

ABSTRACT
This study was undertaken to examine the activities and levels of major antioxidants/oxidants in cultured human fibroblasts incubated with a sublethal dose of Echis coloratus venom (EcV).
Glutathione peroxidase (GPx), catalase (CAT), superoxide dismutase (SOD) and glutathione reductase (GR) activities and gene expression levels as well as reduced glutathione (GSH) levels, and the concurrent hydrogen peroxide (H2O2), superoxide anions (SOA), lipid peroxides (LPO) and oxidized glutathione (GSSG) generation rates were assayed in fibroblast cultures and sonicates incubated with 0.5 µg ml–1 medium EcV for 4 h at 37°C.
Data indicated that the activities of all antioxidant enzymes were significantly decreased and their corresponding transcripts downregulated in EcV-incubated cells compared to controls (p < 0.001). In contrast, there were parallel equally significant increases in H2O2, SOA and LPO generation rates in venom-incubated cells compared to controls (p < 0.001). Additionally, GSH levels were significantly decreased and those of GSSG were equally significantly increased in venom-incubated cultures compared to controls (p < 0.001) leading to a lowered GSH/GSSG ratio.
In conclusion, incubation of fibroblast cultures with EcV resulted in a shift towards oxidative metabolism causing severe OS. This correlated with significant downregulation in the expression levels of all investigated antioxidant genes.
KEYWORDS: Echis coloratus, venom, oxidative stress, cultured fibroblasts
Introduction
Echis coloratus (the painted saw-scaled viper) belongs to the Viperidae family which comprises Echis carinatus, Echis pyramidum, Cerastes cerastes, Pseudo cerates and Bitis arientans.[1,2] Echis coloratus is found in north-east Africa and throughout Arabia. In Saudi Arabia, it is found in the north and east of the central region and is spread on a large scale in mountainous areas.[3]
Snake venoms are mixtures of enzymes, peptides, toxins, nerve growth factors, carbohydrates, lipids, metal ions and organic compounds.[4] The biological activities of snake venoms are primarily a function of their protein components rather than the non-proteins.[5,6] The enzymes include phospholipase A2, amino acid oxidase, phosphodiesterase, proteolytic enzymes and arginine ester hydrolase. Clinical effects produced by venoms include neurotoxic effects causing sensory, motor, cardiac and respiratory difficulties. Cytotoxic effects are directed at erythrocytes, blood vessels, heart, muscle, liver, kidney, lungs and defects in coagulation caused by the local release of enzymes. Some enzymes have been reported to increase vascular permeability and induce inflammatory reactions [7] primarily caused by metalloproteinases, which can be hemorrhagic.[8]
The pathologic manifestations of viper envenomation are numerous and varied. Venoms contain proteins that interfere with the coagulation cascade, the normal hemostatic system and tissue repair.[9,10] Viperdae venoms contain over 100 proteins including the enzymes serine proteinases, Zn+2- metalloproteinases, L-amino acid oxidase and group II phospholipase A2 which are unique for Viperdae venoms.[11] Human envenomation is often characterized by clotting disorders, hypofibrinogenemia and local tissue necrosis. Those related to Echis coloratus include pronounced local swelling, pain and inflammation.[12] Renal failure and death due to respiratory failure have been shown to occur.[13] Several studies have investigated the effect of viper venoms on hematological, cardiovascular and metabolic parameters in rats and cultured human cells.[14–18] Novel disintegrins were purified from the venom of Echis pyramidum,[14] and different fractions of the venom were shown to either increase or decrease the plasma levels of alanine transaminase, aspartate transaminase, glucose, triglycerides, cholesterol, urea and creatinine [15] indicating hepatotoxic and nephrotoxic effects. In addition, hemorrhagic metalloproteinase was isolated from the venom of three vipers of Egypt and its activity on rabbit skin was demonstrated.[16] In one study, we were able to show that different fractions of Echis coloratus venom significantly decreased the activities of key glucose catabolic enzymes, but enhanced those of glycogen phosphorylase, alanine and aspartate transaminases in human fibroblasts cultured in the presence of the venom proteins.[17] In addition, we showed that certain fractions of Cerastes cerastes venom significantly lowered the activity of key enzymes of the mitochondrial respiratory chain.[18]
During normal oxidative metabolism, mitochondria utilize oxygen, reducing it by sequential reactions of the respiratory chain to water. A small fraction of this oxygen is converted to highly reactive and toxic derivatives known as reactive oxygen species (ROS). Such species include superoxide anions (SOA), hydrogen peroxide (H2O2), lipid peroxides (LPO) and hydroxyl radicals.[19] Limited ROS concentrations are important regulators of many cellular functions through their action as secondary messengers that cause the phosphorylation and activation of specific transcription factors, and as mediators of signaling transduction pathways in cell growth, proliferation and apoptosis.[19] However, excessive production of ROS, also known as oxidative stress (OS), causes damage involving cellular organelles and alteration of the structure of membrane lipids, nuclear and mitochondrial DNA and proteins.[20] Cytotoxicity occurs as a result of alteration in the expression of genes including those related to apoptosis.[21] Cells have developed an extensive defense system to neutralize the harmful oxidative effects of excessive ROS generation. This system includes the antioxidant enzymes superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) which reduce ROS to water.[19] Apart from being involved in damaging cellular components, ROS seem to play a major role in venom-induced toxicity. It has been reported that excessive ROS production takes place during envenomation, causing toxicity in experimental mice.[22] Echis pyramidum venom, due to its phospholipase A2 activity, has been shown to produce free oxygen radicals leading to the formation of highly reactive LPO and OS in different organs of mice.[23] More recently, the same venom was shown to significantly lower hepatic CAT and SOD activities and to increase levels of TBARS (a marker of lipid peroxidation) in rats.[24] Echis ocellatus envenomed mice were also shown to exhibit marked decreases in serum SOD, CAT and GPx activities.[25] Similarly, it was demonstrated that the crude venom of the cobra Naja haje caused significant increases in hepatic and renal levels of H2O2, LPO, carbonyl proteins and nitric oxide, and concurrent decreases in CAT and SOD activities.[26]
In spite of the emerging findings that associate envenomation with OS and toxicity, comprehensive data related to the biochemical and molecular changes in the antioxidant capacity of human tissue subjected to Echis coloratus venom is not available. This study was undertaken to examine and relate the activities and levels of the major antioxidants SOD, GPx, GR, CAT and GSH, to those of the corresponding oxidants including SOA, H2O2, LPO and GSSG in cultured human fibroblasts incubated with a sub-lethal dose of crude Echis coloratus venom. In addition, the study examines the mRNA transcriptional levels of all the investigated antioxidant enzyme genes in venom-incubated cultures compared to controls.
Materials and methods
Echis coloratus crude venom (EcV) was purchased from Latoxan, Rosans, France. All chemicals and fine biochemicals were obtained from Sigma Chemical Company, Poole, UK. Eagle’s Minimum essential medium (MEM), Hanks buffered salt solution (HBSS), fetal calf serum, penicillin, streptomycin, trypsin, L-glutamate and tissue culture flasks were purchased from Flow Laboratories, Mclean, VA, USA.
Cultivation of human fibroblasts
The procedure for obtaining and processing of human skin biopsies was approved by the Ethics Committee /IRB of the College of Medicine, King Khalid University Hospital, King Saud University (CM IRB-KKUH-KSU). Primary human fibroblast cultures were established from 12 forearm skin biopsies (~20 mg in weight) taken from healthy adult volunteers with an average age of 27.1 ± 1.35 years (range, 25.3–29.1). Monolayer confluent cultures were obtained by growing cells in MEM (20 ml) containing 10% fetal calf serum and harvested by trypsinization. Details regarding the culture process, contents and preparation of MEM and the trypsinization and harvesting media, are as extensively documented by us elsewhere.[27] Cells were normally cultured in 75 cm2 flasks in a Gelaire BSB4 laminar flow cabinet (Sydney, Australia) at 37°C in an atmosphere containing 18% O2 and 5% CO2. Cells at the early passage 5 of subculture were used for investigation throughout the study.
Incubation of EcV with fibroblast monolayers
Crude EcV was dissolved in HBSS (pH 7.4), and 10 µg/200 µl aliquots were added to the culture medium (20 ml) of triplicate 75 cm2 confluent passage 5 fibroblast cultures (n = 12), thus constituting a 0.5 µg ml–1 venom concentration, and incubated for 4 h at 37°C. Cell cultures not incubated with EcV were run in parallel and considered as controls. At the end of the incubation period cells were harvested by trypsinization, resuspended in harvesting medium, thoroughly washed and centrifuged at 2000 g for 5 min. The pellets were kept on ice and quickly sonicated for 25 s in 0.1 M phosphate buffer pH 7.0 (0.5 ml) using a Thermo-Fisher Sonic Dismembranator Model 150 (Waltham, MA, USA) at 50% of the power output equivalent to 10000 Hz frequency. Appropriate aliquots of the sonicates were used for the assay of various antioxidant enzymes and related metabolites, and intact cell pellets (~50 mg) were used for gene expression profiling of enzymes as described later.
In a separate experiment, triplicate sonicates of the 12 cell cultures (1.5 ml) were incubated with EcV (0.5 µg ml–1 at 37°C for 4 h). Aliquots of each incubation mixture were then assayed for the enzymes and metabolites. Control sonicates (not incubated with EcV) were run in parallel.
Determination of cell viability of EcV-incubated cells
To establish a venom dose and incubation period at which cells maintain in vitro metabolic viability and exhibit normal proliferation, a modified MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide)] assay was performed according to Mosmann [28] and as modified by Southen and LaFontaine.[29,30] The assay is based on the cleavage of MTT, a yellow tetrazolium salt, by the mitochondrial enzyme succinate dehydrogenase of metabolically viable cells and results in the formation of blue formazan crystals, the amount of which is proportional to the number of viable cells. Briefly, triplicate passage 5 fibroblasts were grown for 24 h in 96-well microplates with 8 × 104 cells ml–1 initial concentration. Post-culture, cells were treated with increasing amounts of EcV equivalent to 0.05, 0.1, 0.2, 0.4, 0.5, 1.0, 1.5 and 2.5 µg ml–1 (100 µl aliquots) added to the culture medium and incubated for 4 h at 37°C. In a separate experiment another set of cultured cells were incubated with the same EcV concentrations for 10 h at 37°C. The venom containing medium from all cells was then removed and replaced with phosphate buffered saline (pH 7.2) containing filtration sterilized MTT (Sigma, 2.4 mM, 400 µl). After incubation of the plate at 37°C for 2 h, the MTT solution was removed and cellular formazan crystals were solubilized using acidified isopropanol (300 µl/well). Finally, absorbance of all samples was monitored at 570 nm using an EIA plate reader (Model 2550-Bio-Rad, Hercules, CA, USA) and a background absorbance at 690 nm was subtracted. Cell viability of EcV-incubated cultures was then expressed as mean ± SD percentages for each venom concentration compared to venom-free control cultures, the absorbance value of which was set at 100%.
Biochemical assays
Antioxidant enzyme assays
GPx, CAT, SOD and GR specific activities were assayed in fibroblast sonicates as documented by us elsewhere.[31–33] For GPx fibroblast sonicate (100 µl) was added to an assay reaction mixture (800 µl) containing 50 mM tris-HCl buffer pH 7.6, 1 mM EDTA, 1 IU GR, 0.25 mM GSH and 0.2 mM NADPH. After a 5-min incubation at room temperature, the reaction was initiated by the addition of 15 mM H2O2 (100 µl) and the decrease in absorbance reflecting oxidation of NADPH was monitored at 340 nm. Activity was expressed as the amount of enzyme that catalyzes the oxidation of 1 µmole GSH/minute. CAT activity was determined by measuring the decomposition of H2O2 (µmole/mg protein) directly followed by monitoring the decrease in absorbance at 240 nm. The assay was initiated by the addition of fibroblast sonicate (50 µl) or an appropriately diluted H2O2 stock solution. GR activity was assayed by adding fibroblast sonicate (100 µl) to a reaction mixture (2 ml) containing potassium phosphate buffer (100 mM, pH 7.4), 80 mM EDTA, 2 mM NADPH and 0.3 mM flavine adenine dinucleotide. After a 2-min incubation, the reaction was initiated by the addition of 7.5 mM GSSG and the change in absorbance was monitored at 340 nm. One unit of GR activity was equal to the amount of enzyme responsible for the reduction of 1 µmole NADPH/minute at 25°C. Total SOD activity was measured by addition of fibroblast sonicate (50 µl) to xanthine (25 µl, 1.142 mg ml–1), hydroxyl ammonium chloride (25 µl), water (125 µl) and xanthine oxidase (0.1 U ml–1, 75 µl). The mixture was then incubated at 25°C for 20 min and sulphonilic acid (0.5 ml, 3.3 mg ml–1) and α-naphthylamine (0.5 ml, 1 ng ml–1) were added, further incubated at room temperature for 20 minutes and absorbance was read at 530 nm.
GSH and GSSG assays
GSH was measured by the glutathione reductase-DTNB (5,5 dithiobis-2-nitrobenzoic acid) recycling procedure according to Gherghel et al. [34] and as modified by us.[33] The assay mixture contained sodium phosphate buffer (150 µl, pH 7.4), 8 mM EDTA (50 µl), DTNB solution (50 µl) and 2 mM NADPH (100 µl). GSH standards (20–80 µM) and fibroblast sonicates (50 µl) were then added and the tubes incubated at 37°C for 3 min. Finally, GR (1 IU, 25 µl) was added and absorbance monitored at 410 nm. For the GSSG assay, the reaction mixture was the same as above and additionally contained triethanolamine to prevent a high local pH and oxidation, and 2-vinyl pyridine for GSH derivitization. GSSG standards (0–10 µM) were run in parallel.
H2O2 assay
The rate of H2O2 generation by fibroblasts was assayed according to Zhou et al. [35] and as modified by us.[33] The assay mixture containing peroxidase dissolved in Kreb’s Ringer buffer (10 IU ml–1, 100 µl), sodium phosphate reaction buffer (50 mM, pH 7.4) and appropriately diluted standards and fibroblast sonicates (50 µl) was incubated for 30 min at room temperature. The reaction was initiated by addition of ARR (Amplex Red Reagent, 10-acetyl-3,7-dihydrophenoxazine, 10 mM, 50 µl) which reacts with H2O2 to form the red fluorescent oxidation product resorufin. Fluorescence was then measured using an excitation λ of 530 and an emission λ of 590. Standard H2O2 solutions ranged in concentration from 0 to 15 µM.
SOA assay
SOA generation in fibroblasts was determined according to Johnston et al. [36] and as modified by us.[37] Fibroblast sonicates (50 µl) were incubated for 5 min at 37°C with phosphate buffered saline (1 ml, pH 7.4) containing glucose (2 g l–1) and fatty acid-free bovine serum albumin (2 g l–1) with or without SOD (30 µg, 50 µl). To initiate the reaction, ferricytochrome C solution (1.2 mM, 100 µl) was added to the incubation mixture, and the increase in absorbance was monitored at 550 nm in a recording thermostated spectrophotometer (Shimatzu model UV-2401 PC, Dubai, UAE). SOA generation was determined by calculating the difference between the sample without SOD and that with added SOD and expressed as the amount of reduced ferricytochrome C in µmol min–1 mg–1 protein.
LPO assay
Lipid peroxidation was assayed by determining the level of malondialdehyde using thiobarbituric acid according to Saleh et al. [38] and as modified by us.[33] Fibroblast sonicate (100 µl) was added to trichloroacetic acid (1 ml, 17.5%), thiobarbituric acid (1 ml, 0.6%) and the mixture incubated at 100°C for 15 min and allowed to cool. Trichloroacetic acid (1 ml, 70%) was then added to the mixture for 20 min at room temperature and the mixture centrifuged at 2000 rpm for 15 min. The supernatant was removed, absorbance monitored at 535 nm and malondialdehyde concentration calculated using an extinction coefficient of 1.56 × 105/M cm–1.
Gene expression profiling of hsGPx, hsCAT, hsSOD and hsGR using real-time quantitative PCR (RT-qPCR)
Freshly collected fibroblast pellets were stored in RNAlater® RNA Stabilization Solution (Qiagen, Hilden, Germany) at −80°C. Pellets were then homogenized using a Tissue Lyser LT (Qiagen) in 1.0 ml TRIzol® Reagent (Invitrogen, Paisley, UK) and total RNA was extracted according to standard procedures. After genomic DNA elimination, cDNA was synthesized from RNA (1 µg) in a final reaction volume (20 µl) using the QuantiTect Reverse Transcriptase Kit (Qiagen, QuantiTect®) according to the manufacturer’s instructions. RT-qPCR was subsequently performed using a QuantiTect SYBR Green PCR Kit (Qiagen) with the following gene primers. SOD Qiagen Primer Assay (Hs_SOD_1_SG QuantiTect Primer assay, QTO1664327); CAT (Hs_CAT1_SG QuantiTect Primer assay, QT000796764), GPx (Hs_GPx1_SG QuantiTect Primer assay, QT00203392) and GR (Hs_GR_SG QuantiTect Primer assay, QT00038325) in a final reaction volume (25 µl) containing the diluted cDNA sample (5 µl), 2 × SYBR Green PCR Master Mix (12.5 µl), each forward and reverse primer (10 µM stock, 2.5 µl) and RNAase-free water (2.5 µl). The amplification program and PCR amplicon specificity were performed and assessed according to our previous report.[37] Each fibroblast tissue sample was represented by biological replicas and three technical replicas, with the inclusion of a no-template control (NTC).
Raw data were analyzed using the Rotor-Gene cycler software version 2.3 to calculate the threshold cycle (Ct) using the second derivative maximum method. The fold change in each gene was determined after normalization to the expression levels of 18S as a house-keeping gene. The relative gene expression level (fold change) was calculated using the 2−ΔΔCt method.[39] Thus, the relative expression level for each gene in the control venom-free cells was designated as 1.0. Next, the relative level of each gene transcript normalized to the 18S gene was calculated using the following equation: Fold change = 2 Ct target (control) – Ct target (EcV-treated)/2 Ct 18S (control) − Ct 18S (EcV-treated). The relative gene expression data were subjected to Student’s t-test in order to identify significant differences between EcV-treated samples compared to venom-free controls. Treatments were considered statistically significant when at p < 0.05.
Other assays and statistical analysis
Total protein content of fibroblast sonicates (20 µl) was assayed according to Bradford.[40] Analysis of variance was performed to evaluate statistical differences between mean ± SD values of all parameters assayed in control fibroblast cultures and sonicates incubated with EcV against those recorded for samples not incubated with the venom. This was done using the statistical package for social sciences software (SPSS version 17.0, IBM, Armonk, NY, USA). Values of p < 0.05 were considered statistically significant.
Results
Cell viability of EcV-incubated fibroblasts
As is evident from Figure 1, the MTT assay results indicated that cells incubated with EcV at 0.05, 0.1, 0.2, 0.4 and 0.5 µg ml–1 MEM for 4 h did not undergo any significant loss of cell viability compared with venom-free control cultures. Percentage cell viability values equaled 98.6 ± 2.48%, 102.1 ± 2.63%, 101.7 ± 2.71%, 98.4 ± 2.31% and 97.9 ± 2.16% for 0.05, 0.1, 0.2, 0.4 and 0.5 µg ml–1 EcV respectively against 100% cell viability set for controls. Moreover, very similar results were obtained when cultures were incubated with the same EcV concentrations for a prolonged 10 h period. In contrast, Figure 1 data showed that cells incubated with EcV at 1.0, 1.5 and 2.5 µg ml–1 MEM for 4 h underwent significant progressive loss of cell viability relative to the venom concentration. Percentage cell viabilities equaled 59.5 ± 1.54%, 36.2 ± 0.92%, and 17.0 ± 0.42% for 1.0, 1.5 and 2.5 µg ml–1 EcV respectively against 100% cell viability set for controls (p < 0.001 for all comparisons). In addition, very similar results were seen when the cultures were incubated with the same venom concentrations for a period of 10 h. The above results indicated that loss of fibroblast viability has more than likely started to occur at EcV concentrations above 0.5 µg ml–1 MEM in a dose-dependent fashion. Furthermore, this must have been achieved in less than 4 h. Hence, in all subsequent experiments, 75 cm2 flask fibroblast cultures were incubated with 0.5 µg ml–1 MEM for a period of 4 h prior to being harvested for investigation.
Figure 1.
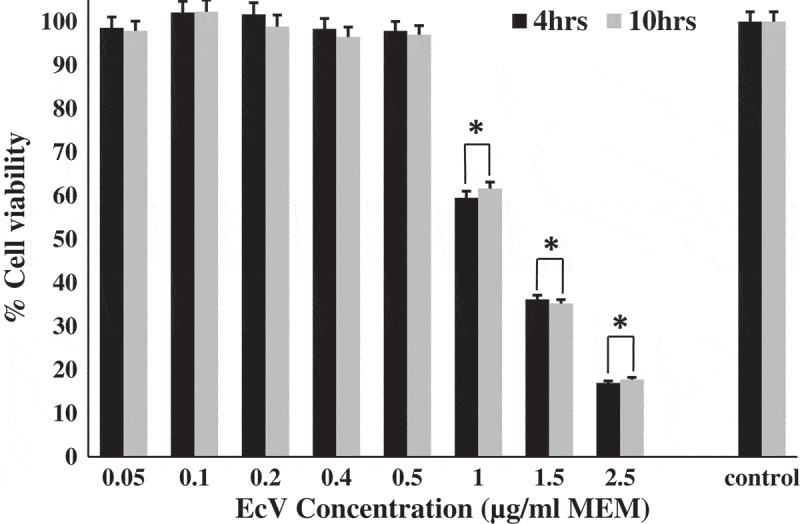
Cell viability of fibroblast cultures (n = 12) after 4 and 10 h incubation with 0.05–2.5 µg ml–1 MEM crude EcV. Confluent passage 5 cultures were used. Data expressed are means ±SD of triplicate determinations for 12 cultures. Control cultures were incubated with vehicle only and were considered to be 100% viable.*p < 0.001 when values for EcV-incubated cells were compared with those of venom-free controls.
Effect of incubation of fibroblast cultures and sonicates with EcV on the activities and levels of enzymatic and non-enzymatic antioxidants and pro-oxidants
Effect on antioxidant enzymes
As is clearly demonstrated in Table 1, the specific activities of all the antioxidant enzymes underwent very significant reductions in fibroblast cultures incubated with 0.5 µg ml–1 EcV for 4 h. Activities equaled 1.14 ± 0.11, 2.22 ± 0.20, 17.1 ± 1.48 and 1.36 ± 0.13 µmole min–1 mg–1 protein for GPx, CAT, SOD and GR respectively when compared with activities recorded for venom-free controls which equaled 1.93 ± 0.16, 3.58 ± 0.29, 26.1 ± 2.29 and 2.48 ± 0.23 µmole min–1 mg–1 protein (p < 0.001 for all comparisons). Such activity decreases were similar in magnitude for all the enzymes and approximately ranged between 35 and 45% of control values. Specific percentage reductions equaled 41.6 ± 3.81%, 38.3 ± 3.26%, 35.6 ± 3.25% and 45.8 ± 4.03% of control activities for GPx, CAT, SOD and GR respectively. In contrast, additional Table 1 data showed no significant alterations in any of the enzyme activities when EcV at the same concentration and for the same period was incubated with fibroblast sonicates compared to venom-free sonicates.
Table 1.
Effect of incubation of fibroblast cultures and sonicates with EcV (0.5 µg ml–1 MEM for 4 h) on activities of antioxidant enzymes.
| Samples assayed (n = 12) | Enzymatic parameters (µmole min–1 mg–1 protein) |
|||
|---|---|---|---|---|
| GPx | CAT | SOD | GR | |
| Control cultures | 1.93 ± 0.16 | 3.58 ± 0.29 | 26.1 ± 2.29 | 2.48 ± 0.23 |
| Cultures + EcV | 1.14 ± 0.11* | 2.22 ± 0.20* | 17.1 ± 1.48* | 1.36 ± 0.13* |
| Control sonicates | 1.88 ± 0.15 | 3.46 ± 0.30 | 24.8 ± 2.10 | 2.53 ± 0.22 |
| Sonicates + EcV | 1.91 ± 0.16 | 3.41 ± 0.28 | 25.6 ± 2.31 | 2.46 ± 0.23 |
Confluent passage 5 cultures were used. The values shown are means ± SD for triplicates of the 12 cultures and sonicates. EcV = Echis coloratus crude venom, GPx = glutathione peroxidase, CAT = catalase, SOD = superoxide dismutase, GR = glutathione reductase. *Significant at p < 0.001 upon comparison of the values obtained for all assayed parameters in EcV-incubated cultures against those recorded for venom-free controls.
Effect on GSH and GSSG
As is evident from Table 2 data, incubation of the 12 fibroblast cultures with 0.5 µg ml–1 EcV for 4 h resulted in highly significant lowering of the cellular GSH levels when compared with those recorded for control venom-free cultures. Levels equaled 30.7 ± 2.42 nmole mg–1 protein for EcV-incubated cells against 45.2 ± 3.61 nmole mg–1 protein obtained for non-incubated controls (p < 0.001). In contrast, cellular GSSG levels were very significantly increased in EcV-incubated cultures upon comparison with non-incubated cultures (1.01 ± 0.08 against 0.76 ± 0.05 nmole mg–1 protein respectively (p < 0.001). The above results indicated a very significant decrease in the GSH/GSSG ratio from 58.3 ± 4.92 for control cultures to 29.2 ± 2.38 for EcV-incubated cultures (p < 0.001). However, no significant changes in GSH and GSSG levels were noted between EcV-incubated fibroblast sonicates and venom-free control sonicates, thus maintaining normal GSH/GSSG ratios of 59.1 ± 4.82 and 57.5 ± 4.64 respectively.
Table 2.
Effect of incubation of fibroblast cultures and sonicates with EcV (0.5 µg ml–1 MEM for 4 h) on the levels of non-enzymatic antioxidants and oxidants.
| Non-enzymatic parameters |
||||||
|---|---|---|---|---|---|---|
| GSSG |
H2O2 |
SOA |
LPO |
|||
| Samples assayed (n = 12) | GSH | nmole mg–1 protein | GSH/GSSG | pmole min–1 mg–1 protein | µmole min–1 mg–1 protein | pmole min–1 mg–1 weight |
| Control cultures | 45.2 ± 3.62 | 0.76 ± 0.05 | 58.3 ± 4.92 | 1.61 ± 0.16 | 0.58 ± 0.05 | 30.3 ± 2.91 |
| Cultures + EcV | 30.7 ± 2.4* | 1.01 ± 0.0* | 29.2 ± 2.38* | 2.45 ± 0.2* | 0.89 ± 0.0* | 43.6 ± 4.1* |
| Control sonicates | 47.6 ± 3.80 | 0.81 ± 0.06 | 57.5 ± 4.64 | 1.70 ± 0.18 | 0.60 ± 0.06 | 29.1 ± 2.83 |
| Sonicates + EcV | 46.1 ± 3.69 | 0.78 ± 0.05 | 59.1 ± 4.82 | 1.65 ± 0.17 | 0.63 ± 0.06 | 30.8 ± 3.02 |
Confluent passage 5 cultures were used. The values shown are means ± SD for triplicates of the 12 cultures and sonicates. GSH = reduced glutathione, GSSG = oxidized glutathione, H2O2 = hydrogen peroxide, SOA = superoxide anion, LPO = lipid peroxidation.*Significant at p < 0.0001 upon comparison of the values obtained for all assayed parameters in EcV-incubated cultures against those recorded for venom-free controls.
Effect on H2O2, SOA and LPO generation
As also seen from Table 2, incubation of fibroblast cultures with EcV (0.5 µg ml–1 MEM for 4 h) resulted in very significant increases in the generation rates of the oxidants H2O2 and SOA as well as LPO levels. Values equaled 2.45 ± 0.23 pmole min–1 mg–1 protein, 0.89 ± 0.08 µmole min–1 mg–1 protein and 43.6 ± 4.11 pmole mg–1 weight for H2O2, SOA and LPO respectively in EcV-incubated cells compared to 1.61 ± 0.16 pmole min–1 mg–1 protein, 0.58 ± 0.05 µmole min–1 mg–1 protein and 30.3 ± 2.91 pmole mg–1 weight respectively recorded for controls (p < 0.001 for all comparisons). In contrast, no significant changes in all the above markers of oxidative stress were noted between EcV-incubated fibroblast sonicates and the venom-free control sonicates.
Effect of incubation of fibroblast cultures with EcV on relative gene expression of antioxidant enzymes
As is clear from Figure 2, cellular hsGPx, hsCAT, hsSOD and hsGR transcripts were very significantly downregulated by 57.2 ± 3.2%, 48.1 ± 2.6%, 51.3 ± 3.0% and 53.1 ± 3.1% respectively in EcV-incubated cultures compared to the respective transcripts recorded for venom-free control cultures (p < 0.001 for all comparisons).
Figure 2.
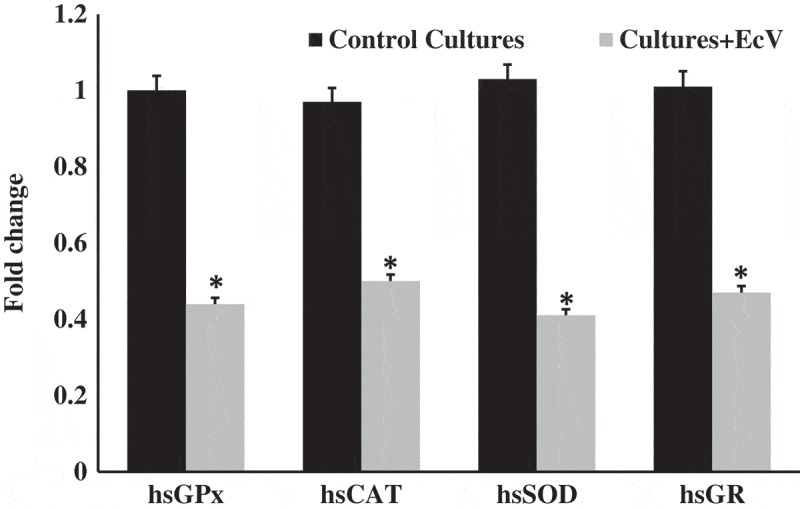
Relative gene expression of hsGPx, hsCAT, hsSOD and hsGR in fibroblast cultures incubated with EcV (0.5 µg ml–1 MEM for 4 h) compared to controls. Confluent passage 5 cultures were used. Fold change values shown are means ± SD for triplicates of the 12-fibroblast cultures. EcV = Echis coloratus crude venom, hsGPx = Homo sapiens glutathione peroxidase, hsCAT = Homo sapiens catalase, hsSOD = Homo sapiens superoxide dismutase, hsGR = Homo sapiens glutathione reductase. *p < 0.001 upon comparison of the fold change in the gene expression levels of each enzyme in EcV-incubated cells relative to venom-free controls.
Discussion
Human primary fibroblast cultures have been used in our laboratory for the study of changes in intracellular metabolism under a variety of experimental conditions, including incubation of the cells with crude or purified venom proteins of different snake species.[17,18,27] From an ethical perspective and increasing public concern for animal welfare, this in vitro model system offers a very feasible alternative for the assessment of venom toxicity and its effect on the metabolic and molecular activity of human tissue. Fibroblasts were chosen since they are relatively easy to establish and maintain in culture and are considered to be rich and versatile in metabolic activity. Furthermore, results obtained can be safely extrapolated to other tissues. In this study, it was possible to investigate the effect of crude EcV on the antioxidant capacity of fibroblasts by incubating confluent cells with the venom which was added to the culture medium. It was very important to choose an EcV concentration and an incubation period that would not affect metabolic and proliferative viability of the cells. Hence, any observed changes in the antioxidant capacity of the venom-incubated cells will not be related to loss of cellular viability, but can be truly attributed to the presence of the venom. This was done by incubating confluent 75 cm2 flask cultures with increasing EcV concentrations in a range of 0.05–2.5 µg ml–1 MEM for either 4 or 10 h, and monitoring the concentration of solubilized formazan crystals using the MTT assay. Figure 1 results showed that there was no significant loss of cell viability when cultures were incubated with 0.05, 0.1, 0.2, 0.4 and 0.5 µg ml–1 EcV for either 4 or 10 h. However, cultures incubated with 1.0, 1.5 and 2.5 µg ml–1 EcV underwent very significant progressive loss of viability in a dose-dependent fashion, the magnitudes of which were very similar at 4 and 10 h. These data indicated that loss of cell viability must have occurred at EcV concentrations above 0.5 µg ml–1 MEM in less than 4 h post-incubation. Hence, to minimize kinetic errors, fibroblast cultures were always incubated with 0.5 µg EcV ml–1 MEM for a period of 4 h prior to harvesting and investigation. In addition, primary passage 5 cultures were used throughout the study as cells beyond passage 10 were previously shown by us to be at an early phase of senescence. Such cells exhibited significant variations in the activities of many key cytosolic and mitochondrial enzymes including antioxidant enzymes, lowered rates of protein synthesis, growth and replication, as well as changes in size and morphology.[31,32,41,42] Other optimal culture conditions were also provided to ensure maximal rates of fibroblast growth, multiplication and metabolism. These included provision of sufficient MEM (20 ml/75cm2 flask) which was frequently changed, addition of Hepes buffer pH 7.4 in both culture and trypsinization media and streptomycin and penicillin to avoid bacterial contamination.
As evident from the results of the present study, incubation of confluent fibroblast cultures with EcV produced very significant decreases of similar magnitude in the specific activities of GPx, CAT, SOD and GR when compared to those recorded in control cultures (Table 1). Although the enzyme activities were expressed in terms of total cellular protein, very similar results were obtained in terms of DNA (data not shown). The ratio of cellular protein to DNA remained relatively constant regardless of EcV incubation (mean = 15.8 µg protein/µg DNA ± 1.43SD for the 12 cultures. Incubation of confluent fibroblasts with 0.5 µg EcV ml–1 MEM for 4 h did not alter the protein yield, which amounted to 693.7 ± 43.8 µg/75cm2 flask of cells, indicating no proteolytic activity of the venom at this concentration and incubation time. To this end, although proteolytic activity has been associated with some venom proteins, those of Viperdae exhibited weak activity and none of the purified Echis coloratus fractions possessed any such activity.[17] In addition, the absence of EcV proteolytic activity could have been due to the fact that cells were cultured in MEM containing 10% (v/v) fetal calf serum; a source of protease inhibitors. Moreover, none of the studied antioxidant enzymes were detected in the venom or the culture medium before or after incubation of the cells with the venom. Hence, the possibility of cellular membrane degradation caused by the venom’s phospholipase A2 activity is ruled out. However, incubation of the fibroblast cultures with higher EcV concentrations than used in the present study (>7.5 µg ml–1 MEM) resulted in rounding, rupture and lysis of cells (unpublished observation). Table 1 data also showed that the decreases in all antioxidant enzyme activities of EcV incubated cultures were not noted when the same dose of the venom was incubated with fibroblast sonicates. This observation coupled with the fact that all enzyme activities underwent reductions of similar magnitude ranging from 30–45% of control values, suggests that EcV does not directly act at the enzyme molecules but rather at the cellular level and may be of mediated nature. Similar findings that further support this hypothesis were previously demonstrated by us where all mitochondrial TCA cycle enzyme activities underwent 50–60% reductions of control values upon incubation of fibroblast cultures with Walterinnesia aegyptia venoms,[27] and where cytosolic phosphofructokinase and mitochondrial citrate synthase activities were decreased by ~62% upon incubation of the cells with crude EcV.[17] In yet another previous study,[43] we showed that incubation of fibroblast cultures with increasing concentrations of crude Cerastes gasperetti venom for different time periods caused inhibition of citrate synthase and stimulation of creatine kinase activities. These effects exhibited a saturation kinetics phenomenon with Km values of the venom required to produce half maximal effects equal to 0.22 and 1.3 µg for creatine kinase and citrate synthase respectively, and were achieved in ~1 h. In the present study, Figure 1 data revealed that incubation of the cultures with EcV concentrations equivalent to 1.0, 1.5 and 2.5 µg ml–1 MEM caused loss of cell viability in a dose dependent fashion and was achieved in less than 4 h. Furthermore, data regarding the kinetics of the inhibiting effect of EcV on antioxidant enzyme activities (submitted separately for publication) also indicated a saturation kinetics phenomenon. All the above findings further point to the possibility that venom proteins could execute their effects by binding to cellular and/or mitochondrial receptors, the mechanisms of which need elucidation.
In addition to the significant reductions in antioxidant enzymes activities, results of the present study showed that EcV-incubated cultures exhibited equally significant concurrent increases in the generation rates of H2O2, LPO and SOA; all of which are potent oxidants (Table 2). Moreover, whereas the levels of the antioxidant polypeptide GSH in the venom-incubated cells were significantly decreased, there were parallel equally significant increases in the levels of GSSG, leading to a lowered GSH/GSSG ratio. The oxidant level increases were similar in magnitude to the decreases noted in antioxidant enzyme activities and approximately equaled ~52, 44, 54 and 33% of control levels for H2O2, LPO, SOA and GSSG respectively (p < 0.0001 for all comparisons). Similarly, the GSH/GSSG ratio was lowered by ~50% of the control cultures value. Although the above findings are in broad agreement with those obtained by other workers,[22–26] such reports investigated few antioxidant and oxidant parameters using venoms of Echis pyramidum, Echis ocellatus and the cobra Naja haje injected into mice. In contrast, the present study investigated the effect of EcV on a comprehensive range of antioxidants and their corresponding oxidants using in vitro maintained human tissue. To the best of our knowledge such a study has not been undertaken before. What further characterizes our study in that the observed decreases in antioxidant activities and levels and the concurrent increases in the generation rates of the corresponding oxidants were of the same magnitude. Other results unique to the present study (Figure 2) revealed that the expression levels of the hsGPx, hsCAT, hsSOD and hsGR genes, encoding antioxidant enzyme activity in EcV-incubated fibroblast cultures, underwent highly significant downregulation by ~56, 48, 58 and 53% for each gene respectively compared to the expression levels documented in control cultures (p < 0.001 for all comparisons). Furthermore, there was close correlation between the level of reduction in each of the investigated antioxidant enzyme activities and the percentage downregulation of its respective gene expression. This finding further indicates that EcV does not act directly on the enzyme molecules but rather at the cellular or compartmental levels. The observed depletion of all antioxidant enzyme activities in EcV-incubated cells and their lowered gene expression levels could have been a result of DNA damage caused by the increased generation of H2O2, SOA and LPO, thus leading to the accumulation of enzyme substrates and downregulation of transcription and translation processes. In this context increased levels of SOA have been shown to activate key cellular hallmark events including mitochondrial alterations and DNA damage,[21,44] thus triggering apoptosis. Furthermore, SOD loss has been reported to induce the phosphorylation of a DNA damage marker (γ – H2AX), and upregulation of p21, a target gene of p 53 in fibroblasts.[45] Equally important is the noted increased generation of H2O2 in the EcV incubated cells, which interacts with SOA leading to the formation of the more reactive hydroxyl radicals [46] that react with purines and pyrimidines causing DNA damage [44] and subsequent lowered gene expression of the antioxidant enzymes.
Antioxidant enzyme activity and oxidant generation rates have been postulated to be useful in estimating the level of oxidative damage.[19,21] It is evident from the present results that the fine balance normally maintained between antioxidant activity and oxidant generation has shifted in favor of oxidative reactions, causing OS in EcV-incubated fibroblasts. It is also known that cells recycle GSH via the redox cycle, thus making the latter an essential defense mechanism for the maintenance of homeostasis and redox balance and the prevention of lipid peroxidation and depletion of cellular thiols.[47] In this cycle, GPx catalyzes the decomposition of H2O2 and lipid peroxides to water and results in the production of GSSG which is reduced back to GSH by GR, thus maintaining a normally high GSH/GSSG ratio. The present results indicated very significant decreases in GSH and parallel equally significant increases in GSSG of EcV-incubated cells compared to controls (p < 0.001), thus producing a significantly lowered GSH/GSSG ratio (Table 2) and causing pronounced OS. Furthermore, there was a very significant reduction in GR activity (Table 1). The drop in GSH levels of the venom-incubated cells could have been caused by the pronounced decrease in GR activity or as a result of GSH reacting directly with excessively generated H2O2 thus leading to increased GSSG production. It has also been documented that GSH levels can be regulated by its de novo synthesis catalyzed by γ-glutamate cysteine ligase and glutathione synthase.[19] Hence, the noted drop in GSH levels of EcV-incubated cells could have been a result of the venom acting to lower the gene expression levels and consequently the activities of these enzymes.
Conclusion
The present study provided evidence of significant decreases in antioxidant enzyme activities in EcV-incubated cells. The concurrent significantly increased generation of major oxidants and subsequent OS, also demonstrated here, could have incurred downregulation in the expression levels of the investigated antioxidant genes.
Responsible Editor Amin Bredan, VIB Inflammation Research Center & Ghent University, Belgium.
Funding Statement
This project was financially supported by King Saud University Vice Deanship of Research Chairs
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Tilmisany AK, Osman H. Snake, Scorpion venom and envenomation. A review. JKAUH Med Sci. 1993;3:11–10. [Google Scholar]
- Al- Asmari A, Khan AA, Manthiri RA. Chemical fingerprinting of Saudi Arabian snake venoms using gel filtration chromatography. Biomed Res. 2014;25:138–140. [Google Scholar]
- Al-Sadoon MK. Survey of the reptilian fauna of the Kingdom of Saudi Arabia. I. The snakes fauna of the Central Region. J King Saud Univ 1A Science. 1989;1:53–61. [Google Scholar]
- Cherifi F, Laraba-Djebrai F. Isolated biomolecules of pharmacological interest in hemostasis from Cerastes cerastes venom. J Venom Anim Toxins Incl Trop Dis. 2013;19:11–18. doi: 10.1186/1678-9199-19-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmy TR, Hemmaid KZ. Histological and histochemical alterations in the liver following intramuscular injection with a sublethal dose of the Egyptian cobra venom. J Nat Toxins. 2000;9:21–32. [PubMed] [Google Scholar]
- Al-Sadoon MK, Abdel-Moneim AE, Bauomy AA. Histochemical and Biochemical effects induced by LD50 of Cerastes cerastes gasperetti crude venom in mice. Life Sci J. 2013;10:810–817. [Google Scholar]
- Zuliani JP, Fernandes CM, Zamuner SR. Inflammatory events induced by Lys-49 and Asp-49 phospholipase A2 isolated from Bothops aseper snake venom: role of catalytic activity. Toxicon. 2005;45:335–346. doi: 10.1016/j.toxicon.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Gutierrez JM, Rucavado A. Snake venom metalloproteinases: their role in the pathogenesis of local tissue damage. Biochimie. 2000;82:841–850. doi: 10.1016/s0300-9084(00)01163-9. [DOI] [PubMed] [Google Scholar]
- Fox JW, Serrano SMT. Snake toxins and haemostasis. Toxicon. 2005;45:951–1181. [Google Scholar]
- Serrano SMT, Shannon JD, Wang D. A multifaceted analysis of Viper snake venoms by two-dimensional gel electrophoresis: an approach to understanding venom proteomics. Proteomics. 2005;5:501–510. doi: 10.1002/pmic.200400931. [DOI] [PubMed] [Google Scholar]
- Mackessy SP. Handbook of venoms and toxins of reptiles. Boca Raton (FL): CRC Press; 2008. [Google Scholar]
- Annobil SH. Complications of Echis Coloratus snake bites in the Asir region of Saudi Arabia. Ann Trop Paediatr. 1993;13:39–44. doi: 10.1080/02724936.1993.11747623. [DOI] [PubMed] [Google Scholar]
- Schulchyaska-Castle H, Dvilanskg A, Keynau A. Echis Coloratus bites: clinical evaluation of 42 patients. A Retrospective Study. Isr J Med Sci. 1986;22:880–884. [PubMed] [Google Scholar]
- Okuda D, Nozaki C, Sekiya F. Comparative biochemistry of disintegrins isolated from snake venom: consideration of the taxonomy and geographical distribution of snakes in the genus Echis . J Biochem. 2001;129:615–620. doi: 10.1093/oxfordjournals.jbchem.a002898. [DOI] [PubMed] [Google Scholar]
- Al-Shammari AM, Khan SU, Al-Sadoon MK. Biochemical characterisation of the Pyramid Viper, Echis pyramidum venom. Pakistan J Zool. 2013;45:1741–1749. [Google Scholar]
- Wahby AF, Abdel-Aty AM, El-Kady EM. Purification of hemorragic SVMPs from venom of three vipers of Egypt. Toxicon. 2012;59:329–331. doi: 10.1016/j.toxicon.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Al-Saleh SS, Ghneim HK, Khan SU. Separation and purification of Echis Coloratus venom and some biological and biochemical effects of the proteins. Cell Biochem Funct. 2002;20:153–162. doi: 10.1002/cbf.962. [DOI] [PubMed] [Google Scholar]
- Al-Saleh SS, Ghneim HK, Khan SU. The effect of crude and purified Cerastes vipera venom protein fractions on respiratory chain function in cultured human fibroblasts. Cell Physiol Biochem. 2003;13:315–320. doi: 10.1159/000074547. [DOI] [PubMed] [Google Scholar]
- Volko M, Leiter D, Moncol J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- Aboul-Soud MAM, Al-Othman AM, El-Desoky GE. Hepatoprotective effect of vitamin E/selenium against malathion-induced injuries on the antioxidant status and apoptosis-related gene expression in rats. J Toxicol Sci. 2011;36:285–296. doi: 10.2131/jts.36.285. [DOI] [PubMed] [Google Scholar]
- Dousset E, Carrega L, Steinberg JG. Evidence that free radical generation occurs during scorpion envenomation. Comp Biochem Physiol C. 2005;140:221–226. doi: 10.1016/j.cca.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Al- Asmari A, Al Moutaery K, Manthari RA. Time-course of lipid peroxidation in different organs of mice treated with Echis pyramidum snake venom. J Biochem Mol Toxicol. 2006;20:93–95. doi: 10.1002/jbt.20121. [DOI] [PubMed] [Google Scholar]
- Al- Asmari A, Khan HA, Banah FA. Serum biomarkers of acute hepatotoxicity of Echis pyramidum snake venom in rats. Int J Clin Exp Med. 2015;8:1376–1380. [PMC free article] [PubMed] [Google Scholar]
- Onyeama HP, Ebong PE, Eteng MU. Evaluation of the effects of Calliandra portoricensis extracts on oxidative stress enzymes in Wistar rats challenged with venom of Echis ocellatus . J App Pharmaceut Sci. 2012;2:199–202. [Google Scholar]
- Tohamy AA, Mohamed AF, Abdel- Moneim AE. Biological effects of Naja haje crude venom on the hepatic and renal tissues of mice. J King Saud Univ Science. 2014;26:205–212. [Google Scholar]
- Ghneim HK, Al-Sheikh YA, Aboul- Soud MAM. The effect of Walterinnesia aegyptia venom proteins on TCA cycle activity and mitochondrial NAD+-redox state in cultured human fibroblasts. Biomed Res Int. 2015:10. doi: 10.1155/2015/738147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to protiferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Southon A, Burke R, Norgate M. Copper homeostasis in Drosophila melanogaster S2 cells. Biochem J. 2004;383:303–309. doi: 10.1042/BJ20040745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFontaine S, Firth SD, Lockhart PJ. Functional analysis and intracellular localisation of the human menkes protein (MNK) stably expressed from a cDNA construct in Chinese hamster ovary cells (CHO-K1) Hum Mol Genet. 1998;7:1293–1300. doi: 10.1093/hmg/7.8.1293. [DOI] [PubMed] [Google Scholar]
- Ghneim HK, Al-Sheikh YA. Effect of selenium supplementation on glutathione peroxidase and catalase activities in senescent cultured human fibroblasts. Ann Nutr Metab. 2011;59:127–138. doi: 10.1159/000334069. [DOI] [PubMed] [Google Scholar]
- Al-Sheikh YA, Ghneim HK. The effect of micronutrients on superoxide dismutase in senescent fibroblasts. Cell Biochem Funct. 2011;29:384–393. doi: 10.1002/cbf.1761. [DOI] [PubMed] [Google Scholar]
- Ghneim HK, Alshebly MM. Biochemical markers of oxidative stress in Saudi women with recurrent miscarriage. J Korean Med Sci. 2016;31:98–105. doi: 10.3346/jkms.2016.31.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherghel D, Mroczkowska S, Quin L. Reduction in blood glutathione levels occurs similarly in patients with primary-open angle or normal tension glaucoma. Invest Ophthalmol Vis Sci. 2013;54:3333–3339. doi: 10.1167/iovs.12-11256. [DOI] [PubMed] [Google Scholar]
- Zhou M, Diwu Z, Panchuk-Voloshina N. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide; application in detecting the activity of phagocyte NADH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- Johnston RB, Jr, Keele BB, Jr, Misra HP. The role of superoxide venom generation in phagocytic bactericidal activity. Studies with normal and chronic granulomatous disease leukocytes. J Clin Invest. 1975;55:1357–1372. doi: 10.1172/JCI108055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghneim HK, Al-Sheikh YA, Alshebly MM. Superoxide dismutase activity and gene expression levels in Saudi women with recurrent miscarriage. Mol Med Rep. 2016;13:2606–2612. doi: 10.3892/mmr.2016.4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh SA, Maraga AD, Ali ME. Regional distribution of superoxide dismutase activity in human placenta and its correlation with lipid peroxidation. Jordan J Biol Sci. 2010;3:125–132. [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Ghneim HK. Enzymatic variations related to glucose and glycogen catabolism in serially subcultured human fibroblasts. Cell Physiol Biochem. 1994;4:44–56. [Google Scholar]
- Ghneim HK, Al-Sheikh YA. The effect of aging and increasing ascorbate concentrations on respiratory chain activity in cultured human fibroblasts. Cell Biochem Funct. 2010;28:283–292. doi: 10.1002/cbf.1653. [DOI] [PubMed] [Google Scholar]
- Ghneim HK, Al-Shammary FJ. The kinetics of the effect of Cerastes cerastes gasperetti venom on citrate synthase and creatine kinase activities. Med Sci Res. 1993;21:229–231. [Google Scholar]
- Agarwal A, Allamaneni S. Oxidants and antioxidants in human fertility. Middle East Fertility Soc J. 2004;9:187–197. [Google Scholar]
- Lei XG, Zhu JM, McClung JP. Mice deficient in Cu, Zn-superoxide dismutase are resistant to acetaminophen toxicity. Biochem J. 2006;399:649–654. doi: 10.1042/BJ20060784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer JP. The haber weiss reaction and mechanism of toxicity. Toxicol. 2000;149:43–50. doi: 10.1016/s0300-483x(00)00231-6. [DOI] [PubMed] [Google Scholar]
- Couto N, Malys N, Gaskell SJ. Partition and turnover of glutathione Reductase from Saccharomyces cerevisiae: a proteomic approach. J Proteom Res. 2013;12:2885–2894. doi: 10.1021/pr4001948. [DOI] [PubMed] [Google Scholar]