

SUMMARY
Classically, G protein-coupled receptor (GPCR) stimulation promotes G protein signaling at the plasma membrane, followed by rapid β-arrestin-mediated desensitization and receptor internalization into endosomes. However, it has been demonstrated that some GPCRs activate G proteins from within internalized cellular compartments, resulting in sustained signaling. We have used a variety of biochemical, biophysical, and cell-based methods to demonstrate the existence, functionality, and architecture of internalized receptor complexes composed of a single GPCR, β-arrestin, and G protein. These super-complexes or “megaplexes” more readily form at receptors that interact strongly with β-arrestins via a C-terminal tail containing clusters of serine/threonine phosphorylation sites. Single-particle electron microscopy analysis of negative-stained purified megaplexes reveals that a single receptor simultaneously binds through its core region with G protein and through its phosphorylated C-terminal tail with β-arrestin. The formation of such megaplexes provides a potential physical basis for the newly appreciated sustained G protein signaling from internalized GPCRs.
In Brief
Megaplexes containing a GPCR simultaneously engaged with a G protein and β-arrestin sustain G protein signaling following internalization into endosomes.
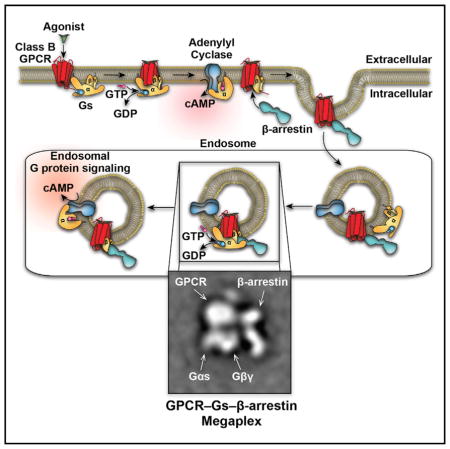
INTRODUCTION
G protein-coupled receptor (GPCR) signaling ensues when an agonist binds to and stabilizes an active receptor conformation. This agonist bound GPCR, acting through its transmembrane core, promotes interaction with heterotrimeric G proteins (Gαβγ), thus stimulating guanine nucleotide exchange and separation of the Gα subunit from the Gβγ subunits (Gilman, 1987). G protein subunits then interact with a variety of effectors, such as enzymes and ion channels, to initiate downstream responses (Gilman, 1987; Pierce et al., 2002).
To terminate G protein signaling, cells have devised a specialized desensitization mechanism that includes phosphorylation of receptors by GPCR kinases (GRKs) and subsequent recruitment of β-arrestins (βarrs) to the phosphorylated receptor (Moore et al., 2007). βarrs engage both the phosphorylated C-tail and the transmembrane core of the receptor. The latter interaction overlaps with the G protein-binding site and thus sterically hinders further G protein activation (Kang et al., 2015; Shukla et al., 2014; Szczepek et al., 2014). Additionally, βarr binding initiates receptor internalization by interaction with the endocytic machinery (i.e., clathrin, adaptin-2, etc.) (Goodman et al., 1996; Laporte et al., 1999). Depending on the strength of the GPCR-βarr interaction, the receptor may either undergo transient internalization, followed by recycling to the plasma membrane for weak interactions (class A GPCRs), or sustained internalization into endosomes for stronger interactions (class B GPCRs) (Oakley et al., 1999, 2000). Furthermore, βarrs themselve serve as an alternative signaling system by acting as adaptors and scaffolds to interact with numerous signaling molecules (Pierce et al., 2002).
Thus, our current understanding features G protein signaling originating at the cell surface, followed by rapid βarr-mediated quenching of G protein signaling, both by competition with G proteins for receptor interaction and by internalization of the receptors. However, recent findings have begun to challenge these paradigms. A number of GPCRs have been reported to engage in sustained G protein signaling, rather than being desensitized after initial agonist stimulation (Calebiro et al., 2009; Feinstein et al., 2013; Ferrandon et al., 2009; Irannejad et al., 2013; Mullershausen et al., 2009). Interestingly, this newly appreciated sustained phase of G protein activation appears to be mediated by internalized receptors in endosomes, where they modulate effectors, such as adenylyl cyclase (Calebiro et al., 2009; Feinstein et al., 2011, 2013; Ferrandon et al., 2009; Irannejad et al., 2013; Mullershausen et al., 2009). These findings cannot be accommodated within the traditional model of GPCR signaling systems since prolonged residence of a GPCR in endosomes requires a persistent “class B” interaction of βarr with the receptors, which should prevent G protein activation (Feinstein et al., 2011, 2013; Wehbi et al., 2013).
Recent X-ray crystallographic findings of the β2-adrenergic receptor (β2AR) in complex with Gs have indicated that the receptor forms a highly engaged complex with Gs that involves interactions of both the N-terminal and C-terminal domains of the Gαs subunit with intracellular loop 2, transmembrane domain 5 (TM5), and TM6 of the β2AR (Rasmussen et al., 2011). A negative-stain electron microscopy (EM) study of GPCR-βarr complexes using a β2V2R receptor chimera (β2AR where the C-terminal tail was exchanged for the vasopressin type 2 receptor [V2R] C-terminal) revealed that βarr assumes two different conformations (Shukla et al., 2014); one in which the βarr is bound only to the phosphorylated receptor C-terminal tail and appears to hang from the receptor (“tail” conformation) and a second more fully engaged conformation, in which, in addition to the tail interaction, a flexible loop in βarr, termed the fingerloop, inserts into the transmembrane core of the receptor (“core” conformation). An arrangement similar to this “core” conformation was recently described in a crystal structure of a rhodopsin-visual arrestin complex (Kang et al., 2015).
The observation of the “tail” conformation of the GPCR-βarr complex, in which the entire receptor cytoplasmic surface encompassing intracellular loops 1, 2, and 3 is exposed, raises the question of whether it might be possible for both Gs and βarr to simultaneously interact with the receptor and thus provide a molecular basis for sustained G protein signaling by GPCRs from endosomes. Accordingly, here we set out to test this hypothesis by using a variety of cellular, biochemical, and biophysical approaches.
RESULTS
Real-Time Cyclic AMP Kinetic Studies of Class A and Class B GPCRs
Sustained G protein signaling by internalized GPCRs has been demonstrated for a number of receptors. Interestingly, this feature has been more commonly observed in class B GPCRs, including the parathyroid hormone type 1 receptor (PTHR) (Ferrandon et al., 2009), V2R (Feinstein et al., 2013), and thyroid-stimulating hormone receptor (TSHR) (Calebiro et al., 2009). These receptors form more stable complexes with βarr compared to class A GPCRs, which only transiently interact with βarr (Oakley et al., 2000). Given that class B receptors bind βarr more tightly, and given the well-characterized role of βarr in desensitizing GPCRs, it seems paradoxical that class B receptors, rather than class A receptors, promote sustained G protein signaling.
To directly assess the relative propensity of class A and B GPCRs to promote sustained signaling, we monitored agonist-stimulated Gs signaling, measured here as cyclic AMP (cAMP) accumulation, in HEK293 cells stably expressing ICUE2, a fluorescence resonance energy transfer (FRET) biosensor detecting cytoplasmic cAMP (Violin et al., 2008). This ICUE2 biosensor measures cAMP concentration in real-time and thus represents an equilibrium between production and degradation of cAMP. These cells were transiently transfected with the β2AR as a prototypical class A GPCR or the V2R as a prototypical class B GPCR. In addition, we used a modified version of the β2AR, the β2V2R, in which the β2AR C-terminal tail has been exchanged with the V2R C-terminal tail. The β2V2R maintains the pharmacological properties of the β2AR, but has significantly higher affinity for βarr than for β2AR, and this increase in affinity manifests predominantly as a change in the receptor internalization pattern from class A to B (Oakley et al., 1999, 2000). In addition, we have successfully purified stable and functional GPCR-βarr complexes using the β2V2R. Therefore, the β2V2R was used to study both the cellular and biophysical basis for sustained G protein signaling by internalized class B GPCR-βarr complexes.
Within the first 5 min of agonist challenge β2AR, β2V2R, and V2R all stimulated cAMP production to a similar extent. Beyond 5 min, the cAMP responses were attenuated to different levels among these receptors and most prominently for the wild-type β2AR (Figure 1A). In addition, the agonist-stimulated cAMP response was diminished slightly more at the β2V2R compared to the V2R (Figure 1A). These results suggest that class B GPCRs promote sustained G protein signaling to a greater degree than does a prototypical class A GPCR.
Figure 1. Sustained Gs Signaling from Internalized Compartments by β2AR, β2V2R, and V2R.

(A) Real-time cAMP measurements, using ICUE2-expressing HEK293 cells, in response to agonist stimulation of β2AR (red), β2V2R (blue), and V2R (black). For β2AR and β2V2R, 1 μM ISO was used to stimulate cells. For V2R, 100 nM AVP was used to stimulate cells. Surface expression of all GPCRs was matched. Data represent the mean ± SE of N = 3 experiments and n ≥ 90 cells. Area under the curve (AUC) was used to calculate the total cAMP response for each GPCR, and one-way ANOVA was performed to determine statistical differences relative to β2AR (**p < 0.01; ****p < 0.0001) and β2V2R (##, p < 0.01) responses.
(B) Schematic representation of the experimental design used to demonstrate sustained Gs activation and signaling from internalized GPCRs.
(C) Real-time cAMP measurements utilized to demonstrate intracellular Gs signaling by GPCRs. Agonist-stimulated cAMP responses (100 nM ISO for β2AR and β2V2R or 100 nM of desmopressin [DESMO] for V2R) was antagonized at 10 min by the addition of 10 μM of cell-membrane-impermeable antagonist (CGP-12217 for β2AR and β2V2R, or H-3192 for V2R; shown in blue). The impact of cell-membrane-impermeable antagonists was measured relative to total antagonism caused by cell-membrane-permeable antagonists (ICI-118551 for β2AR and β2V2R or SR121463 for V2R). Data represent the mean ± SE of N = 3 experiments and n ≥ 87 cells. AUC was used to calculate the total cAMP response for each GPCR after the respective treatments. One-way ANOVA was performed to determine statistical differences among the antagonists compared to DMSO (*p < 0.05; ***p < 0.001; ****p < 0.0001) or compared to cell-membrane-impermeable antagonists (#, p < 0.05; ####, p < 0.0001).
See also Figure S1.
To investigate whether sustained G protein signaling of class B GPCRs arises from internalized compartments β2AR, β2V2R, or V2R-expressing cells were agonist-stimulated for 10 min to allow for internalization to occur. Then Gs signaling arising from only the cell membrane was inhibited by the addition of 10 μM of a membrane-impermeable antagonist (CGP-12177 for β2AR and β2V2R, or (d(CH2)51,D-Tyr(Et)2,Val4,Arg8,des-Gly9)-Vasopressin (H-3192) for V2R; Figure 1B) (Jard et al., 1986; Staehelin et al., 1983). To inhibit Gs signaling arising from both the cell membrane and the internalized compartments, 10 μM of a cell-permeable antagonist (ICI-118551 for β2AR and β2V2R or SR121463 for V2R; Figure 1B) was used (Morello et al., 2000; O’Donnell and Wanstall, 1980).
Under these conditions, for the β2AR, both antagonists quickly inhibited almost all Gs signaling (Figure 1C). However, for the β2V2R, only ICI-118551 fully blocked Gs signaling, whereas CGP-12177 only partially inhibited it (Figure 1C). These results indicate that a significant fraction of the β2V2R-stimulated cAMP originates from internalized compartments. In a similar fashion, the V2R-stimulated cAMP response was only partially antagonized when exposed to the cell-membrane-impermeable antagonist, H-3192, but fully antagonized following the addition of the cell-membrane-permeable antagonist, SR121463 (Figure 1C). Therefore, V2R-mediated Gs signaling at 10 min post-agonist-stimulation is, in part, due to internalized receptors. These results demonstrate that both the β2V2R and V2R stimulate Gs signaling from internalized compartments, whereas the β2AR does not seem to exhibit such behavior using this method.
Monitoring Heterotrimeric Gs Activation at Internalized Compartments by Class A and Class B GPCRs Using Bioluminescence Resonance Energy Transfer Biosensors
To confirm that the receptor-stimulated cAMP response from internalized compartments results from Gs activation, and not from other signaling cascades, we directly monitored agonist-stimulated heterotrimeric Gs activation by bioluminescence resonance energy transfer (BRET), which detects the proximity between two proteins within a 10 nm range (Marullo and Bouvier, 2007). Following Gs activation, the Gαs subunit separates from the Gβγ subunits. This separation was detected following agonist challenge of β2AR, β2V2R, or V2R-expressing cells as a decrease in BRET between the functionally validated BRET pair RlucII-117-Gαs and GFP10-Gγ1 (Figures 2A, 2B, and S1). Such BRET-based assays have been developed as sensors of G protein activation (Galés et al., 2006).
Figure 2. Sustained Gs Activation from Internalized Compartments by β2AR, β2V2R, and V2R Assessed by BRET.

(A) Schematic representation of the experimental design used to monitor agonist-promoted Gs activation, which leads to the separation of Gαs and Gβγ subunits, measured by BRET between RlucII-117-Gαs and GFP10-Gγ1.
(B) BRET titration curves obtained using a constant amount of RlucII-Gαs and with increasing amounts of GFP10-Gγ1. BRET was measured 35 min following the addition of agonist or vehicle. Data are pooled from N = 4 experiments.
(C) Relationship between the duration of agonist stimulation time and Gs activation response 20 min after agonist washout. Gs activity was determined by assessing the reduction in BRET signal between RlucII-117-Gαs and GFP10-Gγ1. Surface expression of all GPCRs was matched. Data are shown as a percent of BRET decrease observed in the unwashed condition (i.e., in the continuous presence of agonist) and represents the mean ± SE of N = 4–5 experiments. One-way ANOVA was performed to assess significant differences in Gs response by increasing agonist stimulation time versus pulse stimulation (0.5 min) (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
See also Figure S1.
To specifically monitor Gs activation/separation at internalized compartments, we developed an agonist washout protocol. In short, agonists were allowed to stimulate receptors for varying time intervals (0.5 to 14 min), followed by a washout of the agonists. Next, the cells were incubated for 20 min in agonist-free media, and responses were recorded (Figures 1B and 2C). Since washout only removes agonist from the extracellular environment, but not from the intracellular environment, which contains internalized receptors, the receptors insensitive to agonist washout must be activating the Gs from within internalized compartments (Figure 1B).
As shown in Figure 2C, the agonist washout protocol blunted Gs activation in β2AR, β2V2R, and V2R-expressing cells. However, by increasing the duration of stimulation prior to agonist washout, and thereby allowing more receptors to internalize, a substantially diminished signal reduction by agonist washout was observed (Figure 2C). This dampening in signal reduction was most prominent, and statistically significant, for β2V2R and V2R, which still maintained 54% and 64% of their Gs activity, respectively, when cells were pre-stimulated for 14 min followed by agonist washout, as compared to the unwashed control conditions (Figure 2C). For the β2AR, increasing the duration of stimulation prior to agonist washout did not result in a statistically significant difference in remaining Gs activity when compared to 0.5 min pulse stimulation (Figure 2C). In this setup, the class B GPCRs, β2V2R and V2R, seem to activate heterotrimeric Gs from internalized compartments, whereas the class A GPCR, β2AR, does not to any significant degree.
Co-localization of GPCR, βarr, and Gαs in Endosomes by Confocal Microscopy
Receptor internalization from the plasma membrane into endosomes is driven by formation of a GPCR-βarr complex. Therefore, to investigate whether G protein may interact with GPCR-βarr complexes in endosomes, we began by tracking the cellular localization of functionally validated N-terminal SNAP-tagged GPCRs (SNAP-β2AR, SNAP-β2V2R, or SNAP-V2R) pre-labeled with SNAP-Surface 649 fluorescence substrate, mStrawberry-βarr2, and mEmerald-67-Gαs expressed in HEK293 cells following agonist treatment using confocal microscopy (Figure S1).
Prior to agonist stimulation, SNAP-tagged receptors (β2AR, β2V2R, or V2R) and mEmerald-67-Gαs were predominantly located at the plasma membrane, whereas mStrawberry-βarr2 was evenly distributed throughout the cytoplasm (Figures 3A, S2, and S3A). Following agonist stimulation, mEmerald-67-Gαs rapidly translocates from the plasma membrane to the cytoplasm, and after 5 min of receptor stimulation mEmerald-67-Gαs was predominantly distributed within the cytoplasm (Figures 3A, S2, and S3A). In contrast, following agonist stimulation, mStrawberry-βarr2 was recruited from the cytoplasm to the activated SNAP-tagged receptors (5 min post-stimulation) at the plasma membrane followed by GPCR-βarr complex internalization into endosomes (>20 min post-stimulation) (Figures 3A, S2, and S3A). At longer agonist exposure times (>20 min post-stimulation), increased mEmerald-67-Gαs intensity can be visualized in β2V2R-βarr2 and V2R-βarr2 containing endosomes (Figures 3 and S3), but this was not observed for the β2AR, which is likely because it only forms transient complexes with βarr2 (Figure S2). Line-scan analysis of all three fluorophores within these endosomes demonstrates co-localization of β2V2R/V2R, βarr2, and Gαs, supporting the hypothesis that “megaplexes” of class B GPCRs, βarr, and heterotrimeric Gs form in endosomes (Figures 3B, 3C, S2B, and S3C).
Figure 3. Cellular Localization of SNAP-β2V2R Pre-labeled with SNAP-Surface 649 Fluorescent Substrate, mStrawberry-βarr2, and mEmerald-67-Gαs Visualized by Confocal Microscopy.

(A) Cellular localization of SNAP-β2V2R (649), mStrawberry-βarr2, and mEmerald-67-Gαs prior to agonist addition (0 min) or 5 min and >20 min after 10 μM ISO treatment (100× objective, N = 4 experiments, n = 49 cells).
(B) Representative endosome (orange dotted box) demonstrating co-localization of SNAP-β2V2R (649), mStrawberry-βarr2, and mEmerald-67-Gαs at >20 min post-ISO addition.
(C) Line-scan analysis of representative endosomal fluorophore intensities.
See also Figures S1, S2, and S3.
Agonist-Stimulated Interaction between βarrs and Gs Subunits
To confirm close molecular proximity between different megaplex components following receptor stimulation, we utilized BRET. If megaplexes form, G protein and βarr will simultaneously interact with a single active receptor, and thus, measurement of BRET between functionally validated Gs subunits (Gαs or Gγ2) and βarr1/2 following agonist stimulation can serve to detect these complexes (Figures 4A and S1). In BRET titration experiments, agonist-stimulation for 20 min of either β2V2R or V2R increased the BRET signal between RlucII-67-Gαs and GFP10-βarr1/2 (Figures 4B and 4D). A significant agonist-promoted BRET increase between RlucII-Gγ2 and GFP10-βarr1/2 was also detected, indicating that the agonist-promoted recruitment of βarr1/2 to the β2V2R or V2R brings βarr1/2 into close proximity to both the Gαs and Gβγ subunits. In contrast, no change in the BRET signal was detected between GFP10-βarr1/2 and either RlucII-67-Gαs or RlucII-Gγ2 following ISO treatment of β2AR-transfected HEK293 cells (Figures 4B and 4D). Interestingly, in BRET kinetic experiments, which are slightly more sensitive than titration experiments, a weak ISO-promoted BRET signal was observed between GFP10-βarr1/2 and RlucII-67-Gαs but not RlucII-Gγ2 in cells expressing β2AR. (Figures 4C and 4E). In BRET kinetic experiments, for both β2V2R and V2R, the agonist-mediated signals between βarr1/2 and both RlucII-67-Gαs and RlucII-Gγ2 were pronounced and rapidly increased until plateauing ~10 min after agonist treatment.
Figure 4. Interaction between βarr1/2 and Either Gαs or Gγ2 following Agonist Stimulation of β2AR, β2V2R, or V2R.

(A) Schematic representation of the experimental design used to monitor agonist-promoted BRET between RlucII-67-Gαs (1), RlucII-Gγ2 (2), or RlucII-CD8 (3) and GFP10-βarr1/2.
(B and D) BRET titration curves using a constant amount of RlucII-67-Gαs, RlucII-Gγ2, or RlucII-CD8 and increasing amounts of GFP10-βarr1 (B) or GFP10-βarr2 (D) monitored 20 min after agonist stimulation. Data are expressed as net BRET absolute values and represent the mean ± SE and are pooled from N = 3–5 experiments. Surface expression of all GPCRs was matched.
(C and E) Kinetics of agonist-promoted BRET between GFP10-βarr1 (C) or GFP10-βarr2 (E) and RlucII-Gαs, RlucII-Gγ2, or RlucII-CD8 obtained for all three GPCRs. Each kinetic point represents the mean ± SE of ΔBRET between agonist-stimulated and vehicle-treated conditions (N = 3–10 experiments). Two-way ANOVA was performed to determine significant differences between CD8 condition and Gαs or Gγ2 for each time point (a p < 0.05; b p < 0.01; c p < 0.001; d p < 0.0001).
See also Figures S1 and S5.
In the resting state heterotrimeric Gs is initially located at the cell membrane and βarr is dispersed within the cytoplasm, thus, we tested whether the BRET signals detected following agonist stimulation could have arisen from random collisions between the plasma membrane-translocated βarr and any membrane proteins. To control for this possibility, BRET was measured between the membrane protein RlucII-CD8 and GFP10-βarr1/2 following agonist-stimulation of β2AR, β2V2R or V2R (Figure 4A). In this setup, no BRET response was observed following agonist stimulation of any of the receptors indicating that the BRET detected between βarr and both Gαs and Gγ2 reflects molecular proximity consistent with the formation of megaplexes (Figures 4B–4E). Taken together, these experiments provide further evidence that megaplexes containing receptor, βarr and Gs form robustly at both the β2V2R and V2R.
GPCR-βarr Fusion Proteins Activate Heterotrimeric G Protein following Agonist Stimulation
The BRET and confocal data shown thus far support the existence of megaplexes and suggest that these complexes occur more readily for the class B GPCRs, β2V2R and V2R, and minimally for the class A β2AR. However, these results do not demonstrate whether GPCR-βarr complexes can directly activate G protein. To investigate whether GPCR–βarr complexes can interact with, and activate, G protein in a cellular environment we generated fusion proteins of GPCR–βarr and investigated their ability to activate Gs in HEK293 cells. We used the β2V2R as our model class B GPCR to be consistent with the biophysical experiments in this study where we assessed the ability of a purified β2V2R-βarr1 complex to interact with, and activate, Gs (see below). To date, we have been unable to purify biochemically functional GPCR-βarr complexes using other class B GPCRs, such as the V2R. The β2V2R has been rigorously characterized both herein and in previous studies; it displays similar biological properties to the V2R, as well as to other class B GPCRs, and has been routinely used as a robust model class B GPCR (Lee et al., 2016; Oakley et al., 1999, 2000; Tohgo et al., 2003).
Initially, we tested the functionality and stability of the β2V2R–βarr1/2 fusions. Immunoprecipitation (IP) of the FLAG-tagged fusion proteins, expressed in HEK293 cells, confirmed that the fusions were intact and not subjected to cellular cleavage (Figures S4A and S4B). When compared with β2V2R, ISO-promoted displacement of 125I-cyanopidolol (CYP) binding for both β2V2R–βarr1/2 fusions revealed a biphasic curve reflecting a higher affinity state for agonist (Figure 5A): β2V2R–βarr1 dissociation constant logKiHi = −7.11 ± 0.03 (41%) and logKiLo = 6.01 ± 0.03 (59%); and β2V2R–βarr2 dissociation constant logKiHi = −8.29 ± 0.03 (49%) and logKiLo = −6.47 ± 0.03 (51%). These results are characteristic of the allosteric effect of βarr on receptor binding properties, as previously reported (Gurevich et al., 1997), and confirm the functional interaction between the two moieties of the fusions. Fusion to βarr had no effect on the affinity of the antagonist ICI-118551 (Figure 5A). When expressed in HEK293 cells both fusions constitutively internalize resulting in a relatively low amount of β2V2R–βarr1/2 fusions being present at the cell membrane (Figure 5B). This internalization pattern further shows that βarr, as a fusion partner, retains its functionality to promote βarr-mediated receptor endocytosis. Furthermore, when stimulated with ISO, the β2V2R–βarr1/2 fusions promote ERK1/2 phosphorylation at 10 min post-stimulation (Figures 5C and S4C). These results indicate that both individual proteins, of the β2V2R–βarr1/2 fusions, are functional when expressed in cells.
Figure 5. Functionality and Capability of β2V2R-βarr1/2 Fusions to Activate Gs in HEK293 Cells.

(A) Functional assessment of β2V2R-βarr1/2 fusions using radioligand competition binding experiments. Both agonist (ISO) and antagonist (ICI-118551) successfully competed off [125I]-CYP at β2V2R, β2V2R-βarr1 and β2V2R-βarr2. Data represent the mean ± SE of N = 3–4 experiments.
(B) Cellular localization of SNAP-β2V2R and SNAP-β2V2R-βarr1/2 fusions pre-labeled with SNAP-Surface 549 fluorescent substrate (549) using confocal microscopy (100× objective, N = 3 experiments, and n ≥ 16 cells).
(C) Characterization of 1 μM ISO-stimulated ERK1/2 phosphorylation response at 10 min post-stimulation in mock, β2V2R, β2V2R-βarr1, and β2V2R-βarr2-transfected cells (N = 6 experiments).
(D) ISO-stimulated Gs activation in mock (gray), β2V2R (black), β2V2R-βarr1 (red), and β2V2R-βarr2 (blue) transfected cells determined by BRET titration curves 30 min after stimulation (N = 4 experiments).
(E) Real-time cAMP measurement, utilizing HEK293-ICUE2 cells, in response to ISO-stimulation of β2V2R (black), β2V2R-βarr1 (red), and β2V2R-βarr2 (blue). Data represent the mean ± SE of N = 3 experiments and n ≥ 93 cells. Surface expression of GPCRs was matched in all experiments.
See also Figures S1 and S4.
To test whether β2V2R, as part of the β2V2R–βarr fusions, retains its ability to activate Gs, changes in BRET were measured between RlucII-117-Gαs and GFP10-Gγ1 in response to ISO stimulation as compared to vehicle treatment. As shown in Figure 5D, ISO stimulation of β2V2R–βarr1/2 induced a decreased BRET between RlucII-117-Gαs and GFP10-Gγ1, reflective of the Gs subunits separation and activation, although to a lesser extent than non-fused β2V2R. These results demonstrate that both β2V2R–βarr1/2 fusions can activate Gs to some degree, following agonist-mediated receptor stimulation, despite their constant coupling to functional βarr1/2.
To further confirm the ability of the β2V2R–βarr1/2 fusions to activate Gs, real-time kinetic studies of ISO-stimulated cAMP production were undertaken. Both β2V2R–βarr1/2 fusions were able to initiate Gs signaling (Figure 5E), providing further support that G proteins are capable of being activated by GPCR–βarr complexes.
In Vitro Formation and Isolation of Megaplexes
To further investigate whether GPCRs can form megaplexes by simultaneously interacting with βarr and heterotrimeric G protein, we attempted to form such megaplexes in vitro and to then isolate them by co-immunoprecipitation (coIP). For this study, we used the FLAG-β2V2R expression construct to form stable complexes with βarr1 in sf9 insect cells, as previously described (Shukla et al., 2014). Following stimulation with the high-affinity agonist, BI-167107 (BI), β2V2R forms appreciable amounts of complex with βarr1 that can be purified by affinity purification and size-exclusion chromatography (SEC). To obtain highly stable β2V2R–βarr1 complexes, that remain intact throughout the purification, we added the conformationally active antibody binder, Fab30, which binds to and stabilizes active βarr1 conformations (Shukla et al., 2013, 2014). We were unable to obtain monodispersed and functional V2R–βarr1 complexes using this approach (data not shown).
To test whether this Fab30-stabilized BI-occupied β2V2R–βarr1 complex (Fab30 complex) interacts with the heterotrimeric Gs, we added purified Gs in excess to the Fab30 complex and pulled-down the FLAG-β2V2R using M1 anti-FLAG beads. As shown in Figure 6A, βarr1 and all three components of the heterotrimeric Gs (Gαs, Gβ1, and Gγ2) were pulled-down together with FLAG-β2V2R in a stoichiometric fashion. A similar result was observed when using protein A/G agarose beads, which bind Fab30, to pull-down the individual components of the megaplex, confirming that it forms in vitro (Figure 6A).
Figure 6. In Vitro Formation and Functional Characterization of the Megaplex.

(A) Coomassie-stained gels of representative coIP experiments of the megaplex by either M1 anti-FLAG beads (to pull-down FLAG-β2V2R; left) or protein A/G agarose beads (to pull-down Fab30; right) (N = 4 experiments).
(B) Schematic presentation of the biochemical steps in G protein activation in the megaplex: (1) heterotrimeric G protein is recruited to the GPCR-βarr “tail” conformation to form the megaplex in an agonist-dependent manner; (2) activated receptor in the megaplex stimulates GDP-GTP exchange in the heterotrimeric G protein, causing activation and separation of the Gαs subunit; and (3) activated Gαs subunit has intrinsic GTPase activity causing hydrolysis of GTP to GDP and inorganic phosphate (Pi).
(C) M1 anti-FLAG coIP experiment of BI-occupied β2V2R, Fab30 complex, or Cz-occupied β2V2R both with and without heterotrimeric Gs present. Gs binding was determined and quantified by western blot using an anti-Gαs antibody. Data represent the mean ± SE of N = 4 experiments. One-way ANOVA was performed with pairwise comparison to BI-β2V2R (****p < 0.0001).
(D) M1 anti-FLAG coIP experiment with either BI-occupied β2V2R-Gs complex or megaplex in presence of control buffer, 20 μM GDP, or 20 μM GTPγS. Gαs subunit separation was determined and quantified by western blot by using an anti-Gαs antibody. Data represent the mean ± SE of N = 4 experiments. Two-way ANOVA was performed to assess significant differences between control buffer (*p < 0.05; **p < 0.01; ****p < 0.0001) and GDP (####, p < 0.0001). There were no statistical differences between the BI-occupied β2V2R-Gs complex and the megaplex.
(E) Characterizing the ability of BI-occupied β2V2R (top) or Fab30 complex (bottom) to modulate GDP-GTP exchange and Gs activity via GTPase activity. Data represent the mean ± SE of N = 5–6 experiments. Two-way ANOVA was performed to test the effect of Gs presence at each receptor/complex concentrations (**p < 0.01; ****p < 0.0001), and one-way ANOVA tests the effect on Gs modulation by different receptor/complex concentrations (#, p < 0.05; ####, p < 0.0001).
See also Figure S5.
Megaplex In Vitro Functionality
To test the functionality of the receptor in the megaplex we first investigated whether binding of the heterotrimeric Gs to the Fab30 complex is agonist-dependent by FLAG-tag coIP (Figure 6C). The Fab30 complex was able to bind Gs to a similar extent as agonist BI-bound β2V2R (Figures 6C and S5A). In contrast, the antagonist carazolol (Cz)-bound β2V2R had almost no ability to bind Gs indicating that the binding of the heterotrimeric Gs to β2V2R in the Fab30 complex is dependent on an agonist-stabilized active receptor conformation (Figures 6C and S5A).
We next investigated whether the β2V2R maintains its guanine nucleotide exchange factor (GEF) functionality with respect to the heterotrimeric Gs when residing in the megaplex. To assess GEF functionality, the megaplex was formed in the presence of either GDP or non-hydrolysable GTPγS. An exchange of GDP to GTPγS in the heterotrimeric Gs causes activation of the Gαs subunit and separation from the Gβγ subunits (Figure 6B). This separation event was followed by a FLAG-tag coIP. The addition of GDP caused a small decrease in Gs binding to the BI-occupied β2V2R and Fab30 complex (Figures 6D and S5B). However, the addition of GTPγS led to a nearly complete separation of the Gαs subunit from both the BI-occupied β2V2R and Fab30 complex (Figures 6D and S5B). This dramatic effect indicates that the receptor retains its GEF functionality while residing in the megaplex and can promote G protein activation and separation. Interestingly, unlike the Gαs subunit, the Gβγ subunits remained in complex with both the Fab30 complex and BI-occupied β2V2R after GTPγS treatment (Figure S5B).
Once activated by the receptor, the Gαs subunits display intrinsic GTPase activity. To further characterize the β2V2R functionality in the megaplex, modulation of Gs, measured as GTPase activity by the Fab30 complex, was investigated. As shown in Figure 6E, the Fab30 complex does indeed positively modulate the GTPase activity of the Gs, and to a similar level as the BI-occupied β2V2R control.
These in vitro experiments clearly show that the receptor, in the megaplex, retains its full capability to both interact with and to activate heterotrimeric Gs.
Structural Studies of the Megaplex Using Electron Microscopy
To investigate the architecture of the megaplex, we formed complexes on a preparative scale and isolated them by SEC (Figures S6A and S6B). To increase the overall stability and homogeneity of the megaplex preparation, we employed two strategies, which were previously utilized to form stable β2AR–Gs complexes (Rasmussen et al., 2011): (1) we removed GDP from the heterotrimeric Gs by addition of apyrase; and (2) we stabilized the nucleotide-free transition state of the Gs, which is known to interact strongly with the β2AR transmembrane core region by adding the conformationally active nanobody binder, Nb35. These two strategies resulted in a stable and monodisperse preparation of megaplexes as assessed by SEC and conventional negative-stain EM (Figures 7 and S6) (Peisley and Skiniotis, 2015). The β2V2R construct used for our in vitro studies was engineered to contain an N-terminal T4-lysozyme fusion, (T4L) β2V2R, which can be used as a marker for receptor orientation in EM studies (Shukla et al., 2014; Westfield et al., 2011).
Figure 7. Single-Particle EM Analysis of the (T4L) β2V2R-Gs-Nb35-βarr1-Fab30 Megaplex.

(A) Representative EM image of negative-stained megaplex.
(B) Representative class averages of the megaplex (135 total particle projections).
(C) Class averages of the previously published (T4L) β2AR-Gs-Nb35 complex and the (T4L)β2V2R-βarr1-Fab30 complex in the “tail” conformation (images reprinted and modified from Shukla et al., 2014; Westfield et al., 2011). Superimposition of these averages results in a density map identical to the one representing the megaplex. The scale bars in (A–C) correspond to 100, 10, and 10 nm, respectively.
See also Figures S6 and S7.
To visualize the structure of the in vitro reconstituted megaplex we applied classification and averaging of the EM particle projections (Figure S6C). The class averages revealed a complex architecture with distinct features that allow domain assignment, with the T4L marking the receptor extracellular face and the heterotrimeric Gs bound diametrically opposite at the intracellular side of β2V2R, in a configuration that appears identical to the previously characterized (T4L) β2AR–Gs (Figures 7B and 7C) (Westfield et al., 2011). Additional density attributed to βarr1-Fab30 is observed on the side of the β2V2R–Gs complex. This configuration of βarr1, which interacts with the phosphorylated β2V2R C-terminal tail, is reminiscent of the ‘tail’ conformation of the (T4L)β2V2R–βarr1-Fab30 complex that we previously reported by EM (Shukla et al., 2014). In fact, overlaying the averages of the (T4L)β2AR–Gs complex and the ‘tail’ configuration of the (T4L)β2V2R–βarr1-Fab30 complex (using receptor and T4L densities as the common features) results in a projection that appears almost identical to the one of the megaplex (Figures 7B and 7C). In this super-complex, βarr1 is positioned adjacent to the Gβγ subunits and several class averages indicate a direct interface between Gβγ and βarr1 (Figure S6C). This possible Gβγ–βarr1 interaction was further explored by glutathione sepharose (GST) pull-down assays, whereby an interaction between the GST-βarr1, in complex with Fab30 and the phosphorylated V2R C-terminal peptide (V2Rpp), and heterotrimeric Gs was observed (Figure S7). Interestingly, when the Gβγ subunits were separated from the Gαs subunit by addition of non-hydrolysable GTP surrogate, GDP·AlF4−, Gβγ binds GST-βarr1 more prominently whereas Gαs subunit association with GST-βarr1 diminishes (Figures S7B and S7C). These results suggest that a direct interaction occurs between βarr1 and Gβγ subunits. Consistent with this finding, we further demonstrated that expression of the Gβγ scavenger, T8-βARKct, significantly reduced the β2V2R-stimulated BRET response between RlucII-Gγ2 and GFP10-βarr1 in HEK293 cells (Figure S7D).
DISCUSSION
Sustained G protein signaling by internalized GPCRs has been difficult to incorporate within the classical understanding of GPCR signaling since receptor internalization is thought to be driven by the formation of GPCR-βarr complexes and because βarr plays a fundamental role in the desensitization of GPCR-mediated G protein signaling. Thus, we found it surprising that class B GPCRs, including PTHR, V2R, and TSHR, which are known to interact tightly with βarr, have been shown to promote sustained G protein signaling from internalized compartments (Calebiro et al., 2009; Feinstein et al., 2013; Ferrandon et al., 2009; Wehbi et al., 2013). In the current study we directly demonstrate that by exchanging the C-terminal tail of the β2AR with the V2R C-terminal tail (β2V2R), the receptor internalization pattern changes from class A to B and that the β2V2R chimera displays behavior similar to the V2R (Figures 3A and S2) (Oakley et al., 1999, 2000). Interestingly, as observed with the V2R, this modification significantly enhances the ability of the β2V2R, relative to β2AR, to promote sustained G protein activation and signaling from internalized compartments (Figures 1 and 2). Previous findings suggest a role for βarr in sustained G protein signaling by the PTHR and V2R. Co-expression of a constitutively active version of βarr1 enhances sustained G protein signaling at the PTHR and V2R, and such signaling was diminished by small interfering RNA (siRNA) knockdown of both βarr1/2 (Feinstein et al., 2011, 2013; Wehbi et al., 2013). In contrast, for the class A GPCR, β2AR, desensitization of Gs signaling is enhanced by a constitutively active βarr1, but diminished by siRNA knockdown of βarr1/2 (Violin et al., 2008; Wehbi et al., 2013).
How is βarr involved in receptor-mediated G protein activation at internalized GPCRs? Several studies with the PTHR, V2R, TSHR, δ opioid, and CCR1 (Audet et al., 2012; Calebiro et al., 2009; Feinstein et al., 2013; Ferrandon et al., 2009; Gilliland et al., 2013; Wehbi et al., 2013) indirectly support our megaplex hypothesis for GPCRs. In the present study, however, we provide direct evidence for formation of megaplexes; super-complexes in which the heterotrimeric Gs subunits come into close proximity with βarr1/2 following stimulation of the β2V2R or V2R (Figure 4). This event seems to occur at internalized receptors (Figure 3). Furthermore, following agonist addition, β2V2R-βarr1/2 fusions retain the ability to activate Gs and promote signaling (Figures 5 and S4). We demonstrated in vitro that the receptor in a purified agonist-occupied β2V2R-βarr1-Fab30 complex interacts strongly with heterotrimeric Gs through its transmembrane core, while it couples simultaneously with βarr1 through the receptor C-terminal tail (Figures 6 and 7). This megaplex architecture allows the receptor to promote GDP-GTP exchange and to activate Gs (Figures 6, S5, and S6) and thus explains how βarr can drive receptor internalization without interfering with G protein coupling to the receptor.
Why does βarr partake in sustained internalized G protein activation at class B GPCRs, while desensitizing it at others, such as the prototypical class A GPCR, β2AR? The current study suggests that a strong interaction between the GPCR C-terminal tail and βarr is required to robustly form a megaplex (Figures 4, 6, and 7). To obtain a highly stable β2V2R-βarr1 complex, used for the aforementioned structural studies, this complex was engineered to have a strong interaction between the receptor C-terminal tail and βarr1, which was accomplished by exchanging the β2AR C-terminal tail with the V2R C-terminal tail and stabilizing the active conformation of βarr1 with Fab30 (Shukla et al., 2014). β2V2R and βarr1 only interact through the V2R C-terminal tail region in the “tail” conformation, which is the arrangement that allows the receptor to interact with Gs. Thus, in order to form megaplexes, it might be a requirement for GPCRs to have a C-terminal tail that promotes a strong interaction with βarr following phosphorylation. Class B GPCRs have been reported to contain highly conserved serine/threonine phosphorylation site clusters in their C-terminal tails, which are critical for the formation of highly stable GPCR–βarr complexes (Oakley et al., 2001; Vilardaga et al., 2002). Therefore, a dependency on a strong C-terminal tail interaction, that promotes the “tail” conformation, could explain why class B GPCRs form megaplexes that lead to G protein signaling from internalized compartments. On the other hand, class A GPCRs lack serine/threonine clusters at their C-terminal tails and, thus, promote a transient interaction between the GPCR C-terminal tail and βarr (Oakley et al., 2001). Therefore, formation of class A GPCR-βarr complexes, might be dependent on the interaction between the receptor transmembrane core and βarr, which would explain why class A GPCRs only form megaplexes to a limited degree.
Overall, our results indicate that megaplexes of a single class B GPCR, βarr, and heterotrimeric G protein exist and may explain the recently appreciated phenomenon of sustained G protein signaling from endosomes.
EXPERIMENTAL PROCEDURES
Real-Time Measurement of cAMP Production
HEK293-ICUE2 cell lines transiently transfected with GPCRs were imaged in the dark, on a 37°C temperature-controlled stage, and for the entire stimulation experiment by using a DeltaVision Deconvolution microscope (GE Healthcare) with a Coolsnap HQ2 CCD camera (Photometrics) controlled by SoftWoRx 6.1 (GE Healthcare). Dual-emission ratio imaging used a CFP/YFP dichroic mirror and 470 ± 24 nm and 535 ± 25 nm emission filters for CFP and YFP, respectively.
BRET Assay
Following agonist stimulation, transfected HEK293 cells were incubated at 37°C and luciferase substrate coelenterazine 400a was added 5 min prior to reading BRET in a Synergy Neo microplate reader (BioTek) equipped with an acceptor filter (515 ± 30 nm) and donor filter (410 ± 80 nm). The BRET signal was determined as the ratio of light emitted by GFP10-tagged biosensors (energy acceptors) and light emitted by RlucII-tagged biosensors (energy donors).
Confocal Microscopy
HEK293 cells transfected with fluorescence protein were fixed with ice-cold 6% formaldehyde diluted in DPBS prior to, or at different time points, during stimulation of the GPCRs. Confocal images were obtained on a Zeiss LSM510 laser-scanning microscope using multi-track sequential excitation (488, 568, and 633 nm) and emission (515–540, 585–615, and 650 nm) filter sets.
Co-immunoprecipitation of In Vitro Complexes
Fab30 complex, BI-β2V2R, or Cz-β2V2R were mixed with Gs in a molar ratio of 1:1.5 in presence of control buffer, 100 nM BI, 100 nM Cz, 20 μM GDP, or 20 μM GTPγS. Next, FLAG-β2V2R was immobilized on M1 anti-FLAG agarose beads followed by extensive wash. Finally, FLAG-β2V2R and associated proteins were eluted by elution buffer containing 1 mg/ml FLAG peptide.
Megaplex Preparation for Structural Studies
To form stable megaplexes, Fab30 complex was incubated with Gs and Nb35 in a molar ratio of 1:1.5:3 for 1 hr at room temperature. Megaplex was treated with 25 mU/ml of apyrase for 1 hr, and the CaCl2 concentration was adjusted to 4 mM. Finally, the megaplex was purified on an SEC column (Superdex 200, 16/600, GE Healthcare).
Electron Microscopy
Megaplexes were prepared for EM using conventional uranyl formate negative staining. The negative-stained sample was imaged at room temperature with a Tecnai T12 electron microscope operated at 120 kV using low-dose procedures. Images were recorded at a magnification of 71,138× and a defocus value of ~1.5 μm on a Gatan US4000 CCD camera. All images were binned (2 × 2 pixels) to obtain a pixel size of 4.16 Å on the specimen level.
See the Supplemental Experimental Procedures for a detailed description of all experimental procedures.
Supplementary Material
Highlights.
Some GPCRs simultaneously interact with both G protein and β-arrestin (βarr)
In these “megaplexes,” G protein binds to the receptor transmembrane core
Concurrent with G protein coupling, βarr binds to the receptor C-terminal tail
G protein activation within megaplexes occurs from internalized compartments
Acknowledgments
We are grateful to L. Barak and B. Kobilka for generous gifts of plasmids encoding mStrawberry-βarr2 and Nb35, respectively. We thank C.-R. Liang, L.-L. Gu, J.-M. Shan, and X. Chen for synthesizing BI-167107. We thank S. Johnson, C. Le Gouill, A. Laperrière, M. Walters, M. DeLong, M. Plue, T. Milledge, D. Capel, X. Jiang, R. Irannejad, and M. von Zastrow for support and discussion. This work received supported from the Danish Council for Independent Research & Lundbeck Foundation (to A.R.B.T.), NIH grants (F30HL129803 to T.J.C.; RO1GM083118 to R.K.S.; T32GM007767 to J.P.M.; DK090165 to G.S.; and HL16037 to R.J.L.); CIHR grants (post-doctoral fellowship to B. Plouffe; SNSF P1EZP3_165219 to F.M.H.; and MOP10501 to M.B.). R.J.L. is a HHMI Investigator. R.J.L. is a co-founder and shareholder of Trevena. M.B. holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, A.R.B.T., B. Plouffe, T.J.C., G.S., M.B., and R.J.L.; Investigation, A.R.B.T., B. Plouffe, T.J.C., A.K.S., J.T.T., A.M.D., A.W.K., R.T.S., B. Pani, and F.M.H.; Resources, A.R.B.T., A.K.S., R.T.S., J.P.M., L.-Y.H., and B.B.; Writing – Original Draft, A.R.B.T., B. Plouffe, T.J.C., G.S., M.B., and R.J.L.; Writing – Review and Editing, A.R.B.T., B. Plouffe, T.J.C., A.K.S., R.T.S., J.P.M., R.K.S., G.S., M.B., and R.J.L.; Visualization, A.R.B.T., B. Plouffe, T.J.C., J.T.T., and G.S.; Funding Acquisition, A.R.B.T., B. Plouffe, T.J.C., J.P.M., R.K.S., G.S., M.B., and R.J.L.; Supervision, R.K.S., G.S., M.B., and R.J.L.
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.07.004.
References
- Audet N, Charfi I, Mnie-Filali O, Amraei M, Chabot-Doré AJ, Millecamps M, Stone LS, Pineyro G. Differential association of receptor-Gβγ complexes with β-arrestin2 determines recycling bias and potential for tolerance of δ opioid receptor agonists. J Neurosci. 2012;32:4827–4840. doi: 10.1523/JNEUROSCI.3734-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, Vilardaga JP. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284. doi: 10.1038/nchembio.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Yui N, Webber MJ, Wehbi VL, Stevenson HP, King JD, Jr, Hallows KR, Brown D, Bouley R, Vilardaga JP. Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem. 2013;288:27849–27860. doi: 10.1074/jbc.M112.445098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742. doi: 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galés C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Gilliland CT, Salanga CL, Kawamura T, Trejo J, Handel TM. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, β-arrestin-mediated internalization. J Biol Chem. 2013;288:32194–32210. doi: 10.1074/jbc.M113.503797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jard S, Gaillard RC, Guillon G, Marie J, Schoenenberg P, Muller AF, Manning M, Sawyer WH. Vasopressin antagonists allow demonstration of a novel type of vasopressin receptor in the rat adenohypophysis. Mol Pharmacol. 1986;30:171–177. [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, Luttrell LM. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature. 2016;531:665–668. doi: 10.1038/nature17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marullo S, Bouvier M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol Sci. 2007;28:362–365. doi: 10.1016/j.tips.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Laperrière A, Bernier V, Arthus MF, Lonergan M, Petäjä-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–434. doi: 10.1038/nchembio.173. [DOI] [PubMed] [Google Scholar]
- O’Donnell SR, Wanstall JC. Evidence that ICI 118, 551 is a potent, highly Beta 2-selective adrenoceptor antagonist and can be used to characterize Beta-adrenoceptor populations in tissues. Life Sci. 1980;27:671–677. doi: 10.1016/0024-3205(80)90008-9. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- Peisley A, Skiniotis G. 2D projection analysis of GPCR complexes by negative stain electron microscopy. Methods Mol Biol. 2015;1335:29–38. doi: 10.1007/978-1-4939-2914-6_3. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehelin M, Simons P, Jaeggi K, Wigger N. CGP-12177. A hydrophilic beta-adrenergic receptor radioligand reveals high affinity binding of agonists to intact cells. J Biol Chem. 1983;258:3496–3502. [PubMed] [Google Scholar]
- Szczepek M, Beyrière F, Hofmann KP, Elgeti M, Kazmin R, Rose A, Bartl FJ, von Stetten D, Heck M, Sommer ME, et al. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Krasel C, Chauvin S, Bambino T, Lohse MJ, Nissenson RA. Internalization determinants of the parathyroid hormone receptor differentially regulate beta-arrestin/receptor association. J Biol Chem. 2002;277:8121–8129. doi: 10.1074/jbc.M110433200. [DOI] [PubMed] [Google Scholar]
- Violin JD, DiPilato LM, Yildirim N, Elston TC, Zhang J, Lefkowitz RJ. beta2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J Biol Chem. 2008;283:2949–2961. doi: 10.1074/jbc.M707009200. [DOI] [PubMed] [Google Scholar]
- Wehbi VL, Stevenson HP, Feinstein TN, Calero G, Romero G, Vilardaga JP. Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. Proc Natl Acad Sci USA. 2013;110:1530–1535. doi: 10.1073/pnas.1205756110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfield GH, Rasmussen SG, Su M, Dutta S, DeVree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, et al. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci USA. 2011;108:16086–16091. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.