

Abstract
A nonmotile, Gram-positive bacterium that shows an elongated and branching cell shape was isolated from soil samples from the botanical garden of Ulm University, Ulm, Germany. Here, the isolation procedure, identification, genome sequencing and metabolic features of the strain are described. Phylogenetic analysis allowed to identify the isolated strain as Oerskovia enterophila. The genus Oerskovia belongs to the family Cellulomonadaceae within the order Actinomycetales. The length of cells of O. enterophila ranges from 1 μm to 15 μm, depending on the growth phase. In the exponential growth phase, cells show an elongated and branching shape, whereas cells break up to round or coccoid elements in the stationary growth phase. The 4,535,074 bp long genome consists of 85 contigs with 3918 protein-coding genes and 57 RNA genes. The isolated strain was shown to degrade numerous complex carbon sources such as cellulose, chitin, and starch, which can be found ubiquitously in nature. Moreover, analysis of the genomic sequence revealed the genetic potential to degrade these compounds.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-017-0244-4) contains supplementary material, which is available to authorized users.
Keywords: Oerskovia, Cellulomonadaceae, Cellulose degradation, Soil bacteria, Phylogenetic analysis
Introduction
Oerskovia enterophila was formerly characterized as Promicromonospora enterophila by Jàger et al. in 1983 [1]. Later, P. enterophila was re-classified as O. enterophila by Stackebrandt et al. [2], since only spore-like elements and no real spores are formed. Furthermore, a phylogenetic tree based on the 16S rRNA gene sequences of strains of the genera Cellulomonas and Promicromonospora shows that O. enterophila did not cluster with the type species of Promicromonospora , Promicromonospora citrea, or Promicromonospora sukumoe [2, 3]. The genus Oerskovia was initially described in 1970 by Prauser et al. [4] and harbors currently four species with O. turbata as type species [2]. Bacteria of the genus Oerskovia belong to the phylum Actinobacteria, which is one of the largest taxonomic units among the domain Bacteria [5]. Bacteria belonging to Actinobacteria show a wide range of G + C-content, from 51% to more than 70% [5–7]. Actinobacteria are widely distributed in terrestrial as well as in aquatic habitats [8, 9]. In general, members of the class Actinobacteria show a high morphological variety, which is also true for species of the genera Oerskovia and Cellulomonas [10]. Furthermore, members of the family Cellulomonadaceae are known for their ability to decompose plant-derived biopolymers such as starch, cellulose or chitin [11]. Due to the close relationship of members of the genera Oerskovia and Cellulomonas [12, 13] it is likely that both share genetic features enabling them to degrade these biopolymers. To investigate the genetic potential for biopolymer degradation, the genome of the isolate was sequenced. Furthermore, a genome wide comparison of the isolated strain with other Oerskovia type strains was performed. Additionally, the isolated strain was aerobically grown on respective carbon sources to validate the functionality of the proposed degradation pathways.
In this contribution, the classification, the metabolic features, and the genome insights of the isolated strain are provided.
Organism information
Classification and features
The isolated strains were identified as Oerskovia enterophila based on 16S rRNA gene sequence identities of more than 99% compared to the type strain of O. enterophila DSM 43852 [14]. All subsequent analyses were performed using the strain designated as O. enterophila VJag. Information regarding the enrichment and isolation procedures as well as identification of Oerskovia strains are described in the Additional files 1 and 2: S1 and S2.
Investigations of the cell morphology of the isolated strain O. enterophila VJag (Table 1) using scanning electron microscopy revealed that cells show different morphologies in exponential and stationary growth stage. In the exponential growth phase, cells show extensive branches with an overall length up to 15 μm, whereas the cells are smaller and less branched in the stationary growth phase (Fig. 1). These different cell morphologies were also previously observed by Stackebrandt et al. [2].
Table 1.
Classification and general features of O. enterophila VJag according to the MIGS recommendations [26]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain: Bacteria | TAS [39] | |
| Phylum: ‘Actinobacteria’ | TAS [5] | ||
| Class: Actinobacteria | TAS [12] | ||
| Order: Actinomycetales | TAS [40–42] | ||
| Family: Cellulomonadaceae | TAS [11, 19] | ||
| Genus: Oerskovia | TAS [4] | ||
| Species: Oerskovia enterophila | TAS [1, 2] | ||
| Strain: VJag (LRIE00000000) | TAS [5, 14] | ||
| Gram stain | Positive | IDA, TAS [4] | |
| Cell shape | Rods | IDA, TAS [4] | |
| Motility | Non-motile | IDA, TAS [4] | |
| Sporulation | Non-sporulating | IDA, TAS [2] | |
| Temperature range | Mesophile | IDA, TAS [4] | |
| Optimum temperature | 28–30 °C | IDA, TAS [4] | |
| pH range, optimum | 3–11, 7 | TAS [1], IDA | |
| Carbon source | glucose, fructose, mannose, galactose, ribose, xylose, cellobiose, maltose, trehalose, saccharose, lactose | IDA, TAS [1, 2, 4] | |
| MIGS-6 | Habitat | Affiliated to gut environments of invertebrates, soil | TAS [11] |
| MIGS-6.3 | Salinity | 5–7% (w/v) | TAS [1] |
| MIGS-22 | Oxygen-requirement | Facultative anaerobe | TAS [4] |
| MIGS-15 | Biotic relationship | Free-living, commensal | IDA, TAS [11] |
| MIGS-14 | Pathogenicity | - | |
| MIGS-4 | Geographic location | Botanical garden of Ulm University, Ulm, Germany | IDA |
| MIGS-5 | Sample collection | January 2013 | IDA |
| MIGS-4.1 | Latitude | 48.42218 °N | IDA |
| MIGS-4.2 | Longitude | 9.95922 °E | IDA |
| MIGS-4.4 | Altitude | - |
aEvidence code - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement; These evidence codes are from the Gene Ontology project [43]. If the evidence is IDA, then the property was directly observed for a live isolate by one of the authors
Fig. 1.
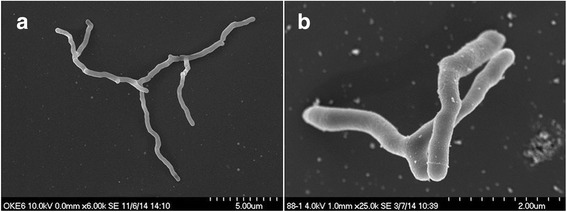
Electron micrograph of O. enterophila VJag, using a Hitachi S-5200 scanning electron microscope. a: cells from the exponential growth phase; scale bar: 5 μm. b: cells from the stationary growth phase; scale bar: 2 μm. Zentrale Einrichtung Elektronenmikroskopie, Universität Ulm
The 16S rRNA gene sequence (OJAG_11220, LRIE01000058.1) of O. enterophila VJag was blasted [15] and used for subsequent phylogenetic analysis. Therefore, 16S rRNA reference sequences of 17 closely related type strains were aligned using MAFFT version 7.215 [16, 17] and was performed using EMBL-EBI web services. The length of the 17 references ranged from 1395 to 1612 bp and had average length of 1486 bp. The phylogenetic tree was reconstructed using the software MrBayes version 3.2.6 [18]. The recommended settings in the manual for tree reconstruction use a generalized time reversible evolutionary model. The quick start instructions were followed to run Bayesian phylogenetic analysis. The run was stopped since the standard deviation of split frequencies was below 0.0042 after 1,000,000 generations.
The resulting phylogenetic tree is shown in Fig. 2. Described species of the genera Oerskovia and Cellulomonas belong to the same family of Cellulomonadaceae . On the other hand, Sanguibacter belongs to the family of Sanguibacteriaceae which is defined as a neighboring group to Cellulomonadaceae [19]. Sanguibacter is the only described genus within the respective family with currently six species [20–24].
Fig. 2.
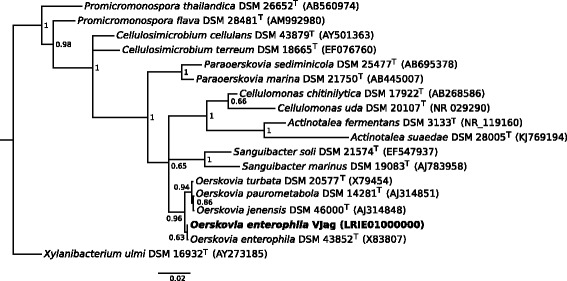
Phylogenetic tree based on the 16S rRNA sequences indicating the genetic relationships between the isolate O. enterophila VJag and other closely related type strains. The scale bar shows 0.02 nucleotide changes per nucleotide position. The phylogenetic tree was created using MrBayes [18] version 3.2.6, sequences were aligned using MAFFT [16, 17]. Numbers at the nodes present the posterior probability
Genome sequencing information
Genome project history
The genome of O. enterophila VJag was sequenced to get insights in the genomic features and the metabolic potential of this strain. Furthermore, no genomes of members of this species were available at the time of writing. A draft sequence is available at NCBI for the species O. turbata NRRL B-8019 (JOFV00000000) [25]. The complete genome of O. enterophila VJag has a size of 4,535,074 bp and consists of 85 contigs. In this contribution the version LRIE01000000 is described. The genome sequencing and gene annotation was performed by Goettingen Genomics Laboratory (Germany). The sequence can be found under the accession number LRIE00000000. Table 2 shows the project information according to MIGS specification [26].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Improved-high-quality draft |
| Two genomic libraries: 454 pyrosequencing shotgun library, Illumina paired-end library | ||
| MIGS-28 | Libraries used | 1 kb insert size |
| MIGS 29 | Sequencing platforms | 454 GS FLX Titanium, Illumina GAII |
| MIGS 31.2 | Fold coverage | 11.46 × 454, 68.28 × Illumina |
| MIGS 30 | Assemblers | MIRA 3.4 and Newbler 2.9 |
| MIGS 32 | Gene calling method | Prodigal |
| Locus Tag | OJAG | |
| GenBank ID | LRIE00000000 | |
| GenBank Date of Release | 20-APR-2016 | |
| GOLD ID | Gp0050669 | |
| BIOPROJECT | PRJNA309230 | |
| MIGS 13 | Source Material Identifier | VJag |
| Project relevance | Investigation of degradation capabilities of O. enterophila VJag |
Growth conditions and genomic DNA preparation
O. enterophila VJag was cultivated in 5 ml TSYE-medium (medium 92, DSMZ) at 28 °C overnight in an orbital shaker at 120 rpm for the isolation of genomic DNA. Genomic DNA was isolated using MasterPure Gram positive DNA Purification kit (Epicentre, Madison, WI, USA) according to the manufacturer’s instructions. DNA concentrations and purity were analyzed using the UV-Vis spectrophotometer NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The genomic DNA yield was 2463 ng/μl. The DNA purity was determined using the UV absorbance ratio 260/208 nm and 260/230 nm and revealed ratios of 2.01 and 2.17, respectively.
Genome sequencing and assembly
A combined approach was used for the whole-genome sequencing of O. enterophila VJag using the 454 GS-FLX TitaniumXL system (titanium GS70 chemistry, Roche Life Science, Mannheim, Germany) and the Genome Analyzer II (Illumina, San Diego, CA). According to the manufacturer’s protocols, the shotgun libraries were prepared, which resulted in 97,681 reads for 454 shotgun sequencing (11.46 × coverage) and 4,756,630 112-bp paired end Illumina reads (68.28 × coverage). Illumina reads were trimmed using Trimmomatic 0.32 [27] to remove sequences with quality scores lower than 20 (Illumina 1.9 encoding) and remaining adaptor sequences, respectively. The initial hybrid de novo assembly was performed using the MIRA 3.4 [28] and Newbler 2.9 (Roche Life Science, Mannheim, Germany) software. The final assembly resulted in 85 contigs with an average coverage of 79.60, an N50 value of 96,617 bp and an N90 value of 28,097 bp, respectively.
Genome annotation
The Prodigal software tool [29] was used for automatic gene prediction [29], rRNA and tRNA gene identification was performed using RNAmmer [30] and tRNAscan [31], respectively. The automatic gene-annotation was performed by using the IMG-ER system [32, 33]. The annotation was manually curated using the Swiss-Prot, TrEMBL, and InterPro databases [34].
Genome properties
The genome of O. enterophila VJag is 4,535,074 bp in length and has an average G + C content of 72.4% (Fig. 3). The genome sequence shows 3975 genes in total, 3918 are protein-coding genes, 57 are RNA genes, of which 6 code for rRNA. The remaining genes code for proteins with unknown function or hypothetical proteins. All statistics and properties are listed in Table 3, the number of protein-coding genes associated with general COG functional categories is shown in Table 4.
Fig. 3.
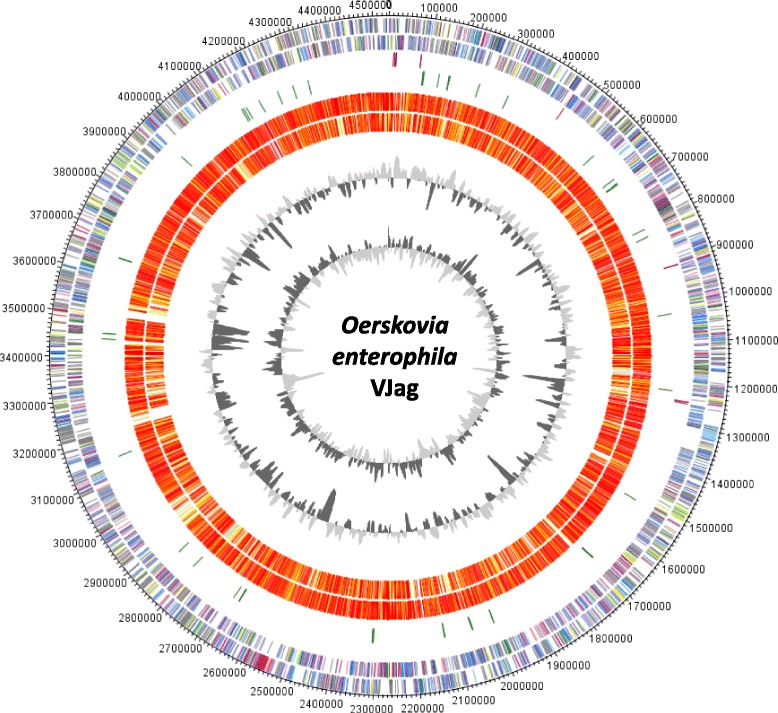
Circular representation of the genome comparison of O. enterophila VJag with other Oerskovia strains. Outer circles 1 and 2, genes (marked in COG colors) encoded by the leading and the lagging strand of O. enterophila VJag; circles 3 and 4, positions of rRNA and tRNA genes, respectively; circle 5, comparison of genes present in O. enterophila VJag and O. enterophila DFA-19T; circle 6, gene comparison of O. enterophila VJag and O. turbata NRRL B-8019; circle 7 represent the GC content circle 8; represent GC skew. Color code of genome comparison: grey: >e-20-1; light yellow: <e-50- > e-20; gold: <e-50- > e-90; light orange: <e-90- > e-100; orange: <e-100- > e-120; red: <e-
Table 3.
Genome statistics
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 4,535,074 | 100 |
| DNA coding (bp) | 4,016,676 | 88.57 |
| DNA G + C (bp) | 3,283,351 | 72.40 |
| DNA scaffolds | 85 | |
| Total genes | 3975 | 100 |
| Protein coding genes | 3918 | 98.57 |
| RNA genes | 57 | 1.43 |
| Pseudogenes | 0 | 0 |
| Genes in internal clusters | 928 | 23.35 |
| Genes with function prediction | 3036 | 76,38 |
| Genes assigned to COGs | 2559 | 64,38 |
| Genes with Pfam domains | 3158 | 79,45 |
| Genes with signal peptides | 332 | 0,84 |
| Genes with transmembrane helices | 1142 | 28,73 |
| CRISPR repeats | 0 | 0 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | %age | Description |
|---|---|---|---|
| J | 199 | 6.92 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03 | RNA processing and modification |
| K | 290 | 10.08 | Transcription |
| L | 94 | 3.27 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 27 | 0.94 | Cell cycle control, cell division, chromosome partitioning |
| V | 87 | 3.02 | Defense mechanisms |
| T | 131 | 4.55 | Signal transduction mechanisms |
| M | 134 | 4.66 | Cell wall/membrane/envelope biogenesis |
| N | 11 | 0.38 | Cell motility |
| U | 19 | 0.66 | Intracellular trafficking, secretion, and vesicular transport |
| O | 103 | 3.58 | Posttranslational modification, protein turnover, chaperones |
| C | 145 | 5.04 | Energy production and conversion |
| G | 320 | 11.12 | Carbohydrate transport and metabolism |
| E | 241 | 8.38 | Amino acid transport and metabolism |
| F | 83 | 2.88 | Nucleotide transport and metabolism |
| H | 168 | 5.84 | Coenzyme transport and metabolism |
| I | 109 | 3.79 | Lipid transport and metabolism |
| P | 204 | 7.09 | Inorganic ion transport and metabolism |
| Q | 62 | 2.16 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 283 | 9.84 | General function prediction only |
| S | 156 | 5.42 | Function unknown |
| - | 1416 | 35.62 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
A circular representation of the O. enterophila VJag genome sequence and comparison to O. enterophila DFA-19T [14] and O. turbata NRRL B-8019 genome sequences is shown in Fig. 3. For O. enterophila VJag the genes encoded by the leading and the lagging strand (outer circles 1 and 2) are marked in COG colors in the artificial chromosome map. The third and fourth circle show the positions of rRNA genes and tRNA genes, respectively. The fifth and sixth circle show comparisons of genes present in the strains O. enterophila VJag and O. enterophila DFA-19T chromosome as well as O. enterophila VJag and O. turbata NRRL B-8019 chromosome, respectively. The red colored regions indicate high similarity, whereas yellow colored regions indicate low similarity (see color code, Fig. 3). The two inner most plots represent the GC content and the GC skew (circle 7-8). Furthermore, a pairwise ANI analysis of the VJag strain and type strain O. enterophila DFA-19 [14] showed a similarity value of 99.36%, whereas a respective analysis of VJag strain and O. turbata NRRL B-8019 resulted in 89.31% similarity.
Insights from the genome sequence
Because of the close relationship to members of the genus Cellulomonas, O. enterophila VJag was expected to use cellulose as carbon source. According to the KEGG pathway, genes coding for enzymes probably responsible for the degradation of cellulose to cellobiose and β-D-glucose were found in O. enterophila VJag. Cellulose is one of the main components of plant material and is one of the most abundant biopolymers in the environment [35]. Plate assays revealed that O. enterophila VJag is able to utilize cellulose [Additional file 3: Figure S1]. The used plates contained CMC as sole carbon source and Congo red to stain CMC. O. enterophila VJag hydrolyzed CMC to glucose whereby the Congo red was eluted, the red color got lost and resulted in formation of bright halos around cell spots.
A gene (OJAG_15690) encoding a cellulose 1,4- β -cellobiosidase is present in genome that converts cellulose to 1,4-β-D-glucan. 1,4-β-D-glucan would be further converted to β-D-glucose through the action of a β-glucosidase. The genome sequence of O. enterophila VJag comprises 13 genes encoding such β-glucosidases (OJAG_01470, OJAG_39370, OJAG_33570, OJAG_33160, OJAG_31620, OJAG_25090, OJAG_25070, OJAG_16840, OJAG_16640, OJAG_15960, OJAG_15000, OJAG_14990, OJAG_11840).
Furthermore, cellulose can be converted to cellobiose, using endoglucanases (encoded by OJAG_04410; OJAG_07660), and can also be converted to β-D-glucose through the action of a β-glucosidase.
Starch is also ubiquitous in nature as it accumulates in plants as storage compound [36]. The genome sequence of O. enterophila VJag harbors genes coding for α-amylases (OJAG_12050; OJAG_09450) and a starch phosphorylase (OJAG_12070). Thus, starch is either converted to glycogen, dextrin, or amylose by O. enterophila VJag. Starch or glycogen could also be degraded to trehalose by respective enzymes (glycogen debranching enzyme encoded by OJAG_00790 or OJAG_12120). Subsequently, trehalose would be further converted to β-D-glucose-1-phosphate or D-glucose via an α-trehalose phosphorylase (encoded by OJAG_12210). Dextrin would be converted to α-D-glucose by an oligo-1,6-glucosidase (encoded by OJAG_08510). A plate assay using Jag-MM-agar plates containing starch (2% w/v) as carbon source showed that starch is utilized during growth of O. enterophila VJag [Additional file 3: Figure S2]. After incubation, starch was stained using Lugol’s solution and bright halos around cell spots showed starch consumption by O. enterophila VJag (see Additional file 3: Figure S2).
Another commonly occurring compound in natural environments besides cellulose and starch is chitin. Chitin is a major structural polymer of the cell walls of fungi and the exoskeletons of invertebrates [37]. Numerous genes which encode enzymes for the degradation of chitin to chitobiose or N-acetylglucosamine were found in the genome sequence of O. enterophila VJag (OJAG_26450; OJAG_36940; OJAG_36950; OJAG_36990; OJAG_38030; OJAG_38450; OJAG_38460). Chitobiose could be converted to N-acetylglucosamine or N-acetylglucosamine-1-phosphate by hexosaminidases and β-N-acetylhexosaminidase (encoded by OJAG_07390; OJAG_13640; OJAG_13650; OJAG_33360; OJAG_35500; OJAG_09950; OJAG_30030; OJAG_15920). Furthermore, one gene (OJAG_13250) was found that encodes a glucosamine-1-phosphate N-acetyltransferase, which converts N-acetylglucosamine-1-phosphate to UDP-acetylglucosamine. This intermediate would be further transformed to N-acetylglucosamine enopyruvate by an UDP-N-acetylglucosamine-1-carboxyvinyltransferase (encoded by OJAG_15040; OJAG_22690). N-acetylglucosamine enopyruvate can subsequently be converted to N-acetylmuramic acid via an UDP-N-acetylmuramate dehydrogenase (encoded by OJAG_01210). N-acetylmuramic acid would be metabolized through the peptidoglycan biosynthesis pathway or the D-glutamin and D-glutamate metabolism (OJAG_14230; _ OJAG 14240).
Additionally, genes encoding enzymes for xylose degradation were found in the O. enterophila VJag genome sequence. D-xylose could be converted to D-xylulose by a xylose isomerase (encoded by OJAG_26770). Furthermore, D-xylulose would be phosphorylated to D-xylulose-5-phosphate via a xylulokinase (OJAG_26780). D-xylulose-5-phosphate would be converted to D-ribulose-5-phosphate by a ribulose-5-phosphate 3-epimerase (OJAG_00210), and then metabolized via the pentose phosphate pathway, or D-xylulose-5-phosphate would be converted to L-ribulose -5-posphate via a L-ribulose-5-phosphate 4-epimerase (OJAG_27380). This also fits into the overall picture since xylose is a main part of hemicellulose and makes up a part of plant materials [38].
Conclusions
The genome of O. enterophila VJag, which was isolated from forest soil, is described. Furthermore, the phylogenetic and phenotypic characteristics of the isolated strain are presented. It has been shown that the isolate belongs to the family of Cellulomonadaceae. Scanning electron micrographs confirmed the variable phenotype in exponential or stationary growth phase. Genome sequences analysis revealed that O. enterophila VJag has the genetic properties to degrade compounds typically abundant in forest soils. Plate assays showed that the isolated strain is able to use starch and cellulose as sole carbon and energy source. The genome sequence of O. enterophila VJag has been deposited at DDBJ/EMBL/GenBank and can be found under the accession number LRIE00000000. The version described in this paper is version LRIE01000000.
Additional files
Enrichment, isolation and selection of bacterial strains; identification of isolated strains (S1). (DOCX 15 kb)
Detailed composition of Jag-MM agar and silica plates (S2). (DOCX 17 kb)
Jag-MM-silica plates with CMC and Congo red; Figure S2. Jag-MM-agar plates with starch. (ZIP 964 kb)
Acknowledgements
Financial support by Ulm University is gratefully acknowledged.
Authors’ contributions
AP and RD planned the genome sequencing, AP did the genome sequencing and the genome annotations, FB and VJ planned the isolation procedure, VJ performed the strain isolation, growth experiments, and the genomic DNA preparation and prepared the electron micrographs and the phylogenetic tree. VJ and AP wrote the manuscript. PD conceived of the study, participated in its design and coordinated and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviation
- CMC
Carboxymethyl cellulose
References
- 1.Jáger K, Márialigeti K, Hauck M, Barabás G. Promicromonospora enterophila sp. nov., a new species of monospore Actinomycetes. Int J Syst Bacteriol. 1983;33:525–531. doi: 10.1099/00207713-33-3-525. [DOI] [Google Scholar]
- 2.Stackebrandt E, Breymann S, Steiner U, Prauser H, Weiss N, Schumann P. Re-evaluation of the status of the genus Oerskovia, reclassification of Promicromonospora enterophila (Jáger et al. 1983) as Oerskovia enterophila comb. nov. and description of Oerskovia jenensis sp. nov. and Oerskovia paurometabola sp. nov. Int J Syst Evol Microbiol. 2002;52:1105–1111. doi: 10.1099/00207713-52-4-1105. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi Y, Tanaka Y, Iwai Y, Omura S. Promicromonospora sukumoe sp. nov., a new species of the Actinomycetales. J Gen Appl Microbiol. 1987;33:507–519. doi: 10.2323/jgam.33.507. [DOI] [Google Scholar]
- 4.Prauser H, Lechevalier MP, Lechevalier H. Description of Oerskovia gen. n., to harbor Ørskov’s motile Nocardia. Appl Microbiol. 1970;19:534. doi: 10.1128/am.19.3.534-534.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodfellow M. Phylum XXVI. Actinobacteria phyl. nov. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s manual of systematic bacteriology, vol. 5, Part A. New York: Springer; 2012. p. 33. [Google Scholar]
- 6.Lightfield J, Fram NR, Ely B. Across bacterial phyla, distantly-related genomes with similar genomic GC content have similar patterns of amino acid usage. PLoS ONE. 2011;6(3):e17677. doi: 10.1371/journal.pone.0017677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill LR. An index to deoxyribonucleic acid base compositions of bacterial species. J Gen Microbiol. 1996;44:419–437. doi: 10.1099/00221287-44-3-419. [DOI] [PubMed] [Google Scholar]
- 8.Ghai R, McMahon KD, Rodriguez-Valera F. Breaking a paradigm: cosmopolitan and abundant freshwater Actinobacteria are low GC. Environ Microbiol Rep. 2012;4:29–35. doi: 10.1111/j.1758-2229.2011.00274.x. [DOI] [PubMed] [Google Scholar]
- 9.Servin JA, Herbold CW, Skophammer RG, Lake JA. Evidence excluding the root of the tree of life from the Actinobacteria. Mol Biol Evol. 2008;25:1–4. doi: 10.1093/molbev/msm249. [DOI] [PubMed] [Google Scholar]
- 10.Embley TM, Stackebrandt E. The molecular phylogeny and systematics of the Actinomycetes. Annu Rev Microbiol. 1994;48:257–289. doi: 10.1146/annurev.mi.48.100194.001353. [DOI] [PubMed] [Google Scholar]
- 11.Stackebrandt E, Schumann P, Prauser H. The family Cellulomonadaceae. New York: Springer; 2006. pp. 983–1001. [Google Scholar]
- 12.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol. 1997;47:479–491. doi: 10.1099/00207713-47-2-479. [DOI] [Google Scholar]
- 13.Stackebrandt E, Häringer M, Schleifer KH. Molecular genetic evidence for the transfer of Oerskovia species into the genus Cellulomonas. Arch Microbiol. 1980;127:179–185. doi: 10.1007/BF00427191. [DOI] [Google Scholar]
- 14.Jag V, Poehlein A, Bengelsdorf FR, Daniel R, Dürre P. Genome sequence of the facultative anaerobe Oerskovia enterophila DFA-19 (DSM 43852T) Genome Announc. 2016 doi: 10.1128/genomeA.00973-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson M, Zaretskaya I, Raytselis Y, Merezhunk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katoh K, Toh H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinformatics. 2008 doi: 10.1186/1471-2105-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katoh K, Standley M. MAFFT sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stackebrandt E, Prauser H. Assignment of the genera Cellulomonas, Oerskovia, Promicromonospora and Jonesia to Cellulomonadaceae fam. nov. Syst Appl Microbiol. 1991;14:261–265. doi: 10.1016/S0723-2020(11)80378-4. [DOI] [Google Scholar]
- 20.Fernández-Garayzábal JF, Dominguez L, Pascual C, Jones D, Collins MD. Phenotypic and phylogenetic characterization of some unknown coryneform bacteria isolated from bovine blood and milk: description of Sanguibacter gen. nov. Lett Appl Microbiol. 1995;20:69–75. doi: 10.1111/j.1472-765X.1995.tb01289.x. [DOI] [PubMed] [Google Scholar]
- 21.Pascual C, Collins MD, Grimont PAD, Dominguez L, Fernandez-Garayzabal JF. Sanguibacter inulinus sp. nov. Int J Syst Bacteriol. 1996;46:811–813. doi: 10.1099/00207713-46-3-811. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Dai X, He L, Wang YN, Wang BJ, Liu Z, et al. Sanguibacter marinus sp. nov., isolated from coastal sediment. Int J Syst Evol Microbiol. 2005;55:1755–1758. doi: 10.1099/ijs.0.63471-0. [DOI] [PubMed] [Google Scholar]
- 23.Hong SG, Lee YK, Yim JH, Chun J, Lee HK. Sanguibacter antarcticus sp. nov., isolated from Antarctic sea sand. Int J Syst Evol Microbiol. 2008;58:50–52. doi: 10.1099/ijs.0.65031-0. [DOI] [PubMed] [Google Scholar]
- 24.Kim MK, Pulla RK, Kim SY, Yi TH, Soung NK, Yang DC. Sanguibacter soli sp. nov., isolated from soil of a ginseng field. Int J Syst Evol Microbiol. 2008;58:538–541. doi: 10.1099/ijs.0.65399-0. [DOI] [PubMed] [Google Scholar]
- 25.Doroghazi JR, Albright JC, Goering AW, Ju K-S, Haines RR, Tchalukov KA, et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10:963–968. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequence information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics. 1999;99:45–56. [Google Scholar]
- 29.Hyatt D, Chen G-L, LoCascio P, Land M, Larimer F, Hauser L. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(1):119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 33.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zdobnov EM, Apweiler R. InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 35.Leschine SB. Cellulose degradation in anaerobic environments. Annu Rev Microbiol. 1995;49:399–426. doi: 10.1146/annurev.mi.49.100195.002151. [DOI] [PubMed] [Google Scholar]
- 36.Britz SJ, Hungerford WE, Lee DR. Rhythms during extended dark periods determine rates of net subsequent light periods in leaves of Sorghum. Planta. 1987;171:339–345. doi: 10.1007/BF00398679. [DOI] [PubMed] [Google Scholar]
- 37.Reguera G, Leschine SB. Chitin degradation by cellulolytic anaerobes and facultative aerobes from soils and sediments. FEMS Microbiol Lett. 2001;204:367–374. doi: 10.1111/j.1574-6968.2001.tb10912.x. [DOI] [PubMed] [Google Scholar]
- 38.Loewus FA, Kelly S, Neufeld EF. Metabolism of myo-inositol in plants: conversion to pectin, hemicellulose, D-xylose, and sugar acids. Proc Natl Acad Sci USA. 1962;48:421–425. doi: 10.1073/pnas.48.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchanan RE. Studies in the nomenclature and classification of bacteria. II. The primary subdivisions of the Schizomycetes. J Bacteriol. 1917;2:155–164. doi: 10.1128/jb.2.2.155-164.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Evol Microbiol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [Google Scholar]
- 42.Zhi XY, Li WJ, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol. 2009;59:589–608. doi: 10.1099/ijs.0.65780-0. [DOI] [PubMed] [Google Scholar]
- 43.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Enrichment, isolation and selection of bacterial strains; identification of isolated strains (S1). (DOCX 15 kb)
Detailed composition of Jag-MM agar and silica plates (S2). (DOCX 17 kb)
Jag-MM-silica plates with CMC and Congo red; Figure S2. Jag-MM-agar plates with starch. (ZIP 964 kb)