

Abstract
Background
Among viruses, bacteriophages are a group of special interest due to their capacity of infecting bacteria that are important for biotechnology and human health. Composting is a microbial-driven process in which complex organic matter is converted into humus-like substances. In thermophilic composting, the degradation activity is carried out primarily by bacteria and little is known about the presence and role of bacteriophages in this process.
Results
Using Pseudomonas aeruginosa as host, we isolated three new phages from a composting operation at the Sao Paulo Zoo Park (Brazil). One of the isolated phages is similar to Pseudomonas phage Ab18 and belongs to the Siphoviridae YuA-like viral genus. The other two isolated phages are similar to each other and present genomes sharing low similarity with phage genomes in public databases; we therefore hypothesize that they belong to a new genus in the Podoviridae family. Detailed genomic descriptions and comparisons of the three phages are presented, as well as two new clusters of phage genomes in the Viral Orthologous Clusters database of large DNA viruses. We found sequences encoding homing endonucleases that disrupt a putative ribonucleotide reductase gene and an RNA polymerase subunit 2 gene in two of the phages. These findings provide insights about the evolution of two-subunits RNA polymerases and the possible role of homing endonucleases in this process. Infection tests on 30 different strains of bacteria reveal a narrow host range for the three phages, restricted to P. aeruginosa PA14 and three other P. aeruginosa clinical isolates. Biofilm dissolution assays suggest that these phages could be promising antimicrobial agents against P. aeruginosa PA14 infections. Analyses on composting metagenomic and metatranscriptomic data indicate association between abundance variations in both phage and host populations in the environment.
Conclusion
The results about the newly discovered and described phages contribute to the understanding of tailed bacteriophage diversity, evolution, and role in the complex composting environment.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3729-z) contains supplementary material, which is available to authorized users.
Keywords: Bacteriophages, Composting, Homing endonucleases, tRNA genes, Genomics, Metagenomics, Pseudomonas aeruginosa, Siphoviridae, Podoviridae
Background
Viruses present remarkable diversity regarding morphology, genomes, and proteins [1]. Among viruses, bacteriophages (or simply phages) are a group of special interest, given their interactions with bacteria that are important for biotechnology and human health. As new phage genomes are characterized, unusual features are found, including new genes and novel genome architectures [2–4]. Thus, the study of phage diversity can contribute to the understanding of their evolution and their influence on any microbial community.
Composting is a diverse microbial environment in which complex organic molecules such as lignocellulose are converted into humus-like substances suitable for use as a soil amendment [5]. The study of composting microbial communities is important for elucidating the pathways of biomass degradation, and has contributed to the discovery of novel microorganisms and valuable enzymes for biotechnological applications [6, 7]. Aside from the great diversity of bacteria and fungi species in this environment [8–11], phages have also been identified in composting material [12–14]. Recent studies have reported novel phage genomes in composting [13] and interesting features, such as phage thermostable enzymes [14].
In this work, we describe three new phages isolated from a composting operation at the Sao Paulo Zoo Park (Brazil) using Pseudomonas aeruginosa PA14 as host. This reference strain is a clinical and highly virulent isolate that represents the most common clonal group worldwide [15]. Along with the characterization of these phages, this work also presents results concerning transcribed phage genes and phage abundance variation from a three-month time-series sampling of the composting process. To our knowledge, this is the first study to present such results concerning the complex microbial context in which these phages live.
Results and Discussion
Phage isolation and sequencing
We screened composting samples from the São Paulo Zoo Park (São Paulo, Brazil) [7] for phages infecting Pseudomonas aeruginosa PA14, in order to access a slice of the cultivable phage diversity in this complex microbial community. Three new phages were isolated, which we named Pseudomonas phage ZC01, ZC03 and ZC08. Their genomes were fully sequenced, assembled, and annotated. Overall characteristics of these phage genomes are summarized in Table 1. A final genome of 57,061 bp was obtained for phage ZC01, which was linearized following phage YuA reference and close genomes [16]. Assemblies for isolates ZC03 and ZC08 resulted in two slightly different sequences with 69,844 bp and 70,774 bp, respectively. For all three phage genomes, coverage was above 8000x and uniform through the entire contig. Metrics about the assembly process are available in the Additional file 1: Table S1.
Table 1.
New phages genomic features
| Feature | Phage ZC01 | Phage ZC03 | Phage ZC08 |
|---|---|---|---|
| Accession Number | KU356689 | KU356690 | KU356691 |
| Genome size (bp) | 57,061 | 69,844 | 70,774 |
| GC content (%) | 63 | 42 | 42 |
| Genes predicted | 78 | 85 | 83 |
| tRNA genes | None | 10 | 9 + 1 Pseudogene |
Genomic features of the three new Pseudomonas phages described in this work
Genomic and functional characterization of phage ZC01
The majority of the ZC01 genome consists of coding sequences, with the exception of three main non-coding regions: 370 bp at the 5’ end, 249 bp around the 8 kbp position and 933 bp around the 31 kbp position. A common characteristic for these non-coding regions is their lower GC content (41%) compared to the average GC content for the entire ZC01 genome (63%, Table 1). This variation is due to an increase of T nucleotides from a mean value of 16% in the genome to up to 37% in these non-coding regions.
We have searched the National Center for Biotechnology Information (NCBI) nt and microbial RefSeq genomes databases [17] for genomes similar to phage ZC01. The most similar genomes include phage Ab18 (98% coverage and 96% identity), phage PaMx11 (80% coverage and 72% identity) and phage YuA (31% coverage and 69% identity) [16, 18, 19]. Phage YuA belongs to the YuA-like virus genus of the Siphoviridae family [20]. Based on this information, we have created a new cluster of viral genomes in the Viral Orthologous Clusters database (VOCs) [21], which was named Siphoviridae YuA-like and is publicly available through the VOCs Java client [22]. Genomes in the YuA-like cluster were selected using BLASTN [23] results and the Jaccard index of similarity based on shared genes, as detailed in the Methods section. Clusters created in this study are not meant to reflect strict taxonomy groups, but similarity through shared genes only. This cluster contains 14 different genomes (listed in Table 2), 932 genes and 401 ortholog groups (OGs).
Table 2.
Phage genomes assigned to Siphoviridae YuA-like and Podoviridae N4-like VOCs clusters
| Siphoviridae YuA-like clustera | Podoviridae N4-like clustera | ||
|---|---|---|---|
| Phage species | Accession number | Phage species | Accession number |
| Burkholderia phage BcepGomr | NC_009447 | Enterobacter phage EcP1 | NC_019485 |
| Phage phiJL001 | NC_006938 | Enterobacteria phage N4 | NC_008720 |
| Pseudomonas phage 73 | NC_007806 | Erwinia phage vB_EamP-S6 | NC_019514 |
| Pseudomonas phage Ab18 | LN610577 | Escherichia phage vB_EcoP_G7C | NC_15933 |
| Pseudomonas phage B3 | NC_006548 | Pseudomonas phage LIT1 | NC_013692 |
| Pseudomonas phage D3112 | NC_005178 | Pseudomonas phage LUZ7 | NC_013691 |
| Pseudomonas phage DMS3 | NC_008717 | Pseudomonas phage ZC03b | KU356690 |
| Pseudomonas phage M6 | NC_007809 | Pseudomonas phage ZC08b | KU356691 |
| Pseudomonas phage MP22 | NC_009818 | Roseophage DSS3P2 | NC_012697 |
| Pseudomonas phage MP29 | NC_011611 | Roseophage EE36P1 | NC_012696 |
| Pseudomonas phage MP38 | NC_011611 | ||
| Pseudomonas phage PaMx11 | NC_0028770 | ||
| Pseudomonas phage YuA | NC_010116 | ||
| Pseudomonas phage ZC01b | KU356689 | ||
aClusters created in this work are not meant to reflect strict taxonomy groups as defined by the ICTV. bNew phages described in this study. Data is available in the VOCs database of large DNA viruses
All ten most conserved genes in the YuA-like cluster have orthologous representatives in phage ZC01 (Table 3). Likewise, conserved DNA helicase, RecD-like protein, deoxyuridylate hydroxymethyltransferase and DNA polymerase A were annotated and assigned to ortholog groups with six or more orthologous genes. Due to the high similarity among phages ZC01, Ab18 and PaMx11, there are 36 ortholog groups shared by them only. These include well-known proteins such as a holin and a DNA ligase, but also several hypothetical proteins. Moreover, phages Ab18 and ZC01 share two specific ortholog groups (VOCs ID: 18968, 18971), which were annotated as hypothetical and have no similarity with anything else in the NCBI nr database. Their function remains to be discovered. In addition, we found in the ZC01 genome a gene coding for a protein that is the only member of the VOCs ortholog group 18954. The predicted protein is the Rz1 smaller lipoprotein. It was manually annotated through the inspection of the Rz larger lipoprotein ORF (Rz1 is nested within Rz). This annotation was based on similar findings for the lambda phage genome, where Rz1 was experimentally isolated and characterized [24]. We verified that ZC01, Ab18 and PaMx11 share orthologous proteins to the Rz larger lipoprotein (VOCs ID: 22052); therefore, we conclude that the Rz1 smaller nested lipoprotein is also encoded in Ab18 and PaMx11 genomes, but it was not annotated (see Additional file 2 for the tblastn alignment of this genomic region).
Table 3.
List of the 20 most conserved ortholog groups in the Siphoviridae YuA-like cluster
| VOCs IDa | Ortholog Group | Number of genes | Number of genomes |
|---|---|---|---|
| 18328 | Putative tail assembly protein (PSP-YuA-073) | 12 | 12 |
| 18331 | Putative tail protein (PSP-YuA-076) | 12 | 12 |
| 18325 | Tail fiber protein (PSP-YuA-070) | 12 | 11 |
| 18326 | Structural phage protein (PSP-YuA-071) | 11 | 11 |
| 18327 | Tail assembly protein (PSP-YuA-072) | 11 | 11 |
| 18330 | Conserved tail assembly protein (PSP-YuA-075) | 11 | 11 |
| 18306 | Terminase large subunit (PSP-YuA-051) | 8 | 8 |
| 18323 | Virion structural protein (PSP-YuA-068) | 8 | 8 |
| 18278 | Putative deoxycytidylate deaminase (PSP-YuA-023) | 7 | 7 |
| 18318 | Structural phage protein (PSP-YuA-063) | 7 | 7 |
| 18257 | DNA helicase (PSP-YuA-002) | 6 | 6 |
| 18258 | Hypothetical protein (PSP-YuA-003) | 6 | 6 |
| 18259 | Hypothetical protein (PSP-YuA-004) | 6 | 6 |
| 18260 | Hypothetical protein (PSP-YuA-005) | 6 | 6 |
| 18261 | RecD-like DNA helicase (PSP-YuA-006) | 6 | 6 |
| 18269 | Hypothetical protein (PSP-YuA-014) | 6 | 6 |
| 18272 | Deoxyuridylate hydroxymethyltransferase (PSP-YuA-017) | 6 | 6 |
| 18273 | Hypothetical protein (PSP-YuA-018) | 6 | 6 |
| 18275 | Bacteriophage conserved protein (PSP-YuA-020) | 6 | 6 |
| 18276 | DNA Polymerase I (PSP-YuA-021) | 6 | 6 |
aData is available in the VOCs database of large DNA viruses
Functional annotation of the predicted proteins based on VOCs clusters context and additional tools showed that 41% of ZC01 proteins have unknown function. “DNA metabolism and replication” and “structural proteins” are the functional categories with most genes. Few protein products were annotated as involved in host interaction pathways, e.g., membrane or cell wall interaction and metabolism regulation. ZC01 genome annotation is summarized in Fig. 1. Detailed annotation of the ZC01 genome and genes is presented as Additional file 3: Table S2.
Fig. 1.
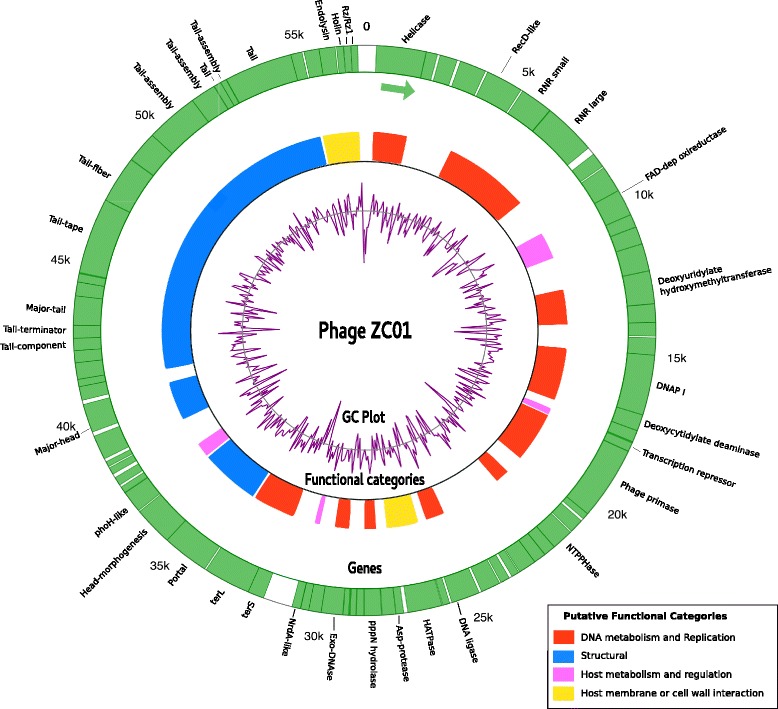
Phage ZC01 genome plot. Circular representation of the Pseudomonas phage ZC01 genome. The outer circle represents genes (all genes are on the plus strand, as indicated by the arrow). Putative functional categories were defined according to annotation and are represented by colors. Gaps in the functional block circle represent proteins with unknown function. The central graph (in purple) shows genomic GC content variation computed in 100 bp windows
Genomic and functional characterization of phages ZC03 and ZC08
Phages ZC03 and ZC08 present very similar genomes, with 95% of their nucleotide sequences aligned averaging 98% identity. Differences are mainly due to two indels at the 47 kbp and 51 kbp positions. The first indel consist of a unique sequence (~800 bp) present in phage ZC03, while the second indel consists of a unique sequence (~1000 bp) present in phage ZC08.
ZC03 and ZC08 non-coding regions also present variation on the GC content. However, in contrast to ZC01, the ZC03 and ZC08 non-coding regions display an increase in the GC content (~60%) with respect to the average CG content of the genome (42%, Table 1). The average GC content from phages ZC03 and ZC08 significantly diverges from the GC content of the assumed host genome (66%), suggesting that P. aeruginosa may not be the optimal host for these two phages [25].
BLASTN searches of ZC03 and ZC08 genomes against the NCBI RefSeq database returned hits with low genomic coverage and identity. The best hits included Enterobacteria phage N4 (10% coverage and 70% identity), Erwinia phage Ea9-2 (8% coverage and 70% identity) and Enterobacteria phage IME11 (7% coverage and 71% identity). These results strongly indicate that phages ZC03 and ZC08 are rather different from known phage species and that they probably belong to a new genus in the Podoviridae family. For this reason, creating a cluster of similar genomes in this case was challenging due to the shortage of similar genomes. Phage N4 is the most similar known genome and also the only officially representative of a genus recognized by the International Committee on Taxonomy of Viruses (ICTV), the N4-like-virus [20]. Notwithstanding, several phages have been reported as strongly related to this genus, including Pseudomonas, Escherichia and Achromobacter phages [26]. Thus, considering BLASTN results and the Jaccard index of similarity to select phage genomes, we created a new cluster of viruses in the VOCs database, which we named Podoviridae N4-like cluster. Ten different genomes were selected to be part of this cluster (Table 2), comprising 876 genes and 491 ortholog groups. Data for the YuA-like, N4-like and a third model cluster for T4-like myophages are publicly available through the VOCs Java client at the Viral Bioinformatics Resource Center (VBRC) web platform [22].
The ZC03 and ZC08 genomes harbor representatives from all the 15 core genes found for the N4-like cluster, or 14 core genes for the N4-like genus according to the literature [27]. These genes include RNA polymerases 1 and 2, DNA helicase, DNA polymerase, primase, exonuclease, terminase small and large subunits and coat proteins (Table 4).
Table 4.
List of the 15 most conserved ortholog groups in the Podoviridae N4-like cluster
| VOCs IDa | Ortholog Group | Number of genes | Number of genomes |
|---|---|---|---|
| 18472 | RNAP1 (EBP-N4-015) | 10 | 10 |
| 18473 | RNAP2 (EBP-N4-016) | 10 | 10 |
| 18481 | AAA ATPase containing protein (EBP-N4-024) | 12 | 10 |
| 18494 | DNA helicase (EBP-N4-037) | 10 | 10 |
| 18496 | DNAP (EBP-N4-039) | 10 | 10 |
| 18499 | Putative exonuclease (EBP-N4-042) | 10 | 10 |
| 18500 | Putative primase (EBP-N4-043) | 10 | 10 |
| 18501 | gp44 (EBP-N4-044) | 10 | 10 |
| 18502 | Single-stranded DNA-binding protein (EBP-N4-045) | 10 | 10 |
| 18512 | gp55 (EBP-N4-055) | 10 | 10 |
| 18513 | Major coat protein (EBP-N4-056) | 10 | 10 |
| 18514 | gp57 (EBP-N4-057) | 10 | 10 |
| 18516 | Putative portal protein (EBP-N4-059) | 10 | 10 |
| 18525 | Terminase large subunit (EBP-N4-068) | 10 | 10 |
| 18526 | gp69 (EBP-N4-069) | 10 | 10 |
aData is available in the VOCs database of large DNA viruses
Forty-four genes were assigned to specific ZC03/ZC08 ortholog groups, corresponding to more than half of the full set of genes in each genome. ZC03 and ZC08 genes are very different from gene sequences available in public databases, indicating the high degree of novelty of these genomes. Among these 44 specific genes, only two products could be annotated: a putative peptidoglycan hydrolase gp181 (VOCs ID: 18902) and a putative homing endonuclease (VOCs ID: 18919). ZC03 genome contains six unique genes and an additional tRNA gene that is not present in the ZC08 genome (probably a pseudogene, as we discuss later). On the other hand, ZC08 presents five unique genes that are not present in the ZC03 genome. Given the evidence, we hypothesize that these two phages have diverged in the recent evolutionary past.
Functional annotation of the predicted proteins for phages ZC03 and ZC08 based on VOCs clusters context and other tools showed that approximately 50% of the predicted proteins have unknown function. ZC03/ZC08 genome annotation and features are summarized in Fig. 2. For a complete list of evidence and annotation, see Additional file 4: Table S3.
Fig. 2.
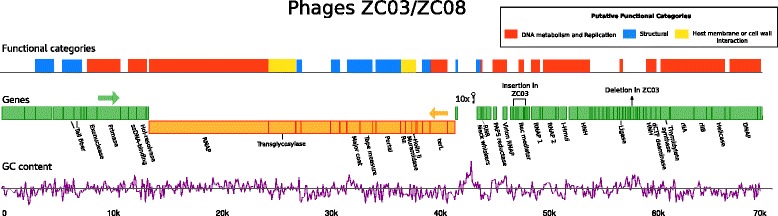
Phage ZC03 genome plot. Linear representation of the Pseudomonas phage ZC03 genome. The two central bands represent genes being codified by the plus strand (green) or minus strand (orange). Putative functional categories were defined according to annotation and are represented by colors in the top strand. Gaps in the functional blocks band represent proteins with unknown function. The bottom purple graph shows GC content variation computed in 100 bp windows. Hairpin symbol shows genome region where tRNA genes were predicted. Ins/del regions are shown comparing ZC03 and ZC08 phages
ZC03 and ZC08 specific genes and differences
Given their overall similarity, the differences between the ZC03 and ZC08 genomes can help in the understanding of viral evolution. Major differences include one gene region (ZC03_002 and ZC08_002) with lower nucleotide identity than its neighborhood (48 to 94% identity, respectively) and three indel regions encoding several genes (Table 5).
Table 5.
Phages ZC03 and ZC08 specific genes
| Gene | Protein Size (aa) | Annotation | BLAST and CDD weak hitsa |
|---|---|---|---|
| ZC03_002 | 331 | Hypothetical protein | No significant hits |
| ZC03_047 | 143 | Hypothetical protein | Conserved Hypothetical protein (CDD:DUF2461), Cellulase-like domain (pfam12876) |
| ZC03_051 | 60 | Hypothetical protein | SH3 Proline recognition superfamily (CDD:cl17036) |
| ZC03_052 | 61 | Hypothetical protein | Metallophosphatase superfamily (CDD:cl13995), Conserved hypothetical protein (CDD:DUF1501) |
| ZC03_053 | 166 | DrpA-like recombination mediator (InterPro:IPR003488) | --- |
| ZC03_054 | 32 | Hypothetical protein | Flagellar basal body-associated protein FliL (COG1580) |
| ZC03_055 | 37 | Hypothetical protein | No significant hits found |
| ZC03_073 | 65 | Hypothetical protein | Metallochaperone hypA superfamily (pfam01155) |
| --- | --- | --- | --- |
| ZC08_002 | 337 | Hypothetical protein | Tail assembly protein Xyllela phage Salvo (AHB12240.1) |
| ZC08_048 | 82 | Hypothetical protein | Borrelia lipoprotein (pfam00820) |
| ZC08_055 | 209 | Putative HNH homing endonuclease | --- |
| ZC08_073 | 47 | Hypothetical protein | No significant hits found |
| ZC08_074 | 132 | Hypothetical protein | Phage head-tail joining protein (COG5614) |
| ZC08_075 | 129 | Hypothetical protein | Metal-responsive transcriptional regulator (pfam15611) |
aHits were considered weak hits if the alignment presented coverage >= 0.3 and identity >= 20% through HMM or PSSM searches
The genes ZC03_002 and ZC08_002 were annotated as hypothetical proteins, although BLASTP results show similarity (coverage 41%, identity 29%) with a tail assembly protein of Xylella phage Salvo (AHB12240.1). Genes ZC03_002 and ZC08_002 were assigned to separate individual VOCs ortholog groups, likely because their encoded amino acid sequences present only 29% identical residues in a full alignment. Multiple alignment of the genomes indicates a syntenic relationship between these genes, providing additional evidence for the hypothesis that they are distant orthologs.
ZC03 presents a cassette of genes that might have been originated from a horizontal gene transfer event. Five genes (ZC03_051 to ZC03_055) are encoded by this region, and most of them were annotated as hypothetical. Weak hits suggest functions for the genes as shown in Table 5, but such evidence was not considered enough for a robust annotation. The only annotated gene in the cassette is a DrpA-like DNA recombination mediator. Other two indels events consist of unique sequences in ZC08 that encode one putative HNH homing endonuclease (ZC08_055) and three hypothetical proteins, respectively (Table 5). Details of the homing endonuclease insertion region will be discussed later in this work.
tRNAs and codon bias
Most phages in the N4-like group present one to three tRNAs genes that are not encoded by the host genome. In this group, there are Pseudomonas phages without any tRNA genes while some Salmonella phages harbor 10 or more genes for several tRNAs [26]. In this regard, phages ZC03 and ZC08 are the first Pseudomonas N4-related phages to carry tRNAs genes. Analysis of the ZC03 and ZC08 tRNA genes revealed anti-codons for seven different amino acids, with proline and leucine present twice. The prediction software could not accurately assign the anti-codon for one ZC03 tRNA gene, which also happens to be the one missing in ZC08 genome. It seems that the equivalent region to this tRNA gene in ZC03 and ZC08 genomes may have accumulated enough substitutions to produce a pseudo tRNA gene (see Additional file 5: Figure S1).
We analyzed codon usage for phage ZC03 and the host P. aeruginosa PA14 proteins. Table 6 shows predicted codons concerning ZC03 and ZC08 tRNAs and usage bias for each codon among all the codons for the same amino acid. The results show that tRNAs carried by the phage correspond to codons rarely used by the host. There is even an extreme case for the codon UUA (Leu) whose tRNA is not encoded by P. aeruginosa PA14 genome. This data corroborates reports in the literature, suggesting that a selective recruitment of tRNAs compensates for the compositional differences between phage and host genomes [28]. The only exceptions to this pattern seem to be the tRNAs for asparagine and methionine, which do not present any detectable bias.
Table 6.
Codon usage for phage ZC03 and P. aeruginosa PA14
| Codon | UUA | CUA | AUG | AAC | CCA | AGA | UCA | ACA | GUA |
|---|---|---|---|---|---|---|---|---|---|
| Amino Acid | Leu | Leu | Met | Asn | Pro | Arg | Ser | Thr | Val |
| tRNAs encoded by phage genome | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1a |
| tRNAs encoded by host genome | 0 | 1 | 4 | 2 | 1 | 1 | 1 | 1 | 2 |
| Random usage | 0.166 | 0.166 | 1 | 0.5 | 0.25 | 0.166 | 0.166 | 0.25 | 0.25 |
| Phage ZC03 usage | 0.132 | 0.15 | 1 | 0.532 | 0.3 | 0.242 | 0.154 | 0.275 | 0.3 |
| P. aeruginosa PA14 usage | 0.003 | 0.013 | 1 | 0.85 | 0.047 | 0.007 | 0.014 | 0.025 | 0.059 |
aThe anti-codon for one tRNA could not accurately be predicted, and, therefore, this might be a tRNAVal pseudogene
We have investigated proteins with high frequency of proline/leucine and proteins with high usage of the nine codons corresponding to the tRNAs carried by the phages. These were an Rz lipoprotein (ZC03_025), a putative class II holin (ZC03_027), two putative homing endonucleases (ZC03_062 and ZC08_055), one hypothetical protein containing a cellulase-like domain (ZC03_047), plus some hypothetical proteins (for a complete list of genes and codon composition, see Additional file 6: Table S4). Some studies have shown that highly translated mRNAs encoding important proteins to the organism are less susceptible to codon negative bias and wobble base-pairing, since the translation on those cases could be less efficient [29, 30]. In this context, the presence of tRNA genes may be related with phage virulence to ensure optional translation of late genes and faster lytic cycle, as previously suggested in [28]. Thus, it seems that the genes listed above might be especially important for phage lytic activity. By all means, a more detailed investigation is necessary to corroborate the linkage between presence of tRNA genes in phage genomes and their virulence.
Homing endonucleases insertion region
Homing Endonucleases (HEs) are site-specific DNA endonucleases encoded by genes inside mobile elements such as self-splicing introns and inteins (auto-processing protein domains). These mobile elements can insert themselves within conserved genes without altering their function due to their posterior self-splicing activity at the RNA or protein level [31]. They undergo a life cycle that starts with the invasion of a population, continues with the spreading through individuals, and ends when the element is fixed and is no longer under positive selection. At this point, the homing endonuclease gene (HEG) sequence degenerates and loses its function through random processes [32].
HEs are commonly found in phage genomes, with reports indicating up to 15 genes in phage T4 [33]. Although, phage T4 is thought to be an outlier, since many T4-like viruses have been studied and they do not have as many HEs. Moreover, it remains a challenge to understand the influence of HEs in producing phenotypes and in the mosaic evolution of phage genomes [34].
At least two different HEGs were identified within phages ZC03 and ZC08 genomes, both resembling endonucleases from the HNH family [31]. The homing endonucleases insertion region and HE-containing genes in phages ZC03 and ZC08 are listed in the Additional file 7: Table S5. Figure 3 shows a genome plot of this region, where one can observe a common element equally inserted within an ATPase-domain-containing protein (ADCP) of 350 aa in ZC03 and ZC08 genomes. Because of the insertion, this protein was predicted as two separated pieces, and multiple alignment of orthologous proteins in the N4-like cluster (VOCs ID: 18481) indicates that ZC03 and ZC08 are the only genomes to present an HE within this ORF. Searches of the fusion protein against the Reference Proteome HMMER database of HMMs [35] suggest that the protein is a ribonucleotide reductase. Similar cases of HE disrupting conserved ribonucleotide reductase genes were reported for phage Aeh1 and Twort [36, 37].
Fig. 3.

HNH endonuclease insertion region. Comparison of the HNH endonuclease insertion region in ZC03 and ZC08 genomes. Blue lines in the top strand are substitutions, the red block represents the HE element insertion in ZC08 only and blank spaces are identical aligned regions. ADCP: ATPase Domain Containing Protein
The second HE is inserted within the RNA polymerase (RNAP) subunit 2 gene in ZC08 genome only (Fig. 3). ZC08 specific HE was assigned to the ortholog group VOCs ID:18862, which also contains two other proteins from phages G7C (YP_004782150.1) and EE36P1 (YP_002898939.1). However, this homolog HE element is inserted in a different location inside phages G7C and EE36P1 genomes, more specifically between the genes RNAP1 and RNAP2 (YP_004782141.1 and YP_004782143.1 in phage G7C, respectively), which encode a two-subunits RNAP.
A closer investigation about the RNAP genes in phages from the Podoviridae family shows three organization types: (I) One single-unit protein of about 880 aa (T7-type), (II) two adjacent genes encoding for a two-subunits RNAP (N4-type), and (III) two genes spaced by one or more non-related canonical genes encoding a two-subunits RNAP (G7C-type). Multiple alignment of the RNAP protein sequences from T7-type, N4-type and G7C-type strongly suggests that the two subunits are actually non-overlapping pieces from the larger T7-type RNAP (see Additional file 8 for the alignment). Thus, the HE insertion within the RNAP2 gene in ZC08 genome may provide an insight to understand the evolutionary history of RNA polymerases in phages.
Altogether these findings suggest that single-unit and two-subunits RNAPs in Podoviridae may be linked by a common evolutionary pathway, as previously suggested in [38]. Our hypothesis is that a single-unit RNAP was present in the common ancestor of T7-like and N4-like phages. After these two lineages diverged, this single-unit protein was probably disrupted by the insertion of an HE element in the lineage that originated the N4-like phages. Since then, random events have led to two-subunits RNAP genes that continue to present affinity to assemble the complex required for transcription, as was experimentally demonstrated [38]. Although this hypothesis needs more supporting data, it is also corroborated by the comparative analysis of phage genomes from the T7-like and N4-like groups. Sequence inspection shows size variations in the spacing region between RNAP1 and RNAP2 genes. For instance, phage G7C presents only one putative HNH endonuclease (YP_004782142.1) between the RNAP subunits genes, while phage EE36P1 presents one putative HNH endonuclease (YP_002898939.1) plus seven predicted hypothetical proteins. There are examples from zero up to eight spacing genes between the RNAP1 and RNAP2 genes in N4-like phages.
Phylogenetic analyses
We performed phylogenetic analyses based on the Terminase Large Subunit gene (terL) of each cluster of phages. The YuA-like cluster presents three different ortholog groups for this gene (VOCs ID:18306, 19671, 19804) which may indicate non-orthologous or distant orthologous proteins. A maximum likelihood phylogenetic tree was generated (Fig. 4a). The presence of three distinct groups, which correspond exactly to the VOCs ortholog groups assignment, suggests that these sequences have long undergone separate evolutionary pathways. Phages Lambda and N15 belong to an external group, since they are phages from the Siphoviridae family but from a different and closely related group (Lambda-like phages).
Fig. 4.
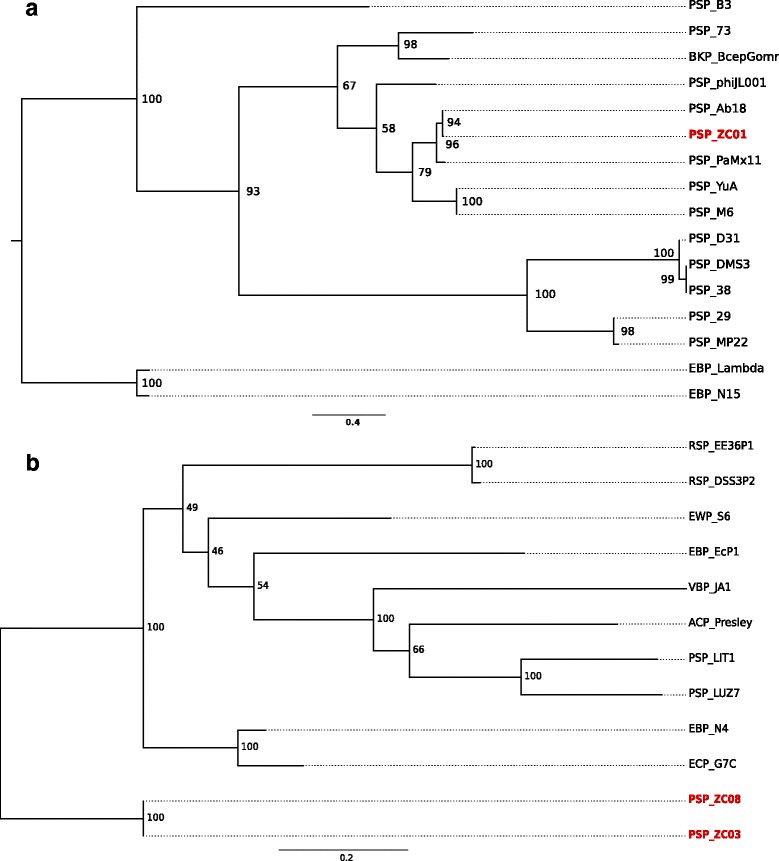
YuA-like and N4-like phylogenetic trees. a Maximum likelihood phylogenetic tree based on the terL gene for phages in the YuA-like VOCs cluster. The tree was rooted by two external groups represented here by Enterobacteria phage Lambda (EBP-Lambda) and Enterobacteria phage N15 (EBP-N15). b Maximum likelihood unrooted phylogenetic tree based on the terL gene for phages in the N4-like VOCs cluster. Bootstrap values are shown close to the nodes in percentages
All terL genes from the N4-like cluster were assigned to the same ortholog group (VOCs ID: 18525), which was also part of the N4-like core-genome. Fig. 4b shows an unrooted maximum likelihood tree for phages from the N4-like group. Internal nodes display weak bootstrap support, indicating that the relationships among phages inside the N4-like cluster are unresolved. However, the bootstrap values strongly support the existence of two clades, one grouping ZC03 and ZC08 and another grouping N4-like and related phages. This data supports the claim that phages ZC03 and ZC08 constitute a new genus inside the Podoviridae family.
It is worth mentioning that building phylogenetic trees for phages remains a tough challenge due to the non-existence of marker genes present in all species. The terL gene represents a candidate for this purpose in the order Caudovirales, but it is clearly limited by its lack of identifiable homology among families inside the order.
Host range and phage morphology
As previously mentioned, the three phages under study were isolated from composting samples using P. aeruginosa PA14 as host. In infection assays of P. aeruginosa PA14, phage ZC01 exhibits large lysis plaques with well-defined borders and diameter of 2.0-2.5 mm. ZC03 and ZC08 present much smaller lysis plaques (0.5-1 mm) than ZC01 (see Additional file 9: Figure S2). The three phages formed clear plaques, which is typical for lytic (virulent) phages. Given that ZC03/ZC08 probably belong to a new genus in the Podoviridae family, we performed one-step growth curve experiments for ZC03 as its archetype. The curve revealed a latent period of ~50 min with the number of phage particles reaching a peak at 240 min after infection and a calculated burst size of 10 phage particles per infected cell (see Additional file 10: Figure S3).
Phage host range was evaluated using 30 different strains, including bacteria from well-studied genera (e.g. Escherichia, Enterococcus, Bacillus), as well as several clinical P. aeruginosa isolates besides the reference strains PA14 and PAO1. Out of these, only four P. aeruginosa isolates were susceptible to phage lysis (Table 7). Higher lysis efficiency was observed only for strains PA14 and H6044, where clear plaques appeared even in more diluted titers for all the three phages. PAO1 strain was not susceptible to lysis. Additional file 11: Figure S4 shows images of the drop test for P. aeruginosa PA14 and PAO1 reference strains. These results indicate that the new phages present a narrow host range for P. aeruginosa strains, but other yet-to-be discovered potential hosts cannot be ruled out.
Table 7.
Assessment of ZC01, ZC03 and ZC08 host range
| Species/strain | Phage | ||
|---|---|---|---|
| ZC01 | ZC03 | ZC08 | |
| Bacillus subtilis PY79 | - | - | - |
| Chromobacterium violaceum ATCC 124721 | - | - | - |
| Chromobacterium violaceum isolated from Rio Negro | - | - | - |
| Escherichia coli MG1655 | - | - | - |
| Enterococcus faecalis ATCC 29212 | - | - | - |
| Klebsiella pneumoniae ATCC 13883 | - | - | - |
| Pseudomonas aeruginosa PA14 | C | C | C |
| Pseudomonas aeruginosa PAO1 | - | - | - |
| Pseudomonas aeruginosa 442 | - | - | - |
| Pseudomonas aeruginosa U456 | - | - | - |
| Pseudomonas aeruginosa H6086 | C | - | - |
| Pseudomonas aeruginosa 95291 | - | - | - |
| Pseudomonas aeruginosa 5172 | - | - | - |
| Pseudomonas aeruginosa U3554 | - | - | - |
| Pseudomonas aeruginosa H6044 | C | C | C |
| Pseudomonas aeruginosa 5757 | T | T | - |
| Pseudomonas aeruginosa 5728 | - | - | - |
| Pseudomonas aeruginosa 5031 | - | - | - |
| Pseudomonas aeruginosa 438 | - | - | - |
| Pseudomonas aeruginosa 426C | - | - | - |
| Pseudomonas aeruginosa 5728NF | - | - | - |
| Pseudomonas aeruginosa 5833 | - | - | - |
| Pseudomonas aeruginosa U514 | - | - | - |
| Pseudomonas aeruginosa PHB64 | - | - | - |
| Pseudomonas aeruginosa DE01 | - | - | - |
| Pseudomonas aeruginosa 48.1997A | - | - | - |
| Serratia marcescens isolated from Rio Negro | - | - | - |
| Staphylococcus aureus ATCC 29213 | - | - | - |
| Stenotrophomonas maltophilia ATCC 13637 | - | - | - |
| Xanthomonas axonopodis pv. citri 306 | - | - | - |
C (clear phage plaque); T (turbid phage plaque); - (no phage plaque). See Additional file 12: Table S6 for references of these strains
Morphology features for the three new phages virions were assessed by transmission electron microscopy. Phage ZC01 has the typical morphology for phages of the Siphoviridae family and more specifically for phages of the YuA-like group [16] (Fig. 5a–c). We identified a prolate and more elongated head of ~80 nm by ~58 nm (morphotype B2). Tail is ~150 nm long, cross-banded, flexible and non-contractile, with a terminal structure resembling short fibers. As predicted from genomic comparisons, phages ZC03 and ZC08 belongs to Podoviridae and as such, exhibit morphological characteristics of phages from this family (Fig. 5 d and e, respectively). The electron micrographs show their icosahedral head of ~72 nm by ~59 nm and a short tail ~21 nm long with terminal fibers.
Fig. 5.
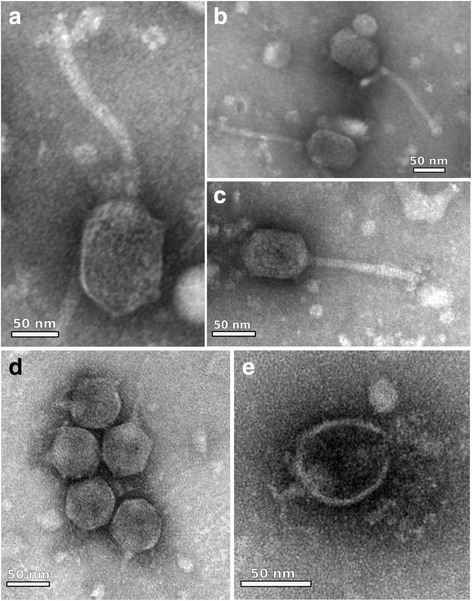
Electron micrographs of phages ZC01, ZC03 and ZC08. Transmission electron micrographs of negatively stained Pseudomonas phages virions found in composting: a–c Pseudomonas phage ZC01 with typical morphology of members of the Siphoviridae family; (d, e) Pseudomonas phages ZC03 and ZC08, full virions and empty shelled, respectively, with typical morphology of members of the Podoviridae family. Note the short tail
Putative cell lysis associated proteins
We have screened phage genomes for genes possibly involved in pathways of cell lysis and biomass degradation, since the lysis and turnover of bacterial cell components constitute important steps in the process of nutrients recycling [39]. Most genes involved in cell lysis and biomass degradation in phages are responsible for breaking cell wall peptidoglycan components and making pores in the lipid membrane, which are important steps in infection or in the release of phage progeny [40]. Nine proteins were found, including peptidoglycan hydrolases, N-acetylmuramidases, Rz lipoproteins, holins and an endolysin (Table 8). For example, a peptidoglycan hydrolase gp181-like (831 aa) is present in both ZC03 and ZC08 genomes. These proteins were assigned to the ortholog group VOCs ID: 18902, which contains only these two genes and no other homologs in the N4-like cluster. We identified a central lysozyme-like domain (pfam1464) and N-acetyl-D-glucosamine binding sites in the protein. Previous reports indicate a similar architecture for Pseudomonas phage phiKZ gp181 (Uniprot Q8SCY1) [41].
Table 8.
Putative cell lysis associated proteins encoded by phages ZC01, ZC03 and ZC08
| Phage | Gene name | Annotation | Size (aa) | Additional information |
|---|---|---|---|---|
| Phage ZC01 | ZC01_075 | Putative endolysin | 168 | TIGR02594 family protein |
| Phage ZC01 | ZC01_076 | Putative holin | 70 | Two transmembrane domains found with TMHMM |
| Phage ZC01 | ZC01_078 | Rz/Rz1 lipoprotein | 182 | Bacteriophage Rz lysis protein (pfam03245) |
| Phage ZC03 | ZC03_016 | Peptidoglycan hydrolase gp181-like | 831 | N-acetyl-D-glucosamine binding site; lysozyme-like domain (pfam01464) |
| Phage ZC03 | ZC03_025 | Putative RZ/Rz1 lipoprotein | 164 | Similar to Rz/RzI spanin protein in phage EC1-UPM (AGC31575.1) |
| Phage ZC03 | ZC03_026 | N-acetylmuramidase | 194 | Glycosyl hydrolase 108 (pfam05838); Pepitidoglycan binding domain |
| Phage ZC08 | ZC08_016 | Peptidoglycan hydrolase gp181-like | 831 | N-acetyl-D-glucosamine binding site; lysozyme-like domain (pfam01464) |
| Phage ZC08 | ZC08_025 | Putative Rz/Rz1 lipoprotein | 164 | Similar to Rz/RzI spanin protein in phage EC1-UPM (AGC31575.1) |
| Phage ZC08 | ZC08_026 | N-acetylmuramidase | 194 | Glycosyl hydrolase 108 (pfam05838); Pepitidoglycan binding domain |
Additional information include significant predictions of binding sites, transmembrane regions, domain regions and significant hits to similar proteins
Pseudomonas aeruginosa biofilm degradation
To investigate the ability of phages to mediate biofilm degradation, we challenged 24/48 hours P. aeruginosa PA14 biofilms with the three different phages isolated in this study. Exposure to the three phages strongly reduced biofilm cell densities, mainly for phage ZC01 (Fig. 6). These results indicate a promising degradation potential for these three new phages against P. aeruginosa PA14 biofilms. This strain is highly virulent in susceptible animal hosts and known to form a biofilm structure resistant to currently available antibiotics [42]. In these assays we used lower phage titers (~5 × 105 PFU ml-1) than the titers normally used in biofilm degradation assays (1 × 106 PFU ml-1 to × 1010 PFU ml-1) [43–45] highlighting the antibiofilm effectiveness of these phages.
Fig. 6.
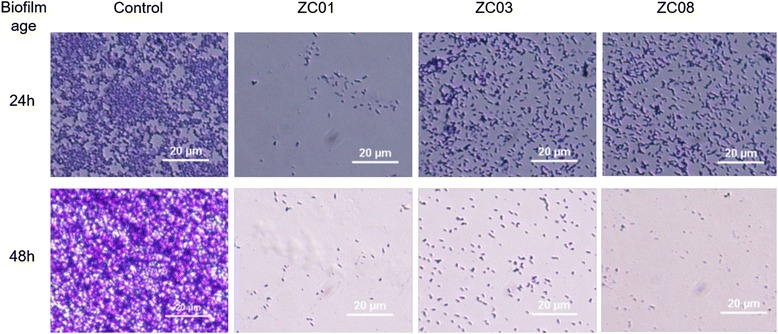
Biofilm degradation assay. Biofilms of 24 h and 48 h were exposed to phages ZC01, ZC03, and ZC08 for 24 h. Image shows the results after exposure for each of the phages compared to the control, which was exposed to a buffer solution only. Images are representative of n=4 replicates
Phages in action: metagenomic and metatranscriptomic analyses in the composting process
This work is part of a project that aims to understand the composting process at the microbial and molecular levels [7, 9]. In this project, time-series samples of a composting unit were obtained and corresponding DNA and mRNA sequence datasets were generated. As the samples from this composting unit were used for both the metagenomic and phage isolation studies, it was feasible to verify the presence of phages/host in the DNA and mRNA sequence datasets for each sampling day. We indeed found sequences that correspond to these genomes in all datasets and the relative abundance was inferred for phage-host populations (Fig. 7). Phages ZC03 and ZC08 relative abundance variation parallels that of P. aeruginosa but with an apparent delay, which is consistent with mathematical models that have been proposed for phage-host variation [46, 47]. We calculated a correlation score applying the Local Similarity Analysis (LSA) technique for time-series samples [48], and a positive LS score of 0.71 was obtained for P. aeruginosa and phages ZC03/ZC08 abundances (p-value < 0.02). This data suggests that, in this environment, P. aeruginosa and phages ZC03/ZC08 may present a mutualistic relationship that is characteristic of lysogenic phages, as discussed in [49]. We emphasize that our experimental results show that ZC01, ZC03 and ZC08 are lytic phages in the conditions we used for cultivation in PA14. Additionally, we also performed lysogeny experiments and the results showed negligible frequency of lysogeny (<1%) for the three phages. However, we cannot rule out the possibility that these phages could establish a mutualistic relationship with their host in different environmental conditions.
Fig. 7.
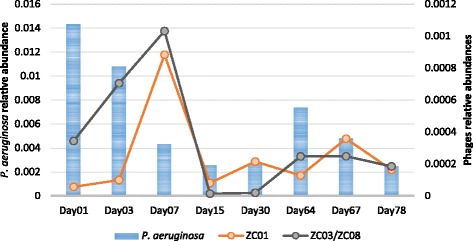
Phage-Host relative abundance in the composting metagenome. Relative abundance of metagenomic reads through the composting process. Raw reads count for phages and host was divided by the total number of reads in each sample and normalized by the genome size of the organism (given in percentage)
We investigated active phage-related functions in the composting process through the identification of metatranscriptomic reads mapped to phage genes. Fig. 8 shows the proportion of mRNA reads identified for each day in the respective function. “Structural” and “DNA metabolism and replication” are the predominant phage functions expressed through the days of the composting process. We identified mRNA for host lysis in the sample of day 7, more specifically mRNA reads for a class II phage holin (ZC03_027). It is interesting to note that a spike in phage abundance is also observed on day 7, as well as a marked decrease in host abundance (Fig. 7). This observation suggests a cause-effect relationship, but additional studies are necessary to gather additional evidence for this hypothesis.
Fig. 8.
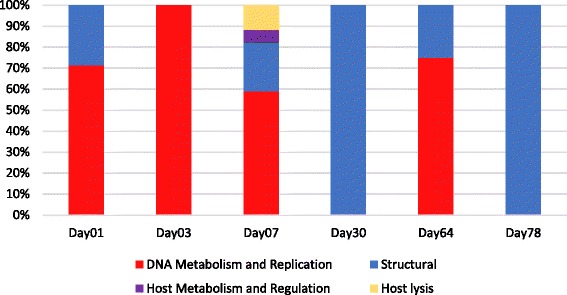
Phage-related functions in the composting microbial community. Phage-related protein functions through the composting process assessed by the identification of mRNA sequences from phages and the host P. aeruginosa in time-series metatranscriptomic samples. Values are shown in percentage of the total of proteins with known function
Conclusions
In this work, three new Pseudomonas phages have been characterized in terms of genomic structure, genes, and the putative proteins encoded by their genomes. Two of the three phages present remarkable novelty at the genomic level and may be members of a new genus in the Podoviridae family. Comprehensive comparative analyses of the new phages in a context with phages from YuA-like and N4-like clusters provided insights about the evolution and diversity of tailed phages. Moreover, infectivity and biofilm degradation experiments suggest a narrow host range and a potential as anti-microbial agents against P. aeruginosa PA14 infections, warranting further studies to explore this promising application. Finally, metagenomic and metatranscriptomic analyses provided data to situate phages ZC01, ZC03, and ZC08 in the microbial community to which they belong, yielding interesting clues about phage population dynamics and phage transcript presence in this complex environment.
Methods
Bacterial strains and growth conditions
Pseudomonas aeruginosa PA14 cells were grown at 37 °C in LB-medium. Solid LB medium contained 1.5% (w/v) of Bacto agar (Difco) and the soft agar top-layer contained 0.7% of Bacto agar. All strains were subcultured once and glycerol stocks were done and stored frozen at -80 °C until further use.
Phages isolation and propagation
Composting sample for phage isolation was collected from the composting facility in the São Paulo Zoo Park, São Paulo, Brazil following the procedure previously described [9] upon 67 days after completion of the composting pile. The procedure for phage isolation was adapted from [50]. The compost sample (~75 g) was suspended in 300 mL of SM buffer (10 mM MgSO4; 50 mM Tris-HCl, pH 7.5) containing 3% NaCl (w/v), dispensed into 50 mL centrifuge tubes and incubated for 60 min at 4 °C. Suspensions were homogenized for 5 min at maximum speed using the Tissuelyser II (Qiagen) and centrifuged at 3000 xg for 10 min. The supernatants were filtered through a 0.2 μm membrane and immediately used for infection by P. aeruginosa PA14 using the soft-agar overlay method [51]. After overnight incubation at 37 °C, several individual lytic plaques were collected, suspended in 100 μL of SM buffer and used for a new round of infection to warrant phage purification. The genomes of seven phage isolates were fully sequenced and out of them, three were found to be distinct (ZC01, ZC03 and ZC08). The other five isolates were identical to one of these three selected phages.
Phages were propagated using the soft-agar overlay method [51] using P. aeruginosa PA14 as the host strain. Briefly, 10 μL of isolated phage lysate were mixed with overnight bacterial culture and 3-5 mL of top-agar LB, and then added onto a LB Petri dish. After incubation, the lysate from a clear Petri dish was eluted with SM buffer and stored at 4 °C for further use. High titer phage suspensions were prepared using CsCl gradient centrifugation using standard protocols.
Phage titration and one-step growth curve
Bacteriophage titer was determined as described by [51]. Briefly, 100 μL of diluted phage suspension, 100 μL of a P. aeruginosa PA14 overnight culture, and 5 mL of LB top agar were mixed in a tube and poured into a LB agar-containing Petri dish. After incubation for 18 h at 37 °C, plaque forming units (PFU) were enumerated.
For one-step growth curve, a phage suspension was added to P. aeruginosa PA14 culture at multiplicity of infection (MOI) of 0.01. After incubation at 37 °C for 10 min to allow phages adsorption, the mixture was centrifuged for 30 s at 12,000 xg. The supernatant was collected and further centrifuged for 2 min 12,000 xg for evaluation of the fraction of non-adsorbed phages. Pellet was resuspended in 30 mL of LB, incubated at 37 °C without shaking and 300 μL samples were collected every 10 min and diluted for PFU enumeration.
Phage DNA extraction and Illumina MiSeq sequencing
For DNA extraction, phages were propagated in P. aeruginosa PA14 strain and collected after complete bacterial lysis, using 10 mL of SM buffer per Petri dish. The phage suspension was filtered through a 0.2 μm membrane and viral particles were precipitated with 10% polyethylene glycol (PEG) 8000 (w/v) and 1 M NaCl overnight at 4 °C. Viral particles were collected by centrifugation at 3,000 × g for 5 min. The pellet was suspended in 1 mL of SM buffer and treated with DNAse (TURBO DNA-free, Life Technologies) as a way attempt to reduce contamination with P. aeruginosa DNA. The intact viral particles suspension were treated with phenol:chloroform:isoamylalchool and phage DNA was extracted using MoBio PowerMax Soil DNA kit (MoBio Laboratories). Purified phage DNA was subjected to a final clean-up step using QIAamp mini spin columns (Qiagen, USA) and stored at -80 °C.
DNA purity and concentration were evaluated on a ND-1000 spectrophotometer (Nano Drop Technologies, USA) at 260 nm, 280 nm and 230 nm. Further quantification was performed with Quant-iT Picogreen dsDNA assay kit (Life Technologies, USA). DNA integrity was examined with DNA 7500 chip using 2100 Bioanalyzer and were mostly enriched in fragments higher than 10 kbp. Shotgun genomic libraries were prepared using an Illumina Nextera DNA library preparation kit (Illumina, Inc., USA) with total DNA input of 20-35 ng. The resulting DNA fragment libraries were cleaned up with Agencourt AMPure XP beads (Beckman Coulter, Inc., USA) and fragment size within the range of 400-700 bp was verified by running in the 2100 Bioanalyzer using Agilent High Sensitivity DNA chip. Quantification of Illumina sequencing libraries with KAPA Library Quantification Kit, normalization, and pooling were performed following standard protocols for sequencing in the Illumina MiSeq platform. Pooled libraries were subjected to one run using the MiSeq Reagent kit v2 (500-cycle format, paired-end (PE) reads). On average, Illumina PE read1 and read2 presented, respectively, >80% and >75% of bases with quality score at least 30 (Q30).
Genome assembly
Raw reads were subject to host DNA contamination removal with Deconseq [52] followed by a three-way protocol for digital normalization of high coverage libraries using KHMER Perl scripts [53]. Resulting reads were assembled with MIRA 4 (mode: genome, accurate, others parameters default) [54] and final genomes were assessed by manual inspection of coverage and mapping on IGV [55].
Clusters of phages
In order to define clusters of similar genomes to be implemented in the VOCs database, we counted the number of shared genes between two phage genomes according to Phage Ortholog Groups (POGs) data available by FTP [56]. Then, numbers of shared genes were used to calculate the Jaccard index (or Jaccard coefficient of similarity) for each pair of genomes according with the following expression:
| 1 |
Where A and B are the set of genes from A and B, respectively.
We consider that this index reflects similarity with more reliability than only using an absolute number of shared genes, since the J-index also consider the similarity between the total number of genes in the two phages being assessed. Lastly, we selected one reference genome for each one of the clusters and grouped genomes with a pairwise J(ref, X) ≥ 0.1.. Phages YuA and N4 were chosen as reference genomes due to their close relationship (attested by BLASTN searches to the nt NCBI database in Jan 2016) with ZC01 and ZC03/ZC08, respectively. The J-index cutoff was defined based on exploratory analyses of our data.
Ortholog assignment methodology and VOCs implementation were made as previously described in [21]. All VOCs data and genomic information about phage genomes in the two clusters used in this work are public available through a Java client in the VBRC web platform [22].
Genomic and functional characterization
Genes were predicted by GenMarkS [57] and Prodigal [58] using models for phage genes and t-RNA predictions were performed by Aragorn [59]. Proteins were automatically annotated by ProKKA [60] followed by additional manual characterization using CDD-Search [61], HMMER-Search [35] and BLAST searches [23] (Jan 2016). Hits were considered robust and significant for annotation when above the following alignment thresholds: E-value: 10E-5, alignment coverage: 60% and identity: 50%. VOCs ortholog groups and embedded tools were used for transitive annotation and comparative analyses [21, 62, 63].
Phylogenetic analyses
We identified ortholog groups for the Terminase Large Subunit gene (terL) by similarity in each of the clusters and performed multiple alignments through the VOCs GUI interface using MAFFT 7 (L-INS-i iterative algorithm and others parameters default) [64]. Guidance [65] was used to test the robustness of the multiple alignments and columns with confidence score below 0.4 were removed. The evolutionary history was inferred by using the maximum likelihood method based on the Whelan-Goldman model [66] and Le-Gascuel model [67] for the Siphoviridae cluster and Podoviridae cluster, respectively. Discrete Gamma distributions with 5 categories were used to model evolutionary rate differences among sites. Robustness of branches were tested by 1000 interactions of bootstrap [68]. Best fitting models and evolutionary analyses were conducted in RAxML version 8 [69].
Analyses of phages abundance in composting samples
Metagenomics and metatranscriptomics datasets from composting time-series used in the analyses were generated from a composting unit at the Sao Paulo Zoo Park (Brazil) and are publicly available in MG-RAST (see [7] for sampling details and accession numbers). Sequences were subject to mapping using Bowtie2 [70] (default parameters) against the isolated phage genomes and P. aeruginosa PA14. Reads mapping were considered as belonging to the new phages or the host and counted. Relative abundances were calculated dividing the number of reads by the total number of sequencing reads generated for the sample being analyzed. We applied genome size normalization in order to compare relative abundance of phages and the host P. aeruginosa.
Phage host range assay
Host range of the isolated phages was assessed by drop test against 30 bacterial strains (Additional file 12: Table S6), including the reference P. aeruginosa strains PA14 and PAO1. Bacterial lawns of the different strains were propagated in LB agar plates by plating 100 μL of overnight cultures and 10 μL drops of phages suspension at 107, 108, 109 and 1010 PFU mL-1. The plates were incubated for 18 h and then checked for presence of lysis plaques.
Lysogeny assay
Phage suspensions (100 μL) diluted to 1010 PFU mL-1 were seeded in LB plates. Overnight culture of P. aeruginosa PA14 at OD600nm = 1.0 was serially diluted (10-4 to 10-7) and 100 μL of each dilution were mixed with 4.5 mL LB top agar and added to phage seeded plates. Plates were incubated at 37 ° C for 3 days for CFU (colony forming unit) enumeration. Unseeded plates were used as control.
Study of bacteriophages effects on biofilm formation
Biofilms were allowed to form on 8-well chamber stainless slides for 24 h or 48 h as described in [71]. Briefly, bacterial culture (200 μl) with an OD600 of 0.05 - 0.1, which corresponds to approximately 1-2 × 107 cells was added to each well. The slide was incubated at 37 °C for 24 h or 48 h without shaking. The medium was replaced once a day during the whole experiment. Afterwards, the slides were washed twice with LB medium and the biofilms were challenged with 100 μl of LB and 100 μl of phage solution with a concentration of 5 x 105 PFU ml-1 during 24 h at 37 °C. Control experiments were performed at the same conditions with the slides incubated with 100 μl of LB and 100 μl of SM buffer. Biofilms attached to slides before and after phage infection were stained with 1% of crystal violet solution in ethanol 96% and analyzed in the microscope.
Transmission electron microscopy
For transmission electron microscopy, copper grids covered with carbon-coated Formvar films were floated, membrane side down, on a drop of phage suspension for about 10 min. After eliminating excess liquid and washing with distilled water, grids were floated on a drop of 1% (w/v) uranyl acetate for 5 min. After eliminating excess liquid, dried grids were examined in a JEOL JEM 1011 or Philips EM 300 transmission electron microscope and the images registered digitally. At least 10 virions were examined for each phage preparation.
Additional files
Assembly details and benchmarks for phages ZC01, ZC03 and ZC08 genomes. (XLSX 9 kb)
tblastn search output from ZC01 Rz1 lipoprotein against the YuA-like cluster of genomes. (TXT 4 kb)
Detailed information about ZC01 annotation. (XLSX 38 kb)
Detailed information about ZC03 and ZC08 annotation. (XLSX 45 kb)
tRNA genes in ZC03 and ZC08 genomes. (PDF 21 kb)
ZC03 and ZC08 genes list and codon usage for amino acids of interest. (XLSX 28 kb)
List of the genes present in the Homing endonuclease insertion region for phages ZC03 and ZC08. A list of gene features is also presented. (XLSX 11 kb)
Multiple sequence alignment of the T7-like RNA polymerase and N4-like RNA polymerases subunits 1 and 2. (TXT 12 kb)
Phages ZC01, ZC03 and ZC08 lysis plaques morphology. (PDF 127 kb)
One-step growth curve for phage ZC03. (PDF 88 kb)
Phage drop test for P. aeruginosa PA14 and PAO1 reference strains. (PDF 260 kb)
Source of strains used in host range assays shown in Table 7. (XLSX 13 kb)
Acknowledgements
The authors would like to thank Dr. João Batista da Cruz and Dr. Paulo Bressan from the São Paulo Zoo Park for continued support for this project and for making the composting operation available for sampling. We express our gratitude to Dr. Regina Lucia Baldini for providing the Pseudomonas aeruginosa PA14 and PAO1 strains and for suggestions in phage infection assays. We are indebted to Drs. Beny Spira, Nilton E. Lincopan, Rodrigo S. Galhardo and Chuck S. Farah for providing various bacterial strains for host range assays. We also thank Carlos Morais for help in computational analyses, Luiz Thiberio Rangel for fruitful discussions, and the Viral Bioinformatics Resource Center team for computational tool support.
Funding
This work was supported by grant 2011/50870-6 from the São Paulo State Research Foundation (FAPESP). DA was supported by fellowships from FAPESP (2014/16450-8 and 2015/14334-3) and from the Coordination for the Improvement of Higher Education Personnel (CAPES). AMDS, JCS and KCL received Research Fellowship Awards from the National Council for Scientific and Technological Development (CNPq). The funders had no role in study design, data collection, analysis, decision to publish or preparation of the manuscript.
Authors’ contributions
AMDS, DA and JCS conceived the study and designed the experiments. DA performed genomic characterization and bioinformatics analyses. KCL and LPA isolated the new phages from composting. KCL, LPA and LFM performed phage DNA isolation and sequencing. APSS, LFM and RBQ performed phage infection, virions purification, biofilm assays and one-step growth curve experiments. GGN contributed to the design of experiments and acquisition of data of P. aeruginosa PA14 phage infection assays. EWK and APSS generated electron microscopy images. CU guided the work with the VOCs database. AMDS, DA, and JCS wrote the manuscript.
Availability of data and materials
All the new phage genomes here described have been deposited in GenBank. Accession numbers are: KU356689 (ZC01), KU356690 (ZC03), and KU356691 (ZC08). The VOCs database of viral genomes and orthologs groups (clusters YuA-like, N4-like) used for comparative analyses are publicly available in the Viral Bioinformatics Resource Center online platform [22].
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
No ethics approval was required for the study. Sampling at the composting facility in the São Paulo Zoo Park was performed with the consent of the Sao Paulo Zoo Park Foundation (FPZSP) under the license 02001.000693/2013-89 issued by the Brazilian Institute of the Environment and Renewable Natural Resources (IBAMA).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ADCP
ATPase domain containing Protein
- HE
Homing Endonuclease
- HEG
Homing endonuclease gene
- ICTV
International Committee for Taxonomy of Viruses
- NCBI
National Center for Biotechnology Information
- OG
Ortholog group
- POGs
Phage Ortholog Groups
- RNAP
RNA Polymerase
- terL
Terminase Large Subunit
- VOCs
Viral Orthologous Clusters
Contributor Information
Deyvid Amgarten, Email: deyvid.amgarten@usp.br.
Layla Farage Martins, Email: laylabiol@gmail.com.
Karen Cristina Lombardi, Email: karen.lombardi@usp.br.
Luciana Principal Antunes, Email: lucianaprincipal@gmail.com.
Ana Paula Silva de Souza, Email: anapss@iq.usp.br.
Gianlucca Gonçalves Nicastro, Email: nicastro@iq.usp.br.
Elliott Watanabe Kitajima, Email: ewkitaji@usp.br.
Ronaldo Bento Quaggio, Email: rquaggio@iq.usp.br.
Chris Upton, Email: cupton@uvic.ca.
João Carlos Setubal, Email: setubal@iq.usp.br.
Aline Maria da Silva, Email: almsilva@iq.usp.br.
References
- 1.Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005;13:278–84. doi: 10.1016/j.tim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Rohwer F. Global phage diversity. Cell. 2003;113:141. doi: 10.1016/S0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 3.Hendrix RW. Bacteriophage genomics. Curr Opin Microbiol. 2003;6:506–11. doi: 10.1016/j.mib.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, et al. Uncovering Earth’s virome. Nature. 2016;536:425–30. doi: 10.1038/nature19094. [DOI] [PubMed] [Google Scholar]
- 5.Ryckeboer J, Mergaert J, Vaes K, Klammer S, Clercq D, Coosemans J, et al. A survey of bacteria and fungi occurring during composting and self-heating processes. Ann Microbiol. 2003;53:349–410. [Google Scholar]
- 6.Jurado M, López MJ, Suárez-Estrella F, Vargas-García MC, López-González JA, Moreno J. Exploiting composting biodiversity: Study of the persistent and biotechnologically relevant microorganisms from lignocellulose-based composting. Bioresour Technol. 2014;162:283–93. doi: 10.1016/j.biortech.2014.03.145. [DOI] [PubMed] [Google Scholar]
- 7.Antunes LP, Martins LF, Pereira RV, Thomas AM, Barbosa D, Lemos LN, et al. Microbial community structure and dynamics in thermophilic composting viewed through metagenomics and metatranscriptomics. Sci Rep. 2016;6:38915. doi: 10.1038/srep38915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Partanen P, Hultman J, Paulin L, Auvinen P, Romantschuk M, Epstein E, et al. Bacterial diversity at different stages of the composting process. BMC Microbiol. 2010;10:94. doi: 10.1186/1471-2180-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martins LF, Antunes LP, Pascon RC, de Oliveira JCF, Digiampietri LA, Barbosa D, et al. Metagenomic analysis of a tropical composting operation at the São Paulo Zoo Park reveals diversity of biomass degradation functions and organisms. PLoS One. 2013;8:e61928. doi: 10.1371/journal.pone.0061928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neher DA, Weicht TR, Bates ST, Leff JW, Fierer N. Changes in bacterial and fungal communities across compost recipes, preparation methods, and composting times. PLoS One. 2013;8:e79512. doi: 10.1371/journal.pone.0079512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.López-González JA, Suárez-Estrella F, Vargas-García MC, López MJ, Jurado MM, Moreno J. Dynamics of bacterial microbiota during lignocellulosic waste composting: Studies upon its structure, functionality and biodiversity. Bioresour Technol. 2015;175:406–16. doi: 10.1016/j.biortech.2014.10.123. [DOI] [PubMed] [Google Scholar]
- 12.Marks TJ, Hamilton PT. Characterization of a thermophilic bacteriophage of Geobacillus kaustophilus. Arch Virol. 2014;159:2771–5. doi: 10.1007/s00705-014-2101-8. [DOI] [PubMed] [Google Scholar]
- 13.Lima-junior JD, Viana-niero C, Conde Oliveira DV, Machado GE, da Silva Rabello MC, Martins-Junior J, et al. Characterization of mycobacteria and mycobacteriophages isolated from compost at the São Paulo Zoo Park Foundation in Brazil and creation of the new mycobacteriophage Cluster U. BMC Microbiol. 2016;16:111. doi: 10.1186/s12866-016-0734-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheepudom J, Lee CC, Cai B, Meng M. Isolation, characterization, and complete genome analysis of P1312, a thermostable bacteriophage that infects Thermobifida fusca. Front Microbiol. 2015;6:959. doi: 10.3389/fmicb.2015.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosquera-Rendón J, Rada-Bravo AM, Cárdenas-Brito S, Corredor M, Restrepo-Pineda E, Benítez-Páez A, et al. Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genomics. 2016;17:45. doi: 10.1186/s12864-016-2364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceyssens P-JJ, Mesyanzhinov V, Sykilinda N, Briers Y, Roucourt B, Lavigne R, et al. The genome and structural proteome of YuA, a new Pseudomonas aeruginosa phage resembling M6. J Bacteriol. 2008;190:1429–35. doi: 10.1128/JB.01441-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I. RefSeq microbial genomes database: New representation and annotation strategy. Nucleic Acids Res. 2014;42:553–9. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Essoh C, Latino L, Midoux C, Blouin Y, Loukou G, Nguetta SPA, et al. Investigation of a large collection of Pseudomonas aeruginosa bacteriophages collected from a single environmental source in Abidjan Côte d’Ivoire. PLoS One. 2015;10:1–25. doi: 10.1371/journal.pone.0130548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sepúlveda-Robles O, Kameyama L, Guarneros G. High diversity and novel species of Pseudomonas aeruginosa bacteriophages. Appl Environ Microbiol. 2012;78:4510–5. doi: 10.1128/AEM.00065-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King AMQ, Adams MJ, Carsten EB, Lefkowitz EJ. Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses. Oxford: Elsevier Inc.; 2012.
- 21.Ehlers A, Osborne J, Slack S, Roper RL, Upton C. Poxvirus Orthologous Clusters (POCs) Bioinformatics. 2002;18:1544–5. doi: 10.1093/bioinformatics/18.11.1544. [DOI] [PubMed] [Google Scholar]
- 22.Upton C et al. Viral Bioinformatics Resource Center. http://virology.uvic.ca. Accessed 14 Oct 2016.
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Zhang N, Young R. Complementation and characterization of the nested Rz and Rz1 reading frames in the genome of bacteriophage λ. Mol Gen Genet. 1999;262:659–67. doi: 10.1007/s004380051128. [DOI] [PubMed] [Google Scholar]
- 25.Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol Syst Biol. 2009;5:311. doi: 10.1038/msb.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wittmann J, Dreiseikelmann B, Rohde M, Meier-Kolthoff JP, Bunk B, Rohde C. First genome sequences of Achromobacter phages reveal new members of the N4 family. Virol J. 2014;11:14. doi: 10.1186/1743-422X-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan JZM, Millard AD, Mann NH, Schäfer H. Comparative genomics defines the core genome of the growing N4-like phage genus and identifies N4-like roseophage specific genes. Front Microbiol. 2014;5:506. doi: 10.3389/fmicb.2014.00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17:1486–95. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulmer M. Coevolution of codon usage and transfer RNA abundance. Nature. 1987. p. 728–30. [DOI] [PubMed]
- 30.Stadler M, Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 2011;17:2063–73. doi: 10.1261/rna.02890211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 32.Gogarten JP, Hilario E. Inteins, introns, and homing endonucleases: recent revelations about the life cycle of parasitic genetic elements. BMC Evol Biol. 2006;6:94. doi: 10.1186/1471-2148-6-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgell DR, Gibb EA, Belfort M. Mobile DNA elements in T4 and related phages. Virol J. 2010;7:290. doi: 10.1186/1743-422X-7-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol. 2011;1:298–303. doi: 10.1016/j.coviro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedrich NC, Torrents E, Gibb EA, Sahlin M, Sjöberg B-M, Edgell DR. Insertion of a homing endonuclease creates a genes-in-pieces ribonucleotide reductase that retains function. Proc Natl Acad Sci U S A. 2007;104:6176–81. doi: 10.1073/pnas.0609915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landthaler M, Begley U, Lau NC, Shub DA. Two self-splicing group I introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucleic Acids Res. 2002;30:1935–43. doi: 10.1093/nar/30.9.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willis SH, Kazmierczak KM, Carter RH, Rothman-Denes LB. N4 RNA polymerase II, a heterodimeric RNA polymerase with homology to the single-subunit family of RNA polymerases. J Bacteriol. 2002;184:4952–61. doi: 10.1128/JB.184.18.4952-4961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004. p. 127–81. [DOI] [PubMed]
- 40.Moak M, Molineux IJ. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol Microbiol. 2004;51:1169–83. doi: 10.1046/j.1365-2958.2003.03894.x. [DOI] [PubMed] [Google Scholar]
- 41.Briers Y, Miroshnikov K, Chertkov O, Nekrasov A, Mesyanzhinov V, Volckaert G, et al. The structural peptidoglycan hydrolase gp181 of bacteriophage phiKZ. Biochem Biophys Res Commun. 2008;374:747–51. doi: 10.1016/j.bbrc.2008.07.102. [DOI] [PubMed] [Google Scholar]
- 42.Pawar V, Komor U, Kasnitz N, Bielecki P, Pils MC, Gocht B, et al. In vivo efficacy of antimicrobials against biofilm-producing Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2015;59:4974–81. doi: 10.1128/AAC.00194-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cerca N, Oliveira R, Azeredo J. Susceptibility of Staphylococcus epidermidis planktonic cells and biofilms to the lytic action of staphylococcus bacteriophage K. Lett Appl Microbiol. 2007;45:313–7. doi: 10.1111/j.1472-765X.2007.02190.x. [DOI] [PubMed] [Google Scholar]
- 44.Doolittle MM, Cooney JJ, Caldwell DE. Lytic infection of Escherichia coli biofilms by bacteriophage T4. Can J Microbiol. 1995;41:12–8. doi: 10.1139/m95-002. [DOI] [PubMed] [Google Scholar]
- 45.Doolittle M, Cooney J, Caldwell D. Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J Ind Microbiol. 1996;16:331–41. doi: 10.1007/BF01570111. [DOI] [PubMed] [Google Scholar]
- 46.Gourley SA, Kuang Y. A delay reaction-diffusion model of the spread of bacteriophage infection. SIAM J Appl Math. 2004;65:550–66. doi: 10.1137/S0036139903436613. [DOI] [Google Scholar]
- 47.Krysiak-Baltyn K, Martin GJO, Stickland AD, Scales PJ, Gras SL. Computational models of populations of bacteria and lytic phage. Crit Rev Microbiol. 2016;42:1–27. doi: 10.3109/1040841X.2015.1114466. [DOI] [PubMed] [Google Scholar]
- 48.Xia LC, Steele JA, Cram JA, Cardon ZG, Simmons SL, Vallino JJ, et al. Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Syst Biol. 2011;5:S15. doi: 10.1186/1752-0509-5-S2-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Obeng N, Pratama AA, van Elsas JD. The significance of mutualistic phages for bacterial ecology and evolution. Trends Microbiol. 2016;24:440–9. doi: 10.1016/j.tim.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Yoshida M, Takaki Y, Eitoku M, Nunoura T, Takai K. Metagenomic analysis of viral communities in (hado)pelagic sediments. PLoS One. 2013;8:e57271. doi: 10.1371/journal.pone.0057271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams MH, others. Bacteriophages. Bacteriophages (1959).
- 52.Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One. 2011;6:e17288. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown CT, Howe A, Zhang Q, Pyrkosz AB, Brom TH. A Reference-Free Algorithm for Computational Normalization of Shotgun Sequencing Data. arXiv. 2012;1203.4802:1–18
- 54.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–59. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kristensen DM, Waller ASAS, Yamada T, Bork P, Mushegian AR, Koonin EV. Orthologous gene clusters and taxon signature genes for viruses of prokaryotes. J Bacteriol. 2013;195:941–50. doi: 10.1128/JB.01801-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33:451–4. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Access O, Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–6. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 61.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, et al. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41:D348–52. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Upton C, Hogg D, Perrin D, Boone M, Harris NL. Viral genome organizer: a system for analyzing complete viral genomes. Virus Res. 2000;70:55–64. doi: 10.1016/S0168-1702(00)00210-0. [DOI] [PubMed] [Google Scholar]
- 63.Hillary W, Lin S-H, Upton C. Base-By-Base version 2: single nucleotide-level analysis of whole viral genome alignments. Microb Inform Exp. 2011;1:2. doi: 10.1186/2042-5783-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Penn O, Privman E, Ashkenazy H, Landan G, Graur D, Pupko T. GUIDANCE: A web server for assessing alignment confidence scores. Nucleic Acids Res. 2010;38:W23–8. doi: 10.1093/nar/gkq443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–9. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- 67.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–20. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 68.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 69.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jurcisek JA, Dickson AC, Bruggeman ME, Bakaletz LO. In vitro biofilm formation in an 8-well chamber slide. J. Vis. Exp. 2011 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Assembly details and benchmarks for phages ZC01, ZC03 and ZC08 genomes. (XLSX 9 kb)
tblastn search output from ZC01 Rz1 lipoprotein against the YuA-like cluster of genomes. (TXT 4 kb)
Detailed information about ZC01 annotation. (XLSX 38 kb)
Detailed information about ZC03 and ZC08 annotation. (XLSX 45 kb)
tRNA genes in ZC03 and ZC08 genomes. (PDF 21 kb)
ZC03 and ZC08 genes list and codon usage for amino acids of interest. (XLSX 28 kb)
List of the genes present in the Homing endonuclease insertion region for phages ZC03 and ZC08. A list of gene features is also presented. (XLSX 11 kb)
Multiple sequence alignment of the T7-like RNA polymerase and N4-like RNA polymerases subunits 1 and 2. (TXT 12 kb)
Phages ZC01, ZC03 and ZC08 lysis plaques morphology. (PDF 127 kb)
One-step growth curve for phage ZC03. (PDF 88 kb)
Phage drop test for P. aeruginosa PA14 and PAO1 reference strains. (PDF 260 kb)
Source of strains used in host range assays shown in Table 7. (XLSX 13 kb)
Data Availability Statement
All the new phage genomes here described have been deposited in GenBank. Accession numbers are: KU356689 (ZC01), KU356690 (ZC03), and KU356691 (ZC08). The VOCs database of viral genomes and orthologs groups (clusters YuA-like, N4-like) used for comparative analyses are publicly available in the Viral Bioinformatics Resource Center online platform [22].