

Abstract
Background
Autoinflammatory diseases in dogs are characterized by complex disease processes with varying clinical signs. In Shar-Pei, signs of inflammation including fever and arthritis are known to be related with a breed-specific predisposition for Shar-Pei Autoinflammatory Disease (SPAID).
Results
Clinical and histopathological examinations of two severely SPAID-affected Shar-Pei revealed signs of inflammation including fever, arthritis, and perivascular and diffuse dermatitis in both dogs. A multifocal accumulation of amyloid in different organs was found in one SPAID-affected case. Whole genome sequencing resulted in 37 variants, which were homozygous mutant private mutations in SPAID-affected Shar-Pei. Nine SNVs with predicted damaging effects and three INDELs were further investigated in 102 Shar-Pei affected with SPAID, 62 unaffected Shar-Pei and 162 controls from 11 different dog breeds. The results showed the missense variant MTBP:g.19383758G > A in MTBP to be highly associated with SPAID in Shar-Pei. In the region of this gene a large ROH (runs of homozygosity) region could be detected exclusively in the two investigated SPAID-affected Shar-Pei compared to control dog breeds. No further SPAID-associated variant with predicted high or moderate effects could be found in genes identified in ROH regions. This MTBP variant was predicted to affect the MDN2-binding protein domain and consequently promote proinflammatory reactions. In the investigated group of Shar-Pei older than six years all dogs with the mutant genotype A/A were SPAID-affected whereas SPAID-unaffected dogs harbored the homozygous wildtype (G/G). Shar-Pei with a heterozygous genotype (G/A) were shown to have a 2.13-fold higher risk for disease development, which gave evidence for an incomplete dominant mode of inheritance.
Conclusions
The results of this study give strong evidence for a variant in MTBP related with proinflammatory processes via MTBP-MDM2 pathway. Thus, these results enable a reliable detection of SPAID in Shar-Pei dogs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3737-z) contains supplementary material, which is available to authorized users.
Keywords: Shar-Pei, Autoinflammatory disease, Whole genome sequencing, MTBP
Background
High-throughput technologies have been shown to play an increasingly important role in clinical research and diagnostics [1–4]. In whole genome sequencing data various causative variants for disease traits have been detected in human as well as other mammals including cattle, horse and dogs [5–8]. It has been shown that disease causing mutations can be breed or population specific and are often linked or promoted by characteristic features or phenotypic traits supported in breeding programs [9, 10]. In Shar-Pei dogs, the development of a characteristically strongly wrinkled skin stabilized by deposits of hyaluronic acid were suggested to be involved in inflammatory processes resulting in a periodic fever [11]. It was proposed that a 16.1 Kb duplication upstream of the hyaluronan synthase 2 (HAS2) on chromosome (CFA) 13, which was detected in Shar-Pei with a padded muzzle (meatmouth type), was associated with skin wrinkling and periodic fever in Shar-Pei dogs [11, 12]. Therefore, this variant was assumed to affect both fever and thick and folded skin [11]. However, a direct relation of specific copy numbers detected by real-time PCR to Shar-Pei fever as an individual phenotype with no regard to other inflammatory signs could not be confirmed [13]. Further investigations of affected Shar-Pei showed that clinical manifestation was much more variable and recurrent bouts of fever were only one of various clinical signs designated now as a syndrome termed Shar-Pei autoinflammatory disease (SPAID) [14]. Additional signs of inflammation could be shown to be arthritis, dermatitis, otitis and systemic amyloidosis [14]. It was proposed that genetic risk factors on CFA13 and CFA14 might trigger these inflammatory processes [14]. Further droplet digital PCR (ddPCR) analysis of the 16.1 Kb copy number variation (CNV) upstream of HAS2 revealed stable CNV alleles in this region in contrast to continuous alleles previously detected by real-time PCR and subsequently suggested an association with SPAID [15]. This breed specific CNV was suggested to be inherited in a bi-allelic way and proposed to explain 9.7% of the genetic variance whereas the influence of further potential causative variants and triggers for SPAID were still unclear [15]. The complexity of the disease and the variable onset of signs of inflammation showed a high similarity to human autoinflammatory diseases like the Familial Mediterranean Fever [16, 17]. Various genes have been shown to be involved in human autoinflammatory processes and were also found to interact with each other in triggering recurrent episodes of inflammation [18–23].
In our study, we investigated genetic variants derived from next generation sequencing (NGS) data to identify potential causative mutations which might be involved in the development of autoinflammatory processes and thus play a significant role in triggering SPAID in Shar-Pei.
Results
Phenotype
Clinical investigations and patient histories recorded by a questionnaire resulted in 102 Shar-Pei with clinical signs consistent with SPAID and 62 unaffected dogs. The study included Shar-Pei of different breed types including horse, brush and bear coats, modern types with a meatmouth and traditional types with a bonemouth as well as Shar-Pei with a strongly wrinkled skin and others with few wrinkles (Table 1). In total, 46 of the SPAID-affected and 11 SPAID-unaffected Shar-Pei were older than six years and were therefore considered to be a reliable set for the SPAID-phenotype. First signs of the disease could be observed in a range of one to twelve years in the individual dogs whereas the average onset of the disease was 2.7 years (median 1.0). Only three Shar-Pei developed first signs of inflammation at an age of older than six years. Specific investigations of SPAID signs revealed all combinations of the inflammatory types fever, arthritis, erythema, thickened and pasty skin, otitis, eye inflammation and recurrent intestinal inflammations (Fig. 1, Additional file 1: Table S1).
Table 1.
Phenotypes of 164 Shar-Pei investigated for SPAID
| Age | ≤6 years | >6 years | |||
|---|---|---|---|---|---|
| Suspected SPAID- type | SPAID-affected | SPAID-unaffected | SPAID-affected | SPAID-unaffected | |
| Breed specific phenotype | |||||
| Type | bonemouth | 10 (17.86%) | 10 (19.61%) | 7 (15.23%) | 5 (45.46%) |
| meatmouth | 46 (82.14%) | 41 (80.39%) | 39 (84.78%) | 6 (54.54%) | |
| Coat | horse | 10 (17.86%) | 9 (17.65%) | 11 (23.91%) | 5 (45.46%) |
| brush | 40 (71.49%) | 31 (60.78%) | 28 (60.87%) | 4 (36.36%) | |
| bear | 1 (0.56%) | 1 (0.51%) | 2 (4.35%) | 0 (0.00) | |
| unknown | 5 (8.93%) | 10 (19.61%) | 5 (10.87%) | 2 (18.18%) | |
| Wrinkles | few | 11 (19.64%) | 25 (49.02%) | 17 (36.96%) | 10 (90.91%) |
| moderate | 40 (71.49%) | 25 (49.02%) | 23 (50%) | 1 (0.11%) | |
| pronounced | 5 (8.93%) | 1 (0.51%) | 6 (13.04%) | 0 (0.00) | |
| Total number of individuals | 56 | 51 | 46 | 11 | |
Shar-Pei of different types concerning coat, mouth and degree of wrinkles in the age groups ≤6 years and >6 years used for this study are shown
Fig. 1.

Exemplary illustration of clinical signs for SPAID. Affected Shar-Pei can show skin erythema in the region of wrinkles (a) or signs of inflammation at the tarsal joints (b)
Histopathologic examination
Due to bad general condition and poor prognosis two severely SPAID-affected Shar-Pei were euthanized and underwent histopathological examination. In addition, an SPAID-unaffected dog with no signs of inflammation was used as control. That dog was euthanized because of poor prognosis due to metastasizing tumors. In both SPAID-affected dogs, the skin revealed a perivascular and diffuse dermatitis with lymphocytes, plasma cells, eosinophilic granulocytes and mast cells (Fig. 2). In addition, one SPAID-case revealed a multifocal accumulation of amyloid in the salivary gland, liver, pancreas, kidneys (Fig. 3), adrenal glands, thyroid glands, spleen and in the lamina propria mucosae of stomach, small and large intestine. In all three examined Shar-Pei a generalized cutaneous mucinosis was present (Fig. 4).
Fig. 2.
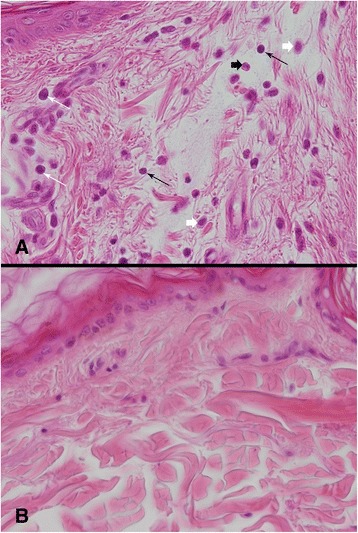
Inflammatory processes in the skin. Perivascular and diffuse infiltration with lymphocytes (long black arrows), plasma cells (long white arrows), eosinophilc granulocytes (short white arrow) and mast cells (short black arrows) in the skin of a SPAID-affected Shar-Pei (a). Normal skin of a SPAID-unaffected Shar-Pei (b). HE, magnification 400x
Fig. 3.
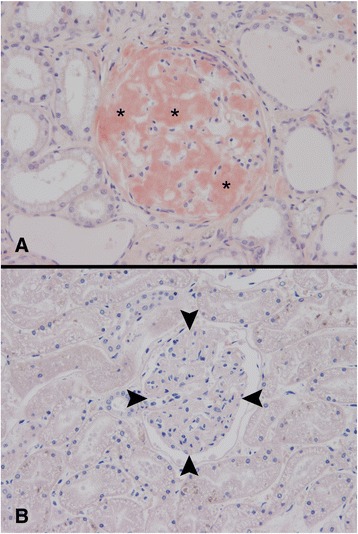
Histopathologic examination of the kidney. Accumulation of amyloid (asterisks) in a glomerulum of a SPAID-affected Shar-Pei (a). Normal glomerulum (arrow heads) of a SPAID-unaffected Shar-Pei (b). Congo red, magnification 200x
Fig. 4.
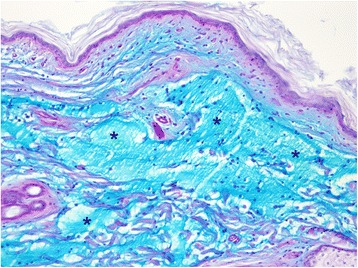
Cutaneous mucinosis. Marked dermal accumulations of alcian blue positive material (asterisks) in a SPAID-affected Shar-Pei. AB/PAS, magnification 100x
Whole genome sequencing
The whole genome of the two severely SPAID-affected Shar-Pei with recurrent bouts of fever and acute joint inflammations confirmed by careful examination of an accredited veterinarian was sequenced in order to compare genetic variants for an association with SPAID using data from five reference dogs of different breeds from sequence read achieve (SRA). Due to the high incidence of SPAID-affected progeny and parents, these two investigated Shar-Pei were expected to be very likely homozygous carriers of a potential causative mutation for SPAID. Whole genome sequencing resulted in a coverage of 11X (Shar-Pei 1) and 12X Shar-Pei 2 (Additional file 2: Table S2). In total, 31 missense variants, two disruptive in-frame insertions and deletions, one noncoding transcript exon variant, two frameshift variants and one splice acceptor variant could be detected in filtering analysis for homozygous mutant genotypes exclusively present in SPAID-affected Shar-Pei (Table 2). Six SNVs that were predicted to be deleterious (SIFT) as well as probably damaging (PolyPhen-2) and three insertions/deletions (INDELs) were chosen for further investigation of their association with SPAID (Table 3).
Table 2.
Results from filtering analysis for SPAID-associated genotypes
| CFA | Position | Base change | Amino acid change | Consequence | Genotype (5 reference dogs) | Genotype (2 affected Shar-Pei) | Gene (transcript) | SIFT | PolyPhen-2 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 8186638 | A > G | K > R | missense variant | 0/0 | 1/1 | RTTN (ENSCAFT00000000055) | tolerated (0.3) | benign (0.036) |
| 1 | 112413056 | T > C | K > E/K > E | missense variant | 0/0 | 1/1 | CD79A (ENSCAFT00000037106/ENSCAFT00000008000) | tolerated low confidence | benign (0.227) |
| 1 | 112413114 | C > CGTGATG | I67_T68dup/I67_T68dup | disruptive inframe insertion | 0/0 | 1/1 | CD79A (ENSCAFT00000037106/ENSCAFT00000008000) | NA | NA |
| 3 | 14027517 | A > C | L > R | missense variant | 0/0 | 1/1 | ARSK (ENSCAFT00000012646) | deleterious low confidence | probably damaging (0.993) |
| 4 | 6374801 | G > A | C > Y | missense variant | 0/0 | 1/1 | PCNXL2 (ENSCAFT00000018397) | deleterious low confidence | NA |
| 4 | 19114717 | GT | D > E/D > E | missense variant | 0/0 | 1/1 | DNAJC12 (ENSCAFT00000021116/ENSCAFT00000049485) | tolerated (0.1/0.28) | benign/possibly damaging (0.023/0.579) |
| 4 | 75351270 | A > G | V > A | missense variant | 0/0 | 1/1 | PDZD2 (ENSCAFT00000047348) | tolerated (0.33) | benign (0.000) |
| 5 | 21123033 | C > T | R > Q/R > Q/R > Q | missense variant | 0/0 | 1/1 | DIXDC1 (ENSCAFT00000022334/ENSCAFT00000022333/ENSCAFT00000035298) | tolerated (1/1/1) | benign (0.000/0.003/0.001) |
| 5 | 48648175 | G > C | - | non coding transcript exon variant, non coding transcript variant | 0/0 | 1/1 | ENSCAFG00000018860 (novel gene; ENSCAFT00000029943) | NA | NA |
| 5 | 60223074 | G > T | P > Q | missense variant | 0/0 | 1/1 | ACOT7 (ENSCAFT00000049704) | deleterious low confidence | possibly damaging (0.694) |
| 6 | 36821482 | G > T | S > Y | missense variant | 0/0 | 1/1 | C16orf96 (ENSCAFT00000048346) | deleterious (0) | probably damaging (0.995) |
| 6 | 38937765 | C > T | A > V | missense variant | 0/0 | 1/1 | ZNF598 (ENSCAFT00000030940) | tolerated (0.62) | benign (0.017) |
| 6 | 40648375 | C > T | P > S/P > S/P > S | missense variant | 0/0 | 1/1 | ENSCAFG00000024344 (novel gene; ENSCAFT00000037586/ENSCAFT00000037216/ENSCAFT00000037580) | deleterious (0/0/0) | probably damaging (1.00/1.00/1.00) |
| 6 | 56637047 | TACAA > T | F76fs | frameshift variant | 0/0 | 1/1 | RPAP2 (novel gene; ENSCAFT00000032095) | NA | NA |
| 6 | 56760354 | T > C | K > E | missense variant | 0/0 | 1/1 | KIAA1107 (ENSCAFT00000032101) | tolerated (0.26) | benign (0.053) |
| 6 | 56786359 | G > C | D > E | missense variant | 0/0 | 1/1 | KIAA1107 (ENSCAFT00000032101) | tolerated (1) | benign (0.000) |
| 6 | 57204844 | A > G | Y > C | missense variant | 0/0 | 1/1 | TGFBR3 (ENSCAFT00000032134) | deleterious (0) | probably damaging (0.998) |
| 9 | 3074837 | G > C | H > Q/H > Q/H > Q | missense variant | 0/0 | 1/1 | TNRC6C (ENSCAFT00000048770/ENSCAFT00000050041/ENSCAFT00000008443) | tolerated (0.12/0.8/0.72) | probably damaging (0.999/0.988/0.999) |
| 11 | 62457942 | G > A | A > T | missense variant | 0/0 | 1/1 | ZNF462 (ENSCAFT00000004432) | NA | benign (0.000) |
| 13 | 19383758 | G > A | E > K | missense variant | 0/0 | 1/1 | MTBP (ENSCAFT00000001477) | deleterious (0.01) | probably damaging (0.979) |
| 13 | 58949521 | C > G | L > V/L > V | missense variant | 0/0 | 1/1 | UGT2B31 (ENSCAFT00000022724/ENSCAFT00000004520) | tolerated (1/1) | benign (0.000/0.000) |
| 13 | 58949524 | G > A | V > I/V > I | missense variant | 0/0 | 1/1 | UGT2B31 (ENSCAFT00000022724/ENSCAFT00000004520) | tolerated (0.49/0.48) | benign (0.005/0.001) |
| 13 | 58949536 | A > G | T > A/T > A | missense variant | 0/0 | 1/1 | UGT2B31 (ENSCAFT00000022724/ENSCAFT00000004520) | tolerated (0.66/0.65) | benign (0.000/0.000) |
| 15 | 42583230 | A > C | N > H/N > H | missense variant | 0/0 | 1/1 | HCFC2 (ENSCAFT00000011975/ENSCAFT00000011972) | tolerated (0.09)/deleterious (0.04) | possibly damaging (0.583/0.616 |
| 17 | 58777715 | G > A | S > L | missense variant | 0/0 | 1/1 | TXNIP (ENSCAFT00000018124) | tolerated (0.2) | benign (0.126) |
| 18 | 40794747 | C > T | R > H | missense variant | 0/0 | 1/1 | OR10H12 (ENSCAFT00000037956) | tolerated (0.37) | benign (0.000) |
| 20 | 54717650 | G > A | T > M | missense variant | 0/0 | 1/1 | KDM4B (ENSCAFT00000030040) | tolerated (0.05) | possibly damaging (0.934) |
| 20 | 54729635 | A > G | - | splice acceptor variant | 0/0 | 1/1 | KDM4B (ENSCAFT00000030040) | NA | NA |
| 22 | 30574626 | C > T | T > M | missense variant | 0/0 | 1/1 | CLN5 (ENSCAFT00000008156) | deleterious (0.01) | probably damaging (1.000) |
| 24 | 46507770 | C > T | R > W | missense variant | 0/0 | 1/1 | ENSCAFG00000030917 (novel gene; ENSCAFT00000046966) | NA | NA |
| 27 | 31792218 | T > C | I > V | missense variant | 0/0 | 1/1 | C12orf60 (ENSCAFT00000020587) | tolerated (0.39) | benign (0.009) |
| 28 | 34998871 | G > A | V > I | missense variant | 0/0 | 1/1 | BCCIP (ENSCAFT00000020430) | tolerated (0.24) | possibly damaging (0.992) |
| 30 | 29440908 | C > T | E > K | missense variant | 0/0 | 1/1 | RASL12 (ENSCAFT00000027120) | tolerated (0.08) | benign (0.034) |
| 30 | 29986720 | G > A | E > K | missense variant | 0/0 | 1/1 | SLC24A1 (ENSCAFT00000027314) | tolerated (0.09) | benign (0.223) |
| 32 | 22124585 | GTCTTT > G | K64fs | frameshift variant (microsatellite) | 0/0 | 1/1 | ENSCAFG00000031054 (novel gene; ENSCAFT00000047190) | NA | NA |
| 37 | 502225 | CCTTGTGCAA > C | L4_K6del | inframe deletion | 0/0 | 1/1 | OSGEPL1 (ENSCAFT00000014928) | NA | NA |
| 38 | 20346455 | T > A | V > D | missense variant | 0/0 | 1/1 | C1orf111 (ENSCAFT00000036623) | tolerated (0.94) | benign (0.000) |
In total 37 variants with predicted high or moderate effects (SNPEff) could be exclusively found homozygous for the mutant allele in SPAID-affected Shar-Pei. SIFT and PolyPhen-2 variant effect estimations are shown for each position
Table 3.
Case–control test results for candidate variants for SPAID
| CFA | Polymorphism | MAF total | Wild type allele | Minor allele | MAF controls | MAF cases | Allele odds ratio | Chi-Square Genotype (Probability) | Chi-Square Allele (Probability) | Chi-Square Trend (Probability) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CD79A:g.112413114insGTGATG | 0.149 | C | del | 0.137 | 0.152 | 1.128 | 2.287 (P = 0.319) | 0.136 (P = 0.712) | 0.156 (P = 0.693) |
| 6 | C16orf96:g.36821482G > T | 0.149 | G | C | 0.169 | 0.142 | 1.230 | 2.445 (P = 0.295) | 0.442 (P = 0.507) | 0.438 (P = 0.508) |
| 6 | ENSCAFG00000024344:g.40648375C > T | 0.334 | C | C | 0.387 | 0.305 | 0.695 | 4.450 (P = 0.108) | 2.311 (P = 0.128) | 2.470 (P = 0.116) |
| 6 | RPAP2:g.56637047delACAA | 0.301 | TACAA | ins | 0.307 | 0.299 | 1.036 | 0.436 (P = 0.804) | 0.020 (P = 0.887) | 0.020 (P = 0.887) |
| 6 | TGFBR3:g.57204844A > G | 0.455 | A | G | 0.436 | 0.465 | 0.886 | 6.180 (P = 0.050) | 0.276 (P = 0.599) | 0.267 (P = 0.605) |
| 13 | MTBP:g.19383758G > A | 0.247 | G | G | 0.395 | 0.157 | 3.517 | 25.671 (P = 2.664E-06) | 23.550 (P = 1.217E-06) | 23.551 (P = 1.217E-06) |
| 15 | HCFC2:g.42583230A > C | 0.408 | A | C | 0.395 | 0.422 | 0.896 | 0.193 (P = 0.908) | 0.222 (P = 0.637) | 0.192 (P = 0.661) |
| 22 | CLN5:g.30574626C > T | 0.214 | C | T | 0.266 | 0.177 | 1.692 | 3.787 (P = 0.151) | 3.732 (P = 0.053) | 3.633 (P = 0.057) |
| 37 | OSGEPL1:g.502225delCTTGTGCAA | 0.485 | CCTTGTGCAA | del | 0.500 | 0.466 | 0.872 | 1.341 (P = 0.511) | 0.364 (P = 0.546) | 0.388 (P = 0.533) |
SPAID-associated variants with predicted high or moderate effects (SNPEff) were genotyped for 102 SPAID-affected and 62 SPAID-unaffected Shar-Pei
Validation of genetic variants
Validation of all nine potentially deleterious genetic variants detected in whole genome sequencing data in 102 SPAID-affected and 62 SPAID-unaffected Shar-Pei using Kompetitive Allele Specific PCR (KASP) and gel electrophoresis revealed a borderline significant P-value for TGFBR3:g.57204844A > G located in TGFBR3 (transforming growth factor beta receptor 3; P = 0.050), but a highly significant P-value for MTBP:g.19383758G > A (P = 2.664E-06) located in MTBP (Mdm2, transformed 3 T3 cell double minute 2, p53 binding protein). Further validation of all nine variants in 162 dogs of 11 different breeds showed that none of the variants could be found in any other tested dog breed (Additional file 3: Table S3). The missense mutation MTBP:g.19383758G > A was predicted to result in a deleterious (0.01; SIFT) as well as probably damaging (0.979; PolyPhen-2) substitution of glutamic acid to lysine located in the MDN2-binding protein, C-terminal domain.
Genotypic distribution of MTBP:g.19383758G > A in Shar-Pei of all age groups (1–14 years) revealed none of the investigated SPAID-affected Shar-Pei harboring a homozygous wild type genotype. In total, 68.63% showed the A/A genotype whereas 31.37% had a heterozygous genotype (G/A). In the validated SPAID-unaffected Shar-Pei we found the G/G genotype in 16.13%, G/A in 45.16% and A/A in 38.71%. Further investigation of the genotypes in a subgroup of Shar-Pei older than six years revealed the disease associated genotype A/A exclusively in SPAID-affected dogs as well as the wild type genotype G/G only in SPAID-unaffected Shar-Pei (Table 4). In 25 Shar-Pei with a heterozygous genotype, 32% were not affected at the date of examination whereas 68% revealed signs of SPAID and therefore showed a 2.13-fold higher risk for disease development. The distribution of genotypes by phenotypes suggested an incomplete dominant mode of inheritance. Complex segregation analysis supported this mode of inheritance because a significant difference based on a log-likelihood ratio test (differences between −2 log-likelihoods) among a model with arbitrary Mendelian factors and a model accounting for environmental variation (χ 2 = 11.58, df = 5, p = 0.040) could be found, whereas other models (recessive or polygenic models) did not result in significant differences. With regard to the breed specific types, the modern type with a meatmouth was found to be most frequent among SPAID-affected Shar-Pei (83.3%) although all three genotypes were detected in meatmouth as well as bonemouth Shar-Pei.
Table 4.
Genotypic distribution of SPAID-associated missense mutation MTBP:g.19383758G > A
| Phenotype | Number of Shar Pei (n) | Genotype G/G | Genotype G/A | Genotype A/A |
|---|---|---|---|---|
| Age group (>6 years) | ||||
| SPAID affected Shar-Pei | 46 | 0 | 17 | 29 |
| SPAID unaffected Shar-Pei | 11 | 3 | 8 | 0 |
| All age groups (1–14 years) | ||||
| SPAID affected Shar-Pei | 102 | 0 | 32 | 70 |
| SPAID unaffected Shar-Pei | 62 | 10 | 28 | 24 |
| Muzzle types (all age groups) | ||||
| meatmouth | 132 | 3 | 41 | 88 |
| bonemouth | 32 | 7 | 19 | 6 |
None of the affected Shar-Pei older than six years showed the wild type genotype G/G whereas none of the unaffected Shar-Pei of this group harboured the disease associated genotype A/A. Regardless of the muzzle type both genotypes G/G or A/A could be found in meatmouth and bonemouth Shar-Pei types
ddPCR
MTBP:g.19383758G > A was located 1 Mb proximal to the copy number variation CNV_16.1 on CFA13. Further detection of CNV_16.1 by ddPCR revealed stable alleles of one or five copies. These copy numbers were in perfect cosegregation with the genotypes of MTBP:g.19383758G > A (Table 5).
Table 5.
Comparison of MTBP:g.19383758G > A genotypes with CNV_16.1
| MTBP:g.19383758G > A | A/A | A/G | G/G |
|---|---|---|---|
| All age groups (1–14 years) | |||
| SPAID affected Shar-Pei | 17 | 3 | 0 |
| SPAID unaffected Shar-Pei | 3 | 6 | 2 |
| CNV mean (RT-PCR) | 10.05 | 5.22 | 2.0 |
| CNV (ddPCR) | 10 | 6 | 2 |
| MTBP:g.19383758G > A | A/A | A/G | G/G |
| Age group >6 years | |||
| SPAID affected Shar-Pei | 8 | 3 | 0 |
| SPAID unaffected Shar-Pei | 0 | 3 | 1 |
| CNV mean (RT-PCR) | 9.5 | 5.0 | 2.0 |
| CNV (ddPCR) | 10 | 6 | 2 |
The results of CNV detection by digital droplet PCR show perfect cosegregation with MTBP:g.19383758G > A
Candidate gene sequencing
Sanger sequencing results of MTBP (ENSCAFT00000001477) in one SPAID-affected Shar-Pei and two control dogs confirmed the candidate SNV MTBP:g.19383758G > A (ss2136554981) in the complementary DNA in the affected Shar-Pei (Additional file 4: Table S4) and the MTBP gene model predicted by Ensembl. In addition, we detected four SNVs which could not be exclusively found in the affected Shar-Pei.
Expression analysis
To investigate the potential functional effect of MTBP:g.19383758G > A variant on MTBP we studied TP53 (cellular tumor antigen p53) mRNA expression as a bioindicator for MTBP. MTBP was shown to form a complex with MDM2 and thereby stabilizes this protein [24]. MDM2 is an inhibitor of p53, encoded by TP53 [25]. Thus, MTBP indirectly inhibits p53 by stabilizing MDM2 in the p53-MDM2-MTBP pathway [25].
Quantification of TP53 levels performed in skin and hair samples of all three MTBP:g.19383758G > A genotypes G/G, G/A and A/A revealed a significant decrease of relative TP53 expression in dogs with a heterozygous genotype (p < 0.0001) as well as in dogs with a homozygous mutant genotype (p < 0.0001) in relation to the wild type genotype (Fig. 5). In-between G/A and A/A genotypes the differential expression was also significant (p = 0.0034). It could be observed that relative expression levels of G/A-samples showed the highest variation ranging from low 2-ΔCt (min 2-ΔCt = 0.23), which could also be found in A/A samples, to higher 2-ΔCt (max 2-ΔCt = 0.94) found in G/G samples. Skin samples showed a similar distribution of relative expression levels and confirmed a significant decrease in G/A and an even higher decrease in A/A samples.
Fig. 5.
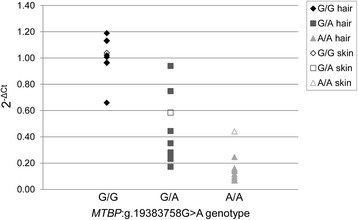
Relative expression levels of TP53. Relative expression of TP53 and its relation to MTBP:g.19383758G > A genotypes are shown. The ΔΔCT method was used to compute expression levels (2-ΔCt) for skin and hair tissues. Compared to samples with a homozygous wild type genotype (G/G), the relative expression levels of samples with a heterozygous genotype (G/A; p < 0.0001) or homozygous mutant genotype (A/A; p < 0.0001) were significantly decreased
Runs of homozygosity (ROH)
Analysis for ROH regions was performed to investigate homozygosity in the region of MTBP and to identify further potential signatures of selection in the same SPAID-affected Shar-Pei which were studied in whole genome sequencing analysis. In total, 12 shared ROH regions could be detected in canine high density bead chip data of the two Shar-Pei, which could not be found in the investigated eight control dogs of different breeds (Additional file 5: Table S5). ROHs were identified on CFA6, 13, 18, 19, 22, 30, 35, 36 and 38. The largest ROH region was found on CFA13 at 18,625,926-19,967,354 bp (CanFam3.1) including MTBP as well as ENPP2 (ectonucleotide pyrophosphatase/phosphodiesterase 2), TAF2 (TATA-box binding protein associated factor 2), DSCC1 (DNA replication and sister chromatid cohesion 1), DEPTOR (DEP domain containing MTOR-interacting protein), COL14A1 (collagen type XIV alpha 1 chain), MRPL13 (mitochondrial ribosomal protein L13), SNTB1 (syntrophin beta 1), SYT17 (synaptotagmin 17), RNA5SP34 (RNA, 5S ribosomal pseudogene 34) and seven novel genes. On CFA6 only one novel gene (ENSCAFT00000051190) could be identified in the ROH region as well as the three genes CNN3 (calponin 3), TF (transferrin) and ABCD3 (ATP binding cassette subfamily D member 3) in close proximity to this region.
Functional annotation for human orthologue genes detected in all ROH regions found exclusively in Shar-Pei showed a particularly high number of genes involved in cellular processes (GO:0009987) and metabolic processes (GO:0008152; Additional file 6: Table S6). In pathway analysis an inflammation mediated by chemokine and cytokine signalling pathway (P00031) was detected for the gene COL14A1. Nevertheless, no SPAID-associated variant with predicted high or moderate effect could be found in this gene and further candidate genes from ROH analysis. Further pathways were transcription regulator (P00023/P00055), metabolic (P02782), ubiquitin proteasome (P00060) and integrin signalling pathways (P00034).
Discussion
Analysis of NGS data in two SPAID-affected Shar-Pei and further validation revealed MTBP:g.19383758G > A on CFA13 to be highly associated with SPAID in Shar-Pei. All 102 SPAID-affected dogs harbored at least one mutant A-allele of the missense mutation predicted to have a damaging effect. In SPAID-unaffected Shar-Pei 24 dogs were found to harbor the homozygous mutant genotype (A/A). These dogs were all younger than six years, whereas none of the older dogs showed the A/A genotype. We suppose that due to the variable onset of the disease SPAID-unaffected Shar-Pei with homozygous mutant genotype, which are younger than six years, might still develop signs of the disease. Shar-Pei at an age of at least seven years with no signs of the disease were proposed to be most likely healthy with regard to SPAID [14].
Under the assumption of a variable age of onset, the results propose an incomplete dominant mode of inheritance for MTBP:g.19383758G > A. This suggestion was supported by the high incidence of SPAID-affected Shar-Pei as much as by the identification of a large ROH region exclusively found in two SPAID-affected Shar-Pei but not in control dogs. ROH detection has been shown to be a successful method for the investigation of disease traits as well as other phenotypic traits [12, 26–29]. Long ROHs were reported to be frequent in genomic regions harboring genes associated with autosomal-dominant diseases [30]. Analysis for signatures of selection specific for Shar-Pei gave evidence for a region on CFA13 harboring HAS2, which was suggested to be associated with breed specific skin wrinkling [12]. Further genome screening for signatures of selection in 50 US-Shar-Pei detected a potential signature of selective sweeps downstream of HAS2 and revealed further reduction of homozygosity on CFA5, 6, 13 and X [11]. The results from present ROH analysis in two SPAID-affected Shar-Pei genotyped on the canine Illumina high density bead chip identified a ROH region proximal of the previously detected ROHs harboring the candidate gene MTBP. Possible reasons for this shift of ROH regions might be the differences in marker density and investigated populations (US and German Shar-Pei) in these studies but might also be the specific testing for SPAID-affected Shar-Pei in our analysis. We assume that the gene locus for a thick and wrinkled skin responsible for the meatmouth Shar-Pei type might be close to the causative variant for SPAID and could explain the assumption that CNV_16.1 near HAS2 is related to affected skin formation simultaneously to periodic fever [11]. Despite the limitation of our analysis due to the low number of tested Shar-Pei, the detection of a highly SPAID-associated variant in MTBP supports this assumption.
The main role of MTBP was shown to be the formation of a stable complex with MDM2 by preventing it from autoubiquitination [24, 25]. MDM2 could be shown to have a pro-inflammatory as well as an epithelial regeneration effect [31]. A blockade of MDM2 investigated in postischemic kidneys resulted in a suppression of a sterile inflammatory response, an enhanced apoptosis and intrarenal expression of proapoptotic p53 target genes [31]. A similar suggestion was made for variants in MFEV resulting in human Mediterranean Fever which did not only increase sensitivity for triggers of inflammatory responses but did also play a role in apoptotic processes [32]. In addition, MTBP was proposed to promote indirectly the degradation of p53 by MDM2-mediated ubiquitination [25]. Due to this strong relationship in the MTBP-MDM2-p53 pathway, TP53 mRNA was used a bioindicator for MTBP-MDM2 activity. It was shown that an increase of MDM2 leads to a decrease of p53 levels and finally also a decrease of MDM2 in a feedback loop [24]. In our analysis for SPAID we detected this effect as well, showing a decrease of TP53 expression in Shar-Pei with a mutant G/A or A/A genotype due to the p53-inhibitory function of MDM2. For this reason, we assume that a modification in the MDM2-binding domain of MTPB protein in SPAID-affected dogs might affect the binding affinity to MDM2 and potentially result in a higher complex stability or reduced complex dissociation [24] and consequently increase MDM2 activity. Due to this functional relation and the distribution of the mutant allele in tested dogs, we assume that the missense mutation MTBP:g.19383758G > A induces proinflammatory effects via MTBP-MDM2 pathway in SPAID-affected Shar-Pei.
This variant is located near CNV_16.1 which has previously been suggested to increase SPAID-risk in Shar-Pei dogs [15]. In our analyses this CNV showed complete linkage with MTBP:g.19383758G > A. Nevertheless, it was suggested that CNV_16.1 might alter the expression of HAS2 and thereby affect both the development of a meatmouth muzzle type and SPAID [11]. The distribution of genotypes actually confirmed the high incidence of SPAID in meatmouth Shar-Pei types but also detected SPAID-affected bonemouth types. In addition, HAS2 mRNA was further shown to be highly expressed in cells of severely folded skin and skin vesicles derived from vesicular hyaluronosis due to an accumulation of hyaluronic acid [33, 34]. Our pathohistologic examinations showed that both SPAID-affected and SPAID-unaffected dogs harboured signs of cutaneous mucinosis as a result of hyaluronic acid accumulation. For this reason we propose that hyaluronic acid levels are not related to SPAID. Thereby, the role of CNV_16.1 and the influence of HAS2 on SPAID remain unclear. In contrast, mutant MTBP is not related to hyaluronic acid accumulation but to inflammatory and apoptotic processes via MDM2 and thus suggests an important role in SPAID development. Nevertheless, due to the complex nature of inflammatory processes we cannot exclude that further variants might play a role in disease development.
Functional analysis of genes detected in potential signatures of selection in SPAID-affected Shar-Pei revealed a high number of genes involved in signaling pathways that might be involved in inflammatory processes. Investigations of inflammatory mediators in alveolar epithelial derived from inflamed lungs in mice proposed that especially genes involved in signal transduction were downregulated in cells affected by inflammation [35]. Particularly COL14A1n which could be found to be involved in the inflammation mediated by chemokine and cytokine signaling pathway in our ROH region analysis, was also reported to be downregulated in inflamed lung cells in mice [35]. The candidate gene DEPTOR was identified as a further proinflammatory gene in this ROH region on CFA13, involved in the regulation of vascular endothelial cell activation and proinflammatory responses [36]. In addition, ABCD3 encoding for a peroxisomeal membrane protein was detected close to a ROH region on CFA6. It was shown that ABCD3 was involved in the protection of cortical neurons from inflammatory mediators [37]. Although no SPAID-associated variants with predicted high or moderate effects could be detected in these genes, it cannot be excluded that further functional variants might affect them and might therefore be involved in the autoinflammatory processes in SPAID-affected Shar-Pei.
Conclusions
In conclusion, in this study we showed a missense variant in MTBP acting via MDM2 for a proinflammatory reaction in SPAID-affected Shar-Pei. These results represent an important advance in the efforts to elucidate genetic mechanisms involved in pre-inflammatory processes in dogs.
Methods
Animals
The genomic DNA of 164 Shar-Pei dogs and 162 dogs from different breeds as controls was isolated from EDTA-blood samples and adjusted to 10 ng/μl. The health status of all Shar-Pei dogs was determined by clinical examination by the authors (28 Shar-Pei), accredited veterinarians and the owners observations recorded in a questionnaire (Additional file 7: Table S7).
In addition, in 132 study participants a supplementary form was filled in by dog owners and their veterinarians to specify the types of inflammatory processes. Specific parameters like the occurrence of fever, arthritis, erythema, thickened and pasty skin, otitis, eye inflammation and recurrent intestinal inflammation were assessed in this form. Two of SPAID-affected Shar-Pei with severe clinical signs including recurrent bouts of fever and arthritis were chosen for whole-genome sequencing.
Histopathologic examination
In two SPAID-affected and one SPAID-unaffected Shar-Pei a full necropsy was carried out. Tissue samples from skin, mamma, lymph nodes (pulmonales and mesenteriales), tonsils, salivary gland, liver, pancreas, kidneys, adrenal glands, parathyroid glands, thyroid glands, spleen, lamina propria mucosae of stomach, small and large intestine were fixed in 10% formalin and embedded in paraffin wax. Paraffin sections (4 μm) were stained with hematoxylin and eosin (HE) for histology. Mucin was stained applying the combined alcian blue (pH 2.5)/periodic acid Schiff reaction (AB/PAS). For detection of amyloid, sections of all organs were stained with Congo red and examined using light microscopy. Presence of amyloid was confirmed by examination of Congo red-stained sections under polarized light. Photographs were taken with a light microscope (Olympus BX51, Olympus, Hamburg, Germany) with an Olympus camera DP72 and Olympus cellSense software.
Whole-genome sequencing
For library preparation, genomic DNA of two Shar-Pei affected with recurrent bouts of fever was isolated from blood samples using Invisorb Spin Blood Mini Kit (STRATEC biomedical, Birkenfeld, Germany). DNA samples were enzymatically tagmented and fragmented using Nextera Library Preparation Kit (Illumina, San Diego, CA) and quality controlled on the Bioanalyzer High Sensitivity DNA Kit (Agilent, Santa Clara, California). NGS was performed on the Illumina MiSeq for 2x300 bp reads in paired-end mode. In total two runs with v3 Reagent Kits (15 Gb, Illumina) were carried out for each SPAID-affected Shar-Pei (BioProject PRJNA327712). We performed quality control using fastqc 0.11.5 [38], mapping to the reference genome CanFam 3.1 (Ensembl) using BWA 0.7.13 [39] and variant calling using SAMtools 1.3.1 [40], Picard tools (http://broadinstitute.github.io/picard/, version 2.3.0) and GATK 3.5 [41]. Five fastq files from a Korean Jindo Dog (DRR001566), an Afghan Hound (SRR1061643), two German Shepherd dogs (SRR1130247, SRR1124304) and a Border Collie (SRR654728) derived from Sequence Read Archive (NCBI) were included in this study as controls. Variants with a read depth of 3–999 and quality values >20 were chosen for further analysis and investigated for their potential effects using SNPEff version 4.1 g [42].
Selection of variants
Variants derived from whole genome sequencing analysis were filtered for homozygous mutant genotypes exclusively found in SPAID-affected Shar-Pei with predicted high or moderate effects using SAS, version 9.4 (Statistical Analysis System, Cary, NC). Variants which could be found in the dbSNP database assigned as known variants or which were predicted to be located in an incomplete region of the reference genome (incomplete transcript) were omitted from analysis. The remaining variants were investigated for their potential effects on the protein using SIFT [43] and PolyPhen-2 [44]. Those SNVs which were predicted to be deleterious by SIFT and also predicted to be probably damaging by PolyPhen-2 as well as three INDEL were chosen for validation.
Validation
All nine variants were genotyped in all 102 SPAID-affected, 62 SPAID-unaffected Shar-Pei and 162 dogs of 11 different breeds. For six SNVs KASP assays (LGC Genomics, Middlesex, UK; [45]) were designed and run on an ABI7300 real-time system for 96 well plates (Additional file 8: Table S8). Further three variants were genotyped using gel electrophoresis on an acrylamide gel and a LI-COR 4300 DNA Analyzer (LI-COR, Lincoln, Nebraska). Allele and genotype distributions and their association with SPAID were investigated applying the CASECONTROL procedure from SAS.
Sanger sequencing
Skin tissues for RNA isolation were available from an SPAID-affected Shar-Pei that was euthanized due to signs of severe renal dysfunction including sickness, diarrhea, fever and swollen joints. In addition skin tissues from an Irish Wolfhound and English Springer Spaniel which were euthanized due to orthopedic problems were used as reference samples. RNA was extracted according to standard protocols for the RNeasy Mini Kit (QIAGEN, MD, USA). Tissues were homogenized in QIAzol Lysis Reagent (QIAGEN) using the Precellys homogenizer (Bertin Technologies, Montigny le Bretonneux, France). The obtained RNA was transcribed into cDNA using the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Fermentas, Waltham, MA, USA). For Sanger sequencing primers (Additional file 9: Table S9) were designed using Primer3 software (version 0.4.0, http://bioinfo.ut.ee/primer3-0.4.0/) and PCR reactions were run on a thermocycler TProfessional 96 (Biometra, Göttingen, Germany).
Segregation analysis
Complex segregation analysis was performed for five pedigrees with 79 individuals using the procedure SEGREG of S.A.G.E. (Statistical Analysis for Genetic Epidemiology, release 6.4, http://darwin.cwru.edu). A regressive logistic model was used to test for the most probable type of inheritance with the age of onset as covariate.
ddPCR
Detection of CNV_16.1 was performed in 31 Shar-Pei using QX200 Droplet Digital PCR (ddPCR) System (BIO RAD, Munich, Germany). Primers, probes and reference gene were used according to previous analysis [11]. The DNA was digested using HaeIII restriction enzyme (New England Bio Labs, Ipswich, MA).
Expression analysis
RNA was isolated from skin and hair samples and transcribed into complementary DNA (cDNA) according to standard protocols [46]. In total, two skin samples from Great Danes with MTBP:g.19383758G > A genotype G/G and two samples from SPAID-affected Shar-Pei with a G/A and a A/A genotype were used for analysis. Hair root samples were derived from nine SPAID-affected Shar-Pei with a homozygous mutant genotype (A/A), nine Shar-Pei with a heterozygous genotype (G/A; four dogs with signs of the disease and five dogs without signs of the disease) and further seven dogs with a G/G genotype including one Shar-Pei and six Great Danes as controls. A FAM-labeled custom assay for RPL13A (primer forward: GCCAAGATCCATTATCGGAAGAAGA, primer reverse: CCACATTCTTTTCGGCCTGTTT, reporter: CCGTAGCCTCATAAGCT) as housekeeping gene, previously shown to be one of the most stably expressed genes in canine whole skin [47], a VIC-labeled gene expression assay for TP53 (ID: Cf02623149_m1, reporter: GAAAACAATGTTCTGTCTTCGGAGC) and TaqMan gene expression master mix (Thermo Fisher Scientific) were used for quantitative real-time (qRT)-PCR on an ABI7300 sequence detection system (Thermo Fisher Scientific, Applied Biosystems instrument). All samples were analyzed in triplicates. Relative expression levels were calculated using the ΔΔCT method [48]. Mean ΔCT of dogs with G/G genotype was assigned as reference.
ROH detection
Two SPAID-affected Shar-Pei and eight control dogs from different breeds (Bernese Mountain dog, Dalmatian, German shepherd, Lagotto Romagnolo, Parson Russell Terrier, Samoyed, Boxer, German Pinscher) were genotyped on the canine Illumina high density bead chip (Illumina) according to manufacturer’s protocols. A genotyping rate per sample >0.70 and a genotyping rate per SNP of >0.90 were applied for quality control and resulted in 157,642 SNPs for ROH analysis. ROHs were detected in sliding windows of 10 SNPs with homozygous regions of >120 kb using PLINK, version 1.90 (www.cog-genomics.org/plink/1.9/). A maximum of two SNPs with missing genotypes and no heterozygous SNPs were admitted in each window. Consensus ROHs regions detected in both Shar-Pei which could not be found in control dogs were investigated for human orthologues using Galaxy intersection tool (https://usegalaxy.org/) [49, 50], g:Profiler [51] and PANTHER gene list analysis for biological processes and pathways (release 2016-07-15, version 11.1, [52]).
Additional files
Phenotypic data of 164 study participants with specified inflammatory process types. The number of Shar-Pei dogs and their individual signs of inflammation is shown (1 = affected; 0 = unaffected). In 62 Shar-Pei no signs of the disease could be found. (PDF 260 kb)
Whole genome sequencing statistics for two SPAID-affected Shar-Pei and five controls. DNA-libraries of two Shar-Pei were sequenced on the Illumina MiSeq whereas further five whole genome sequences from dogs of four different breeds were derived from sequence read archive (NCBI). (DOCX 15 kb)
Mutant allele frequencies for SPAID candidate variants. All nine variants were investigated for their genotypic distribution in SPAID-affected and unaffected Shar-Pei as well as in 162 dogs of 11 different breeds. (DOCX 20 kb)
Variant detection in complementary DNA of MTBP. In total five variants could be detected in cDNA of MTBP including the candidate SNV MTBP:g.19383758G > A. None of the other variants could be exclusively found in the SPAID-affected Shar-Pei. (DOCX 17 kb)
Table S2. Shared runs of homozygosity (ROH) for SPAID-affected Shar-Pei. Two Shar-Pei were investigated for shared ROH regions which could not be detected in eight control dogs of different breeds. The chromosomal position (CanFam2.0 and CanFam3.1) of shared ROH regions, size of shared ROHs, number of SNPs, genes IDs (CanFam3.1) and human orthologues are shown. (DOCX 20 kb)
The proportion of gene hits against total number of process hits for genes detected in runs of homozygosity (ROH) regions shared by SPAID (Shar-Pei Autoinflammatory Disease) affected Shar-Pei, which could not be found in control dogs, is shown. PANTHER gene list analysis for biologic processes and pathways was done for human orthologues. (DOCX 16 kb)
Questionnaire for the assessment of health status in investigated Shar-Pei. A description of the project and information consent for dog owners and veterinarians is included. (PDF 160 kb)
Primer sequences and assays for the validation of candidate variants. Kompetitive Allele Specific PCR (KASP) assays were used for six variants whereas further three variants were genotyped by gel electrophoresis on an acrylamide gel. Primer pairs, validation type, amplicon size, annealing temperature and number of PCR cycles are shown. (DOCX 17 kb)
Primers for sequencing MTBP complementary DNA. The gene regions, product sizes and annealing temperatures are shown (DOCX 14 kb)
Acknowledgements
The authors thank all the dog owners and veterinarians, the 1. Deutscher Shar Pei Club 1985 e.V., the Shar Pei Freunde Deutschland and World of Shar Pei for their exceptional support. We also thank the Club für Exotische Rassehunde e.V. Special thanks to Mrs. Kurz and Mrs. Brusis. Many thanks to J. Wrede for his help in data analysis and M. Drabert for his support in DNA isolation. The authors would like to thank the North-German Supercomputing Alliance (HLRN) for its HPC-resources that contributed to the research results.
Funding
This study was supported by the 1. Deutscher Shar Pei Club 1985 e.V., Shar Pei Freunde Deutschland and World of Shar Pei. The funders had no role in study design, selection of dogs, data analysis and publication.
Availability of data and material
Whole genome sequencing data of the SPAID-affected Shar-Pei can be downloaded at NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra), BioProject PRJNA327712 (SRP077856).
Authors’ contributions
JM, OD designed the study, carried out the experiments and data analysis, drafted and finalized the manuscript. AN, AKU and MHT performed the pathologic and histologic examinations. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have financial competing interests as a European patent for genetic testing of MTBP:g.19383758G > A has been filed. This patent has no influence on data availability. Furthermore SNV validation of the mutation was financially supported by 1. Deutscher Shar Pei Club 1985 e.V., Shar Pei Freunde Deutschland and World of Shar Pei. The funders had no role in study design, selection of dogs, data analysis and publication.
Consent for publication
Written informed consent was obtained from the dog owners to use the EDTA-blood samples for diagnostic purposes additionally for SPAID research. In those cases where images were taken from affected dogs, dog owners confirmed the use of these images for further investigations and publication.
Ethics approval
All animal work has been conducted according to the national and international guidelines for animal welfare. EDTA-blood samples of Shar-Pei dogs and control breeds were taken by accredited veterinarians for diagnostic purposes and tissues were collected immediately after euthanasia from affected dogs so that no ethics board approval was needed. The participation was part of standard care. Written informed consent was obtained from the dog owners and veterinarians for the participation in the questionnaire survey and the use of their samples for SPAID research.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AB
Alcian blue
- ADCY8
Adenylate Cyclase 8
- CNV
Copy number variation
- COL14A1
Collagen Type XIV Alpha 1 Chain
- ddPCR
Droplet digital PCR
- DEPTOR
Domain-Containing Protein 6
- DSCC1
DNA Replication And Sister Chromatid Cohesion 1
- ENPP2
Ectonucleotide Pyrophosphatase/Phosphodiesterase 2
- HAS2
Hyaluronan synthase 2
- HE
Hematoxylin and eosin
- MEFV
Mediterranean Fever
- MRPL13
Mitochondrial Ribosomal Protein L13
- MTBP
MDM2 Binding Protein
- PAS
Periodic acid Schiff reaction
- SNTB1
Syntrophin Beta 1
- SPAID
Shar-Pei Autoinflammatory Disease
- SRA
Sequence read achieve
- TAF2
TATA-Box Binding Protein Associated Factor 2
Contributor Information
Julia Metzger, Phone: +49-511-953-8874, Email: julia.metzger@tiho-hannover.de.
Anna Nolte, Email: anna.wagner1@tiho-hannover.de.
Ann-Kathrin Uhde, Email: ann-kathrin.uhde@tiho-hannover.de.
Marion Hewicker-Trautwein, Email: marion.hewicker-trautwein@tiho-hannover.de.
Ottmar Distl, Phone: +49-511-953-8874, Email: ottmar.distl@tiho-hannover.de.
References
- 1.Berg JS, Khoury MJ, Evans JP. Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet Med. 2011;13(6):499–504. doi: 10.1097/GIM.0b013e318220aaba. [DOI] [PubMed] [Google Scholar]
- 2.Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DV, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 2011;10(3):188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 3.Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, Andraws N, Patterson ML, Krivohlavek LA, Fellis J, Humphray S, Saffrey P, Kingsbury Z, Weir JC, Betley J, Grocock RJ, Margulies EH, Farrow EG, Artman M, Safina NP, Petrikin JE, Hall KP, Kingsmore SF. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4(154):154ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunne WM, Jr, Westblade LF, Ford B. Next-generation and whole-genome sequencing in the diagnostic clinical microbiology laboratory. Eur J Clin Microbiol Infect Dis. 2012;31(8):1719–1726. doi: 10.1007/s10096-012-1641-7. [DOI] [PubMed] [Google Scholar]
- 5.Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz M. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daetwyler HD, Capitan A, Pausch H, Stothard P, Van Binsbergen R, Brøndum RF, Liao X, Djari A, Rodriguez SC, Grohs C. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46(8):858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 7.Metzger J, Tonda R, Beltran S, Agueda L, Gut M, Distl O. Next generation sequencing gives an insight into the characteristics of highly selected breeds versus non-breed horses in the course of domestication. BMC Genomics. 2014;15(1):562. doi: 10.1186/1471-2164-15-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzger J, Pfahler S, Distl O. Variant detection and runs of homozygosity in next generation sequencing data elucidate the genetic background of Lundehund syndrome. BMC Genomics. 2016;17:535. doi: 10.1186/s12864-016-2844-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzger J, Wöhlke A, Mischke R, Hoffmann A, Hewicker-Trautwein M, Kuch EM, Naim HY, Distl O. A Novel SLC27A4 Splice Acceptor Site Mutation in Great Danes with Ichthyosis. PLoS ONE. 2015;10(10):e0141514. doi: 10.1371/journal.pone.0141514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hillbertz NHS, Isaksson M, Karlsson EK, Hellmén E, Pielberg GR, Savolainen P, Wade CM, Von Euler H, Gustafson U, Hedhammar Å. Duplication of FGF3, FGF4, FGF19 and ORAOV1 causes hair ridge and predisposition to dermoid sinus in Ridgeback dogs. Nat Genet. 2007;39(11):1318–1320. doi: 10.1038/ng.2007.4. [DOI] [PubMed] [Google Scholar]
- 11.Olsson M, Meadows JR, Truve K, Rosengren Pielberg G, Puppo F, Mauceli E, Quilez J, Tonomura N, Zanna G, Docampo MJ, Bassols A, Avery AC, Karlsson EK, Thomas A, Kastner DL, Bongcam-Rudloff E, Webster MT, Sanchez A, Hedhammar A, Remmers EF, Andersson L, Ferrer L, Tintle L, Lindblad-Toh K. A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet. 2011;7(3):e1001332. doi: 10.1371/journal.pgen.1001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci. 2010;107(3):1160–1165. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metzger J, Distl O. A study of Shar-Pei dogs refutes association of the ‘meatmouth’duplication near HAS2 with Familial Shar-Pei Fever. Anim Genet. 2014;45(5):763–764. doi: 10.1111/age.12193. [DOI] [PubMed] [Google Scholar]
- 14.Olsson M, Tintle L, Kierczak M, Perloski M, Tonomura N, Lundquist A, Muren E, Fels M, Tengvall K, Pielberg G, Dufaure de Citres C, Dorso L, Abadie J, Hanson J, Thomas A, Leegwater P, Hedhammar A, Lindblad-Toh K, Meadows JR. Thorough investigation of a canine autoinflammatory disease (AID) confirms one main risk locus and suggests a modifier locus for amyloidosis. PLoS ONE. 2013;8(10):e75242. doi: 10.1371/journal.pone.0075242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsson M, Kierczak M, Karlsson A, Jablonska J, Leegwater P, Koltookian M, Abadie J, De Citres CD, Thomas A, Hedhammar A, Tintle L, Lindblad-Toh K, Meadows JR. Absolute quantification reveals the stable transmission of a high copy number variant linked to autoinflammatory disease. BMC Genomics. 2016;17(1):299. doi: 10.1186/s12864-016-2619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Consortium FF. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17(1):25. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 18.Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, Fiore M, Delfino L, Lasiglie D, Ferraris C. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 2011;63(3):830–839. doi: 10.1002/art.30170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Q, Lee G-S, Brady J, Datta S, Katan M, Sheikh A, Martins MS, Bunney TD, Santich BH, Moir S. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91(4):713–720. doi: 10.1016/j.ajhg.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, Hessner MJ, Verbsky J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leslie KS, Lachmann HJ, Bruning E, McGrath JA, Bybee A, Gallimore JR, Roberts PF, Woo P, Grattan CE, Hawkins PN. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Arch Dermatol. 2006;142(12):1591–1597. doi: 10.1001/archderm.142.12.1591. [DOI] [PubMed] [Google Scholar]
- 22.Al-Mosawi ZS, Al-Saad KK, Ijadi-Maghsoodi R, El-Shanti HI, Ferguson PJ. A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum. 2007;56(3):960–964. doi: 10.1002/art.22431. [DOI] [PubMed] [Google Scholar]
- 23.Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA, Kastner DL. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci. 2003;100(23):13501–13506. doi: 10.1073/pnas.2135380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alam MJ, Fatima N, Devi GR, Ravins, Singh RK. The enhancement of stability of p53 in MTBP induced p53-MDM2 regulatory network. Biosystems. 2012;110(2):74–83. doi: 10.1016/j.biosystems.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Brady M, Vlatkovic N, Boyd MT. Regulation of p53 and MDM2 activity by MTBP. Mol Cell Biol. 2005;25(2):545–553. doi: 10.1128/MCB.25.2.545-553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gandin I, Faletra F, Faletra F, Carella M, Pecile V, Ferrero GB, Biamino E, Palumbo P, Palumbo O, Bosco P. Excess of runs of homozygosity is associated with severe cognitive impairment in intellectual disability. Genet Med. 2014;17(5):396–399. doi: 10.1038/gim.2014.118. [DOI] [PubMed] [Google Scholar]
- 27.Shaheen R, Faqeih E, Sunker A, Morsy H, Al-Sheddi T, Shamseldin HE, Adly N, Hashem M, Alkuraya FS. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet. 2011;89(2):328–333. doi: 10.1016/j.ajhg.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nalls M, Guerreiro R, Simon-Sanchez J, Bras J, Traynor B, Gibbs J, Launer L, Hardy J, Singleton A. Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer’s disease. Neurogenetics. 2009;10(3):183–190. doi: 10.1007/s10048-009-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzger J, Karwath M, Tonda R, Beltran S, Agueda L, Gut M, Gut IG, Distl O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genomics. 2015;16(1):764. doi: 10.1186/s12864-015-1977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012;91(2):275–292. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulay SR, Thomasova D, Ryu M, Anders H-J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 2012;81(12):1199–1211. doi: 10.1038/ki.2011.482. [DOI] [PubMed] [Google Scholar]
- 32.Manukyan G, Aminov R, Hakobyan G, Davtyan T. Accelerated apoptosis of neutrophils in familial mediterranean Fever. Front Immunol. 2015;6:239. doi: 10.3389/fimmu.2015.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zanna G, Docampo MJ, Fondevila D, Bardagí M, Bassols A, Ferrer L. Hereditary cutaneous mucinosis in shar pei dogs is associated with increased hyaluronan synthase‐2 mRNA transcription by cultured dermal fibroblasts. Vet Dermatol. 2009;20(5–6):377–382. doi: 10.1111/j.1365-3164.2009.00799.x. [DOI] [PubMed] [Google Scholar]
- 34.Zanna G, Fondevila D, Bardagi M, Docampo MJ, Bassols A, Ferrer L. Cutaneous mucinosis in shar-pei dogs is due to hyaluronic acid deposition and is associated with high levels of hyaluronic acid in serum. Vet Dermatol. 2008;19(5):314–318. doi: 10.1111/j.1365-3164.2008.00703.x. [DOI] [PubMed] [Google Scholar]
- 35.Gereke M, Grobe L, Prettin S, Kasper M, Deppenmeier S, Gruber AD, Enelow RI, Buer J, Bruder D. Phenotypic alterations in type II alveolar epithelial cells in CD4+ T cell mediated lung inflammation. Respir Res. 2007;8:47. doi: 10.1186/1465-9921-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruneau S, Nakayama H, Woda CB, Flynn EA, Briscoe DM. DEPTOR regulates vascular endothelial cell activation and proinflammatory and angiogenic responses. Blood. 2013;122(10):1833–1842. doi: 10.1182/blood-2013-03-488486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gray E, Ginty M, Kemp K, Scolding N, Wilkins A. Peroxisome proliferator-activated receptor-α agonists protect cortical neurons from inflammatory mediators and improve peroxisomal function. Eur J Neurosci. 2011;33(8):1421–1432. doi: 10.1111/j.1460-9568.2011.07637.x. [DOI] [PubMed] [Google Scholar]
- 38.Andrews S. FastQC: A quality control tool for high throughput sequence data. In: Reference Source. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 1 Oct 2016.
- 39.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 44.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He C, Holme J, Anthony J. SNP genotyping: the KASP assay. In: Fleury D, Whitford R, editors. Crop Breeding. New York: Springer New York; 2014. p. 75–86. [DOI] [PubMed]
- 46.Metzger J, Schrimpf R, Philipp U, Distl O. Expression Levels of LCORL Are Associated with Body Size in Horses. PLoS ONE. 2013;8(2):1–9. doi: 10.1371/journal.pone.0056497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wood SH, Clements DN, McEwan NA, Nuttall T, Carter SD. Reference genes for canine skin when using quantitative real-time PCR. Vet Immunol Immunopathol. 2008;126(3–4):392–395. doi: 10.1016/j.vetimm.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blankenberg D, Kuster GV, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J: Galaxy: a web-based genome analysis tool for experimentalists. Current protocols in molecular biology. 2010;89:19.10. 11–19.10. 21. [DOI] [PMC free article] [PubMed]
- 51.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g: Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35(suppl 2):W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(D1):D377–386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypic data of 164 study participants with specified inflammatory process types. The number of Shar-Pei dogs and their individual signs of inflammation is shown (1 = affected; 0 = unaffected). In 62 Shar-Pei no signs of the disease could be found. (PDF 260 kb)
Whole genome sequencing statistics for two SPAID-affected Shar-Pei and five controls. DNA-libraries of two Shar-Pei were sequenced on the Illumina MiSeq whereas further five whole genome sequences from dogs of four different breeds were derived from sequence read archive (NCBI). (DOCX 15 kb)
Mutant allele frequencies for SPAID candidate variants. All nine variants were investigated for their genotypic distribution in SPAID-affected and unaffected Shar-Pei as well as in 162 dogs of 11 different breeds. (DOCX 20 kb)
Variant detection in complementary DNA of MTBP. In total five variants could be detected in cDNA of MTBP including the candidate SNV MTBP:g.19383758G > A. None of the other variants could be exclusively found in the SPAID-affected Shar-Pei. (DOCX 17 kb)
Table S2. Shared runs of homozygosity (ROH) for SPAID-affected Shar-Pei. Two Shar-Pei were investigated for shared ROH regions which could not be detected in eight control dogs of different breeds. The chromosomal position (CanFam2.0 and CanFam3.1) of shared ROH regions, size of shared ROHs, number of SNPs, genes IDs (CanFam3.1) and human orthologues are shown. (DOCX 20 kb)
The proportion of gene hits against total number of process hits for genes detected in runs of homozygosity (ROH) regions shared by SPAID (Shar-Pei Autoinflammatory Disease) affected Shar-Pei, which could not be found in control dogs, is shown. PANTHER gene list analysis for biologic processes and pathways was done for human orthologues. (DOCX 16 kb)
Questionnaire for the assessment of health status in investigated Shar-Pei. A description of the project and information consent for dog owners and veterinarians is included. (PDF 160 kb)
Primer sequences and assays for the validation of candidate variants. Kompetitive Allele Specific PCR (KASP) assays were used for six variants whereas further three variants were genotyped by gel electrophoresis on an acrylamide gel. Primer pairs, validation type, amplicon size, annealing temperature and number of PCR cycles are shown. (DOCX 17 kb)
Primers for sequencing MTBP complementary DNA. The gene regions, product sizes and annealing temperatures are shown (DOCX 14 kb)