

ABSTRACT
A modular and adaptive mass spectrometry (MS)-based platform was developed to provide fast, robust and sensitive host cell protein (HCP) analytics to support process development. This platform relies on one-dimensional ultra-high performance liquid chromatography (1D UHPLC) combined with several different MS data acquisition strategies to meet the needs of purification process development. The workflow was designed to allow HCP composition and quantitation for up to 20 samples per day, a throughput considered essential for real time bioprocess development support. With data-dependent acquisition (DDA), the 1D UHPLC-MS/MS method had excellent speed and demonstrated robustness in detecting unknown HCPs at ≥ 50 ng/mg (ppm) level. Combining 1D UHPLC with sequential window acquisition of all theoretical spectra (SWATH) MS enabled simultaneous detection and quantitation of all HCPs in single-digit ng/mg range within 1 hour, demonstrating for the first time the benefit of SWATH MS as a technique for HCP analysis. As another alternative, a targeted MS approach can be used to track the clearance of specific known HCP under various process conditions. This study highlights the importance of designing a robust LC-MS/MS workflow that not only allows HCP discovery, but also affords greatly improved process knowledge and capability in HCP removal. As an orthogonal and complementary detection approach to traditional HCP analysis by enzyme-linked immunosorbent assay, the reported LC-MS/MS workflow supports the development of bioprocesses with optimal HCP clearance and the production of safe and high quality therapeutic biopharmaceuticals.
KEYWORDS: Bioprocess development, host cell protein, mass spectrometry, SWATH
Abbreviations
- HCP
Host cell protein
- 1D UHPLC
One-dimensional ultra-high performance liquid chromatography
- DDA
Data-dependent acquisition
- SWATH
Sequential window acquisition of all theoretical spectra
- ELISA
Enzyme-linked immunosorbent assay
- LC
Liquid chromatography
- MS
Mass spectrometry
- MRM
Multiple reaction monitoring
- PRM
Parallel reaction monitoring
- DIA
Data-independent acquisition
- 2D-LC
Two-dimensional liquid chromatography
- PLBL2
Phospholipase B-like 2
Introduction
Host cell protein (HCP) impurities derived from biotherapeutic production processes can pose potential safety risks for patients. Therefore, clearance of HCPs to levels deemed safe is required by regulatory agencies.1 While a multi-analyte enzyme-linked immunosorbent assay (ELISA) is most commonly used to deliver an aggregate sum measurement of HCPs,2,3 it does not provide the identity of HCPs, which would be required for risk assessment and could be helpful for downstream process optimization. Some HCPs have poor immunogenicity in the animals used to generate the immunoassay reagent, and therefore are not likely to be detected by ELISA if they are present in final product or in-process pools.4,5 Some may be present at levels that exceed the specific anti-HCP antibody available for binding (antigen excess),5 in which case the use of a total ELISA may underestimate the amount of those HCPs in the sample.4 Additionally, ELISA does not discriminate whether several HCPs are present at low levels or an individual HCP has accumulated at higher levels. To address these challenges, methods using advanced liquid chromatography (LC) and mass spectrometry (MS) techniques have been developed, most of which focus on detecting low level HCPs in the final monoclonal antibody (mAb) products.6-11 However, there is currently an unmet need to provide high throughput and robust LC-MS/MS analysis of HCPs in various in-process pools to help design and optimize purification processes toward optimal clearance.
As an orthogonal and complementary approach to immunoassay detection, a sensitive, MS-based method is highly desirable to ensure the identification of any HCPs in the final product that may have escaped ELISA detection and are not reflected in the total ELISA measurement value. This benefit is especially important when such co-purifying HCPs are at levels that may be considered potentially unsafe. Equally important for the identification is a robust and high throughput LC-MS/MS method capable of quantifying HCPs. We believe the ultimate goal of HCP analytics is to understand the effect of chromatographic conditions on HCP clearance, and to provide quick and confident guidance on process modifications that result in the removal or reduction of these HCPs to acceptable levels. In our experience, a desired throughput is 10–20 samples per day, which would allow assessment of up to 10 different process conditions in either one or two replicates. An ideal workflow should have the ability to provide sensitive detection and accurate quantitation of all (including a priori unknown) HCPs present in both final product and in-process pools. It can be used to “declare” the absence of HCP above a certain level, and to support the removal of identified HCP with high analytical throughput without the need of method optimization for individual HCPs.
When applicable, protein-specific ELISA or targeted MS approaches such as multiple reaction monitoring (MRM)12 or parallel reaction monitoring (PRM)13,14 could provide highly sensitive and accurate quantitation to monitor a limited number of known HCPs during process development. However, the HCPs that need to be monitored in the in-process pools are frequently unknown a priori due to the fact that their co-purification with the product is often dictated by product-specific binding or interactions.15 Producing peptide/protein standards and developing reagents for ELISA is time-consuming and unsuitable to satisfy the desire of tracking HCPs and their clearance within days of discovery. MS with data-independent acquisition (DIA)16-19 can be used for sensitive and simultaneous quantitation of all identified HCPs as a universal method, thus eliminating the need for targeted method development. One DIA method,19 MSE, has been coupled with two dimensional liquid chromatography (2D-LC) to identify and quantify low abundance HCPs, using the “Top 3″ label-free approach (MS signal response for the three most intense tryptic peptides) for protein quantitation.7,8,10,20,21 However, 2D-LC analysis may have reproducibility challenges and the nano-LC often used in the second dimension for improved sensitivity is not always robust. In addition, its 10–20 hours analysis time/sample makes it challenging to analyze multiple samples from in-process pools using the 2D-LC-MS/MS approach to provide real time support and quick response for addressing potential HCP concerns.
As an alternative DIA strategy, sequential window acquisition of all theoretical spectra (SWATH) MS performs sequential MS/MS of multiple smaller acquisition windows and relies on a targeted data extraction and analysis strategy using a comprehensive peptide fragment ion spectral library.18,21 Compared to MSE, which uses MS/MS of a single large m/z window and standard protein sequence database searching tools for data mining, SWATH MS provides higher fragment ion specificity and potential for improved number of protein identifications and quantitation accuracy.18 It has the potential to provide large-scale MRM with increased throughput, and to quantify all identified proteins without a priori knowledge.18,22,23Moreover, SWATH MS quantitation at fragment ion spectra level was shown to offer a 2-8-fold gain in sensitivity compared to the limit of detection based on precursor ion signals detected in the full MS scans.18
In this study, we developed a modular and adaptive 1D UHPLC-MS/MS-based workflow to support fast bioprocess development with optimal HCP clearance. This workflow takes advantage of the fact that HCPs are present at higher levels in the upstream pools and therefore can be detected more readily than in the final pools. This knowledge of HCPs that are potentially present, coupled with the high protein detection sensitivity and quantitation accuracy of SWATH MS,18 provides improved detection of low-level HCPs in the final product using 1D UHPLC separation. Our workflow can be tailored to match different HCP detection sensitivities from single digit to ≥ 50 ppm (ng HCP per mg of product) while accommodating the needs of purification process development. As a result, sensitive and simultaneous quantitation of all identified HCPs can be achieved while maintaining high throughput and robustness.
Results
Overview of the 1D UHPLC-MS/MS workflow
Biotherapeutic antibodies are typically purified from harvested cell culture fluid (HCCF) by performing an initial step of affinity chromatography (e.g., Protein A binding), followed by two or three additional (and orthogonal) chromatography steps to remove HCPs and other process/product related impurities. Fig. 1 shows approximate HCP level commonly seen using proprietary Chinese hamster ovary (CHO) cell lines and purification processes. In HCCF, the mass ratio of HCPs to mAb is around 1:1 ratio (∼106 ppm). The majority of HCPs are largely removed following Protein A affinity purification (∼103 ppm in the pool). After additional purification steps, HCPs are often present at a low level (∼101 ppm or less) in the final product.
Figure 1.
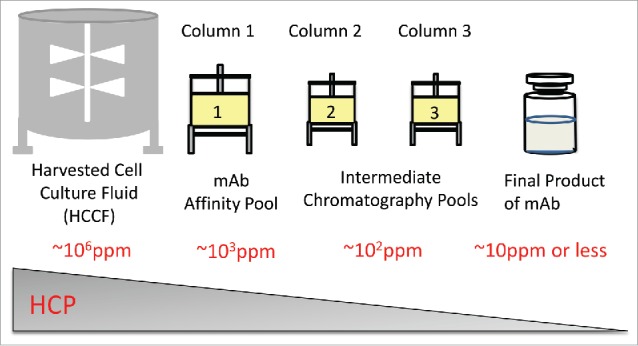
Typical host cell protein level during purification process for mAb biologics.
Fig. 2 illustrates our 1D UHPLC-MS/MS based workflow. The use of 1D UHPLC coupled to MS in DDA mode, as the first option for process development support, provides sufficient throughput and robustness to detect relatively high level HCPs (≥ 50 ppm) in all mAb in-process pools, including the final product. In addition, the data-dependent MS/MS analysis of upstream pools can provide HCP list/peptide fragmentation information to facilitate the use of DIA and targeted MS methods such as MRM or PRM to quantify and identify HCPs with higher sensitivity in the downstream pools. DIA provides quantitation data for all detected HCPs and can simultaneously monitor their clearance during purification process with higher sensitivity (∼10 ppm). To track specific, known HCPs with single digit (∼2) ppm sensitivity, targeted MS method can be applied. This strategy is further demonstrated in the following sections.
Figure 2.
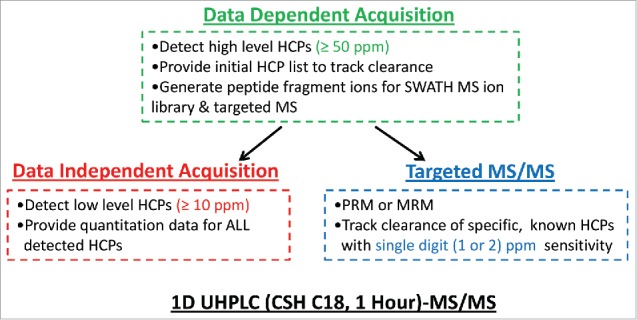
Summary of the modular and adaptive 1D UHPLC-MS-based workflow to support fast bioprocess development for optimal host cell protein clearance.
Sensitivity and robustness of the 1D UHPLC-MS/MS using data-dependent acquisition
The workflow developed in this study was based on a fast (1-hour) and robust LC-MS /MS method that used charge-surface hybrid (CSH)7,24 C18 UHPLC interfaced with a mass spectrometer with fast scanning speed (MS/MS at 20Hz). The average relative standard deviation for retention time of 5 different peptides from 5 LC-MS analyses collected over 2 months was 1.1%, indicating good reproducibility of the LC separation.
To assess method capability for detecting unknown residual HCPs in biopharmaceutical products, 1D LC-MS/MS analyses with DDA were performed using a purified antibody product (mAb-1) spiked with known levels of 48 protein Universal Proteomics Standard (UPS-1; Sigma Aldrich) proteins to simulate trace level HCPs in a product. The levels of 48 UPS-1 proteins ranged from 8 to 110 ppm in the spiked mAb-1 (5 mg/mL). The number of unique peptides identified for each of the 48 simulated HCPs and their respective levels (in ppm) are illustrated in Fig. 3A. The results demonstrate that, while there is clear variation in the level of detection for individual proteins as unknowns, positive identification (≥ 2 unique peptides, used throughout this study) was achieved with consistency when levels of the UPS-1 proteins were ≥ 50 ppm. Reproducibility of identification for each of the UPS-1 proteins spiked into mAb-1 at > 50 ppm is shown by averaging the number of unique peptides found over six independent LC-MS/MS analyses (Fig. S2).
Figure 3.
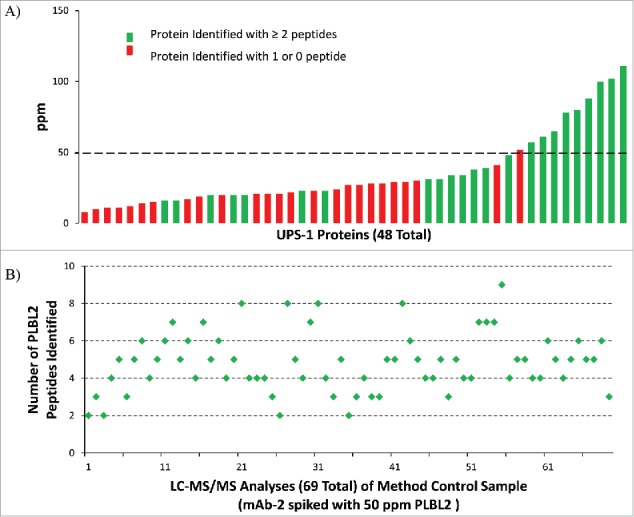
Proteins are consistently identified with ≥ 2 peptides above 50 ppm using 1D UHPLC-DDA. (A)UPS-1 (48) standard proteins ranging from 8 to 110 ppm were spiked into mAb-1. Dashed line indicates level of method sensitivity. Results from six replicate analyses showed 80–100% detection of ten proteins spiked in at above 50 ppm level. (B) Analysis of the mAb-2 sample spiked with 50 ppm PLBL2 (method control sample) in 69 test sessions over 18 months.
To further illustrate the use of this strategy in demonstrating HCP clearance in the final product, and to demonstrate the robustness of our method, a method control sample was generated, which was a mAb-2 sample spiked with 50 ppm of a purified recombinant CHO protein, Cricetulus griseus phospholipase B-like 2 (PLBL2).5 A summary of the test results is provided for the trypsin digest of this method control sample from 69 test sessions conducted in a period of 18 months (Fig. 3B). Positive identifications by database search were returned for PLBL2 in all 69 test sessions. These results also suggested that it has the potential to provide the confidence to “declare” the absence of any individual HCPs at or above a particular ppm level when no HCPs are detected by this method, coupled with a positive finding using an appropriate method control sample. The results of both UPS-1 and PLBL2 spiking studies described here are consistent in that a 50 ppm detection sensitivity should be expected with this method.
To identify HCP candidates that can potentially be present in the final products due to insufficient removal or co-purification, we used this 1D UHPLC-MS/MS DDA method to analyze a tryptic digest of CHO cell supernatant from a null cell line that does not express any product-coding genes. The supernatant contains a mixture of secreted proteins along with proteins released following cell lysis, and is representative of the HCP impurities that can be present in downstream product pools. Using 1D UHPLC-MS/MS DDA and just one hour of acquisition time, we identified more than 1,300 proteins and 19,000 peptides. More importantly, this list contained all HCP peptides and proteins that have been identified in Protein A affinity and other downstream pools (data not shown). Fragment ions for the HCP peptides identified in the null cell supernatant were used to build the spectral ion library for additional HCP analysis using DIA SWATH MS, described below in more detail. Taken together, the UPS-1 and PLBL2 spiking data, combined with the CHO proteomic analysis data demonstrate that the 1D UPLC-MS/MS DDA method can be used successfully to identify unknown HCPs in downstream product pools.
1D UHPLC-DIA SWATH MS for sensitive detection and simultaneous relative quantitation of all HCPs
The 1D LC-MS/MS DDA method served as the first step to examine whether there are high levels of HCPs present in the final pool that may require process change or optimization. To readily enable more sensitive detection of proteins that may be present in final pools, 1D LC-MS/MS DDA was performed to identify residual HCPs in affinity pools of 16 mAbs. As shown in Fig. 4, the trend observed for these affinity purification pools shows a wide range (1 to 52) of identified HCPs, dependent on the specific molecule15 as well as the affinity column wash step conditions used before antibody (product) elution. These results also showed that the vast majority of HCPs present in cell culture fluid were removed after the first affinity chromatography step, consistent with previous observations.21 In the second step of the process, most products had no remaining detectable HCPs (at ≥ 50 ppm level), while a few had between 1 and 4 HCPs per product. Note that the number of observed HCPs here are associated with the 1D UHPLC-MS/MS DDA method with a detection sensitivity of 50 ppm. Therefore, tracking these HCPs in downstream pools requires a method with higher detection sensitivity than 1D LC-MS/MS DDA. Toward this end, the 1hr 1D UHPLC method was coupled to a universal DIA method, using SWATH MS.
Figure 4.
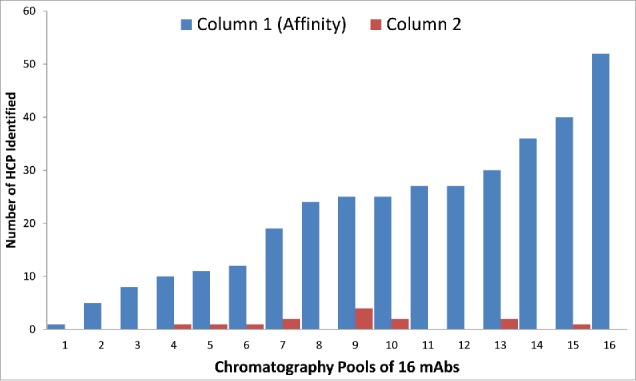
The number of HCPs identified by 1D UHPLC-DDA in the Column 1 (Affinity) and Column 2 in-process pools of 16 mAb products. Many of the HCPs were common, as they were found in multiple mAb products. There were also significant number of HCPs that were unique to a specific mAb product.
To assess the enhanced detection sensitivity18 achieved by DIA versus DDA methods, we analyzed a mAb-2 sample spiked with PLBL2 over a range of 5 to 100 ppm. The DDA method showed (Fig. 5A) detection of PLBL2 down to 30 ppm with 2-peptide identifications, but no peptide detection at 20 ppm. Using the DIA approach, 3 PLBL2 peptides were identified at the 1, 2, and 5 ppm level, and >3 peptides were identified at all other tested levels. The results also showed a linear response (R2 > 0.99) within the tested range for DIA SWATH MS and good quantitation accuracy (100–115% recovery) for the 5–100 ppm range (Fig. 5B). Quantitation precision was assessed from 5–50 ppm and observed RSD values ranged from 2.1–5.8%, based on triplicate injections (Fig. S3). Analysis of additional protein standard samples (data not shown) showed that a high sensitivity (10 ppm) was consistently achieved with this 1-hour 1D UHPLC-DIA SWATH MS method. This is comparable to online 2D-LC-MS/MS analysis that would require 10–20 hours of data acquisition time, with a reported detection limit of ∼13 ppm for a specific HCP.10,21
Figure 5.
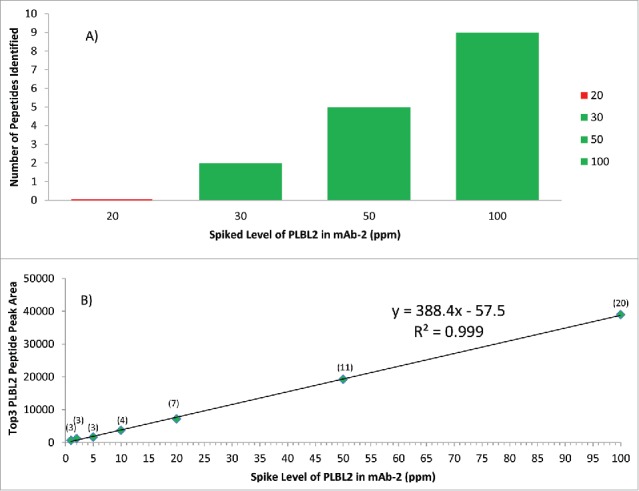
Comparison of DDA (5A) and DIA SWATH MS (5B) methods to detect PLBL2 at different spiked levels in mAb2. Green represents successful detection, while red indicates failure to detect. The detection sensitivity of DDA is shown in 5A, and the sensitivity and linearity of DIA SWATH MS TOP 3 method is shown in 5B, with the number of peptide identified shown in parentheses. PLBL2 peak area measurement by SWATH (Top 3 peptides) at ≥ 5 ppm levels had observed RSD values ranging from 2.1–5.8%, based on triplicate injections.
In addition to the ∼5-fold improved detection sensitivity for specific HCPs vs. DDA, the DIA SWATH MS approach was also able to monitor clearance of multiple HCPs simultaneously through actual in process pools from an antibody purification process. Fig. 6 shows an example of using DIA SWATH MS for relative quantitation of three specific HCPs to monitor their clearance in parallel during the purification process. Note that in the mAb-3 final product, no HCPs were identified using the DDA method (data not shown). In contrast, HCP-3 was detected (Fig. 6) with three peptides identified using DIA SWATH MS method, again confirming higher sensitivity of DIA SWATH method compared to DDA. No peptides were identified for HCP-1 and HCP-2 in the mAb-3 final product, indicating the effective removal of these HCPs.
Figure 6.
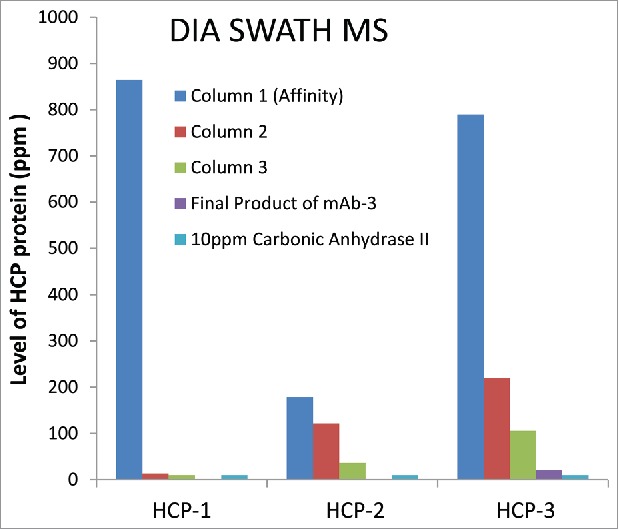
Simultaneous relative quantitation of three HCPs in mAb-3 by 1D UHPLC-DIA SWATH MS (∼10 ppm sensitivity).
1D UHPLC-MS/MS strategies for absolute quantitation of HCPs with SWATH MS or targeted MS methods
While we have demonstrated how SWATH MS can be used for relative quantification of HCP (comparison between samples), there is often a need for absolute quantification of a specific HCP during process development. However, calibration standards, in forms of either recombinantly expressed protein from the same host or stable isotope-labeled HCP peptides, are often not readily available and would require substantial lead-time to obtain. To circumvent this limitation, we employed the previously reported “Top 3″ peptide quantification strategy,19 which relies on the observation that the average signal responses of the three most intense peptides per mole of protein is constant within ± 10%.
Detection of three peptides from HCP-3 in the final product of mAb-3 by DIA SWATH MS (Fig. 6) suggested that its level should be around or above the method detection limit. Using the Top 3 strategy with a single spike of 10 ppm protein standard (bovine carbonic anhydrase II), the level of HCP-3 in mAb-3 final product was estimated at ∼20 ppm by 1D UHPLC-DIA SWATH-MS. The level of HCP-3 (20 ppm) measured by the Top 3 method is ∼1.5-fold higher than 13 ppm, which was obtained from LC-MRM analysis using a synthetic, stable isotope-labeled HCP-3 peptide, a well-accepted protein quantitation method by MS.12 This observation is consistent with the findings reported by Schenauer et al20 that most unknown HCPs could be quantified within a range of 1.5-fold (high) and 1.8-fold (low) of their actual value using the Top 3 method. This example shows that a good quantitative estimate of HCPs using a universal DIA SWATH MS method with Top 3 strategy can be achieved quickly, without the delay required in obtaining specific protein or peptide standards.
DIA SWATH MS was used to obtain absolute quantitation of PLBL2, an HCP that had previously been found to co-purify with a mAb product. PLBL2 was identified and quantified in HCCF, with the Top 3 strategy19 using a 4-point calibration curve of spiked internal standard protein (bovine carbonic anhydrase II). Table 1 shows the quantitation results obtained by DIA SWATH MS and by a PLBL2-specific ELISA25 in five HCCF samples. Results showed that the measured PLBL2 levels by DIA SWATH MS quantification agreed well with those from an orthogonal analysis by PLBL2-specific ELISA (Table 1). Note that quantitative values obtained by MS and multi-analyte HCP ELISA are generally not expected to be directly comparable with each other, due to the use of very different standards and calibration methods.
Table 1.
Agreement of the PLBL2 level (µg/mL) measured by DIA SWATH MS Top 3 approach with that by PLBL2 specific ELISA in HCCF from following expression conditions: CHO host cell lines 1, 2, and 3, all harvested on Day 14; Host cell line 3, harvested on Day 10 and 17. The results were the average of 2 biological replicates. For each biological replicate sample, PLBL2 concentrations were averages of four replicate analyses. Mean RSD (of five samples) was 5.0%, with highest RSD observed at 9.4%.
| Sample (n = 2) | By SWATH Top 3 (μg/mL) | By Specific ELISAa (μg/mL) |
|---|---|---|
| Host 1-Day 14 | 7.7 ± 1.5 | 7.6 |
| Host 2-Day 14 | 4.1 ± 0.35 | 6.3 |
| Host 3-Day 14 | 2.1 ± 0.18 | 1.5 |
| Host 3-Day 10 | 2.3 ± 0.04 | 1.6 |
| Host 3-Day 17 | 1.6 ± 0.04 | 1.6 |
Reference25.
When a specific known HCP causes a safety concern for a mAb product, sensitivity (single digit ppm) similar to ELISA may be needed to measure the absolute level of this HCP in the mAb final product. To meet this need, 1D UHPLC can be coupled with targeted MS approaches such as MRM (low resolution) or PRM (high resolution) to specifically monitor and quantify the amount of HCP with high sensitivity. There are a variety of different commercial MS instruments available for such measurements. In this study, we were able to demonstrate low single digit ppm (by both LC-MRM and LC-PRM) detection sensitivity for PLBL2, which was spiked into mAb-1 (Supplementary Information and Fig. S1).
Discussions
We developed and demonstrated the utility of a modular and adaptive 1D UHPLC-MS/MS-based workflow that provides robust, high-throughput and sensitive HCP detection to meet the needs at different stages of purification development for optimal clearance. This workflow, especially the use of DIA SWATH MS, takes full advantage of the fact that residual HCPs in mAb products are present at higher levels in the upstream pools11 (including HCCF) and can be identified there more readily than in the final product. The HCP peptides/proteins identified in the upstream pool greatly facilitates routine and high throughput detection of residual HCPs at single digit ppm levels (in final product), without requiring lengthy 2D LC separation. Sensitive and simultaneous HCP quantitation was achieved with one-day turnaround for up to 20 samples to track their clearance and generate process knowledge. Such ability to provide results in real time fashion is essential to allow fast process development or optimization. Historically, the biopharmaceutical industry has largely relied on immunoassays to guide purification process development, which typically involves an aggregate measurement of a mixture or total HCPs,4 without knowing the identity and characteristics of specific HCPs that might be of concern. The use of MS represents a significant step forward in applying Quality by Design principles to manufacture high quality biopharmaceutical products. With fast quantification a real possibility, both in having an appropriate method available within days of the HCP discovery and in providing assay results for real time feedback in the evaluation of process parameters and capability, an LC-MS/MS approach is an excellent fit for today's standards in developing an efficient process for biologics. In contrast, the development of a specific ELISA for quantitative measurement of a particular HCP would require additional resources and several months of lead-time.
The 1D UHPLC-MS/MS DDA approach complements the pivotal use of ELISA in HCP removal and control strategy, in that it readily addresses a widely recognized limitation in immuno-detection methods, where some HCPs may co-purify with the product and can be present at high or unsafe levels and yet may not be readily detected by the ELISA assay. As a result, LC-MS/MS has established itself as a powerful orthogonal technique to ELISA for HCP detection. There are, however, several limitations in this technique, such as the quality and completeness of databases. Often there is no control for HCP loss during sample preparation, which can affect downstream sensitivity and quantitation. In addition, detection sensitivity with respect to ELISA, at least for some HCPs, may remain a challenge for MS methodology. Complementary and orthogonal approaches, such as improved sample preparation methods via HCPs enrichment or mAb depletion, can improve HCP detection by DDA MS.
In addition to demonstrating the value of positive findings, our study highlights the importance of valid negative findings in HCP analysis by LC-MS/MS. A good detection method should have an established limit above which a positive detection of an unknown HCP would be expected. A negative finding in a test subject (no detection of any HCP) should be interpreted as indicating the absence of any HCPs at or above the established detection limit. This can be more convincingly demonstrated if or when the negative test result is coupled with a positive finding of a known HCP in a control sample, ideally at the detection limit. Sample results associated with a failed identification of the positive control sample should be carefully scrutinized and likely considered invalid, especially when they may otherwise be interpreted to indicate the absence of any detectable HCP. This is an important aspect in the use of mass spectrometry to support process development, as our study results have shown that most mAb purification processes are able to reduce total HCP to well below 100 ppm (by ELISA) in the final pool, and a negative finding by LC-MS/MS would be expected (using a method with 50 ppm single HCP sensitivity). This is also an important consideration in situations where samples from different purification processes are tested. If LC-MS/MS results from these types of analyses show differences in HCP composition, it is important that appropriate system suitability controls are available to confirm that instrument/system performance is not responsible for changes observed in HCP profiles. Similarly, an appropriate negative control sample could be included as part of the system suitability to further control for potential false positive findings.
SWATH MS has been successfully applied to quantify a limited number of spiked peptides, proteins in less complex systems, as well as larger studies on the proteome scale.18,22, 23,26 Compared to previous studies using SWATH MS, HCP detection and quantitation of mAb in-process pools and final product can be considered more challenging due to protein complexity and large dynamic range (between HCPs and mAb) for the proteins of interest. Our results for the first time demonstrated the value of SWATH in detecting and quantifying HCPs as a key component of the LC-MS/MS platform in support of purification development. The 1 hour 1D UHPLC-DIA SWATH MS can be used for relative or absolute HCP quantitation as a universal method with high sensitivity (∼10 ppm). This is comparable to the previously reported detection sensitivity using 2D-LC MSE,10,21 with the additional benefit of high throughput and robustness. The ∼5-fold sensitivity increase seen in DIA SWATH MS in our study as compared with DDA is consistent with the 2-8-fold sensitivity improvement reported.18
The success of the SWATH MS approach in HCP analysis relies on a comprehensive spectral ion library for peptide identifications. HCPs present in the upstream Protein A pools are most likely carried through to the downstream pools, although at lower levels. The coverage of peptides from these proteins in the spectral library will affect their identification in the downstream pools using DIA SWATH MS. It has been reported that the overall HCP composition is similar between product-producing culture and null cell culture.27 In this study, we found lower peptide coverage in general for the HCPs identified in Protein A pool than the null CHO cell supernatant, presumably due to the lower levels of HCPs and the presence of the overwhelming amount of mAb peptides in the Protein A pool sample. This highlights the importance of using the cell supernatant of null CHO cell line, which does not express the mAb product-coding gene, to build a comprehensive CHO spectral library. This ion library can be further expanded with more comprehensive identifications of peptides found in the null CHO cell supernatants and in the in-process pools of different mAb products to help improve low level HCP identification with a universal ion library. This comprehensive universal CHO (or any other host organism) library can also be shared across academic and industry users to facilitate HCP identifications.28,29
Using the DIA method, scientists can not only quantify residual HCPs in a reliable manner (Fig. 6, Table 1), but also gain process knowledge and generate a historical databank that help understand the efficiency of different chromatography steps for successful clearance of specific HCPs. For example, an HCP data repository can be made from the DIA data, and it can be re-mined in the event that a newly discovered HCP becomes a concern. When combining DIA SWATH MS with the Top 3 approach, absolute HCP quantity by either single point or multi-level calibration can be determined. The HCP quantitation obtained with the 1D UHPLC- DIA SWATH MS approach was comparable to MRM or an HCP-specific ELISA. This approach allows detection and quantification of all, including previously unknown, HCPs in various types of in-process samples as well as final product. It provides important process development information regarding HCP clearance and residual levels in one day, which allows assessment of HCP levels without delay.
In summary, this study demonstrated a workflow that can be used to address a significant need in HCP analytics that is relatively unexplored to date, i.e., rapid and robust analytical support toward optimal HCP clearance during process development of biotherapeutic products. Combined with different MS strategies, a 1-hour UHPLC method was used to create a robust, comprehensive HCP detection platform for real-time process development support, allowing analysis of up to 20 samples per day for HCP composition and quantity with high sensitivity down to single digit ppm. This study highlights the capacity of LC-MS/MS to act as an important factor not only for HCP discovery, but also process control. In addition, the technique will serve as an important complementary tool, both in its ability to identify specific HCPs, and in its ability to support ELISA in declaring the absence of HCPs above a certain threshold. The inherent flexibility of this platform allows the analyst to use one or a combination of these methods to analyze samples quickly and reliably, while giving options for balancing method sensitivity requirements with the desired speed and throughput of analysis.
Materials and methods
Materials
All therapeutic mAb samples (mAbs−1, −2 and −3) were produced at Genentech, South San Francisco, CA. Recombinant PLBL2 was expressed at Genentech using stably transfected CHO cells.5 The CHO ELISA standard reagent was produced at Genentech, and was derived from null (non-product expressing) HCCF. Universal Proteomic Standard 1 (UPS-1) was purchased (Sigma-Aldrich) and contained a mixture of 48 recombinant proteins of known equimolar concentration. Bovine carbonic anhydrase II used to generate a universal response factor for absolute quantitation, was purchased commercially (Sigma-Aldrich #C2522). Recombinant porcine trypsin (proteomics grade) used for protein digestion was from Roche (Product Number 03708985001).
Host cell protein ELISA
Total immunoreactive HCP content was measured with an ELISA assay developed at Genentech using polyclonal antibodies raised against concentrated HCCF from non-transfected CHO cells as described previously.5,19
Sample preparation
To test the sensitivity and robustness of 1D UHPLC-MS/MS method using DDA, UPS-1 was spiked into mAb-1 with the 48 protein levels ranging from 110 ppm to 8 ppm relative to the mAb-1 concentration. PLBL2 was spiked in mAb-2 at 1, 2, 5, 10, 25, 50 and 100 ppm to compare the sensitivity of DDA and DIA SWATH MS. All ppm values of spiked proteins were calculated by use of the respective molecular weights of the individual proteins.
To generate the universal response factor for absolute quantitation experiments with DIA SWATH MS,19 bovine carbonic anhydrase II was spiked at a single level of 10 ppm in mAb-3, or at multiple levels to cover a wide range of expected HCP concentrations (0, 3, 6, and 9 pmol) in Cricetulus griseus null HCCF samples (500 μg) as protein internal standard.
Each sample (500 µg, 5 mg/mL) was denatured with 6 M guanidine hydrochloride in 1 mM sodium acetate (pH 5), then reduced with 10 µL of Bondbreaker tris(2-carboxyethyl) phosphine) (ThermoFisher Scientific) for 15 min at 37 °C. Following desalting with a NAP-5 gel permeation column, the pH of the solution was adjusted to 7.2 by addition of 100 µL of 0.5 M (3-(N-morpholino)propanesulfonic acid). Porcine trypsin (10 µg) was added to the samples, followed by incubation at 37 °C for 1 hour. The digestion was quenched with 30 µL of 10% (v/v) trifluoroacetic acid and 40 µg was injected for each LC-MS/MS analysis.
1D UHPLC-MS/MS
Liquid chromatography was performed using a Waters (Milford, MA) Acquity H-class Bio UHPLC® System that was interfaced with a TripleTOF® 5600+ mass spectrometer (Sciex, Concord, Ont, CAN) with DuoSpray™ Ion Source. The CSH C18 stationary phase was based on the charge-surface modification of the ethylene-bridged hybrid (BEH) C18 stationary phase, and allowed up to a 5-fold increase7 in sample load without the peak tailing and column saturation challenges commonly observed in HCP applications using the unmodified C18 stationary phase when high sample load was used to improve HCP detection sensitivity. The optimized 1D CSH C18 LC we used enabled high throughput and robustness for the LC-MS analysis with a 40 µg sample load and 300 µL/min flow rate. All separations were performed on a Waters CSH130 C18 (1.7 µm, 2.1 mm x 150 mm) column. Mobile phases consisted of 0.1% formic acid in water (Mobile Phase A) or acetonitrile (Mobile Phase B). The flow rate was 0.3 mL/min and column temperature was maintained at 60 °C. The total 1 hour LC gradient included a linear gradient of 0–40% B in 40 minutes, a column wash (5 minutes at 90% B), and column re-equilibration (15 minutes at 0% B) steps.
DDA method parameters were set to acquire MS/MS spectra, 50 milliseconds (ms) each, on the top 20 most abundant ions from every MS survey scan (250 ms) over a mass range of 350–1250 m/z, with inclusion of charge states +2 to +5. A precursor ion dynamic exclusion setting of 10 sec after 1 occurrence, and a precursor ion threshold of 150 counts per second for MS/MS selection were used. DIA was performed using 25 sequential m/z windows of 25 amu (atomic mass units) (1 amu overlap on each end of each mass window; 50 ms acquisition time per window) ranging from m/z 400-1000 interleaved with a single 100 ms full scan time-of-flight MS spectrum per cycle to give a total cycle time of 1.38 sec.
Peptide and protein identification/quantitation by 1D UHPLC-MS/MS
LC-MS/MS DDA data files were searched against a database containing sequences of all Genentech biotherapeutic products, plus those of all spiked protein standards, concatenated to the CHO Canonical and Isoform database from Uniprot.org, using the ProteinPilot™ (Version 4.5.0.0; Sciex) software. Protein is required to have a false discovery rate of < 1% with 2 unique peptides (at 95% confidence) for positive identification.
Fragment ions from all peptides identified at > 95% confidence by LC-MS/MS DDA analysis of null CHO cell supernatant were imported into the SWATH™Microapp (Version 2.0; Sciex) within Peakview™ (version 2.1; Sciex) to build the HCP spectral ion library for DIA SWATH MS data analysis. Identification of peptides and protein quantitation using DIA SWATH acquisition was then performed by retention time calibration followed by searching of the DIA data files against the fragment ion spectral library. Protein is required to have 3 unique peptides identified (at 95% confidence and with 5 transitions per peptide) for protein quantitation.
Quantitative analysis of data independent SWATH MS and PRM data
After SWATH data processing, the peak areas of the five most abundant fragment ions for the three highest intensity peptides were summed using Markerview™ software (Version 1.2; Sciex) to generate the Top 3 peak area for relative quantitation. To determine the absolute level of HCP-3 in mAb-3 using a single point calibration, the Top 3 peak areas for the HCP-3 and the spiked bovine carbonic anhydrase II were compared. This molar concentration was then normalized by the MW ratio of the HCP to the bovine carbonic anhydrase II to calculate the ppm level of these HCPs. Similarly, PLBL2 molar concentration was determined using the 4-point standard curve generated from the Top 3 peak area data of bovine carbonic anhydrase II, which was then converted to the µg/mL to compare with ELISA25 results. Carbonic anhydrase II peptides used for quantification were unique to the bovine sequence of the protein.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Alice Zhang for assistance in sample preparation and data analysis; Ben Tran, Vanessa Grosskopf, Paul McDonald, Ailen Sanchez, Susan Fisher, Inn Yuk for their collaboration and providing various bioprocess samples; Judy Zhu-Shimoni, Martin Vanderlaan, Denise Krawitz, John Stults, Pat Rancatore, and Reed Harris for many insightful discussions.
References
- 1.ICH ICH harmonised tripartite guideline. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products Q6B. 1999. [Google Scholar]
- 2.Shukla AA, Jiang C, Ma J, Rubacha M, Flansburg L, Lee SS. Demonstration of robust host cell protein clearance in biopharmaceutical downstream processes. Biotechnol Prog 2008; 24:615-22; PMID:18410156; http://dx.doi.org/ 10.1021/bp070396j [DOI] [PubMed] [Google Scholar]
- 3.Tscheliessnig AL, Konrath J, Bates R, Jungbauer A. Host cell protein analysis in therapeutic protein bioprocessing - methods and applications. Biotechnol J 2013; 8:655-70; PMID:23436780; http://dx.doi.org/ 10.1002/biot.201200018 [DOI] [PubMed] [Google Scholar]
- 4.Zhu-Shimoni J, Yu C, Nishihara J, Wong RM, Gunawan F, Lin M, Krawitz D, Liu P, Sandoval W, Vanderlaan M. Host cell protein testing by ELISAs and the use of orthogonal methods. Biotechnol Bioeng 2014; 111:2367-79; PMID:24995961; http://dx.doi.org/ 10.1002/bit.25327 [DOI] [PubMed] [Google Scholar]
- 5.Vanderlaan M, Sandoval W, Liu P, Nishihara J, Tsui G, Lin M, Gunawan F, Parker S, Wong RM, Low J, et al.. Hamster phospholipase b-like 2 (PLBL2): A host cell proten impurity in therapeutic monoclonal antibodies derive from Chinese hamster ovary cells. Bioprocess Int 2015; 13:18-22. [Google Scholar]
- 6.Thompson JH, Chung WK, Zhu M, Tie L, Lu Y, Aboulaich N, Strouse R, Mo WD. Improved detection of host cell proteins (HCPs) in a mammalian cell-derived antibody drug using liquid chromatography/mass spectrometry in conjunction with an HCP-enrichment strategy. Rapid Commun Mass Spectrom 2014; 28:855-60; PMID:24623688; http://dx.doi.org/ 10.1002/rcm.6854 [DOI] [PubMed] [Google Scholar]
- 7.Doneanu CE, Anderson M, Williams BJ, Lauber MA, Chakraborty A, Chen W. Enhanced detection of low-abundance host cell protein impurities in high-purity monoclonal antibodies down to 1 ppm using ion mobility mass spectrometry coupled with multidimensional liquid chromatography. Anal Chem 2015; 87:10283-91; PMID:26266576; http://dx.doi.org/ 10.1021/acs.analchem.5b02103 [DOI] [PubMed] [Google Scholar]
- 8.Farrell A, Mittermayr S, Morrissey B, Mc Loughlin N, Navas Iglesias N, Marison IW, Bones J. Quantitative host cell protein analysis using two dimensional data independent LC-MS(E). Anal Chem 2015; 87:9186-93; PMID:26280711; http://dx.doi.org/ 10.1021/acs.analchem.5b01377 [DOI] [PubMed] [Google Scholar]
- 9.Madsen JA, Farutin V, Carbeau T, Wudyka S, Yin Y, Smith S, Anderson J, Capila I. Toward the complete characterization of host cell proteins in biotherapeutics via affinity depletions, LC-MS/MS, and multivariate analysis. MAbs 2015; 7:1128-37; PMID:26291024; http://dx.doi.org/ 10.1080/19420862.2015.1082017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doneanu CE, Xenopoulos A, Fadgen K, Murphy J, Skilton SJ, Prentice H, Stapels M, Chen W. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography/mass spectrometry. MAbs 2012; 4:24-44; PMID:22327428; http://dx.doi.org/ 10.4161/mabs.4.1.18748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reisinger V, Toll H, Mayer RE, Visser J, Wolschin F. A mass spectrometry-based approach to host cell protein identification and its application in a comparability exercise. Anal Biochem 2014; 463:1-6; PMID:24949901; http://dx.doi.org/ 10.1016/j.ab.2014.06.005 [DOI] [PubMed] [Google Scholar]
- 12.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Hüttenhain R, Koomen JM, et al.. Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics 2014; 13:907-17; PMID:24443746; http://dx.doi.org/ 10.1074/mcp.M113.036095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rauniyar N. Parallel reaction monitoring: A targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int J Mol Sci 2015; 16:28566-81; PMID:26633379; http://dx.doi.org/ 10.3390/ijms161226120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tong L, Zhou XY, Jylha A, Aapola U, Liu DN, Koh SK, Tian D, Quah J, Uusitalo H, Beuerman RW, et al.. Quantitation of 47 human tear proteins using high resolution multiple reaction monitoring (HR-MRM) based-mass spectrometry. J Proteomics 2015; 115:36-48; PMID:25529431; http://dx.doi.org/ 10.1016/j.jprot.2014.12.002 [DOI] [PubMed] [Google Scholar]
- 15.Sisodiya VN, Lequieu J, Rodriguez M, McDonald P, Lazzareschi KP. Studying host cell protein interactions with monoclonal antibodies using high throughput protein A chromatography. Biotechnol J 2012; 7:1233-41; PMID:22623327; http://dx.doi.org/ 10.1002/biot.201100479 [DOI] [PubMed] [Google Scholar]
- 16.Purvine S, Eppel JT, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics 2003; 3:847-50; PMID:12833507; http://dx.doi.org/ 10.1002/pmic.200300362 [DOI] [PubMed] [Google Scholar]
- 17.Panchaud A, Jung S, Shaffer SA, Aitchison JD, Goodlett DR. Faster, quantitative, and accurate precursor acquisition independent from ion count. Anal Chem 2011; 83:2250-7; PMID:21341720; http://dx.doi.org/ 10.1021/ac103079q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 2012; 11:O111.016717; PMID:22261725; http://dx.doi.org/ 10.1074/mcp.O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol Cell Proteomics 2006; 5:144-56; PMID:16219938; http://dx.doi.org/ 10.1074/mcp.M500230-MCP200 [DOI] [PubMed] [Google Scholar]
- 20.Schenauer MR, Flynn GC, Goetze AM. Identification and quantification of host cell protein impurities in biotherapeutics using mass spectrometry. Anal Biochem 2012; 428:150-7; PMID:22640604; http://dx.doi.org/ 10.1016/j.ab.2012.05.018 [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Goetze AM, Cui H, Wylie J, Trimble S, Hewig A, Flynn GC. Comprehensive tracking of host cell proteins during monoclonal antibody purifications using mass spectrometry. MAbs 2014; 6:659-70; PMID:24518299; http://dx.doi.org/ 10.4161/mabs.28120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert JP, Ivosev G, Couzens AL, Larsen B, Taipale M, Lin ZY, Zhong Q, Lindquist S, Vidal M, Aebersold R, et al.. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat Methods 2013; 10:1239-45; PMID:24162924; http://dx.doi.org/ 10.1038/nmeth.2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Q, Yang L, Luo J, Guo L, Wang Z, Yang X, Jin W, Fang Y, Ye J, Shan B, et al.. SWATH enables precise label-free quantification on proteome scale. Proteomics 2015; 15:1215-23; PMID:25560523; http://dx.doi.org/ 10.1002/pmic.201400270 [DOI] [PubMed] [Google Scholar]
- 24.Lauber MA, Koza SM, McCall SA, Alden BA, Iraneta PC, Fountain KJ. High-resolution peptide mapping separations with MS-friendly mobile phases and charge-surface-modified C18. Anal Chem 2013; 85:6936-44; PMID:23772755; http://dx.doi.org/ 10.1021/ac401481z [DOI] [PubMed] [Google Scholar]
- 25.Yuk IH, Nishihara J, Walker D Jr., Huang E, Gunawan F, Subramanian J, Pynn AF, Yu XC, Zhu-Shimoni J, Vanderlaan M, et al.. More similar than different: Host cell protein production using three null CHO cell lines. Biotechnol Bioeng 2015; 112:2068-83; PMID:25894672; http://dx.doi.org/ 10.1002/bit.25615 [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Huttenhain R, Surinova S, Gillet LC, Mouritsen J, Brunner R, Navarro P, Aebersold R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 2013; 13:1247-56; PMID:23322582; http://dx.doi.org/ 10.1002/pmic.201200417 [DOI] [PubMed] [Google Scholar]
- 27.Jin M, Szapiel N, Zhang J, Hickey J, Ghose S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): Implications for downstream process development. Biotechnol Bioeng 2010; 105:306-16; PMID:19739084; http://dx.doi.org/ 10.1002/bit.22532 [DOI] [PubMed] [Google Scholar]
- 28.Lim UM, Yap MG, Lim YP, Goh LT, Ng SK. Identification of autocrine growth factors secreted by CHO cells for applications in single-cell cloning media. J Proteome Res 2013; 12:3496-510; PMID:23763710; http://dx.doi.org/ 10.1021/pr400352n [DOI] [PubMed] [Google Scholar]
- 29.Kumar A, Baycin-Hizal D, Wolozny D, Pedersen LE, Lewis NE, Heffner K, Chaerkady R, Cole RN, Shiloach J, Zhang H, et al.. Elucidation of the CHO super-ome (CHO-SO) by proteoinformatics. J Proteome Res 2015; 14:4687-703; PMID:26418914; http://dx.doi.org/ 10.1021/acs.jproteome.5b00588 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.