F. Elevated circulating cytotoxic T lymphocyte‐associated antigen‐4 levels are related to lung function and inflammation in chronic obstructive pulmonary disease. Respirology 2013; 18:885–6. [DOI] [PubMed] [Google Scholar]
- 22. Domagala‐Kulawik J, Hoser G, Dabrowska M, Safianowska A, Chazan R. CD4+ /CD25+ cells in systemic inflammation in COPD. Scand J Immunol 2011; 73:59–65. [DOI] [PubMed] [Google Scholar]
- 23. Smyth LJ, Starkey C, Vestbo J, Singh D. CD4‐regulatory cells in COPD patients. Chest 2007; 132:156–63. [DOI] [PubMed] [Google Scholar]
- 24. Kalathil SG, Lugade AA, Pradhan V, Miller A, Parameswaran GI, Sethi S et al T‐regulatory cells and programmed death 1+ T cells contribute to effector T‐cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 190:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P et al Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176:532–55. [DOI] [PubMed] [Google Scholar]
- 26. Jin Y, Wan Y, Chen G, Chen L, Zhang MQ, Deng L et al Treg/IL‐17 ratio and Treg differentiation in patients with COPD. PLoS One 2014; 9:e111044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA‐4 and focal localization towards sites of TCR engagement. Immunity 1996; 4:535–43. [DOI] [PubMed] [Google Scholar]
- 28. Esposito L, Hunter KM, Clark J, Rainbow DB, Stevens H, Denesha J et al Investigation of soluble and transmembrane CTLA‐4 isoforms in serum and microvesicles. J Immunol 2014; 193:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xing Z. Current understanding of macrophage type 1 cytokine responses during intracellular infections. Histol Histopathol 2000; 15:199–205. [DOI] [PubMed] [Google Scholar]
- 30. Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin‐6 induces early γ interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun 2000; 68:3322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV et al CTLA‐4 and PD‐1 receptors inhibit T‐cell activation by distinct mechanisms. Mol Cell Biol 2005; 25:9543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grosso JF, Jure‐Kunkel MN. CTLA‐4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun 2013; 13:5. [PMC free article] [PubMed] [Google Scholar]
- 33. McCoy K, Camberis M, Gros GL. Protective immunity to nematode infection is induced by CTLA‐4 blockade. J Exp Med 1997; 186:183–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kirman J, McCoy K, Hook S, Prout M, Delahunt B, Orme I et al CTLA‐4 blockade enhances the immune response induced by mycobacterial infection but does not lead to increased protection. Infect Immun 1999; 67:3786–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pedicord VA, Montalvo W, Leiner IM, Allison JP. Single dose of anti‐CTLA‐4 enhances CD8+ T‐cell memory formation, function, and maintenance. Proc Natl Acad Sci USA 2011; 108:266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Plumb J, Smyth LJ, Adams HR, Vestbo J, Bentley A, Singh SD. Increased T‐regulatory cells within lymphocyte follicles in moderate COPD. Eur Respir J 2009; 34:89–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gating strategies used to identify CTLA‐4+ T‐cell subsets. (a) Singlets were defined by forward scatter‐area (FCS‐A) and FCS‐Height. (b) Dead cells were then excluded and (c) lymphocytes were gated based on side scatter (SSC)‐A and FSC‐A. (d) CD4+ and (e) CD8+ T cells were identified by co‐expression of CD3 with CD4 or CD8, respectively. (f) Co‐expression of CTLA‐4 and Foxp3 was assessed in CD4+ T cells.