

Abstract
Acute lung injury due to sulfur mustard (SM) inhalation causes formation of airway fibrin casts that obstruct airways at multiple levels, leading to acute respiratory failure and death. These pathophysiological effects are seen in rodent models of acute SM vapor inhalation, as well as in human victims of acute SM inhalation. In rat models, the initial steps in activation of the coagulation system at extravascular sites depend on tissue factor (TF) expression by airway cells, especially in the microparticle fraction, and these effects can be inhibited by TF pathway inhibitor (TFPI) protein. Not only does the procoagulant environment of the acutely injured lung contribute to airway cast formation, but these lesions persist in airways because of the activation of multiple antifibrinolytic pathways, including plasminogen activator inhibitor-1 (PAI-1), thrombin-activatable fibrinolysis inhibitor (TAFI), and α2-antiplasmin (α2AP). Airway administration of tissue plasminogen activator (tPA) can overwhelm these effects and be save lives by preventing fibrin-dependent airway obstruction, gas-exchange abnormalities, and respiratory failure. In human survivors of SM inhalation, fibrotic processes, including bronchiolitis obliterans and interstitial fibrosis of the lung, are among the most disabling chronic lesions. Antifibrotic therapies may prove useful in preventing either or both of these forms of chronic lung damage.
Keywords: lung injury, tissue factor, tissue plasminogen activator, sulfur mustard
Introduction: significance and impact
Sulfur mustard (2,2-dichlorodiethyl(SM)) is a toxic chemical compound best known for its use as a vesicating warfare agent.SM was first synthesized in 1860 by Guthrie, who noted its vesicating property: “small quantities of vapor attack thin parts of the skin between the fingers and around the eyes, destroying the epidermis.” 1 It was put into full-scale military use in 1917, at the height of World War I. By that war’s conclusion, SM had caused more casualties than all other chemical weapons combined. Despite its battlefield introduction nearly 100 years ago, and its prohibition by the Geneva Protocol of 1925, SM has remained a threat into modern times as the most utilized chemical weapon on the planet.2–4 It was used extensively against Iranian soldiers and Kurdish and Iranian civilians between 1983 and 1988, resulting in over 100,000 chemical casualties. In 2015, at least three confirmed incidents of SM use by the Islamic State (ISIL) against Kurdish troops were reported during conflict in Syria.5 As recently as March 25, SM and chlorine were used in combination by ISIL to attack Taza, Iraq, killing three people and causing 35 to be hospitalized.6 These latest deployments occurred despite international efforts to remove chemical weapons from Syria, and serve as reminder that SM remains stockpiled in many nations, is relatively easy to manufacture, and is sought by individuals motivated to cause mass casualties.
Chemical considerations
A fundamental aspect of SM chemistry is the formation of a heterocyclic sulphonium ion. This occurs when unpaired electrons on the central sulfur atom initiate nucleophilic attack upon one of the two terminal chlorines on the molecule (Fig.1). After an exchange of electrons, a single chlorine group leaves the molecule, resulting in formation of an unstable intermediate that is capable of irreversibly alkylating most biological molecules. Sulphonium ion formation is facilitated in aqueous solution, which likely contributes to the sensitivity of certain target organs, such as the eye and lung.7 Structurally, mustard agents are divided into two groups. The first of these are the monofunctional mustards (including 2-chloroethyl ethylsulfide (CEES)), which possess only one chlorine atom in the parent molecule, and, therefore, can only cause a single alkylation reaction. The second group are bifunctional mustards, such as sulfur mustard (SM), having two chlorine atoms, one at either terminal position. Because bifunctional mustards can undergo two alkylation reactions, they can generate molecular crosslinks, and this is believed to be why they are more toxic.
Figure 1.
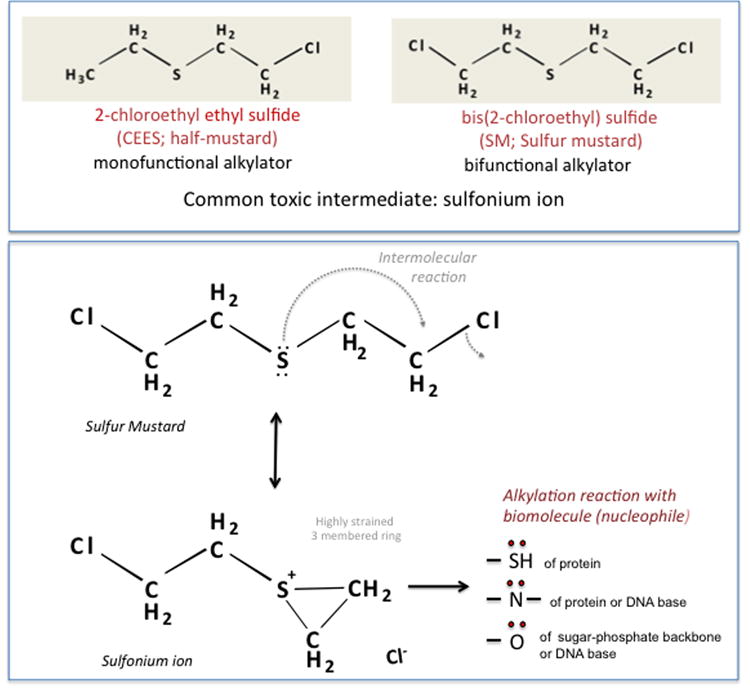
(A) Chemical structures of bis(2-chloroethyl sulfide), also known as sulfur mustard (SM), and its analog 2-chloroethyl ethyl sulfide (CEES). (B) Cyclic sulfonium ion formation from SM and its potential interaction with biomolecules.
Health issues related to sulfur mustard inhalation: the human experience
Acute health effects
Current understanding of the human health effects of SM exposure has developed primarily through clinical investigation of two cohorts of SM victims—those of World War I (1917–1918) and the Iran–Iraq war (1980–1988). Most of these individuals received a single exposure.8
Importantly, airway injury is the principal cause of mortality after SM exposure. In humans, exposure to low or moderate SM concentrations involves acute edema formation and destruction of the airway epithelial lining. Exposure to higher concentrations will elicit injury of greater severity in the larynx, sinuses, trachea, and bronchi, with signs of injury extending to more distal regions. Thorough examination of anatomical injury sites in animal models also has revealed nasopharyngeal, glottis, and subglottic airway injury, edema, mucosal sloughing, and airway obstruction. Similar processes can occur in more distal conducting airways, including an exudative process leading to formation of obstructive airway casts, rich in fibrin content, and a disorder termed “plastic bronchitis.” Lethal exposures cause death by asphyxiation from cardiorespiratory failure, complications due to bronchial obstruction or plastic bronchitis, and/or resulting secondary pneumonia.9
Ghanei and coworkers10 reported a variety of acute effects of SM, including blistering of the skin and blistering and ulceration of the cornea. The timing of the appearance of many of these signs and symptoms appears to be related to dose, such that higher-level exposures result in earlier symptoms and more rapid progression of disease. Initial signs can include palpebral edema with associated conjunctivitis and photophobia, and can occur within 20 min to 4 h after exposure. Later comes a progression of itching and then blistering of the skin, rhinorrhea progressing to nasal obstruction, laryngitis with hoarseness, and tracheitis, evidenced by cough, and followed by expectoration of airway casts. These events can be noted 4–48 h after exposure.
Within the bone marrow, the hematopoietic system can be suppressed. In the gastrointestinal tract, ulceration and epithelial sloughing can result. In the central nervous system, edema can lead to nonspecific neurologic signs and symptoms, such as irritability.
Subacute health effects
Multiple pathological processes stem from the acute phase of SM inhalation injury. Exposed individuals are susceptible to secondary infection due to the loss of barrier function of the eyes, skin, and airways, as well as the suppression of production of various leukocytes in the bone marrow. Infections of the lung and/or skin can progress to sepsis. Airway obstruction caused by leakage of blood plasma constituents can progressively worsen in the days following exposure and may necessitate surgical intervention, including emergency tracheostomy, to remove cast material and/or bypass obstruction. Focal hemorrhage also can occur. Lower airway obstruction by casts can lead to secondary mucus impaction, atelectasis, secondary postobstructive pneumonia, and bronchitis, resulting in acute respiratory failure and a need for mechanical ventilation. In turn, regional air trapping may worsen, potentially resulting in pneumothorax and/or other air-leak syndromes. These complications, including acute respiratory failure, acute airway obstruction, and air-leak syndromes, have been reported to be fatal during hospital care of SM inhalation survivors in the subacute period after exposure.10
In some cases, airway casts have been found at autopsy. In one large cohort of SM-exposed patients referred to European tertiary care centers, 23% (15 of 65) had airway casts visualized. Of those, almost half (7 of 15) died, while an additional 20% (3 of 15) had emergency tracheostomy to relieve acute airway obstruction. In all, approximately 80% of deaths (7 of 9) in this cohort were associated with airway cast formation.9,10 These were patients that were airlifted to European hospitals at days 3–21 following SM exposure during the Iran–Iraq war (1984–1987). Thus, acute/subacute airway obstruction is an important determinant of survival after SM inhalation.
Chronic health effects
A meta-analysis of studies of formerly exposed individuals determined a prevalence of delayed skin disorders of 94.6%, pulmonary complications at 94.5%, and ocular complications at 89.9%.11 The incidence of various cancers varied, but was in the range 1.7–2.2%. Follow-up clinical investigations of SM-exposed individuals have described a number of late-developing respiratory complications after SM exposure, including asthma, emphysema, chronic bronchitis, pulmonary fibrosis, and bronchiolitis obliterans (BO).12–17 Among those exposed in the Iran–Iraq conflict, 75% of survivors were noted to develop tracheal and/or bronchial deformities or strictures, BO, and/or pulmonary fibrosis. Additional airway complications, including chronic bronchitis, recurrent pneumonia, bronchiectasis, and tracheobronchomalacia, also have been reported.2,11 A number of patients have had constrictive bronchiolitis on lung biopsy, even with normal pulmonary function tests and chest high-resolution computed tomography scans (HRCT).2
Other, more subtle forms of lung injury also can occur. A cohort mortality study of 3500 workers at a mustard gas manufacturing plant in England revealed that occupational exposure was associated with significantly higher mortality due to influenza, pneumonia, or asthma.18 Risk of mortality was evident even in workers having less than 3 years of employment at the plant.
Fibrotic disorders arising from SM exposure can also involve other organs. For example, chronic complications in the skin can include hyperpigmentation, hypertrophic scarring, and/or secondary joint deformity. Within the eyes, latent onset of ulcerative keratitis after a decade-long asymptomatic period was described in World War I veterans and in Iranian veterans exposed during the 1980s.3,11 These lesions recur even after corneal transplant and may lead to late-onset blindness.
Epidemiological studies examining the link between SM and cancer after single, high-dose exposure have been inconclusive. Occupational exposure of factory workers (German, Japanese, British)involved in SM production, however, was associated with a greater incidence of malignancies, including leukemia, Bowen’s disease, and laryngeal and bronchial cancer.4,19 SM is rated a group I carcinogen by the International Agency for Research on Cancer (IARC).
Animal models for sulfur mustard inhalation injury
In the rat, nose-only exposure to aerosolized CEES resulted in significant injury to both upper and lower pulmonary tracts, but spared the alveolar regions. 20 However, nose-only exposure to SM vapor resulted in only upper airway injury, causing an acute sclerosing nasopharyngitis, without notable pulmonary airway or parenchymal injury. The lack of lower airway tract injury with SM is thought to be due to the highly reactive nature of SM vapor, and the animal’s “scrubbing” of the SM vapor particles via its extensive nasal passages. For this reason, in the rat, significant tracheobronchial or distal airway injury does not occur unless SM vapor is delivered directly into the trachea by cannula. Both at moderate SM vapor 21or CEES aerosol inhaled doses, airway clots or casts were found lining and occluding the airways, from generations 3–15 in rats within 18 h, exposed via lung microdissection.20 These were associated with marked wheezing, stridor, agonal respirations, hypoxemia, and often death. The airway casts were found to contain mainly fibrin and no mucins, as per immunohistochemical staining. Confocal microscopy and dual vital-staining evaluation of casts showed a mixture of live and dead cells, minimal inflammatory cells, and some sloughed epithelial cells. The casts were also associated with peribronchovascular edema, and they appeared to emanate from the bronchus-associated lymphoid tissue (BALT) regions, as they were contiguous with the epithelial fenestrations of that region. Permeability of the bronchial circulation in these regions also appeared markedly increased, as shown by Monastral blue dye leakage from bronchial blood vessels.
Potential mechanisms of mustard-related tissue injury
Vesicant and mustard agents, including CEES and SM, damage tissues via a variety of mechanisms. Among these is oxidative stress, which is evidenced by 8-oxo-2-deoxyguanosine, 4-hydroxynonenal,22 and 5,5-dimethyl-2-(8-octanoic acid)-1-pyrolline N-oxide (DMPO) protein adduct formation.23 These are markers of damage to diverse cellular targets, including DNA, lipids, and proteins, respectively. Secondarily, DNA damage by SM or other mustard agents may also result from oxidative stress, adduct formation, crosslinking, and/or other related mechanisms.24–27 In addition, acute inflammation28 and related processes, including cyclooxygenase-2, inducible nitric oxide syntheses, and matrix metalloproteinase-9 inductions, also can occur. A variety of cell signaling pathways, including H2A.X phosphorylation;25 activation of MAP kinases Akt, AP-1, and NF-kB;25 and p53 phosphorylation and accumulation24,29 were also activated, potentially further amplifying injury and inflammation. In the skin, these pathways may cause apoptotic cell death, as well as macrophage, mast cell, and neutrophil infiltration and microvesication.30-32 In this rodent model, many of these deleterious effects could be limited or inhibited by catalytic33or flavanone antioxidants.34,35
General mechanisms of acute airway injury due to inhaled mustards
Although both the skin and the eyes can be seriously affected, disease of the respiratory tract is the most critical complication of SM exposure. SM inhalation–induced pulmonary injury is often described as a disease involving virtually all levels of the airway, with distinctive acute, subacute, and late (chronic) phases of injury progression.
Early symptoms of SM-induced pulmonary injury may present at times greater than 12 h after initial exposure, and can include coughing, wheezing, dyspnea, expectoration, progressive hypoxemia and/or cyanosis, and chest radiographic abnormalities. Higher levels of inhaled SM result in a more rapid evolution of lung injury symptoms and a more severe outcome, potentially including death. Patients with milder disease often show only bronchitis-and/or asthma-like symptoms. The European experience in the 1980s demonstrated that airway and ocular lesions progressed over time, often requiring days before patients were deemed to be in a critical state if only moderately or mildly exposed. This often allowed sufficient time to transport victims to tertiary and quaternary care hospitals for treatment. However, there is yet no antidote or treatment for acute SM inhalation toxicity,36 and there is no specific treatment to prevent death due to SM exposure.
Survivors of the acute phase of SM inhalation are susceptible to secondary pulmonary infections, pneumothoraces, acute respiratory distress syndrome (ARDS), and emphysema/bullae in the subacute phase of pulmonary injury. These subacute complications likely result from airway blockages due to partially obstructing casts that were not removed, and air-leak syndromes can occur, especially in those patients requiring mechanical ventilation. In addition, a secondary toxic neutropenia/leukopenia (45%) and/or lymphocytopenia (14%) that causes immunocompromised can occur.9 There is also experimental evidence that fibrin in the airways can facilitate bacterial colonization and/or infection. Chronic sequelae in those who survive the acute and subacute processes include fibrotic airway strictures at any level along the respiratory tree, BO, chronic obstructive pulmonary disease (COPD), and pulmonary fibrosis.37
Activation and derangement of the coagulation system
Acute pulmonary injury resulting from mustard inhalation presents initially as hypoxemia due to acute airway obstruction by fibrin casts, followed by a rapid progression to respiratory failure. A key feature of this syndrome is the shift toward a prothrombotic and antifibrinolytic pulmonary environment (Fig. 2). Several interrelated processes are responsible for this shift. The first of these is the early onset of disruption of airway vascular barrier function. Veress et al. demonstrated that bronchial blood vessels beneath areas of damaged epithelium on distal trachea and central bronchi are anatomical regions of mustard-induced airway vascular leak, and these findings are associated with a lack of alveolar injury.20 Detection of high–molecular weight IgM (900 kDa) in BALF after injury further revealed that fluid inflow from these injured airway sites can persist for several days or more and is relatively nonselective, given that large plasma proteins are entering the airways.20,38
Figure 2.
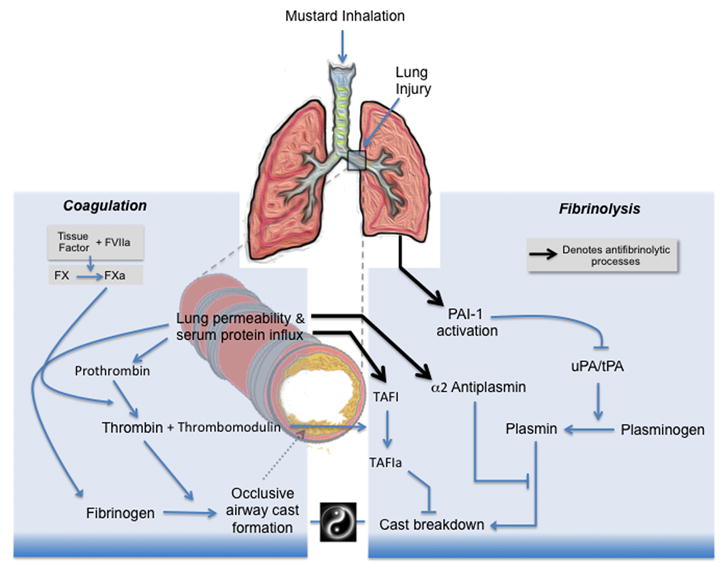
Schematic representation of the initiation of coagulation and recruitment of fibrinolytic inhibitors following mustard inhalation. Mustard inhalation causes lung injury, which facilitates leakage of serum proteins into airways. The presence of airway tissue factor initiates clotting of serum-derived coagulation factors, leading to nascent fibrin cast formation. Plasmin-dependent fibrinolysis is the physiologic mechanism for removal of airway casts and restoration of airway patency. Mustard injury also gives rise to elevated airway levels of antifibrinolytic molecules, which collectively inhibit the plasminogen-activation pathway. Simultaneous involvement of procoagulant and antifibrinolytic pathways within the airways can tip the balance toward cast accrual, leading to impaired gas exchange and respiratory failure.
One of the consequences of fluid intrusion into the airways is the influx of coagulation factors, including fibrinogen, prothrombin, and factor X (FX).20,39 While their initial presence may serve to minimize hemorrhage and provide temporary structural integrity to injured areas,40 continued accumulation of these factors provides raw material for fibrin-dependent cast formation. Thrombin and FXa can also be incorporated into airway casts, where they may exert long-lasting effects, including proinflammatory signaling and proteolytic activity.39
One of the principal ways the body maintains hemostasis is by localizing coagulation to surfaces of cell membranes. Tissue factor (TF) is the only coagulation protein permanently attached to the membrane surface, and it is believed to potentially be the sole initiator of coagulation in vivo.41 Whereas vascular endothelial cells are devoid of TF under normal physiologic situations, epithelial cells lining airways contain high levels of TF.42,43 The presence of TF expression on extravascular epithelium serves as a “hemostatic envelope” to prevent extravasation into the lung in the event of injury. Following mustard-induced airway injury, however, epithelial cells undergo anoikis and apoptotic microparticle formation. These degradative processes increase the TF-dependent procoagulant activity of airway surface liquid, the bulk of which then exists in microparticle form.39 Release and dissemination of procoagulant microparticles is also believed to play a critical role in the development of fibrin-dependent airway obstruction by extending localized zones of thrombin activation at sites of injury further into the central areas of the airway lumen. Despite TF’s potentially harmful role in early airway obstruction,39 it may also have important beneficial roles in airway regeneration, as it can function as a facilitator of survival of airway progenitor cells.44 For this reason, strategies that promote resolution of airway coagulation by promoting fibrinolysis could avoid an impact on TF’s functions in airway epithelial regeneration.
In vascular homeostasis, the effects of TF are counteracted by TF pathway inhibitor (TFPI), which blocks the TF-dependent actions of factors Xa and VIIa. During acute lung injury (ALI), however, elevated TF levels are expressed while lowered TFPI levels are present, causing a shift toward a procoagulant state.43 The importance of the TF/TFPI imbalance in mustard-related pathophysiology has been clearly demonstrated in studies employing TFPI as a rescue therapy.38 In these studies, airway obstruction in the form of fibrin-containing casts was evident in central conducting airways of rats receiving 10% CEES by aerosol inhalation. TFPI decreased cast formation, limited severe hypoxemia, and prevented mortality normally associated with inhalation of CEES at this concentration. Findings of reduced prothrombin consumption and lower thrombin–antithrombin (TAT) complexes in BALF demonstrated that TFPI acted to limit thrombin activation in airways. Also noteworthy was the observation that TFPI did not appreciably correct protein leak, an indication that mustard-induced hypoxemia is not simply a manifestation of lung edema, but is instead caused by fibrin-dependent airway obstruction. Airway delivery of other anticoagulants, including heparin, has been shown to limit mortality in response to half-mustard (CEES), but these had to be given early (1 h) and caused occasional microhemorrhage.45
Following mustard inhalation, plasmin-dependent fibrinolysis is the physiologic mechanism for removal of airway casts and restoration of airway patency. Elevated airway levels of several antifibrinolytic molecules also occur, which collectively inhibit the plasminogen activation pathway. The first of these, plasminogen activator inhibitor-1 (PAI-1), is the principal endogenous inhibitor of tissue plasminogen activator (tPA) and urokinase plasminogen activators (uPA). In addition, thrombin-activatable fibrinolysis inhibitor (TAFI) also restricts plasmin formation by removal of C-terminal lysines from fibrin, which are binding sites for plasminogen. α2-Antiplasmin (α2AP) provides a third level of fibrinolytic suppression by covalently binding any plasmin that is generated. All three antifibrinolytic molecules are present and functionally active in airway surface liquid during the time of nascent cast formation. Reverse transcriptase polymerase chain reaction (RT-PCR) analysis indicated that the lung produces negligible amounts of TAFI or α2AP mRNA, an indication that their increased presence in mustard-damaged airways is due to vascular permeability and leak.46 PAI-1 mRNA expression, however, is greatly increased in the lung after inhalation of mustards, an indication that its presence is independent of leaks, suggesting that PAI-1 is newly synthesized in response to acute lung injury.
Clinical implications
Rescue potential of airway-delivered tissue plasminogen activator in sulfur mustard–mediated plastic bronchitis
Airway injury is the leading cause of mortality in the first few days to weeks after SM inhalation. Exposure to 2.4 mg-min/M3 for 10–60 min, or lesser concentrations for 4–8 h, can cause serious respiratory effects. These doses also cause blistering and may cause incapacitation and even death.47 The lack of efficient postexposure medical countermeasures (MCMs) for victims experiencing respiratory distress, therefore, presents a serious public health risk. Among human victims in the Iran–Iraq war, significant numbers of patients with severe lung injury survived such that they could be transported to tertiary care centers in Europe. Among those patients, airway obstruction by casts (plastic bronchitis) was a prominent cause of acute deterioration and even death.9 Within that group, acute clearance of the conducting airways by bronchoscopy and/or provision of an artificial airway (e.g., tracheostomy) often proved to be lifesaving. Among those that survived the early adverse effects of SM exposure, BO, an irreversible form of airway obstruction, has been the most common severe primary respiratory sequela.16,17
Development of MCMs to reduce the acute massive exudative bronchial obstructive process in the lungs and to preserve lung function after SM exposure is paramount to increasing overall survival in the event of a chemical attack with SM. Alteplase (human recombinant tissue plasminogen activator (tPA)), our proposed MCM to treat victims in the acute phase of pulmonary injury after SM, has several distinct advantages, as outlined here. Among these are (1) a demonstrated safety profile for many years as an intravenous agent; (2) demonstrated efficacy as an airway-administered rescue for SM-related injury in rats; (3) a reasonable logistical burden given the acuity level of the patients that will require it;and (4) anecdotal evidence of efficacy/safety in human patients with non-SM-associated plastic bronchitis. Extravascular airway coagulation due to sulfur mustard vapor inhalation develops over a period of hours to days, a realistic time period in which a complex medical therapy could be implemented. Thus, the concept of operations (ConOps) for this MCM is realistic. Part of our research group’s effort now focuses on development of a large animal model (porcine) of SM-induced lung injury for use in efficacy studies of the Alteplase MCM on its developmental path toward U.S. Food and Drug Administration (FDA)licensure.
Further considerations on mass casualty scenarios and use of tPA treatment
The concept of operations with regard to tPA is that it would not be given indiscriminately to all suspected SM-inhalation victims. Instead, patients eligible for therapy would be selected from individuals that have been transported to field hospitals or referral hospitals with intensive care units. The intention would be that airway-administered tPA would be given to the most severely affected patients. In rodent preclinical studies, animals were required to show a decrease in pulse-oximetry values of at least 5% below normal baseline in association with signs of airway obstruction (stridor, plug expectoration, wheezing), before providing tPA treatment. Similar signs of hypoxemia and respiratory distress would be necessary to identify those human patients that might benefit from tPA MCM. Cutaneous evidence of SM exposure in humans typically appears within 4–12 h, and initial respiratory symptoms are likely to begin to evolve over the same interval. By contrast, ocular signs often occur more rapidly. Although field detectors may give an indication that SM exposure could have occurred, clinically useful biochemical indicators confirming a patient’s exposure promptly enough to influence clinical decisions are not currently available. Thus, clinical findings will be used to indicate which patients would likely benefit from tPA therapy. Drug administration will require directed delivery into the airway by bronchoscopy. Detailed investigation in rats has shown that treatment with airway tPA can be delayed 6–24 h and still be of benefit after inhalation of sulfur mustards. Findings in porcine models have shown clinical signs, and even death, can occur over 6–24 h.48-50
The challenge of prevention of chronic lung disability following sulfur mustard inhalation
To date, our efforts have focused on the prevention of mortality after acute SM exposure in preclinical models. This has included the use of anticoagulants and fibrinolytic agents. The most promising of these involves airway delivery of human recombinant tissue plasminogen activator, an FDA-approved drug present in all hospitals. It remains to be seen if these agents, potentially combined with subsequent antifibrotic drugs, can also be beneficial against the long-term chronic sequelae of SM inhalation exposure, including BO and pulmonary fibrosis.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Guthrie F. On Some derivatives from the olefines. Q J Chem Soc. 1860;12:109–125. [Google Scholar]
- 2.Ghanei M, Tazelaar HD, Chilosi M, et al. An international collaborative pathologic study of surgical lung biopsies from mustard gas-exposed patients. Respir Med. 2008;102:825–830. doi: 10.1016/j.rmed.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Etezad-Razavi M, Mahmoudi M, Hefazi M, et al. Delayed ocular complications of mustard gas poisoning and the relationship with respiratory and cutaneous complications. Clin Experiment Ophthalmol. 2006;34:342–346. doi: 10.1111/j.1442-9071.2006.01220.x. [DOI] [PubMed] [Google Scholar]
- 4.Manning KP, Skegg DC, Stell PM, et al. Cancer of the larynx and other occupational hazards of mustard gas workers. Clin Otolaryngol Allied Sci. 1981;6:165–170. doi: 10.1111/j.1365-2273.1981.tb01527.x. [DOI] [PubMed] [Google Scholar]
- 5.SAMS Hospital Sees Mustard Gas Victims in Mare’e, Aleppo. SAMS Foundation 2015 [Google Scholar]
- 6.Kohnavard N. Iraqi town Taza ’hit in IS chemical attack’ appeals for help. BBC BBC Persian, Taza, Northern Iraq: BBC World News 2016 [Google Scholar]
- 7.Somani SM, Babu SR. Toxicodynamics of sulfur mustard. Int J Clin Pharmacol Ther Toxicol. 1989;27:419–435. [PubMed] [Google Scholar]
- 8.Thorpe E. In: Thorpe’s Dictionary of Applied Chemistry. 4. T E, editor. Vol. 3. London: 1974. p. 8. [Google Scholar]
- 9.Willems J. Clinical management of mustard gas casualties. Ann Med Milit. 1989;3:1–61. [Google Scholar]
- 10.Ghanei MF, Akbari Moqadam M, Mohammad M, et al. Tracheobronchomalacia and air trapping after mustard gas exposure. Am J Respir Crit Care Med. 2006;173:304–309. doi: 10.1164/rccm.200502-247OC. [DOI] [PubMed] [Google Scholar]
- 11.Panahi Y, Gholami N, Ghojazadeh M, et al. Complications and Carcinogenic Effects of Mustard Gas--a Systematic Review and Meta-Analysis in Iran. Asian Pac J Cancer Prev. 2015;16:7567–7573. doi: 10.7314/apjcp.2015.16.17.7567. [DOI] [PubMed] [Google Scholar]
- 12.Graham JS, Schoneboom BA. Historical perspective on effects and treatment of sulfur mustard injuries. Chem Biol Interact. 2013;206:512–522. doi: 10.1016/j.cbi.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Taghaddosinejad F, Fayyaz AF, Behnoush B. Pulmonary complications of mustard gas exposure: a study on cadavers. Acta Med Iran. 2011;49:233–236. [PubMed] [Google Scholar]
- 14.Balali-Mood M, Afshari R, Zojaji R, et al. Delayed toxic effects of sulfur mustard on respiratory tract of Iranian veterans. Hum Exp Toxicol. 2011;30:1141–1149. doi: 10.1177/0960327110389501. [DOI] [PubMed] [Google Scholar]
- 15.Emad A, Rezaian GR. The diversity of the effects of sulfur mustard gas inhalation on respiratory system 10 years after a single, heavy exposure: analysis of 197 cases. Chest. 1997;112:734–738. doi: 10.1378/chest.112.3.734. [DOI] [PubMed] [Google Scholar]
- 16.Ghanei M, Mokhtari M, Mohammad MM, et al. Bronchiolitis obliterans following exposure to sulfur mustard: chest high resolution computed tomography. Eur J Radiol. 2004;52:164–169. doi: 10.1016/j.ejrad.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Saber H, Saburi A, Ghanei M. Clinical and paraclinical guidelines for management of sulfur mustard induced bronchiolitis obliterans; from bench to bedside. Inhal Toxicol. 2012;24:900–906. doi: 10.3109/08958378.2012.725783. [DOI] [PubMed] [Google Scholar]
- 18.Easton DF, Peto J, Doll R. Cancers of the respiratory tract in mustard gas workers. Br J Ind Med. 1988;45:652–659. doi: 10.1136/oem.45.10.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada A. On the Late Injuries Following Occupational Inhalation of Mustard Gas, with Special References to Carcinoma of the Respiratory Tract. Acta Pathol Jpn. 1963;13:131–155. doi: 10.1111/j.1440-1827.1963.tb01918.x. [DOI] [PubMed] [Google Scholar]
- 20.Veress LA, O’Neill HC, Hendry-Hofer TB, et al. Airway obstruction due to bronchial vascular injury after sulfur mustard analog inhalation. Am J Respir Crit Care Med. 2010;182:1352–1361. doi: 10.1164/rccm.200910-1618OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veress LA, Anderson DR, Hendry-Hofer TB, et al. Airway tissue plasminogen activator prevents acute mortality due to lethal sulfur mustard inhalation. Toxicol Sci. 2015;143:178–184. doi: 10.1093/toxsci/kfu225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Neill HC, White CW, Veress LA, et al. Treatment with the catalytic metalloporphyrin AEOL 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free Radic Biol Med. 2010;48:1188–1196. doi: 10.1016/j.freeradbiomed.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pal A, Tewari-Singh N, Gu M, et al. Sulfur mustard analog induces oxidative stress and activates signaling cascades in the skin of SKH-1 hairless mice. Free Radic Biol Med. 2009;47:1640–1651. doi: 10.1016/j.freeradbiomed.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inturi S, Tewari-Singh N, Gu M, et al. Mechanisms of sulfur mustard analog 2-chloroethyl ethyl sulfide-induced DNA damage in skin epidermal cells and fibroblasts. Free Radic Biol Med. 2011;51:2272–2280. doi: 10.1016/j.freeradbiomed.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jain AK, Tewari-Singh N, Gu M, et al. Sulfur mustard analog, 2-chloroethyl ethyl sulfide-induced skin injury involves DNA damage and induction of inflammatory mediators, in part via oxidative stress, in SKH-1 hairless mouse skin. Toxicol Lett. 2011;205:293–301. doi: 10.1016/j.toxlet.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tewari-Singh N, Gu M, Agarwal C, et al. Biological and molecular mechanisms of sulfur mustard analogue-induced toxicity in JB6 and HaCaT cells: possible role of ataxia telangiectasia-mutated/ataxia telangiectasia-Rad3-related cell cycle checkpoint pathway. Chem Res Toxicol. 2010;23:1034–1044. doi: 10.1021/tx100038b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inturi S, Tewari-Singh N, Agarwal C, et al. Activation of DNA damage repair pathways in response to nitrogen mustard-induced DNA damage and toxicity in skin keratinocytes. Mutat Res. 2014:763–764. 53–63. doi: 10.1016/j.mrfmmm.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tewari-Singh N, Rana S, Gu M, et al. Inflammatory biomarkers of sulfur mustard analog 2-chloroethyl ethyl sulfide-induced skin injury in SKH-1 hairless mice. Toxicol Sci. 2009;108:194–206. doi: 10.1093/toxsci/kfn261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar D, Tewari-Singh N, Agarwal C, et al. Nitrogen mustard exposure of murine skin induces DNA damage, oxidative stress and activation of MAPK/Akt-AP1 pathway leading to induction of inflammatory and proteolytic mediators. Toxicol Lett. 2015;235:161–171. doi: 10.1016/j.toxlet.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jain AK, Tewari-Singh N, Orlicky DJ, et al. 2-Chloroethyl ethyl sulfide causes microvesication and inflammation-related histopathological changes in male hairless mouse skin. Toxicology. 2011;282:129–138. doi: 10.1016/j.tox.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tewari-Singh N, Jain AK, Orlicky DJ, et al. Cutaneous injury-related structural changes and their progression following topical nitrogen mustard exposure in hairless and haired mice. PLoS One. 2014;9:e85402. doi: 10.1371/journal.pone.0085402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jain AK, Tewari-Singh N, Inturi S, et al. Histopathological and immunohistochemical evaluation of nitrogen mustard-induced cutaneous effects in SKH-1 hairless and C57BL/6 mice. Exp Toxicol Pathol. 2014;66:129–138. doi: 10.1016/j.etp.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tewari-Singh N, Inturi S, Jain AK, et al. Catalytic antioxidant AEOL 10150 treatment ameliorates sulfur mustard analog 2-chloroethyl ethyl sulfide-associated cutaneous toxic effects. Free Radic Biol Med. 2014;72:285–295. doi: 10.1016/j.freeradbiomed.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tewari-Singh N, Jain AK, Inturi S, et al. Silibinin attenuates sulfur mustard analog-induced skin injury by targeting multiple pathways connecting oxidative stress and inflammation. PLoS One. 2012;7:e46149. doi: 10.1371/journal.pone.0046149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain AK, Tewari-Singh N, Inturi S, et al. Flavanone silibinin treatment attenuates nitrogen mustard-induced toxic effects in mouse skin. Toxicol Appl Pharmacol. 2015;285:71–78. doi: 10.1016/j.taap.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wille T, Worek F, Thiermann H. Translation of experimental research for improved treatment of chemical warfare agents. Chem Biol Interact; Fourteenth International Medical Chemical Defence Conference 2013; 2013. p. 433. [DOI] [PubMed] [Google Scholar]
- 37.Rowell M, Kehe K, Balszuweit F, et al. The chronic effects of sulfur mustard exposure. Toxicology. 2009;263:9–11. doi: 10.1016/j.tox.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 38.Rancourt RC, Veress LA, Ahmad A, et al. Tissue factor pathway inhibitor prevents airway obstruction, respiratory failure and death due to sulfur mustard analog inhalation. Toxicol Appl Pharmacol. 2013;272:86–95. doi: 10.1016/j.taap.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rancourt RC, Veress LA, Guo X, et al. Airway tissue factor-dependent coagulation activity in response to sulfur mustard analog 2-chloroethyl ethyl sulfide. Am J Physiol Lung Cell Mol Physiol. 2012;302:L82–92. doi: 10.1152/ajplung.00306.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown LF, Dvorak AM, Dvorak HF. Leaky vessels, fibrin deposition, and fibrosis: a sequence of events common to solid tumors and to many other types of disease. Am Rev Respir Dis. 1989;140:1104–1107. doi: 10.1164/ajrccm/140.4.1104. [DOI] [PubMed] [Google Scholar]
- 41.Smith SA. The cell-based model of coagulation. J Vet Emerg Crit Care (San Antonio) 2009;19:3–10. doi: 10.1111/j.1476-4431.2009.00389.x. [DOI] [PubMed] [Google Scholar]
- 42.Bastarache JA, Wang L, Geiser T, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62:608–616. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Poll T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit Care. 2008;12(Suppl 6):S3. doi: 10.1186/cc7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmad S, Ahmad A, Rancourt RC, et al. Tissue factor signals airway epithelial basal cell survival via coagulation and protease-activated receptor isoforms 1 and 2. Am J Respir Cell Mol Biol. 2013;48:94–104. doi: 10.1165/rcmb.2012-0189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Houin PR, Veress LA, Rancourt RC, et al. Intratracheal heparin improves plastic bronchitis due to sulfur mustard analog. Pediatr Pulmonol. 2015;50:118–126. doi: 10.1002/ppul.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rancourt RC, Ahmad A, Veress LA, et al. Antifibrinolytic mechanisms in acute airway injury after sulfur mustard analog inhalation. Am J Respir Cell Mol Biol. 2014;51:559–567. doi: 10.1165/rcmb.2014-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.(USACHPPM)., U. A. C. f. H. P. a. P. M. Health Effects Associated With Sulfur Mustard Acute Exposure Guideline Levels (AEGLs)*. [May 13, 2016];2003 https://www.osha.gov/SLTC/emergencypreparedness/chemical/pdf/tier_3_hd_aegl_health_effects_usachppm1_03.pdf.
- 48.Fairhall SJ, Brown RF, Jugg BJ et al. Preliminary studies of sulphur mustard-induced lung injury in the terminally anesthetized pig: exposure system and methodology. Toxicol Mech Methods. 2008;18:355–362. doi: 10.1080/15376510701623383. [DOI] [PubMed] [Google Scholar]
- 49.Fairhall SJ, Jugg BJ, Read RW, et al. Exposure-response effects of inhaled sulfur mustard in a large porcine model: a 6-h study. Inhal Toxicol. 2010;22:1135–1143. doi: 10.3109/08958378.2010.527398. [DOI] [PubMed] [Google Scholar]
- 50.Jugg B, Fairhall S, Smith A, et al. N-acetyl-L-cysteine protects against inhaled sulfur mustard poisoning in the large swine. Clin Toxicol (Phila) 2013;51:216–224. doi: 10.3109/15563650.2013.780208. [DOI] [PubMed] [Google Scholar]