

Abstract
De Sanctis–Cacchione (DSC) syndrome is one of the rarest, most severe forms of xeroderma pigmentosum (XP). These patients with XP are of short stature, have mental disabilities, and develop progressive neurologic degeneration because of a severe inability to repair damaged DNA. Herein, we will present the case of a 9-year-old boy who had DSC syndrome with microcephaly, severe psychomotor retardation, ataxia, and hearing loss. The cutaneous manifestations included giant squamous cell carcinoma (SCC) that covered the eye, multiple facial SCCs, and pigment changes on sun-exposed areas. In addition, we include a review of reported rare cases and a brief discussion of disease management.
Keywords: De Sanctis–Cacchione, Isotretinoin, Neurologic, Squamous cell carcinoma, Xeroderma pigmentosum
Introduction
De Sanctis-Cacchione syndrome (MIM #278800) is characterized by cutaneous photosensitivity, microcephaly, mental retardation, short stature, hypogonadism, spasticity, peripheral neuropathy, and sensorineural deafness (McKusick and Kniffin, 2010).
Case description
A 9-year-old boy was referred to the Otorhinolaryngology-Head and Neck Surgery Department at the University Hospital because of a rapidly growing ulcerative lesion on the face over the past 6 months. He had a neglected case of xeroderma pigmentosum (XP), was the offspring of a consanguineous marriage, and had mental retardation and photosensitivity. An informed consent was obtained from the patient’s parents for this report.
Physical examination
A large, ulcerated tumor (10 × 12 cm) was observed on the left side of the face with involvement of the lower lid, cheek, nose, and upper lip. The left eye was covered by the tumor (Fig. 1). Numerous crusted lesions were noted on the forehead, lower lip, and right cheek. The skin on the face, neck, and limbs of sun-exposed areas appeared dry with freckle-like pigmentary changes including hypopigmentation, hyperpigmentation, and atrophy (poikiloderma). A mucous membrane examination was normal, except for the left eye that was covered by the tumor, so the eye could not be assessed.
Fig. 1.
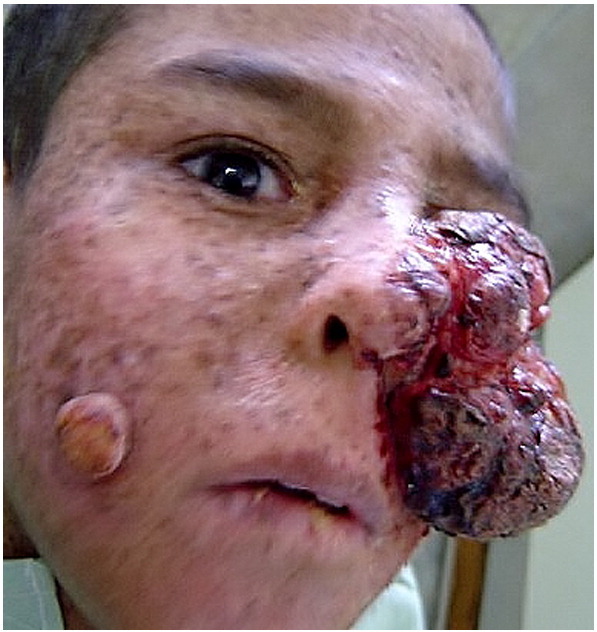
A 9-year-old boy presented with a large ulcerated tumor on the left side of the face with involvement of the nose, cheek, and eye. The skin on the face appeared dry with freckle-like pigmentary changes.
The patient’s general appearance included microcephaly and a short stature. He had bilateral hearing loss. Because of severe psychomotor retardation, ataxia, and spastic movement, he was bedridden and incontinent.
Imaging
A maxillofacial computed tomography (CT) scan demonstrated a large hypodense mass over the left maxilla, with involvement of the soft tissue in this region (Fig. 2). A brain CT scan revealed severe cortical atrophy, but no calcification or mass effect was detected. A chest X-ray revealed no definitive abnormalities.
Fig. 2.
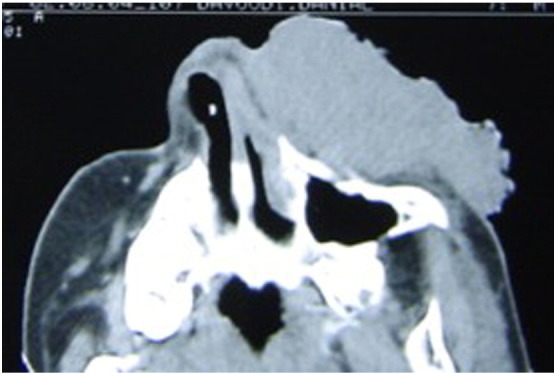
Maxillofacial computed tomography scan demonstrated a large hypodense mass over the left maxilla with the involvement of soft tissue of this region.
Laboratory tests and skin biopsy
A histopathologic examination of the facial skin biopsies indicated a diagnosis of squamous cell carcinoma (SCC). A complete blood cell count, routine blood chemistry, and urinalysis were all within normal limits.
Treatment
The large tumor was resected with a 1-cm margin. The eye had ocular surface lesions and corneal scarring, but there was no tumor involvement. The largest defect was reconstructed using a split-thickness skin graft (Fig. 3). Chemoprevention with oral low-dose isotretinoin was started. We recommended that the patient avoid sun exposure and take vitamin D supplements. No tumor recurrence was detected at an 18-month follow-up.
Fig. 3.
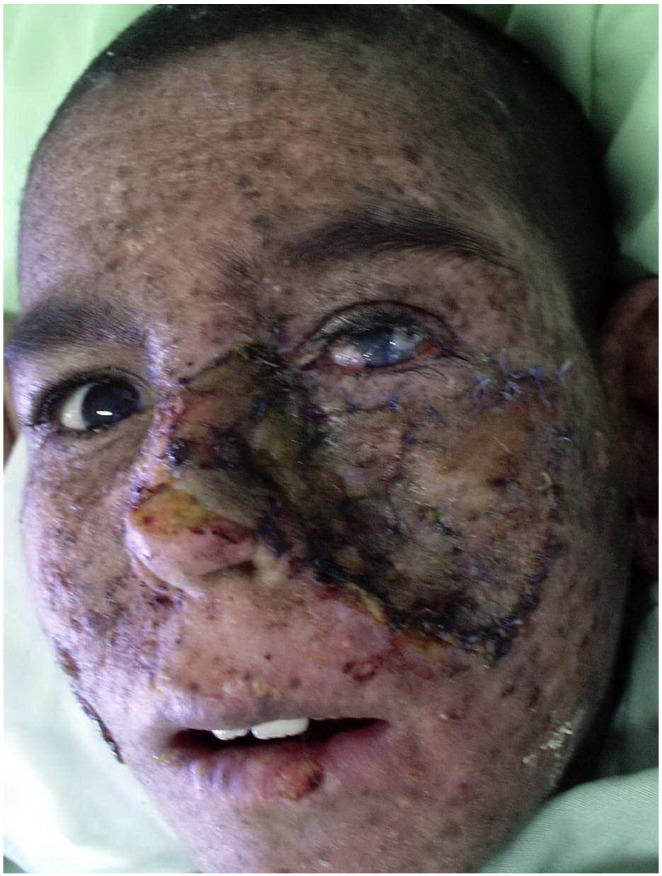
The large squamous cell carcinoma was resected with 1-cm margin, and the defect was repaired with a split-thickness skin graft.
Discussion
XP, or “dry pigmented skin,” is characterized by extreme skin sensitivity to ultraviolet radiation (UVR) and abnormal skin pigmentation, with a high incidence of skin cancers on sun-exposed areas (Kraemer and DiGiovanna, 1993). XP consists of a group of various rare autosomal recessive–inherited conditions with a defective nucleic excision repair (NER) pathway for DNA repair. Major disorders involving defective DNA repair such as XP, Cockayne syndrome (CS), and trichothiodystrophy present with photosensitivity and overlapping genetic and clinical features. However, the development of numerous skin cancers at a young age is characteristic of XP (DiGiovanna and Kraemer, 2012).
Typically, XP patients have a history of sunlight hypersensitivity, freckling, and photophobia before the age of 2 years; develop their first skin cancer prior to the age of 10 years; and develop their first melanoma before age 30 years (DiGiovanna and Kraemer, 2012). The diagnosis is clinical. Confirmatory molecular genetic testing is conducted for research purposes and is not routinely available (DiGiovanna and Kraemer, 2012).
De Sanctis–Cacchione syndrome has been regarded as one of the rarest manifestations of XP with the most severe DNA repair impairment (Cleaver et al., 1984, Cleaver and Thomas, 1988, Mittal et al., 2013). It is recognized by the typical XP skin changes accompanied by progressive neurologic degeneration, a short stature, and delayed gonadal maturation. The presence of progressive neurologic involvement and age at symptom onset correlate with the degree of DNA repair impairment (Caldas and Rodrigues, 2013, Hessel et al., 1992). The median life span of patients with neurologic deterioration is shorter than those without neurological degeneration (29 versus 37 years) (Bradford et al., 2011, DiGiovanna and Kraemer, 2012).
In 1932, de Sanctis and Cacchione derived the term “xerodermic idiocy” for three brothers with XP who had microcephaly, mental deficiency, dwarfism, gonadal hypoplasia, progressive neurologic deterioration, deafness, and ataxia beginning at the age of 2 years (de Sanctis and Cacchione, 1932). This condition, called DSC syndrome, has been rarely reported. Kraemer et al. (1987) performed a review of case reports on XP beginning with 1874, the first time XP was reported, and ending with 1982. According to their review, microcephaly and neurologic deterioration were observed in only 37 and 27, respectively, out of a total of 830 patients. Short stature and delayed secondary sexual development were seen in only 19 patients (Kraemer et al., 1987). Table 1 provides a summary of some recent case reports involving DSC syndrome.
Table 1.
The summary of published case reports of XP with neurologic abnormalities and known De Sanctis-Cacchione syndrome.
| Author, year | Patient’s country | Patients (n) | Age | Sex | Skin change, Skin cancer | Neurologic abnormalities (n, %), comment |
|---|---|---|---|---|---|---|
| (Neisser 1883) | Germany | 2 siblings | 2nd decade | . | XP,. | Progressive neurologic degeneration |
| (de Sanctis C and Cacchione A 1932) | Italy | 3 brothers | . | M | XP,. | ⁎ :MR, dwarfism, gonadal hypoplasia |
| (Kraemer, Lee & Scotto 1987) | Worldwide review (1874 to 1982) |
152 | 12 y (median) | F:74 M:73 |
XP, skin cancer in 77(50%) | MR: 121(80%), Microcephaly: 37(24%), Delayed growth:35(23%), Areflexia:30(20%), Hearing loss:28(18%), Neurologic deterioration:27(18%), Delayed sexual development: 19(12%). |
| (Gupta et al. 1988) | India | 1 | 8 y | F | XP, no cancer | ⁎, but no evidence of ataxia. |
| (Hessel et al. 1992) | USA | 1 | 12 y | M | XP, BCC,SCC | ⁎, but no evidence of ataxia, or hearing loss. |
| (Greenhaw et al. 1992) | Mexico | 3 siblings | 8 y 7 y 10 y |
M F F |
XP(mild), no cancer |
⁎, but biochemical profile of CS. |
| (Niederauer et al. 1992) | Germany | 1 | 6 y | M | XP, . | ⁎ |
| (Itoh et al. 1996) | Japan | 2 sibling | . | . | XP, no cancer | ⁎ , but biochemical profile of CS. |
| (Mishra et al. 1997) | India | 1 | 4 y | M | XP, no cancer | ⁎, also optic atrophy. |
| (Falcon Lincheta et al. 1998) | Cuba | 1 | 5 y | M | XP, skin cancer | ⁎ |
| (Riyaz and Riyaz 1999) | India | 1 | 9 m | F | XP, no cancer | ⁎ |
| (Mazhar and Hannan 2001) | Pakistan | 2 | 6.5 y 13 y |
M M |
XP, no cancer | ⁎ , also congenital glaucoma. |
| (Rosón et al. 2005) | Spain | 1 | 4 y | M | XP, no cancer | ⁎ |
| (Procianoy et al. 2006) | Brazil | 1 | 5 y | F | XP, Conjunctival SCCs | ⁎ , aggravated SCCs after Photodynamic Therapy |
| (Anttinen et al. 2008) | Finland (23y follow) |
11 | 4 y (median) |
XP, skin cancer in 6 (55%) | Progressive neurologic degeneration | |
| (Bradford et al. 2011) | USA, NIH (1971-2009) |
25 | . | . | XP,. | Progressive neurologic degeneration |
| (Viana et al. 2011) | Brazil | 1 | 34 m | M | XP, no cancer | ⁎ , also schizencephaly |
| (Caldas and Rodrigues 2013) | Brazil | 1 | 21 m | F | XP, SCC, AFX | ⁎ |
| (Mittal et al. 2013) | India | 1 | 10 y | M | XP, no cancer | ⁎, but normal gonadal maturation. |
| (Fekete 2014) | USA | 1 | 55 y | F | XP, BCCs | ⁎ |
The classic DSC syndrome defined as XP with mental retardation, short stature and hypogonadism. The neurologic abnormalities included microcephaly, delayed growth, progressive neurologic deterioration, hearing impairment, areflexia, ataxia and abnormal motor activity. Progressive neurologic degeneration: loss of intellectual functioning, deterioration of neurologic status, impaired hearing, abnormal speech, areflexia, ataxia, peripheral neuropathy, and loss of ability to walk and talk. Biochemical profile of Cockayne syndrome (CS): normal DNA-excision repair and a failure of RNA synthesis with UV exposure. AFX, Atypical Fibroxanthoma. Schizencephaly: abnormal clefts in the cerebral hemispheres of the brain that connect the ventricles to the subarachnoid space. .. Not available
Several neurologic abnormalities have been recognized in XP patients. A previous review (Kraemer et al., 1987) reported neurologic abnormalities in 18% of patients, and the most prevalent symptoms were mental retardation, spasticity, ataxia, and microcephaly. Recently, from a 39-year follow-up study involving 106 patients with XP, the National Institutes of Health (NIH) revealed that 24% of patients with XP present with progressive neurologic degeneration (Bradford et al., 2011). Symptoms included loss of intellectual functioning, deterioration of neurologic status, loss of hearing, areflexia, ataxia, peripheral neuropathy, and inability to walk and talk. For our patient, brain imaging revealed brain atrophy with no intracranial calcifications. The severity of neurologic degeneration in patients with XP is correlated with the diffuse cortical, brain stem, and cerebellar atrophy (Anttinen et al., 2008, Fekete, 2014, Mittal et al., 2013), while brain calcification is more suggestive of Cockayne syndrome (Fekete, 2014).
Recently, the NIH reported an increased frequency of different cancer types in patients with XP, as compared with the general population, as follows: tongue cancer (× 100,000), nonmelanoma skin cancer (× 10,000), melanoma (× 2,000), anterior compartment of the eye secondary to sun exposure (× 1,000), and brain and central nervous system (× 50) (DiGiovanna and Kraemer, 2012). Skin cancers were reported in about 50% of patients with XP who have neurologic abnormalities, a rate that was similar to patients who have XP without neurologic abnormalities (Anttinen et al., 2008, Kraemer et al., 1987). In our review, we found a similar skin cancer rate of 42% among recent patients with XP who had neurologic abnormalities (Table 1).
The increased risk of conjunctival SCC, skin cancer on sun-exposed areas, and progressive neurologic degeneration is believed to be caused by an impaired NER pathway in XP cells. NER is a pathway involved in the repair of nucleotide dimerization damage secondary to UVR (DiGiovanna and Kraemer, 2012), psoralen (Cortés et al., 1991), and benzopyrene (carcinogen in cigarette smoke that mimics UVR damage) (Maher et al., 1977). Cumulative mutagenic DNA damage increases the risk of cancer on exposed areas. Although neurons are not exposed to UVR, reactive oxygen species damage DNA. Oxidative processes or reduced superoxide dismutase might induce premature neuron death (DiGiovanna and Kraemer, 2012, Nishigori et al., 1989, Reardon et al., 1997).
Disease management has been mainly supportive. It is highly recommended that the patient’s family restrict the child’s exposure to sunlight and provide the child with skin and eye protection such as sunblock, long-sleeved clothing, and window tinting. Vitamin D supplementation is necessary to prevent vitamin deficiency (DiGiovanna and Kraemer, 2012). Avoidance of cigarette smoke and other environmental carcinogens are recommended. Patients need to be monitored by dermatologists, ophthalmologists, and neurologists for early management of issues (DiGiovanna and Kraemer, 2012, Mittal et al., 2013).
The earliest clinical neurologic findings are frequently an absence of deep tendon reflexes and high-frequency hearing loss (DiGiovanna and Kraemer, 2012). The neurologic symptoms probably start slowly when patients are about 2 years of age and become apparent with cognitive and cerebellar signs in 4-5 years (Anttinen et al., 2008). Therefore, deep tendon reflex testing and audiometry are recommended to screen for the presence of neurologic involvement (DiGiovanna and Kraemer, 2012). Molecular genetic findings have indicated that neurologic abnormalities are more prevalent in patients with certain gene mutations that were previously known as the A (the most frequently group worldwide and in Japan), D, and B complimentary groups for XP (Anttinen et al., 2008, Kraemer et al., 1987, Lai et al., 2013). Neurologic abnormalities are rare in the C complimentary group, which is the most common group in the United States, Europe, and the Middle East (Kraemer et al., 1987). However, molecular genetic studies were not performed for our patient.
Skin neoplasm management involves electrodessication, curettage, surgical excision, Mohs micrographic surgery, and chemoprevention. Mohs microsurgery ensures complete removal and allows maximum skin preservation for future procedures in patients with XP. In this patient, unfortunately, Mohs microsurgery was not available, and the large SCC lesion was surgically resected with a 1-cm margin.
In addition, oral isotretinoin reduces the formation of new skin cancers. Previously, a study by Kraemer et al. (1992) found that chemoprevention with a high oral dose of isotretinoin, 2 mg/kg/day, for 2 years significantly reduced (63%) the number of new skin cancers in patients with XP. We initiated treatment with a lower-dose retinoid because of the potential for the side effect of xerosis in our patient (0.5 mg/kg/day).
Recent studies found that the successful application of topical recombinant DNA repair enzymes (e.g., liposomal encapsulated T4 endonuclease V [Yarosh et al., 2001] and liposomal photolyase with high-protection ultraviolet filters [Giustini et al., 2014]) lowers the rate of development of actinic keratosis, SCC, and basal cell carcinoma in patients with XP.
Conclusion
Patients with XP may have neurologic abnormalities. Early diagnosis of the disorder, appropriate UVR protection, and astute management could help alleviate patients’ symptoms.
Footnotes
Conflicts of interest: The authors have no conflict of interest in publishing this article.
References
- Anttinen A., Koulu L., Nikoskelainen E., Portin R., Kurki T., Erkinjuntti M. Neurological symptoms and natural course of xeroderma pigmentosum. Brain J Neurol. 2008;131:1979–1989. doi: 10.1093/brain/awn126. [DOI] [PubMed] [Google Scholar]
- Bradford P.T., Goldstein A.M., Tamura D., Khan S.G., Ueda T., Boyle J. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48:168–176. doi: 10.1136/jmg.2010.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas A.L.R., Rodrigues M.M. De Sanctis-Cacchione syndrome in a female infant--case report. An Bras Dermatol. 2013;88:979–981. doi: 10.1590/abd1806-4841.20132844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver J.E., Thomas G.H. Rapid diagnosis of sensitivity to ultraviolet light in fibroblasts from dermatologic disorders, with particular reference to xeroderma pigmentosum. J Invest Dermatol. 1988;90:467–471. doi: 10.1111/1523-1747.ep12460917. [DOI] [PubMed] [Google Scholar]
- Cleaver J.E., Charles W.C., Kong S.H. Efficiency of repair of pyrimidine dimers and psoralen monoadducts in normal and xeroderma pigmentosum human cells. Photochem Photobiol. 1984;40:621–629. doi: 10.1111/j.1751-1097.1984.tb05350.x. [DOI] [PubMed] [Google Scholar]
- Cortés F., Morgan W.F., Varcarcel E.R., Cleaver J.E., Wolff S. Both cross-links and monoadducts induced in DNA by psoralens can lead to sister chromatid exchange formation. Exp Cell Res. 1991;196:127–130. doi: 10.1016/0014-4827(91)90464-6. [DOI] [PubMed] [Google Scholar]
- de Sanctis C., Cacchione A. L’idiozia xerodermica. Riv Sper Freniatr Med Leg Alien Ment. 1932;56:269–292. [Google Scholar]
- DiGiovanna J.J., Kraemer K.H. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete R. Xeroderma pigmentosum/de Sanctis-Cacchione syndrome: unusual cause of ataxia. Case Rep Neurol. 2014;6:83–87. doi: 10.1159/000362115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustini S., Miraglia E., Berardesca E., Milani M., Calvieri S. Preventive long-term effects of a topical film-forming medical device with ultra-high UV protection filters and DNA repair enzyme in xeroderma pigmentosum: a retrospective study of eight cases. Case Rep Dermatol. 2014;6:222–226. doi: 10.1159/000368182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessel A., Siegle R.J., Mitchell D.L., Cleaver J.E. Xeroderma pigmentosum variant with multisystem involvement. Arch Dermatol. 1992;128:1233–1237. [PubMed] [Google Scholar]
- Kraemer K.H., DiGiovanna J.J. Xeroderma Pigmentosum. In: Pagon R.A., Adam M.P., Ardinger H.H., Bird T.D., Dolan C.R., Fong C.-T., editors. GeneReviews. University of Washington, Seattle; Seattle: 1993. [Internet] [cited 2015 January 22, Available from: http://www.ncbi.nlm.nih.gov/books/NBK1397/] [Google Scholar]
- Kraemer K.H., Lee M.M., Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- Kraemer K.H., DiGiovanna J.J., Peck G.L. Chemoprevention of skin cancer in xeroderma pigmentosum. J Dermatol. 1992;19:715–718. doi: 10.1111/j.1346-8138.1992.tb03766.x. [DOI] [PubMed] [Google Scholar]
- Lai J.-P., Liu Y.-C., Alimchandani M., Liu Q., Aung P.P., Matsuda K. The influence of DNA repair on neurological degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (X-A, XP-C and XP-D) Acta Neuropathol Commun. 2013;1(1):4. doi: 10.1186/2051-5960-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher V.M., McCormick J.J., Grover P.L., Sims P. Effect of DNA repair on the cytotoxicity and mutagenicity of polycyclic hydrocarbon derivatives in normal and xeroderma pigmentosum human fibroblasts. Mutat Res. 1977;43:117–138. doi: 10.1016/0027-5107(77)90137-3. [DOI] [PubMed] [Google Scholar]
- McKusick V.A., Kniffin C.L. Entry # 278800 - DE SANCTIS-CACCHIONE SYNDROME [Internet] 2010. http://www.omim.org/entry/278800 [cited 2015 January 12] Available from:
- Mittal H., Mehndiratta S., Kaushik J.S., Godbole T. De Sanctis-Cacchione syndrome. Indian J Dermatol Venereol Leprol. 2013;79:849. doi: 10.4103/0378-6323.120760. [DOI] [PubMed] [Google Scholar]
- Nishigori C., Miyachi Y., Imamura S., Takebe H. Reduced superoxide dismutase activity in xeroderma pigmentosum fibroblasts. J Invest Dermatol. 1989;93:506–510. doi: 10.1111/1523-1747.ep12284060. [DOI] [PubMed] [Google Scholar]
- Reardon J.T., Bessho T., Kung H.C., Bolton P.H., Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh D., Klein J., O’Connor A., Hawk J., Rafal E., Wolf P. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet. 2001;357:926–929. doi: 10.1016/s0140-6736(00)04214-8. [DOI] [PubMed] [Google Scholar]