

ABSTRACT
The currently marketed antibody-drug conjugates (ADC) destabilize microtubule assembly in cancer cells and initiate apoptosis in patients. However, few tumor antigens (TA) are expressed at high densities on cancer lesions, potentially minimizing the therapeutic index of current ADC regimens. The peptide/human leukocyte antigen (HLA) complex can be specifically targeted by therapeutic antibodies (designated T cell receptor [TCR]-like antibodies) and adequately distinguish malignant cells, but has not been the focus of ADC development. We analyzed the killing potential of TCR-like ADCs when cross-linked to the DNA alkylating compound duocarmycin. Our data comprise proof-of-principle results that TCR-like ADCs mediate potent tumor cytotoxicity, particularly under common scenarios of low TA/HLA density, and support their continued development alongside agents that disrupt DNA replication. Additionally, TCR-like antibody ligand binding appears to play an important role in ADC functionality and should be addressed during therapy development to avoid binding patterns that negate ADC killing efficacy.
KEYWORDS: Antibody drug conjugate, cancer, duocarmycin, human leukocyte antigen, immunotherapy, TCR-like antibody
Abbreviations
- ADC
antibody drug conjugate
- ADCC
antibody-dependent cell-mediated cytotoxicity
- CDC
complement-dependent cytotoxicity
- CEA
carcinoembryonic antigen
- HLA
human leukocyte antigen
- MIF
macrophage migration inhibitory factor
- SAP
saporin
- TA
tumor antigen
- TCR
T cell receptor
Introduction
Numerous unmodified antibodies are marketed for the treatment of patients with cancer. This antibody format generally requires accessory immune components, such as complement and natural killer cells, to engage the Fc region of a bound antibody and destroy tumor cells through complement-dependent cytotoxicity (CDC) or antibody-dependent cell-mediated cytotoxicity (ADCC), respectively.1 Select antibody molecules may also abrogate downstream signaling cascades that are important in cell growth by interacting with surface-exposed targets such as growth factor receptors.2 However, many external factors govern the clinical success of this targeted approach, including issues such as solid tumor penetrance and the moderate-to-high antibody binding needed to effectively elicit anti-tumor immunologic accessory responses or signaling pathway disruptions.3 Additionally, with respect to CDC and ADCC, distinct Fc receptor polymorphisms in patients have been shown to dictate the degree of responsiveness to an antibody-based strategy.4,5
Engineered molecules such as antibody-drug conjugates (ADC) represent a conceptual improvement over the use of unmodified antibody molecules. Instead of relying on external factors to mediate cancer cell destruction, the antibody itself delivers a cytotoxic payload (i.e., small molecule drug) to tumor cells following endocytosis.6 Cytotoxic drugs are stably attached to the antibody molecule via a short linker sequence and are released during endosome/lysosome fusion pathways, subsequently disrupting microtubule polymerization or DNA replication.7 Several ADCs (brentuximab vedotin, ado-trastuzumab emtansine) are currently marketed and have met with degrees of success in treating malignant disease, albeit with a narrow therapeutic index.8-10 Efforts are now underway to further improve clinical efficacy by optimizing ADC properties such as systemic stability, antibody-linker chemistry, off-target cytotoxicity, and drug potency.11
Tumor antigen (TA) selection (with a preferred internalization profile) represents an additional area for therapeutic index improvement within the ADC field, but the selection is limited because of the need for sufficient target availability on cancer lesions to deliver adequate drug payloads.6,11 Of the more than 50 ADCs in clinical development, none binds a given peptide/human leukocyte antigen (HLA) expressed by tumor cells.8 The peptide/HLA is a unique targeting moiety since it is expressed by nucleated cells within the body and can phenotypically distinguish normal and malignant cells.12 Indeed, considerable work within the area of immunotherapy has incorporated vaccines and genetic engineering to route effector CD8+ T cells to engage and destroy tumor cells through distinct peptide/HLA profiles.13 Therapeutic antibodies (designated T cell receptor [TCR]-like antibodies) can also be raised to specifically bind a given peptide/HLA complex, much like a TCR engages the peptide/HLA.14 Our group and others have successfully developed high affinity TCR-like antibodies against several tumor targets across different HLA alleles and reported favorable anti-tumor properties and protection in preclinical models.15-17 In an extension of this work, we provide here proof-of-principle that TCR-like antibodies effectively function as small molecule drug carriers, particularly when delivering payloads to cancer cells that disrupt DNA replication (via duocarmcyin analogs). The TCR-like ADCs under study were capable of eliciting destruction of breast and colorectal cancer lines and diminishing tumor cell growth at physiologically relevant peptide/HLA surface-bound levels (350–2,000 copies/cell).18,19 Specific TCR-like antibody and peptide/HLA interactions also modulated ADC killing potential, indicating that, regardless of drug conjugate potency, TCR-like antibody docking to defined regions of the peptide/HLA severely dampened ADC cytotoxicity. Taken together, these findings provide support for the continued preclinical development of TCR-like ADCs, with a long-term goal of clinical evaluation in patients with cancer.
Results
TCR-like antibodies deliver an immunotoxin to cancer cells but target copy number affects killing efficacy
Our group has reported on the utility of unmodified TCR-like antibodies to specifically bind peptide/HLA, mediate CDC/ADCC in vitro, and provide protection against tumorigenic growth in vivo.20,21 Interestingly, TCR-like antibodies engage their specific target and become internalized over a short period of time,16,22 but we have not previously focused on the killing potential of TCR-like antibodies conjugated to toxins or small molecule drugs. To initially assess cytotoxicity, the TCR-like antibodies YLL-Ab and FLS-Ab (Supplementary Table 1) were indirectly labeled with the ribosomal inhibiting protein saporin (see Materials and Methods) and incubated alongside breast (MDA-MB-231 (Fig. 1A)) and colon adenocarcinoma (SW620 (Fig. 1B)) cell lines, which express cognate peptide/HLA-A2 ligands (Fig. S1). Although saporin levels remained constant in treated wells, increasing concentrations of YLL-Ab and FLS-Ab enhanced tumor cell death, suggesting a direct correlation with killing efficacy and internalized TCR-like immunotoxin complexes and payload release. These data also confirmed our earlier findings of TCR-like antibody internalization upon peptide/HLA binding.16,22 Tumor cell killing was not a result of unmodified TCR-like antibody binding since no appreciable cell killing was observed with TCR-like antibody concentrations up to 2 µg/ml in vitro (data not shown).
Figure 1.

Surface-bound peptide/HLA-A2 availability affects TCR-like antibody killing potential against breast and colon cancer lines. MDA-MB-231 (A) and SW620 (B) cells (5×103) were cultured in the presence of 100 ng Mab-Zap reagent (goat anti-mouse saporin conjugate) alongside 10-fold dilutions of murine TCR-like (FLS-Ab, YLL-Ab), BB7.2 (anti-HLA-A2 control), or isotype control antibodies. (C) MDA-MB-231 cells were either left unpulsed or loaded with 1 µg/ml and 10 µg/ml KIF peptide for 3 hours at 37 °C. Unbound peptide was washed away and cells co-cultured with 10-fold dilutions of the TCR-like antibody KIF-Ab + 100 ng Mab-Zap. Plates were incubated for 3–5 d at 37 °C with 5% CO2, and target cell viability was determined through conversion of an MTS tetrazolium compound to a soluble formazan product. Specific viability of target cells was calculated by dividing background-corrected specific antibody/Mab-Zap values from isotype control/Mab-Zap absorbance readings at 490 nm. The resulting data was fit to a 4-parameter curve and EC50 values determined for each antibody treatment. In parallel experiments, relevant peptide/HLA-A2 target numbers were assessed on tumor lines using the QIFIKIT assay (Dako) (outlined in the Materials and Methods section) and reported as relative antibody bound per cell. Bars, ± SD.
The MDA-MB-231 and SW620 cell lines were further analyzed for peptide/HLA copy number expression by flow cytometry (QIFIKIT assay) relative to YLL-Ab and FLS-Ab binding (Fig. S1). Interestingly, cell killing for a high target cell line such as MDA-MB-231 appeared linked to endogenous copy number expression. For example, at a 1 µg/ml TCR-like antibody/saporin concentration, cell viability was 39.08% and 51.63% for YLL-Ab (80,000 relative antibody bound/cell) and FLS-Ab (38,000 relative antibody bound/cell), respectively (Fig. 1A). Such high frequencies of FLS-Ab and YLL-Ab positive MDA-MB-231 cells were generally expected since the FLS and YLL peptides represent a significant fraction of surface-exposed peptide/HLA-A2 complexes in cells when detected by mass spectrometry.20,24 The antibody BB7.2 was also used to stain all available HLA-A2 molecules at 475,000 copies per cell, and BB7.2/saporin (1 µg/ml) binding/internalization achieved the highest level of cell killing at 23.48%. SW620 cells were determined to endogenously express low levels of YLL-Ab and FLS-Ab-specific peptide/HLA molecules. Again, BB7.2 staining revealed the highest HLA-A2 copy number expression and killing through saporin conjugation at 45.93% (1 µg/ml) (Fig. 1B). SW620 destruction was not observed using FLS-Ab (1,000 relative antibody bound/cell), while minimal YLL-Ab-mediated killing was attained with target levels at 2,000 (∼80% viability), suggesting a sensitivity threshold for inhibiting ribosomal function and initiating cell death using TCR-like antibody immunotoxins. It is conceivable that the flow-based QIFIKIT method overestimated total peptide/HLA-A2 complexes (via multimerized anti-mouse FITC labeling) in cases of moderate-to-high level expressing cells such as MDA-MB-231, given that typical estimates for surface-exposed HLA-A2 nears 100,000 molcules per cell for some lines.23 Though, other studies have calculated HLA-A2 levels as high as 750,000 copies per cell.23 As low target copy number is a major focus of the current work, we attempted to confirm relative levels of bound FLS-Ab and YLS-Ab using the flow-based BD Quantibrite™ assay (BD Biosciences), which relies upon a distinct phycoerythin (PE)-conjugated anti-mouse antibody (light chain-specific) and PE-conjugated reference standard. Overall, both the QIFIKIT and BD Quantibrite™ methods predicted approximately equal peptide/HLA-A2 values for the SW620 cell line (data not shown).
To further study the role of target copy number and minimize extraneous cell line variability, MDA-MB-231 cells were peptide-pulsed with 1 and 10 µg/ml of the HLA-A2 peptide KIF over a 3-hour period. Unpulsed MDA-MB-231 cells do not exhibit observable reactivity against the KIF-Ab TCR-like antibody (Fig. 1C). However, as detailed in Fig. 1C, KIF-Ab bound tumor cells in a concentration-dependent manner following increasing KIF peptide loading conditions (range of ∼21,000 to 40,000 relative antibody molecules per cell). Saporin was again used as a secondary immunotoxin strategy to peptide-pulsed MDA-MB-231 cells. As more tumor cell peptide/HLA targets were specifically bound by KIF-Ab, cell viability decreased, although EC50 values were similar at 0.130 nM and 0.119 nM for 1 µg/ml KIF and 10 µg/ml KIF peptide-loaded cells, respectively. Under peptide-pulsing conditions of 1 µg/ml KIF, ∼21,000 molcules of KIF-Ab bound MDA-MB-231 cells and reduced viability near 80% through inhibition of protein synthesis (Fig. 1C). Once KIF-Ab engaged 40,000 molcules per tumor cell, target viability dropped to 63.05% (Fig. 1C). Overall, these results demonstrate that TCR-like antibodies can function to directly disrupt tumor cell viability with linked immunotoxins, but killing potential is limited to high surface accessible peptide/HLA.
TCR-like ADCs represent feasible targeted strategies against cancer cell growth
To circumvent the seeming requirement for high target expression, we incorporated a strategy to increase the killing potency of conjugated TCR-like antibodies by using a duocarmycin analog that alkylates DNA. In similar experiments outlined in Fig. 1, the TCR-like antibodies YLL-Ab and FLS-Ab were indirectly labeled with duocarmycin using a secondary anti-mouse ADC reagent (see Materials and Methods) and co-cultured with MDA-MB-231 and SW620 cells for a period up to 5 d in vitro (Panel I, Fig. 2A/2B). Tumor cell viability was generally reduced at EC50 and saturating (1 µg/ml) TCR-like ADC doses (Panel I, Fig. 2A/2B) compared with the killing potential of TCR-like immunotoxin conjugates (Fig. 1). TCR-like ADC EC50 values between MDA-MB-231 and SW620 cell lines were also impressive with concentrations in the sub-nanomolar range, which is typical of ADCs approved by the US Food and Drug Administration (FDA) and under clinical development.25 Additionally, sufficient SW620 cell killing was achieved when targeting low endogenous levels of surface-bound peptide/HLA-A2 molecules (∼1,000–2,000 bound TCR-like antibodies per cell) as documented in Panel I, Fig. 2B, with cancer cell cytotoxicity levels exceeding 30% and 65% using conditions with 1 µg/ml FLS-Ab and YLL-Ab, respectively. The nature of using secondary ADCs as a proof-of-principle approach did not appear to drastically bias tumor cell destruction since primary ADCs using duocarmycin similarly affected the viability of both MDA-MB-231 and SW620 cells (Panel II, Fig. 2A/2B). Yet, secondary FLS and YLL ADCs tended to outperform their primary ADC counterparts when target cell cytotoxicity was directly compared at equivalent log[nM] concentrations. These results may highlight the potential limits of our experimental system that incorporates a secondary ADC approach (which could multimerize TCR-like antibodies and increase target killing) or suboptimal primary ADC conjugation scheme (that produces a TCR-like ADC with heterogeneous duocarmycin distribution). To overcome such variables and achieve maximum benefit, we are currently exploring the cytotoxicity of TCR-like antibodies engineered for site-specific conjugation of potent drugs.
Figure 2.
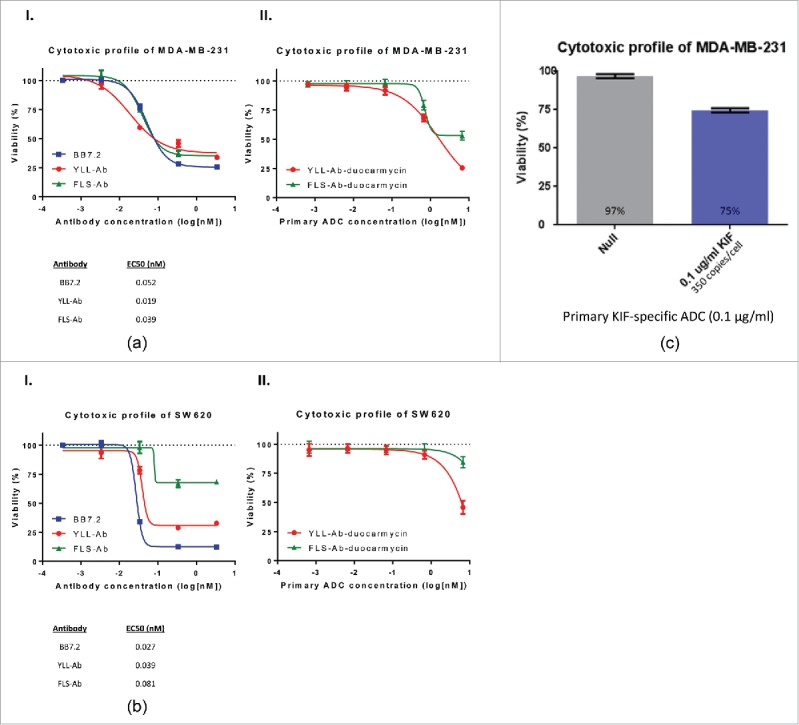
TCR-like ADCs effectively mediate killing of cancer cells expressing high and low peptide/HLA-A2 targets. As described in Fig. 1, plated MDA-MB-231 (high endogenous peptide/HLA-A2 target line) (A) and SW620 (low endogenous peptide/HLA-A2 target line) (B) cells were exposed to 10-fold dilutions of TCR-like, BB7.2, or isotype control antibodies alongside 100 ng of a duocarmycin-conjugated anti-mouse IgG Fc reagent (Panel I). Each tumor cell line was also subjected similarly to co-culture with FLS-Ab and YLL-Ab directly conjugated to duocarmycin (Panel II) (see Materials and Methods). (C) MDA-MB-231 cells were pulsed with 0.1 µg/ml KIF peptide (representing 350 peptide/HLA-A2 copies per cell) and incubated with duocarmycin-conjugated isotype or KIF-Ab antibodies. Percent target viability was calculated after a culture period of 3–5 d and fit to a 4-parameter curve and EC50 values subsequently determined. Bars, ± SD.
The findings in Fig. 2A and B were subsequently extended to determine the minimum surface-bound peptide/HLA-A2 content required to initiate target cell death in vitro using a TCR-like ADC. KIF-Ab was linked to duocarmycin and incubated alongside MDA-MBA-231 cells pulsed with the KIF peptide. As indicated in Fig. 2C, ∼25% of MDA-MB-231 cells harboring as little as 350 KIF/HLA-A2 molecules could be killed in culture. In all, select TCR-like ADCs were adept at minimizing tumor cell growth when targeting physiologic levels of peptide/HLA complexes (< 1,000 copies per cell) routinely observed in clinical specimens.18,19 These data also provide material support for the clinical development of TCR-like antibodies conjugated to potent small molecule drugs such as DNA alkylating agents.
TCR-like antibody target docking affects ADC potential
Although duocarmycin-conjugated TCR-like antibodies facilitated effective tumor cell lysis, target copy number appeared to still affect the overall killing efficacy of TCR-like ADCs, particularly when specific peptide/HLA-A2 expression was endogenously low (see Fig. 2B and C). However, additional factors seemed to play a role in the capacity of duocarmycin-conjugated TCR-like antibodies to mediate tumor cell clearance. For example, although ∼2,000 YLL/HLA-A2 molecules were accessible on the surface of SW620 cells, YLL-Ab-duocarmycin was capable of achieving cytotoxic levels similar to the pan HLA-A2 antibody BB7.2, which binds 37,000 HLA-A2 molecules per cell (Fig. 1B). Likewise, as shown in Fig. 2B, discrepancies were observed in SW620 cytotoxicity between YLL-Ab-duocarmycin and FLS-Ab-duocarmycin, even though the pattern of specific peptide/HLA-A2 expression differed by 2-fold (Fig. 1B). Previous attempts using TCR-like antibody saporin-conjugates (Fig. 1A and B) did not achieve such differences in target killing. We, therefore, sought to evaluate additional TCR-like antibody parameters that could potentially influence ADC function. Our group generated 6 distinct antibody clones (designated YLSG-Ab1–6) against the carcinoembryonic antigen (CEA)-specific HLA-A2 peptide YLS (Supplementary Table 1). To impart TCR-like antibody reactivity, the murine breast cancer line 4T1 was first stably transfected with a single chain trimer construct26 encoding the YLS/HLA-A2 molecule, referred to hereafter as 4T1-CEA (see Materials and Methods). Although parental 4T1 cells did not exhibit YLSG-Ab1–6 binding, 4T1-CEA cells endogenously expressed high YLS/HLA-A2 copies per cell and demonstrated similar mean fluorescent intensity profiles by flow cytometry following YLSG-Ab1–6 staining (data not shown). 4T1-CEA cells were plated in culture alongside various concentrations of each YLSG-Ab clone (0.0001–1 µg/ml) and 100 ng of secondary anti-mouse duocarmycin reagent. Due to an overexpression of YLS/HLA-A2 complexes on 4T1-CEA cells, each YLSG ADC incited target lysis (Fig. 3A). However, YLSG-Ab3,5 failed to adequately elicit 4T1-CEA cell killing as ADCs at both 0.1 and 1 ug/ml concentrations (in comparison to YLSG-Ab1,2,4) (Fig. 3A) even though YLSG-Ab3,5 were functional antibodies based on lysis of 4T1-CEA cells through CDC (Supplementary 2). Additionally, antibody isotype differences could not alone explain ADC potential variability among the YLSG-Ab clones (YLSG-Ab1,2,4,6 = IgG1; YLSG-Ab3,5 = IgG2b) since BB7.2 is an IgG2b antibody and effectively cleared 4T1-CEA cells in culture. We, therefore, determined YLSG-Ab binding affinity to YLS/HLA-A2 via surface plasmon resonance (SPR), but no obvious pattern emerged to explain the killing dynamics of YLSG-Ab3,5 (particularly in reference to YLSG-Ab1,2) as outlined in Table 1. Interestingly, despite levels of > 90% purity, the ADC profile of YLSG-Ab4,6 TCR-like antibodies may be partly described by a combination of reduced antibody Ka and T1/2 properties, respectively, since 4T1-CEA cytotoxicity was directly proportional to antibody concentration.
Figure 3.

TCR-like antibody/target docking influences ADC potency. (A) 4T1-CEA cells endogenously express high copy numbers of the CEA-specific YLS/HLA-A2 molecule, which is specifically reactive to 6 distinct TCR-like antibody clones (designated YLSG-Ab1–6). Five thousand 4T1-CEA cells were incubated for 3 d with various concentrations of YLSG-Ab1–6 antibodies + 100 ng of a duocarmycin-conjugated anti-mouse IgG Fc reagent. Isotype-corrected viability was then determined using an MTS assay as described in Fig. 1. (B) T2-A2 cells were pulsed for 3 hrs with an HLA-A2 CEA-specific peptide library consisting of native and alanine-substituted peptides at indicated positions. Cells were washed and stained with PE-labeled YLSG-Ab and subsequently analyzed for positivity by flow cytometry. The degree of YLSG-Ab binding to altered HLA-A2 peptides was compared with signals achieved with T2-A2 cells bearing the control native YLS peptide as demonstrated by representative flow histograms and summarized in table format. (+): No observable effect on YLSG-Ab clone binding; (+/−): Partial loss of YLSG-Ab clone binding (i.e., ≤ 50% relative to native peptide signal); (−): Substantial loss of YLSG-Ab clone binding (i.e., ≥ 50% relative to native peptide signal). Bars, ± SD.
Table 1.
Specific affinity determination of YLSG-Ab clones to YLS/HLA-A2.
| TCR-like antibody | Ka (M−1S−1) | Kd (M−1S−1) | KD (nM) | T1/2 (sec) |
|---|---|---|---|---|
| YLSG-Ab1 | 1.11×104 | 1.00×10−7 | 34 | 1856 |
| YLSG-Ab2 | 1.20×104 | 6.28×10−4 | 52 | 1103 |
| YLSG-Ab3 | 2.67×103 | 8.04×10−4 | 301 | 862 |
| YLSG-Ab4 | 4.96×103 | 2.80×10−4 | 58 | 2475 |
| YLSG-Ab5 | 1.11×104 | 1.48×10−3 | 134 | 2392 |
| YLSG-Ab6 | 2.96×104 | 1.00×10−7 | 188 | 125 |
COOH chips specific to the SensiQ plasmon resonance system were first activated with EDC/NHS and subsequently immobilized with protein A/G. Chips were then exposed to individual YLSG-Ab clones, washed with Hank's Balanced Saline-Tris EDTA, and subjected to various concentrations of YLS/HLA-A2 folded monomers. Phosphoric acid (10 mM) was used to regenerate chips between YLS/HLA-A2 injection steps. Antibody:peptide/HLA-A2 binding kinetics were plotted and affinity determined using Qdat software. Ka: association rate; Kd: dissociation rate; KD: dissociation rate constant; T1/2: analyte half-life dissociation.
A panel of alanine substituted YLS peptides were next generated at specific residues (1 and 3–8) (see Fig. 3B inset) to analyze whether certain amino acids determined YLSG-Ab TCR-like antibody target binding. Briefly, T2-A2 cells were pulsed with native and altered CEA peptides, incubated with YLSG-Ab clones, and assessed for positive events by flow cytometry using a fluorophore-conjugated secondary antibody. Positions 2 and 9 of the CEA native peptide were left unaltered as they represent anchor residues for stably binding the HLA-A2 molecule.27 As demonstrated in Fig. 3B, alanine substitutions at positions 3 and 8 did not result in observable reductions in YLSG-Ab and CEA peptide/HLA-A2 binding and, therefore, do not likely influence ADC cytotoxicity. YLSG-Ab1,2,3 target specificity was partially reduced when modifying position 1, while YLSG-Ab6 binding was affected to a degree by alanine substitutions at positions 6 and 7. Interestingly, YLSG-Ab3,4,5 experienced substantial reductions in peptide/HLA-A2 binding following T2-A2 altered CEA peptide loading. Positions 1 and 4 were critical to YLSG-Ab4 binding, while position 6 appeared required for YLSG-Ab3,5 staining. Since YLSG-Ab4 adequately performed as a TCR-like ADC (Fig. 3A), native residues at positions 1 and 4 do not likely negatively affect ADC potential. However, the reduced ability of YLSG-Ab3,5 to mediate tumor cell killing as an ADC (Fig. 3A) may be explained by a strict requirement for binding aspartic acid at position 6 of the CEA native peptide (Fig. 3B). Altogether, specific TCR-like antibody/target docking properties appear to correlate with ADC cytotoxicity in our YLSG-Ab-CEA/HLA-A2 model system.
Discussion
During the late 1950s, ADC anti-cancer benefit was first documented in preclinical models using antibodies cross-linked to agents such as methotrexate (anti-metabolite), mitomycin (DNA cross-linker), and vinblastine (anti-microtubule assembly).7 However, early ADC formats had several technical hurdles including antibody immunogenicity and reduced drug potency and drug linker stability. Gradual improvements in these basic design components led to the FDA approval of gemtuzumab ozogamicin (Mylotarg®) in 2000, with approvals of brentuximab vedotin (Adcetris®) and ado-trastuzumab emtansine (Kadcyla®) following in 2011 and 2013, respectively.6 Considerable efforts are now underway to further enhance the therapeutic index of ADCs by improving half-life and drug-linker stability in humans, as well as improving batch-to-batch homogeneity since current linker schemes rely on accessible antibody lysine or cysteine residues, causing heterogeneous preparations with poorly-defined properties.7,11 Yet, it should also be noted that current preclinical in vivo assessments do not accurately predict clinical issues such as dose-limiting toxicities, off-target effects, and efficacy (particularly in unison with an endogenous immune response), underscoring the notion that improvements within the ADC field will be inexorably linked to the reliability of our animal models.8
In addition to the above constraints, ADC strategies are greatly influenced by the tumor microenvironment. Characteristics such as cancer cell target expression, ADC penetrance/payload delivery, and drug resistance all contribute to an ADC's therapeutic index against tumorigenic growth and spread. Most ADCs under preclinical/clinical development use payloads that affect microtubule assembly (i.e., auristatin or maytansinoid classes).28 Additionally, cancer cell sensitivity clearly associates with ADC/target occupancy when correcting for variables such as inter-cell line comparisons. In one experimental system, increasing target levels in a melanoma parental line enhanced monomethylauristatin E (MMAE)-based ADC killing 100-fold.29 It is generally assumed, however, that a minimum target threshold must exist to achieve sufficient tumor cell killing with tubulin-specific ADCs.30,31 For example, Wang and colleagues designed a prostate-specific membrane antigen (PSMA) antibody conjugated to MMAE (designated PSMA-ADC) that is under clinical trial evaluation in individuals with castration-resistant, metastatic prostate cancer.31 In a series of in vitro assays, killing potency was assessed against a panel of human prostate cell lines, and the PSMA-ADC was highly effective against cells expressing more than 100,000 PSMA molecules with lower levels of ADC-specific killing against prostate lines bearing ∼10,000 copies per cell.
The human epidermal growth factor receptor (HER)2/neu has also served as an excellent target for ADCs disrupting microtubule networks within breast cancer cells due in part to its level of distribution. HER2/neu commonly exceeds 1,000,000 copies per cell on metastatic breast lesions, and ADC binding initiates internalization and drug delivery to mediate cancer protection in preclinical models.25,32 These results have extended to the clinic, with the FDA-approved ado-trastuzumab emtansine therapy significantly prolonging median progression-free and overall survival rates in breast cancer patients previously treated with first-line trastuzumab.9 Most surface-exposed TAs, however, are retained at levels several fold lower than model targets such as HER2/neu,33 possibly limiting the anti-tumor functions of ADCs counteracting microtubule polymerization. This presumed higher level requirement for target expression would also constrain the application of ADCs under clinical settings.
A potential alternative strategy against “low-to-moderate” TA expressing cells (i.e., < 10,000 copies/cell) uses drug conjugates that intercalate DNA and initiate cell-cycle arrest and apoptosis in dividing and non-dividing cells.7 Of this class, the highly potent duocarmycin analogs act by binding the minor groove of DNA and alkylating adenine residues. ADC variants linking the duocarmycins have shown excellent anti-tumor profiles in preclinical models of breast cancer,34,35 and a lead anti-HER2/neu duocarmycin-conjugated antibody (SYD985) recently entered clinical development for patients with HER2/neu-expressing tumors (ClinicalTrials.gov Identifier: NCT02277717). Interestingly, SYD985 was explored under preclinical settings using various HER2-expressing breast cancer lines.25 In a direct comparison to ado-trastuzumab emtansine (a maytansinoid ADC), SYD985 sensitization provided enhanced tumor protection against HER2/neu patient-derived xenografts graded 1–3+ by immunohistochemistry (generally 50,000–800,000 copies per cell). Ado-trastuzumab emtansine only achieved significant benefit in vivo against high-expressing HER2/neu 3+ tumors.
Once a drug's potency becomes less reliant on target expression and cell proliferative index (as noted with the duocarmycins), novel targets for ADC development can be identified and explored.28,36 Nucleated cells within the host process and present diverse peptides via the HLA class I molecule. In a series of distinct steps, intracellular proteins are first degraded via the proteasome in the cell cytosol, peptide fragments loaded into the HLA class I complex within the ER lumen, and the fully assembled molecule transported to the outer membrane.12 Under normal settings, surface-expressed peptide/HLA serves as a warning beacon to circulating immune cells such as CD8+ T and NK cells, which maintain the general health of the host. Yet, in diseased conditions such as cancer, tumor sub-types can present distinguishing TA peptide/HLA complexes to potentially “protective” cytotoxic T cells following vaccination or passively administered strategies.13 Antibodies (termed TCR-like antibodies) can also be developed against a given TA peptide/HLA complex, bind with high affinity, and elicit downstream immunologic reactions such as CDC and ADCC, which in turn mediate effective anti-tumor immune responses.17,20 There is currently considerable focus in adapting TCR-like antibodies as Fc and bispecific engineered therapies for patients with cancer.37 However, less attention has been devoted to developing TCR-like ADCs, due in part to concerns for achieving suitable cytotoxicity with low target density27,37 Yoram Reiter's group first reported on the efficacy of incorporating TCR-like antibodies to specifically bind and deliver toxins to tumor cells.15,38,39 In a series of studies, anti-tumor activity was observed using a construct that consisted of a TCR-like single-chain variable fragment fused to the Pseudomonas exotoxin A, although tumor peptide/HLA expression level and TCR-like antibody killing potential were not directly compared. Our efforts linking the ribosomal inhibitor saporin to several TA-specific TCR-like antibodies largely confirmed these previous experiments. The TCR-like immunotoxin approach, however, was generally ineffective against tumor cell lines expressing < 10,000 peptide/HLA copies per cell, limiting their scope of use to tumor lesions with high TA peptide/HLA content. Based on the degree of endogenous peptide/HLA expression18,19 and results from the current report, TA peptide/HLA molecules appear suited for ADC strategies, particularly when incorporating DNA alkylating agents. Our duocarmycin-conjugated TCR-like antibodies were capable of diminishing breast and colorectal tumor cell growth at physiologically relevant peptide/HLA levels between 350–2,000 copies per cell.
Our experimental results also suggest that TCR-like antibody target docking plays a role in ADC function. A series of distinct TCR-like antibodies were developed against folded HLA-A2 harboring the CEA-specific YLS peptide (Fig. 3). Although all TCR-like antibody clones bound the target cancer cell line 4T1-CEA at high density, YLSG-Ab3,5 performed less ideally as ADCs. A number of TCR-like antibody parameters (e.g., isotype, target affinity) (Fig. S2 and Table 1) revealed no obvious insight, but a potential pattern emerged from efforts to understand YLSG-Ab engagement of the peptide/HLA. Alanine substitutions at positions 1, 4, and 7 within the CEA peptide sequence affected the YLSG-Ab binding profiles but failed to overwhelmingly disrupt ADC activity relative to other YLSG-Ab clones (Fig. 3). However, interactions between YLSG-Ab3,5 and YLS/HLA-A2 were abrogated upon altering residue 6 of the CEA peptide, suggesting this particular docking behavior negatively influenced YLSG-Ab ADC killing. To date, several reports have analyzed the structural binding properties between TCR-like antibodies and cognate peptide/MHC ligands, although these studies only assessed similarities/differences to T cell receptor-MHC engagement.40-43 Our data introduce a previously unpublished aspect of TCR-like antibody binding, but the mechanism behind how TCR-like antibody docking influences ADC function is currently unknown and a focus of study in our laboratory. Certainly, it has been previously recognized that ADC candidacy depends to a large degree on antibody internalization kinetics (following surface protein interaction)44 since internalization rates vary even among antibody clones generated against the same target.45 However, the peptide/HLA is unique to other surface-derived receptors since the folded complex retains a short cytoplasmic domain that lacks known amino acid sorting sequences, and, therefore, constitutively internalizes several times every hour via clathrin-/adaptor-protein-independent mechanisms.46,47 Endocytic vesicles containing peptide/HLA molecules subsequently fuse with early endosomes, whereupon, the peptide/HLA is recycled back to the membrane or routed for complete degradation through late endosomes/lysosomes.48 It is not entirely clear how the cell determines the fate of internalized peptide/HLA material, but recent evidence suggests the existence of sorting mechanisms during endosomal transit. In one example, peptide-null MHC-I molecules have reduced surface half-lives (compared with peptide-loaded MHC-I) and are excluded from early endosomal recycling pathways.47
We have previously documented the internalization of KIF-Ab and YLL-Ab in MDA-MB-231 cells using confocal microscopy.16,22 All other TCR-like antibodies under study here also internalize following peptide/HLA binding based on subsequent cancer cell killing, presumably due to cathepsin B-specific release of pro-drug duocarmycin conjugates within lysosomes, which spontaneously rearrange to an active form and travel to the nucleus to mediate irreversible DNA damage.34,49,50 Even in the case of YLSG-Ab3,5 ADCs, some degree of target cytotoxicity (∼25%) was observed following co-culture with 4T1-CEA cells (Fig. 3). Yet, it remains possible that YLSG-Ab3,5 engagement of CEA peptide/HLA-A2 somehow reduces HLA-A2 internalization altogether or precludes nuclear accumulation of duocarmycin by directing internalized material through recycling endosomes.51
The notion that TCR-like antibody target docking can modulate intracellular trafficking patterns of surface-derived HLA is supported by a previous finding from Owen and colleagues.52 The investigators developed a unique anti-HER2 antibody (designated 73JIgG) that bound an extracellular domain of HER2/neu distinct from trastuzumab despite retaining comparable binding specificity/affinity profiles. Although trastuzumab remained associated with HER2 upon internalization and plasma membrane recycling, 73JIgG had an increased propensity to disassociate from the receptor internally and localize within lysosomes in an unbound form. Relatedly, earlier published work has implicated that trastuzumab does not observably influence HER2 cell distribution dynamics, being instead passively bound to recycled material.53 The HER2 molecule itself appears inclined to heterodimerize with other epidermal growth factor receptors (EGFRs), regardless of trastuzumab binding,54,55 enhance growth factor stimulation, and stabilize cell-surface activated EGFRs by promoting recycling or reducing internalization.56,57 HER2 signaling dynamics may also play a decisive role in the potential of anti-HER2 ADCs (such as ado-trastuzumab emtansine or SYD985) to mediate cytotoxicity against tumor cells, particularly in situations where HER2 densities are far below 10,000 copies per cell. We, therefore, performed a pilot comparison of the ability of TCR-like antibodies and the murine anti-HER2 antibody 4D5 (parent molecule of trastuzumab58) to function as ADCs against HER2+ SW620 cells (Fig. S1,59-61), with the hope of gauging ADC efficacy against 2 molcules (i.e., peptide/HLA and HER2/neu) with distinct internalization properties. Fig. S3 indicates observable killing of SW620 cells when FLS-Ab, YLL-Ab, and BB7.2 were linked to duocarmycin, as initially documented in Fig. 2B. However, the 4D5-Ab failed to disrupt SW620 viability when used as an ADC despite yielding identical SW620 binding parameters as FLS-Ab (Fig. S1). In replicate experiments, we were also unable to detect cytotoxicity against low HER2+ MDA-MB-231 cells (Fig. S1,60,61) following co-culture with duocarmycin-conjugated 4D5-Ab (data not shown).
Overall, the larger implications of our work provide compelling evidence that select TCR-like antibodies adequately target and destroy malignant cells (irrespective of peptide/HLA density) using existing ADC strategies approved by the FDA and undergoing clinical development.6,28 Additionally, TCR-like antibodies are not created equal to perform as ADCs, and TCR-like antibody/ligand binding may override potent drug conjugates such as the duocarmycins. It, therefore, seems reasonable to incorporate target docking characteristics in preclinical screening methods designed to identify ADC-compatible TCR-like antibodies for patients with cancer.
Materials and methods
Cell culture
The murine breast 4T1 and human tumor lines MDA-MB-231 (mammary adenocarcinoma; HLA-A2+, HER2+60,61) and SW620 (colorectal adenocarcinoma; HLA-A2+, HER2+59-61) were purchased from ATCC (Manassas, Virginia). All cells were Mycoplasma free and maintained in complete RPMI-1640 media containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin, and 10 µM L-glutamine (all from Thermo Fisher Scientific, Waltham, MA) at 37 °C with 5% CO2. 4T1 cells were stably transfected with a vector encoding a single chain HLA-A2/H-2Db trimer presenting CEA HLA-A2 altered peptide YLSGADLNL (YLS) (designated 4T1-CEA) (kind gift of Dr. Ted Hansen, Washington University).26 The murine 4D5 hybridoma (CRL-10463) was also purchased from ATCC and expanded in serum-free media (Thermo Fisher Scientific) for the purpose of purifying the HER2-specific 4D5 antibody, which is the parent molecule of trastuzumab.58
Peptides
The following HLA-A2 peptides were synthesized to >95% purity by 9-fluorenylmethoxycarbonyl (Fmoc) chemistry (Genscript, Piscataway, NJ) and used for peptide-pulsing experiments in vitro: KIFGSLAFL (KIF), YLS, ALSGADLNL, YLAGADLNL, YLSAADLNL, YLSGAALNL, YLSGADANL, YLSGADLAL.
TCR-like antibodies
The murine TCR-like antibodies FLS-Ab, KIF-Ab, and YLL-Ab were developed after extensive screening for reactivity against specific peptide/HLA-A2 molecules (summarized in Supplementary Table 1).16,20,21 Briefly, 6–8 week-old female BALB/c mice were subcutaneously immunized with specific peptide/HLA-A2 tetramers admixed with Quil-A adjuvant (Sigma-Aldrich, St. Louis, MO), splenocytes fused with a myeloma cell line, and stable hybridoma lines isolated for TCR-like antibody production and specificity. KIF-Ab was raised against a HER2/neu-specific HLA-A2 peptide (i.e., KIF) while YLL-Ab was developed against the HLA-A2-derived YLLPAIVHI (YLL) peptide from the RNA helicase p68. Murine FLS-Ab binds to a macrophage migration inhibitory factor (MIF)-specific HLA-A2 peptide (FLSELTQQL) (FLSL). Both the FLSL and YLL peptides occupy a significant fraction of HLA-A2 molecules on tumor cells.20,24 Additionally, a series of distinct murine clones (designated YLSG [clones 1–6]) were isolated and expanded following peptide/HLA-A2 immunizations against the CEA altered peptide YLS. YLSG-Abs were than purified by protein G affinity chromatography (GE Healthcare, Pittsburgh, PA) and the purity subsequently was determined by HPLC to be greater than 80%. An antibody isotyping kit (Thermo Fisher Scientific) confirmed YLSG-Ab1,2,4,6 are IgG1 antibodies, while YLSG-Ab3,5 are IgG2b antibodies.
Flow cytometry
Tumor cells (1 × 104) were resuspended in 100 µl fluorescence activated cell sorting (FACS) buffer (0.5% bovine serum albumin/0.1% sodium azide in phosphate-buffered saline) containing 2 µg/ml of a primary mouse antibody and incubated for 20 min at 4 °C. Cells were washed once and reconstituted in FACS buffer containing 1:200 of a goat anti-mouse PE-conjugated secondary antibody (Jackson ImmunoResearch [Catalog #115–115–164; Lot #120789], West Grove, PA). Again, cells were incubated in the dark at 4 °C, washed, resuspended in 300 µl FACS buffer, and fluorescence analyzed using a BD LSRFortessa™. All acquired data was analyzed using FlowJo software (FlowJo, LLC, Ashland, OR).
Peptide/HLA-A2 copy number determination
To assess surface-expressed peptide/HLA-A2 on tumor cell lines, the QIFIKIT assay (Dako, Carpinteria, CA) was used according to the manufacturer's guidelines.62 Endogenous peptide/HLA-A2 expression was first determined by harvesting tumor cells and staining with various TCR-like antibodies (1 µg/ml), followed by a QIFIKIT kit-provided anti-mouse FITC antibody (H+L) reagent. MDA-MB-231 cells were also pulsed with various concentrations of KIF HLA-A2+ peptide over the course of 3 hours, washed, stained with the KIF-Ab TCR-like antibody, and indirectly detected with the anti-mouse FITC antibody. The BB7.2 antibody (purified from the BB7.2 hybridoma [ATCC, CRL-HB-82]) was incorporated in all assays to assess overall HLA-A2 copy number. Mean fluorescence intensities were analyzed by flow cytometry and interpolated to a standard curve calculated using FITC signals from distinct bead populations containing known amounts of conjugated mouse antibodies.
Secondary and direct toxin/drug TCR-like antibody conjugation
The toxin saporin disrupts protein synthesis by inhibiting ribosomal activity. TCR-like antibodies were indirectly bound using the Mab-Zap reagent (Advanced Targeting Systems, San Diego, CA), which is a goat anti-mouse saporin conjugate.63 The DNA alkylating drug duocarmycin was also incorporated to indirectly bind TCR-like antibodies using a secondary ADC reactive against the murine Fc antibody region (αMFc-CL-DMDM; Moradec, San Diego, CA). For select killing assays, FLS-Ab was directly conjugated to saporin (Sigma-Aldrich) through exposed amine groups using an SPDP linker as outlined by the vendor (Thermo Fisher Scientific). Duocarmycin SA (Levena Biopharma, San Diego, CA) was also directly conjugated to FLS-Ab, KIF-Ab, AND YLL-Ab via an amine reactive linker using the Safe & Easy Toxin™(SET) kit and purified according to the manufacturer's protocol. Both duocarmycin conjugation schemes comprised a cathepsin B-reactive valine-citruline-PAB linker (Osu-PEG4-vc-PAB).11
TCR-like antibody killing assay
Tumor cells (5×103) were plated in flat-bottom 96-well plates, then Mab-Zap (100 ng) or αMFc-CL-DMDM (100 ng) was added. Various dilutions of isotype control, BB7.2, TCR-like, and 4D5 antibodies were subsequently added to a final volume of 120 µl and plates were incubated for 3–5 d at 37 °C with 5% CO2. In the case of directly-conjugated TCR-like antibodies, reagents were added to tumor cells in culture without secondary conjugated materials. Tumor cell viability was determined by adding 20 µl MTS reagent (CellTiter 96® AQueous One Solution Cell Proliferation Assay; Promega, Madison, WI) and incubating plates for an additional 30 min to 4 hrs at 37 °C. Absorbance values were recorded at 490 nm and background values subtracted from all treated wells. Specific percent viability was determined by dividing TCR-like antibody treatment optical density readings from isotype control exposed wells. Data was fit to 4-parameter curves and half maximal effective concentration (EC50) values determined using the GraphPad Prism program (GraphPad Software, Inc., La Jolla, CA). Tumor cell viability was > 95% when TCR-like antibodies were incubated alongside cells in the absence of toxin/drug conjugates (data not shown).
Complement-dependent cytotoxicity assay
4T1-CEA cells (1×105) were incubated in 96-well round-bottom plates with 4 µg/ml YLSG-Ab and Low-Tox-H rabbit complement (Cedarlane, Burlington, NC) in complete media containing 5% heat-inactivated FBS for 4 hrs at 37 °C with 5% CO2. Specific tumor cell killing was determined by assessing supernatant-derived lactate dehydrogenase (LDH) through absorbance (CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega) using the following calculation: Specific cytotoxicity (%) = (([YLSG-Ab and complement] – [Isotype antibody and complement]) ÷ (4T1-CEA maximum lysis – 4T1-CEA spontaneous release)) X 100. Spontaneous LDH release from 4T1-CEA cells + rabbit complement (in the absence of antibody) did not exceed 20% of maximum 4T1-CEA lysis, determined following one freeze/thaw cycle.
Surface plasmon resonance
Antibody-peptide/HLA-A2 binding interactions were analyzed through SPR using the SensiQ microfluidics system (SensiQ Technologies, Oklahoma City, OK). Carboxylic acid-treated chips were reacted with EDC/NHS followed by injection of protein A/G (10 µg/ml in acetate buffer [pH 4.5]) (all from Thermo Fisher Scientific). Chips were then exposed to 1 M ethanolamine (pH 8.0) to remove unbound or weakly reacted material and block residual reactive NHS esters. YLSG-Ab (25 nM) were then prepared in HEPES-Tris-EDTA buffer and injected over chips followed by exposure to various concentrations of purified YLS/HLA-A2 folded monomers.16 Chips were regenerated between runs using 10 mM phosphoric acid followed by injection of HEPES-Tris-EDTA. Binding kinetics and affinity between YLS/HLA-A2 and each YLSG-Ab clone were determined using the SensiQ-provided Qdat software package.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Dr. Oriana Hawkins for initial discussions and Dr. Raghavender Chivukula for technical support. HPLC analysis of the YLSG-Ab panel was performed by the Shimadzu Center for Advanced Analytical Chemistry at the University of Texas at Arlington.
References
- 1.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol 2010; 10:317-27; PMID:20414205; http://dx.doi.org/ 10.1038/nri2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res 2001; 61:4744-9; PMID:11406546 [PubMed] [Google Scholar]
- 3.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012; 12:278-87; PMID:22437872; http://dx.doi.org/ 10.1038/nrc3236 [DOI] [PubMed] [Google Scholar]
- 4.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, et al.. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008; 26:1789-96; PMID:18347005; http://dx.doi.org/ 10.1200/JCO.2007.14.8957 [DOI] [PubMed] [Google Scholar]
- 5.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 2003; 21:3940-7; PMID:12975461; http://dx.doi.org/ 10.1200/JCO.2003.05.013 [DOI] [PubMed] [Google Scholar]
- 6.Diamantis N, Banerji U. Antibody-drug conjugates–an emerging class of cancer treatment. Br J Cancer 2016; 114:362-7; PMID:26742008; http://dx.doi.org/ 10.1038/bjc.2015.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM. Antibody-drug conjugates: current status and future directions. Drug Discov Today 2014; 19:869-81; PMID:24239727; http://dx.doi.org/ 10.1016/j.drudis.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 8.de Goeij BE, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol 2016; 40:14-23; PMID:26963132; http://dx.doi.org/ 10.1016/j.coi.2016.02.008 [DOI] [PubMed] [Google Scholar]
- 9.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, et al.. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012; 367:1783-91; PMID:23020162; http://dx.doi.org/ 10.1056/NEJMoa1209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 2010; 363:1812-21; PMID:21047225; http://dx.doi.org/ 10.1056/NEJMoa1002965 [DOI] [PubMed] [Google Scholar]
- 11.Jain N, Smith SW, Ghone S, Tomczuk B. Current ADC Linker Chemistry. Pharm Res 2015; 32:3526-40; PMID:25759187; http://dx.doi.org/ 10.1007/s11095-015-1657-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013; 31:443-73; PMID:23298205; http://dx.doi.org/ 10.1146/annurev-immunol-032712-095910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; http://dx.doi.org/ 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weidanz JA, Hawkins O, Verma B, Hildebrand WH. TCR-like biomolecules target peptide/MHC Class I complexes on the surface of infected and cancerous cells. Int Rev Immunol 2011; 30:328-40; PMID:22053972; http://dx.doi.org/ 10.3109/08830185.2011.604880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Epel M, Carmi I, Soueid-Baumgarten S, Oh SK, Bera T, Pastan I, Berzofsky J, Reiter Y. Targeting TARP, a novel breast and prostate tumor-associated antigen, with T cell receptor-like human recombinant antibodies. Eur J Immunol 2008; 38:1706-20; PMID:18446790; http://dx.doi.org/ 10.1002/eji.200737524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain R, Rawat A, Verma B, Markiewski MM, Weidanz JA. Antitumor activity of a monoclonal antibody targeting major histocompatibility complex class I-Her2 peptide complexes. J Natl Cancer Inst 2013; 105:202-18; PMID:23300219; http://dx.doi.org/ 10.1093/jnci/djs521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, Whitten JA, Park SM, Korontsvit T, Zakhaleva V, et al.. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res 2014; 20:4036-46; PMID:24850840; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bossi G, Gerry AB, Paston SJ, Sutton DH, Hassan NJ, Jakobsen BK. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2013; 2:e26840; PMID:24482751; http://dx.doi.org/ 10.4161/onci.26840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, Karbach J, Jäger E, Cameron BJ, Lissin N, et al.. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol 2006; 176:7308-16; PMID:16751374; http://dx.doi.org/ 10.4049/jimmunol.176.12.7308 [DOI] [PubMed] [Google Scholar]
- 20.Hawkins O, Verma B, Lightfoot S, Jain R, Rawat A, McNair S, Caseltine S, Mojsilovic A, Gupta P, Neethling F, et al.. An HLA-presented fragment of macrophage migration inhibitory factor is a therapeutic target for invasive breast cancer. J Immunol 2011; 186:6607-16; PMID:21515791; http://dx.doi.org/ 10.4049/jimmunol.1003995 [DOI] [PubMed] [Google Scholar]
- 21.Verma B, Hawkins OE, Neethling FA, Caseltine SL, Largo SR, Hildebrand WH, Weidanz JA. Direct discovery and validation of a peptide/MHC epitope expressed in primary human breast cancer cells using a TCRm monoclonal antibody with profound antitumor properties. Cancer Immunol Immunother 2010; 59:563-73; PMID:19779714; http://dx.doi.org/ 10.1007/s00262-009-0774-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma B, Jain R, Caseltine S, Rennels A, Bhattacharya R, Markiewski MM, Rawat A, Neethling F, Bickel U, Weidanz JA. TCR mimic monoclonal antibodies induce apoptosis of tumor cells via immune effector-independent mechanisms. J Immunol 2011; 186:3265-76; PMID:21282517; http://dx.doi.org/ 10.4049/jimmunol.1002376 [DOI] [PubMed] [Google Scholar]
- 23.Stevanovic S, Schild H. Quantitative aspects of T cell activation–peptide generation and editing by MHC class I molecules. Semin Immunol 1999; 11:375-84; PMID:10625591; http://dx.doi.org/ 10.1006/smim.1999.0195 [DOI] [PubMed] [Google Scholar]
- 24.Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E, Engelhard VH. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 1992; 255:1261-3; PMID:1546328; http://dx.doi.org/ 10.1126/science.1546328 [DOI] [PubMed] [Google Scholar]
- 25.van der Lee MM, Groothuis PG, Ubink R, van der Vleuten MA, van Achterberg TA, Loosveld EM, Damming D, Jacobs DC, Rouwette M, Egging DF, et al.. The Preclinical Profile of the Duocarmycin-Based HER2-Targeting ADC SYD985 Predicts for Clinical Benefit in Low HER2-Expressing Breast Cancers. Mol Cancer Ther 2015; 14:692-703; PMID:25589493; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-0881-T [DOI] [PubMed] [Google Scholar]
- 26.Yu YY, Netuschil N, Lybarger L, Connolly JM, Hansen TH. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J Immunol 2002; 168:3145-9; PMID:11907065; http://dx.doi.org/ 10.4049/jimmunol.168.7.3145 [DOI] [PubMed] [Google Scholar]
- 27.Dubrovsky L, Dao T, Gejman RS, Brea EJ, Chang AY, Oh CY, Casey E, Pankov D, Scheinberg DA, et al.. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology 2016; 5:e1049803; PMID:26942058; http://dx.doi.org/ 10.1080/2162402X.2015.1049803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullard A. Maturing antibody-drug conjugate pipeline hits 30. Nat Rev Drug Discov 2013; 12:329-32; PMID:23629491; http://dx.doi.org/ 10.1038/nrd4009 [DOI] [PubMed] [Google Scholar]
- 29.Asundi J, Reed C, Arca J, McCutcheon K, Ferrando R, Clark S, Luis E, Tien J, Firestein R, Polakis P. An antibody-drug conjugate targeting the endothelin B receptor for the treatment of melanoma. Clin Cancer Res 2011; 17:965-75; PMID:21245091; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2340 [DOI] [PubMed] [Google Scholar]
- 30.Smith LM, Nesterova A, Alley SC, Torgov MY, Carter PJ. Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol Cancer Ther 2006; 5:1474-82; PMID:16818506; http://dx.doi.org/ 10.1158/1535-7163.MCT-06-0026 [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Ma D, Olson WC, Heston WD. In vitro and in vivo responses of advanced prostate tumors to PSMA ADC, an auristatin-conjugated antibody to prostate-specific membrane antigen. Mol Cancer Ther 2011; 10:1728-39; PMID:21750220; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0191 [DOI] [PubMed] [Google Scholar]
- 32.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RV, Lutz RJ, et al.. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008; 68:9280-90; PMID:19010901; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1776 [DOI] [PubMed] [Google Scholar]
- 33.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14:135-46; PMID:24457417; http://dx.doi.org/ 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 34.Dokter W, Ubink R, van der Lee M, van der Vleuten M, van Achterberg T, Jacobs D, Loosveld E, van den Dobbelsteen D, Egging D, Mattaar E, et al.. Preclinical profile of the HER2-targeting ADC SYD983/SYD985: introduction of a new duocarmycin-based linker-drug platform. Mol Cancer Ther 2014; 13:2618-29; PMID:25189543; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-0040-T [DOI] [PubMed] [Google Scholar]
- 35.Elgersma RC, Coumans RG, Huijbregts T, Menge WM, Joosten JA, Spijker HJ, de Groot FM, van der Lee MM, Ubink R, van den Dobbelsteen DJ, et al.. Design, Synthesis, and Evaluation of Linker-Duocarmycin Payloads: Toward Selection of HER2-Targeting Antibody-Drug Conjugate SYD985. Mol Pharm 2015; 12:1813-35; PMID:25635711; http://dx.doi.org/ 10.1021/mp500781a [DOI] [PubMed] [Google Scholar]
- 36.Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun 2013; 13:15. [PMC free article] [PubMed] [Google Scholar]
- 37.Chang AY, Gejman RS, Brea EJ, Oh CY, Mathias MD, Pankov D, Casey E, Dao T, Scheinberg DA, et al.. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin Biol Ther 2016; 16:979-87; PMID:27094818; http://dx.doi.org/ 10.1080/14712598.2016.1176138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denkberg G, Lev A, Eisenbach L, Benhar I, Reiter Y. Selective targeting of melanoma and APCs using a recombinant antibody with TCR-like specificity directed toward a melanoma differentiation antigen. J Immunol 2003; 171:2197-207; PMID:12928363; http://dx.doi.org/ 10.4049/jimmunol.171.5.2197 [DOI] [PubMed] [Google Scholar]
- 39.Klechevsky E, Gallegos M, Denkberg G, Palucka K, Banchereau J, Cohen C, Reiter Y. Antitumor activity of immunotoxins with T-cell receptor-like specificity against human melanoma xenografts. Cancer Res 2008; 68:6360-7; PMID:18676861; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mareeva T, Lebedeva T, Anikeeva N, Manser T, Sykulev Y. Antibody specific for the peptide.major histocompatibility complex. Is it T cell receptor-like? J Biol Chem 2004; 279:44243-9; PMID:15302863; http://dx.doi.org/ 10.1074/jbc.M407021200 [DOI] [PubMed] [Google Scholar]
- 41.Mareeva T, Martinez-Hackert E, Sykulev Y. How a T cell receptor-like antibody recognizes major histocompatibility complex-bound peptide. J Biol Chem 2008; 283:29053-9; PMID:18703505; http://dx.doi.org/ 10.1074/jbc.M804996200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Messaoudi I, LeMaoult J, Nikolic-Zugic J. The mode of ligand recognition by two peptide:MHC class I-specific monoclonal antibodies. J Immunol 1999; 163:3286-94; PMID:10477598 [PubMed] [Google Scholar]
- 43.Polakova K, Plaksin D, Chung DH, Belyakov IM, Berzofsky JA, Margulies DH. Antibodies directed against the MHC-I molecule H-2Dd complexed with an antigenic peptide: similarities to a T cell receptor with the same specificity. J Immunol 2000; 165:5703-12; PMID:11067928; http://dx.doi.org/ 10.4049/jimmunol.165.10.5703 [DOI] [PubMed] [Google Scholar]
- 44.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol 2006; 6:343-57; PMID:16622479; http://dx.doi.org/ 10.1038/nri1837 [DOI] [PubMed] [Google Scholar]
- 45.Yoshikawa M, Mukai Y, Okada Y, Tsumori Y, Tsunoda S, Tsutsumi Y, Aird WC, Yoshioka Y, Okada N, Doi T, et al.. Robo4 is an effective tumor endothelial marker for antibody-drug conjugates based on the rapid isolation of the anti-Robo4 cell-internalizing antibody. Blood 2013; 121:2804-13; PMID:23365463; http://dx.doi.org/ 10.1182/blood-2012-12-468363 [DOI] [PubMed] [Google Scholar]
- 46.Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic 2009; 10:1745-52; PMID:19761542; http://dx.doi.org/ 10.1111/j.1600-0854.2009.00979.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zagorac GB, Mahmutefendic H, Tomas MI, Kucic N, Le Bouteiller P, Lucin P. Early endosomal rerouting of major histocompatibility class I conformers. J Cell Physiol 2012; 227:2953-64; PMID:21959869; http://dx.doi.org/ 10.1002/jcp.23042 [DOI] [PubMed] [Google Scholar]
- 48.Adiko AC, Babdor J, Gutierrez-Martinez E, Guermonprez P, Saveanu L. Intracellular Transport Routes for MHC I and Their Relevance for Antigen Cross-Presentation. Front Immunol 2015; 6:335; PMID:26191062; http://dx.doi.org/ 10.3389/fimmu.2015.00335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephenson MJ, Howell LA, O'Connell MA, Fox KR, Adcock C, Kingston J, Sheldrake H, Pors K, Collingwood SP, Searcey M. Solid-Phase Synthesis of Duocarmycin Analogues and the Effect of C-Terminal Substitution on Biological Activity. J Org Chem 2015; 80:9454-67; PMID:26356089; http://dx.doi.org/ 10.1021/acs.joc.5b01373 [DOI] [PubMed] [Google Scholar]
- 50.Withana NP, Blum G, Sameni M, Slaney C, Anbalagan A, Olive MB, Bidwell BN, Edgington L, Wang L, Moin K, et al.. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res 2012; 72:1199-209; PMID:22266111; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Endert P. Intracellular recycling and cross-presentation by MHC class I molecules. Immunol Rev 2016; 272:80-96; PMID:27319344; http://dx.doi.org/ 10.1111/imr.12424 [DOI] [PubMed] [Google Scholar]
- 52.Owen SC, Patel N, Logie J, Pan G, Persson H, Moffat J, Sidhu SS, Shoichet MS. Targeting HER2+ breast cancer cells: lysosomal accumulation of anti-HER2 antibodies is influenced by antibody binding site and conjugation to polymeric nanoparticles. J Control Release 2013; 172:395-404; PMID:23880472; http://dx.doi.org/ 10.1016/j.jconrel.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 53.Austin CD, De Maziere AM, Pisacane PI, van Dijk SM, Eigenbrot C, Sliwkowski MX, Klumperman J, Scheller RH. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell 2004; 15:5268-82; PMID:15385631; http://dx.doi.org/ 10.1091/mbc.E04-07-0591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghosh R, Narasanna A, Wang SE, Liu S, Chakrabarty A, Balko JM, González-Angulo AM, Mills GB, Penuel E, Winslow J, et al.. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res 2011; 71:1871-82; PMID:21324925; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hubbard SR. EGF receptor inhibition: attacks on multiple fronts. Cancer Cell 2005; 7:287-8; PMID:15837615; http://dx.doi.org/ 10.1016/j.ccr.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 56.Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: quantitative analysis of HER2 overexpression effects. Cancer Res 2003; 63:1130-7; PMID:12615732 [PubMed] [Google Scholar]
- 57.Ritchie M, Tchistiakova L, Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013; 5:13-21; PMID:23221464; http://dx.doi.org/ 10.4161/mabs.22854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM, et al.. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 1992; 89:4285-9; PMID:1350088; http://dx.doi.org/ 10.1073/pnas.89.10.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu Q, Wu Y, Yan F, Wang N, Wang W, Cao X, Wang Y, Wan T. Efficient induction of a Her2-specific anti-tumor response by dendritic cells pulsed with a Hsp70L1-Her2(341-456) fusion protein. Cell Mol Immunol 2011; 8:424-32; PMID:21785448; http://dx.doi.org/ 10.1038/cmi.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TF Jr, Hudson-Curtis BL, et al.. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif 2007; 40:580-94; PMID:17635524; http://dx.doi.org/ 10.1111/j.1365-2184.2007.00455.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weidanz JA, Nguyen T, Woodburn T, Neethling FA, Chiriva-Internati M, Hildebrand WH, Lustgarten J. Levels of specific peptide-HLA class I complex predicts tumor cell susceptibility to CTL killing. J Immunol 2006; 177:5088-97; PMID:17015692; http://dx.doi.org/ 10.4049/jimmunol.177.8.5088 [DOI] [PubMed] [Google Scholar]
- 62.Porwit-MacDonald A, Janossy G, Ivory K, Swirsky D, Peters R, Wheatley K, Walker H, Turker A, Goldstone AH, Burnett A. Leukemia-associated changes identified by quantitative flow cytometry. IV. CD34 overexpression in acute myelogenous leukemia M2 with t(8;21). Blood 1996; 87:1162-9; PMID:8562943 [PubMed] [Google Scholar]
- 63.Kohls MD, Lappi DA. Mab-ZAP: a tool for evaluating antibody efficacy for use in an immunotoxin. Biotechniques 2000; 28:162-5; PMID:10649788 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.